
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocellulose enriches enantiomers in asymmetric aldol reactions†
Naliharifetra Jessica Ranaivoarimanana‡
 a,
Xin Habaki‡a,
Takuya Utob,
Kyohei Kanomataa,
Toshifumi Yuic and
Takuya Kitaoka*a
a,
Xin Habaki‡a,
Takuya Utob,
Kyohei Kanomataa,
Toshifumi Yuic and
Takuya Kitaoka*a
aDepartment of Agro-Environmental Sciences, Graduate School of Bioresource and Bioenvironmental Sciences, Kyushu University, Fukuoka 819-0395, Japan. E-mail: tkitaoka@agr.kyushu-u.ac.jp
bOrganization for Promotion of Tenure Track, University of Miyazaki, Miyazaki 889-2192, Japan
cDepartment of Applied Chemistry, Faculty of Engineering, University of Miyazaki, Miyazaki 889-2192, Japan
First published on 8th October 2020
Abstract
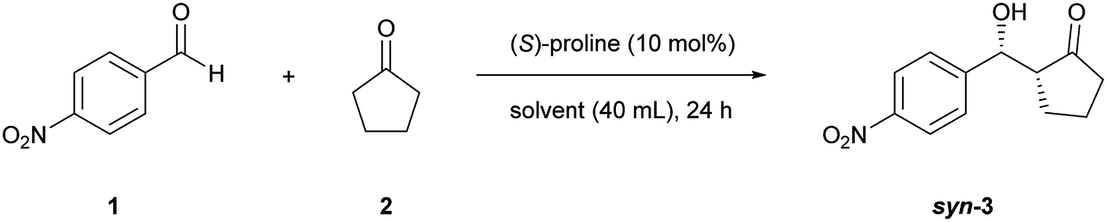
Cellulose nanofibers obtained from wood pulp by TEMPO-mediated oxidation acted as a chiral enhancer in direct aldol reactions of 4-nitrobenzaldehyde and cyclopentanone with (S)-proline as an organocatalyst. Surprisingly, catalytically inactive TEMPO-oxidized cellulose nanofibers enriched the (R,R)-enantiomer in this reaction, affording 89% ee in the syn form with a very high yield (99%). Conversely, nanocellulose-free (S)-proline catalysis resulted in poor selectivity (64% ee, syn form) with a low yield (18%). Green organocatalysis occurring on nanocellulose solid surfaces bearing regularly aligned chiral carbons on hydrophobic crystalline facets will provide new insight into asymmetric synthesis strategies for interfacial catalysis.
Introduction
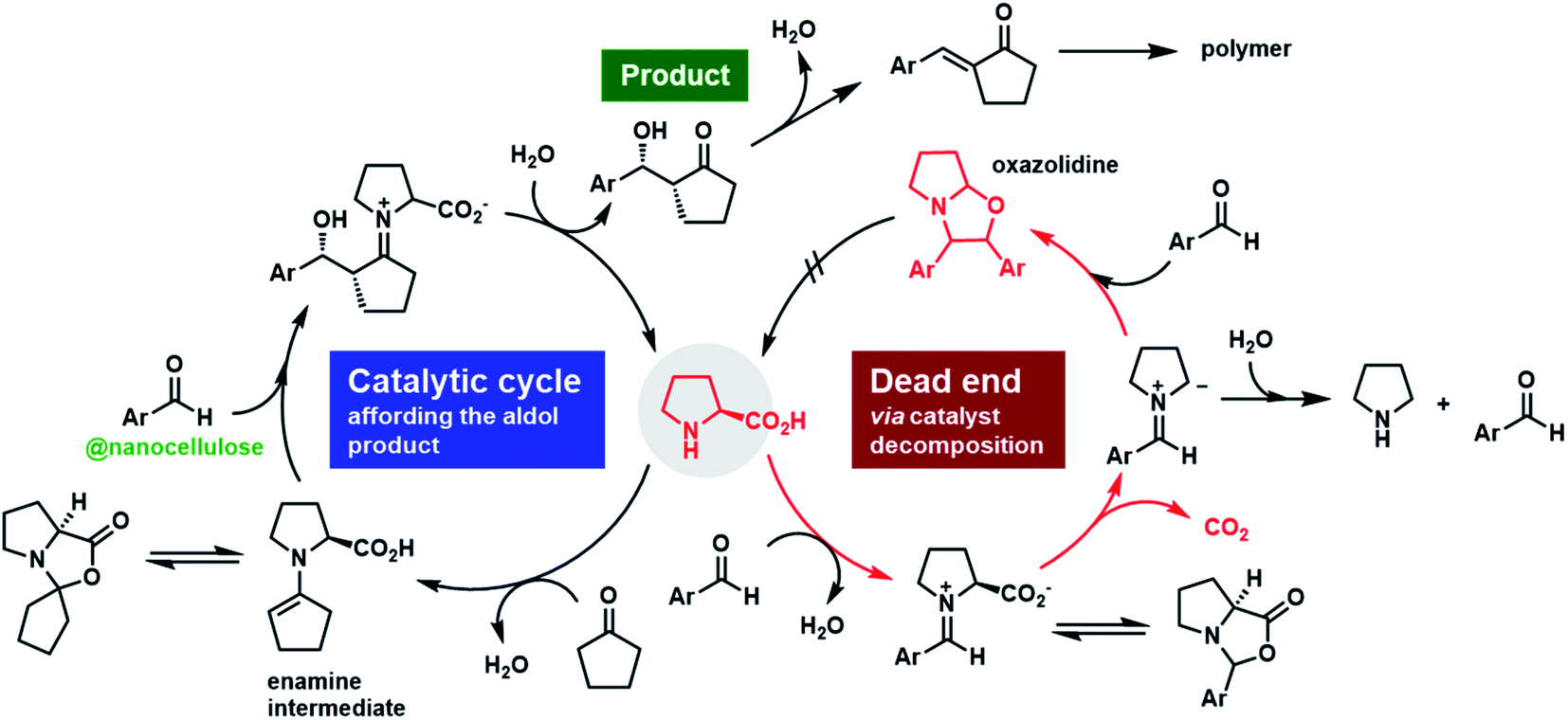
Nanocellulose has emerged as a cutting-edge sustainable nanomaterial in the last two decades.1–3 Native cellulose, found mainly in phytomass such as trees and plants, is a homopolymer of β-1,4-linked D-glucopyranose.4 Biologically synthesized cellulose molecules assemble and then crystallize into nanometer-width fibers during biosynthesis, in which dozens of cellulose chains are regularly packed in parallel.3 Various methods to isolate this naturally occurring nanofiber, known as nanocellulose, have been developed to date, including mechanical, chemical, and enzymatic treatments.5–7 The highly crystalline nature of thin nanocellulose provides steel-level mechanical strength, quartz-level thermal stability, and glass-level transparency. Owing to its superior physicochemical properties, accompanied by huge abundance and carbon-neutral renewability, a wide range of studies are currently underway to develop composite fillers,8 gas-barrier films,9 heat insulators,10 and solid emulsifiers for foods and cosmetics.11,12Despite being a developing field, the use of nanocellulose in catalysis is among its most fascinating applications owing to the high demand for green molecular transformations in fine chemical industries. Metal nanoparticle catalysts immobilized on nanocellulose have been widely studied and demonstrated to have excellent catalytic performance.13,14 The high specific surface area and structural rigidity of nanocellulose are key to enhancing catalytic efficiency in heterogeneous reactions.15 Although nanocellulose has only been reported as a solid support in immobilized catalysts to date, the direct contribution of nanocellulose to heterogeneous catalysis16 will attract much attention in academia and industry because such novel catalytic systems would expand the potential practical applications of nanocellulose and will open up new horizons in nanocellulose engineering and catalysis science. We have recently reported that typical nanocellulose, especially 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO)-oxidized cellulose nanofibers (TOCNFs),17 significantly accelerate catalytic carbon–carbon bond-forming reactions. We discovered that the simple addition of TOCNFs to the reaction media drastically improved the catalytic efficiency of proline, a low-performance organocatalyst, in a direct aldol reaction18 and Michael addition.19 The striking effects of the nanofibrillation and crystallinity of nanocellulose on catalytic performance have clearly indicated that this unique interfacial nanoarchitecture plays an indispensable role in catalytic enhancement.
Some studies have reported that nanosized cellulose materials affect enantioselectivity in catalytic processes. Serizawa and coworkers reported that the hydrolysis of chiral peptides for several days showed different reaction rates for each enantiomer when the hydrolytic reaction was directly catalysed by cellulose nanowhiskers.20 Furthermore, Kaushik and coworkers showed that the hydrogenation of ketones catalyzed by palladium nanoparticles supported on cellulose nanowhiskers afforded the products with some enantioselectivity.21 Although the reaction selectivities were insufficient from the viewpoint of asymmetric synthesis, these two reports clearly indicated that nanocellulose has the potential to induce enantioselectivity, possibly owing to its inherent chiral nanoarchitecture.22–25 As tremendous research effort has been focused on developing enantioselective catalysis, enantiodiscrimination based on naturally-occurring and renewable nanocellulose would be a promising development for a greener fine chemical industry. Herein, we report for the first time that nanocellulose significantly enhanced enantioselectivity and catalytic efficiency in proline-catalyzed aldol reactions, as shown in Fig. 1. This method for synthesizing aldol adducts with up to 89% enantiopurity in the presence of catalytically inactive nanocellulose, TOCNFs, provides an entirely new approach to the development of high-performance proline-based organocatalytic systems among extensive research efforts.26–29 The present conditions afford highly enantioenriched syn-products from cyclic ketones.
 | ||
| Fig. 1 Schematic illustration of the research strategy in this work. | ||
Results and discussion
Optimization of reaction conditions
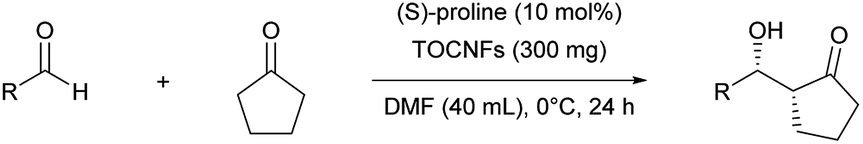
This study was initiated by determining the best conditions for the reaction between 4-nitrobenzaldehyde (1) and cyclopentanone (2) (Table 1) using TOCNFs (Fig. S1–S3†). The amount of proline subjected to the reaction system was set at 10 mol% against 1 instead of the 20 mol% load used in the literature,30 to emphasize the effect of TOCNFs on the reaction outcome and to prevent proline-only catalyzed reactions, which may occur apart from nanocellulose suspended in the reaction medium. The reaction catalyzed by (S)-proline alone resulted in a low yield after 24 h in DMSO at room temperature (entry 1). The major product had a syn-configuration with poor enantioselectivity. TOCNFs alone did not catalyze the aldol reaction (entry 2), indicating that TOCNFs were catalytically inactive in this reaction. However, adding precipitated TOCNFs to (S)-proline in the reaction medium markedly increased the yield from 9% to 55% (entry 3). Water is known to increase the rate of aldol reactions by inhibiting byproduct formation, but also to adversely inhibit the reaction step of the enamine formation from proline and ketone substrates.31,32 Additionally, more than 5 vol% of water resulted in fatal reduction of the ee of the products.30 In the present work, although the TOCNFs used contained the trace amount of residual water (1.2 vol%), the overall activity and enantioselectivity was drastically improved compared with the reaction using (S)-proline alone. To obtain a greater effect at lower temperatures, the reaction solvent was changed to DMF (entries 4 and 5). The reaction without TOCNFs gave a low yield and ee (entry 4), while the product was formed almost quantitatively in the presence of TOCNFs (entry 5). As expected, the ee was improved accordingly (from 84%/71% to 89%/87% for syn/anti). In our previous studies, TOCNF precipitates in the sodium form were sensitive to aggregation in the reaction media without noticeably influencing the selectivity of the aldol reaction.18 To prevent this, standing organogels were used, which enabled superior dispersion in the reaction media compared with that obtained by simple solvent exchange and centrifugation. Using the organogels, the reaction reached completion within 24 h (entry 6). No traces of 4-nitrobenzaldehyde could be detected on stained TLC plates, nor of byproducts. However, the product could not be fully isolated (86% yield), probably owing to its strong adhesion to TOCNF surfaces.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Temperature | TOCNFs | Yieldb (%) | syn/antic | eed (% syn/anti) |
| a Conditions: 1 (2 mmol), 2 (10 mL), (S)-proline (0.2 mmol), TOCNFs (300 mg, when used), solvent (40 mL).b Isolated yield.c Determined by 1H-NMR (400 MHz, CDCl3).d Determined by chiral-phase SFC.e Without (S)-proline. | ||||||
| 1 | DMSO | rt | — | 9 | 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23 :23 |
39/54 |
| 2e | DMSO | rt | Precipitate | — | — | — |
| 3 | DMSO | rt | Precipitate | 55 | 65:35 |
84/71 |
| 4 | DMF | 0 °C | — | 18 | 80:20 |
64/68 |
| 5 | DMF | 0 °C | Precipitate | >99 | 78:22 |
89/87 |
| 6 | DMF | 0 °C | Gel | 86 | 78:22 |
90/86 |
| 7 | DMF | rt | Gel | 85 | 71:29 |
60/79 |
The obtained diastereoselectivity and enantioselectivity were comparable with the reaction using precipitated TOCNFs. The improved enantioselectivity was not solely attributed to the reaction solvent or temperature, as the reaction with TOCNFs at room temperature delivered values comparable to that with proline alone at 0 °C in DMF (entries 4 and 7). Therefore, the stereoselectivity of the reaction with aromatic aldehydes was presumably improved via the TOCNFs, with (R,R)- and (R,S)-formation increasing and (S,S)-formation decreasing. This was possibly due to the aromatic aldehydes being restricted to adsorption to the TOCNF solid surfaces bearing regularly aligned chiral carbons on crystalline facets where hydrophobic and hydrophilic faces are separately located on the nanofiber surfaces.33,34 Cyclohexanone as the ketone substrate also benefited the enhancement via the TOCNFs, from 18% to at least 51% in rough yield, but due to the difficulty in quantitative product extraction, this study focused on cyclopentanone.
Solvent screening indicated that protic solvents (methanol and ethanol) and aprotic solvents (DMAc and dioxane) in which TOCNFs aggregated were unsuitable for delivering the aldol adducts. DMF, which gave the most stable TOCNF dispersion, was the best solvent for the direct aldol reactions. Unless otherwise stated, precipitated TOCNFs in DMF were used in the subsequent experiments.
Investigation of catalytic efficiency
Proline is known to form an oxazolidinone with aldehyde substrates to a certain extent during reactions, which can lead to fatal catalyst deactivation. The proton signals for the deactivated form were clearly detected by 1H-NMR for the reaction in the absence of TOCNFs (Fig. S4†).28,31 Pleasingly, conducting the reaction in the presence of TOCNFs drastically suppressed oxazolidinone formation, indicating effective prevention of proline decarboxylation in the reaction media. Furthermore, the baseline of the NMR spectra was distorted for reactions without TOCNFs, which indicated side polymerization reactions occurring among substrates and/or products (Fig. S5†). Conversely, TOCNFs provided the target product with high purity (Fig. S6†).Several reports on proline-catalyzed aldol reactions showed that despite having complex kinetics, the reaction is often reversible, especially at high catalyst concentrations (>30 mol%) and long reaction times (several days). Albeit the reversibility, a critical control on the transition states can grant higher enantioselectivity within relatively short reaction time.35,36 Fortunately, TOCNFs enabled faster reaction completion and the use of a lower concentration of (S)-proline. Furthermore, upon following the diastereoselectivity determined by SFC analysis until full conversion, no epimerization was detected as the dr stayed constant throughout the reaction (Fig. S15†). As the same was applied to the enantioselectivity, there was no erosion of the ee. Therefore, although there is a possibility for numerous hydroxy and carboxylic acid groups to potentially interact with reaction species, the planar rigid TOCNFs surface probably allowed the stabilization of key transition states as the reaction is kinetically controlled in the presence of the nanocellulose in DMF at 0 °C.
Substrate scope
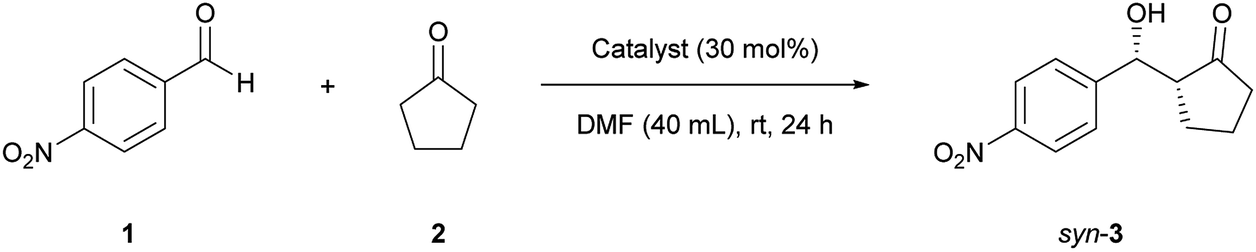
Different aldehyde substrates were tested in the TOCNF/(S)-proline catalyzed aldol reaction (Table 2). The main diastereomer had a syn-configuration (Fig. S7–S14†). Surprisingly, all substrates resulted in enhanced enantioselectivity in the anti-product. The reaction yields gradually decreased with increasing strength of the electron-withdrawing groups on the aldehyde (entries 1 to 3). The bulky naphthaldehyde yielded the anti-product in a low yield with high enantiopurity (entry 4). This effect might be attributed to both electrophilic interaction and steric hindrance between the substrates and the TOCNF solid surface bearing carboxylate groups,39 although a detailed mechanism remains to be determined.
|
|||||
|---|---|---|---|---|---|
| Entry | R | Yieldb (%) | syn/antic | syn eed (%) | anti eed (%) |
| a Conditions: arylaldehyde (2 mmol), cyclopentanone (10 mL), (S)-proline (0.2 mmol), precipitated TOCNFs (300 mg), DMF (40 mL). Each product structure conformed to previous reports.37,38b Isolated yield.c Determined by 1H-NMR (400 MHz, CDCl3).d Determined by chiral-phase SFC. | |||||
| 1 | 4-NO2–C6H4 | >99 | 78:22 |
89 | 87 |
| 2 | 4-Br–C6H4 | 60 | 63:37 |
53 | 88 |
| 3 | 4-Cl–C6H4 | 55 | 64:36 |
55 | 88 |
| 4 | 2-Naphthyl | 24 | 64:36 |
51 | 87 |
Amino acid scope
Several amino acids were also tested to determine whether they benefitted from the assistance of TOCNFs (Table 3). Excluding (S)-proline, no reaction occurred during the screening time period when using amino acids alone. However, when paired with TOCNFs, an appreciable amount of product was formed within 24 h (entries 2, 4, and 6). In general, the ee of the anti-product was high, similar to the case of TOCNFs combined with proline (Table 2). Aliphatic amino acids proved to be a good match with TOCNFs (entries 1–6), while aromatic (S)-phenylalanine and (S)-tyrosine failed to catalyze the reaction (entries 7–10). This was probably due to both hydrophobic catalysts and substrates being competitively adsorbed on the hydrophobic facets of the TOCNF crystals.
|
|||||
|---|---|---|---|---|---|
| Entry | Amino acid | TOCNFs | Yieldb (%) | syn/antic | eed (% syn/anti) |
| a Conditions: 1 (2 mmol), 2 (10 mL), (S)-proline (0.2 mmol), TOCNFs (300 mg, when used), DMF (40 mL).b Isolated yield.c Determined by 1H-NMR (400 MHz, CDCl3).d Determined by chiral-phase SFC. | |||||
| 1 | (S)-Alanine | − | Trace | — | — |
| 2 | + | 60 | 51:49 |
48/89 | |
| 3 | (S)-Valine | − | Trace | — | — |
| 4 | + | 65 | 49:51 |
40/91 | |
| 5 | (S)-Lysine | − | Trace | — | — |
| 6 | + | 54 | 69:31 |
36/82 | |
| 7 | (S)-Phenylalanine | − | Trace | — | — |
| 8 | + | Trace | — | — | |
| 9 | (S)-Tyrosine | − | Trace | — | — |
| 10 | + | Trace | — | — | |
Molecular dynamics simulation
The adsorption of substrates and intermediates onto TOCNFs was probably a key step in these asymmetric aldol reactions. The three-dimensional structure of the product was speculated to be determined by the enantioselective mechanism occurring on the TOCNF surface, rather than asymmetric induction by the catalyst molecule as a unique chiral source. Attempts on the modelization of hexagonal crystals of TOCNFs for the QM/MM-SMD were unfruitful, probably due to the electronegative carboxylate moiety at the C6 position, which may cause the unstable architecture of cellulose chain assembly. On the other hand, the (100)/(200) planes of TOCNF and CNF crystals are regarded as being both hydrophobic.40,41 The adsorption of key intermediates in the TOCNFs-enhanced aldol reaction is expected to occur on the same lattice plane. Despite being less selective, precipitated CNFs devoid of carboxylate groups on their surface also improved the outcome of the reaction with the same conditions as precipitated TOCNFs (90% yield, 54:46 dr, 76/68 ee). Therefore, the adsorption behavior of the substrate and enamine intermediate was evaluated on the CNF surface in DMF solution using conventional molecular dynamics (MD) calculations. The substrate (4-nitrobenzaldehyde) was reversibly adsorbed on the hydrophobic surface of CNF of the (100)/(200) lattice planes of cellulose Iβ (Movie 1†). In particular, the molecular axis of 4-nitrobenzaldehyde preferred being perpendicular to the fiber axis of the cellulose chains, with the aromatic plane of the substrate parallel to the pyranose plane. Fig. 2 shows the distribution of substrate and intermediate on the surface of the CNF model for 2 μs, with the clear distribution of 4-nitrobenzaldehyde obtained by reflecting the aromatic ring.
 | ||
| Fig. 2 Isocounter surfaces of the substrate and intermediate density relative to the structures of the CNF model calculated from the MD trajectories. Red and blue surfaces indicate 0.38 mol L−1 (corresponding to twice the bulk density) for 4-nitrobenzaldehyde and the enamine intermediate, respectively. The density was calculated for a 0.5 × 0.5 × 0.5 Å grid as the number of hits in 2000000 frames. Molecular graphics with isocontour map were analyzed and drawn using PyMOL 1.7.1 (http://www.pymol.org). | ||
The regular and homogeneous high-density distribution of the substrate exclusively appeared over the cellobiose unit, reflecting the two-fold helical symmetry of a cellulose chain, which suggested that the orientation of the substrate was restricted by the surface topology of the CNF. Conversely, the enamine intermediate showed an obscure distribution, indicating less specific adsorption behavior, albeit with a tendency to be located around the substituted groups of the adsorbed substrate. Therefore, substrate 4-nitrobenzaldehyde on the CNF surface was especially constrained by carbohydrate–aromatic CH/π and hydrophobic interactions, which possibly led to nucleophilic attack by the enamine intermediate at the aldehyde group of the adsorbed substrate from a specific direction.
This MD simulation successfully provided pictorial elucidation of the enantioselectivity in the presence of CNF observed in the present aldol reaction. Our next target was to verify whether the asymmetric aldol reaction proceeded on the substrate adsorbed on the CNF surface in the manner proposed by the MD simulation. A quantum mechanics (QM)/molecular mechanics (MM)-steered molecular dynamics (SMD) simulation was conducted to evaluate the aldol reaction on the CNF surface in a DMF solution. The QM region consisted of the aldehyde substrate and the enamine intermediate involved in the reaction. The MM region included the CNF model, DMF solvent and the surrounding substrates/intermediates that are not directly taking part in the new bond formation (Fig. S25†).
The QM/MM-SMD method showed that C–C bond formation was selected as the reaction coordinate leading to enantioselectivity, such that the enamine intermediate carbon (Cα) was pulled toward the carbonyl carbon (Cδ) of the substrate, giving the (R,R)-product. As shown in Fig. 3 and Movie 2† (2a and 2b are the same in different drawings), proton transfer occurred from the carboxy group of the enamine intermediate to the carbonyl oxygen of the substrate, accompanied by Cα–Cδ bond formation. Fig. 4 shows the nonequilibrium work and potential of mean force (PMF) for the reaction coordinate of the Cα–Cδ distance. The C–C bond forming reaction pathway of the present aldol reaction was exothermic (ΔGr = −10.0 kcal mol−1) and involved the transition state at a Cα–Cδ distance of 1.95–2.00 Å, in which the eight-membered ring structure with half-chair form contributed to stabilizing the transition state structure.42–44 The activation energy (ΔG‡) was estimated to be 3.7 kcal mol−1. Therefore, the asymmetric aldol reaction was reasonably concluded to proceed on the CNF surface. Subsequent hydrolysis provided the reaction products, accompanied by recovery of the (S)-proline catalyst (Fig. 5).
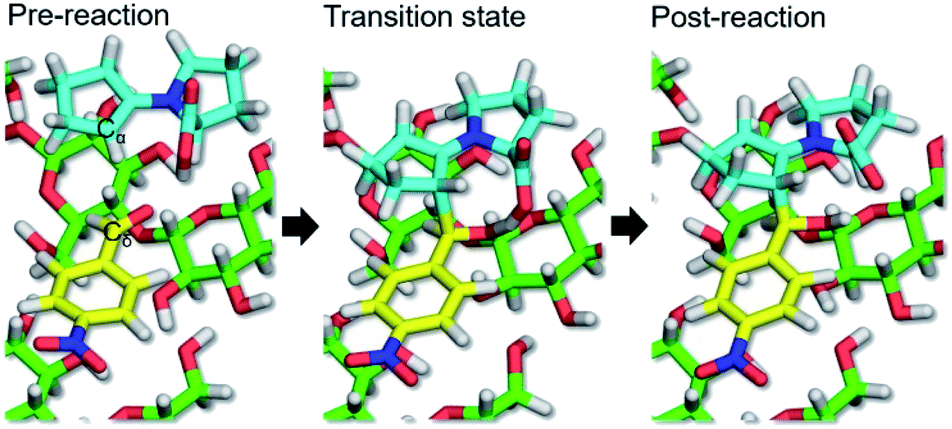 | ||
| Fig. 3 Cα–Cδ bond forming reaction progression on CNF surface observed by QM/MM-SMD simulation. All structure diagrams were generated using PyMOL 1.7.1 (http://www.pymol.org). | ||
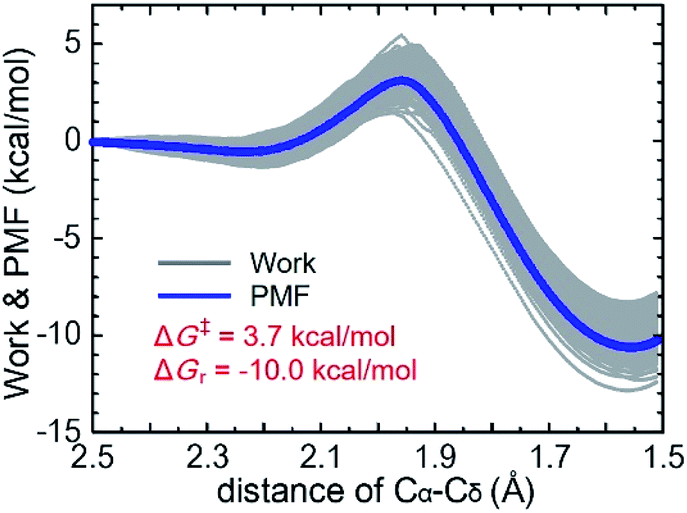 | ||
| Fig. 4 Work distribution and potential mean force (PMF) obtained for the forced approach between the substrate (Cδ) and enamine intermediate (Cα) in DMF solvents, displayed for 0.01 Å ps−1 for 1000 trajectories in an SMD pulling simulation. | ||
 | ||
| Fig. 5 Proposed catalytic pathway for enantioselective asymmetric aldol reactions occurring on nanocellulose surfaces in this study. | ||
Conclusions
In conclusion, green natural nanofibers obtained from wood pulp by aqueous TEMPO-mediated oxidation substantially enriched enantiomers and enhanced catalytic efficiency in direct asymmetric aldol reactions. The proposed catalytic pathway for enantioselective asymmetric aldol reactions occurring on nanocellulose surfaces is shown in Fig. 5. TOCNFs drastically suppressed oxazolidinone formation, which indicates effective prevention of proline decarboxylation in the reaction media and avoidance of undesirable side polymerization reactions among substrates and/or products. MD calculations proved that the aldehyde substrate orientation was controlled by carbohydrate–aromatic CH/π and hydrophobic interactions on the CNF surfaces. The interfacial interaction was assumed to exert considerable enantiocontrol on the aldol reaction. Furthermore, the reaction proceeded in the direct vicinity of the cellulose surface, as suggested by QM/MM-SMD calculation results and proposed in Fig. 1. This extraordinary development of highly efficient, enantioselective, and stereoregulated organocatalysis via nanocellulose provides a new direction for green sustainable chemistry and industry.Experimental
Materials
TEMPO-oxidized cellulose nanofiber (TOCNFs; crystallinity index 72%, crystallite size 2.6 nm, carboxylate content = 1.45 mmol g−1) was kindly supplied by Nippon Paper Industries Co., Ltd. (Tokyo, Japan). 4-Nitrobenzaldehyde and (S)-proline were purchased from Sigma-Aldrich Japan Ltd. (Tokyo, Japan) and used as received. Other chemicals and organic solvents were purchased from Sigma-Aldrich Japan Ltd. (Tokyo, Japan), Wako Pure Chemical Industries, Ltd. (Osaka, Japan), and Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan), and used as received. Water used in this study was purified using an Arium Ultrapure Water System (Sartorius Co., Ltd., Tokyo, Japan). The water content of the precipitated TOCNFs was measured by Karl Fischer titration method on an MKH-700 titrator (Kyoto Electronics Manufacturing Co., Ltd., Kyoto, Japan). Reaction progress was monitored by thin-layer chromatography (TLC) on glass-backed plates precoated with silica gel (silica gel 60 GF254, 0.25 mm Merck, Tokyo, Japan). Purification by column chromatography was performed using an automated flash chromatography system (Smart Flash EPCLC-AI-580S, Yamazen, Osaka, Japan). All product structures were in agreement with previously reported data from nuclear magnetic resonance (NMR) analysis.37,38 1H-NMR spectra were recorded on a JNM-ECZ400 spectrometer (JEOL, Tokyo, Japan) at the Center of Advanced Instrumental Analysis, Kyushu University. The enantioselectivity and diastereomeric ratios of products were measured by supercritical fluid chromatography (SFC; ACQUITY UPC2, Waters, Tokyo, Japan) using chiral stationary phases.Methods
000 × g, 10 min). The supernatant was then renewed with deionized water (25 mL) and the mixture was centrifuged again. This washing cycle was repeated until the supernatant pH reached 4.5 (3 times). To remove water, the protonated TOCNFs were washed with the type of solvent to be used in the respective aldol reaction using the same centrifugation procedure (12000 × g, 10 min, 7 cycles). After the last cycle, the solvent was removed, and the TOCNF precipitate was recovered and used in reactions. Each sample had less than 1.2 wt% (15 eq. of aldehyde 1) water content upon Karl Fischer titration of the dispersed nanomaterial as a reaction solvent.:5; 40 °C; 264 nm).Furthermore, a multiscale simulation combining quantum mechanics (QM) and molecular mechanics (MM) was conducted to reproduce the aldol reaction on the CNF surface. The substrate was appropriately arranged on the CNF model based on the results obtained by MD calculation. The substrate and enamine intermediate involved in the aldol reaction were included as the QM region, and the rest of the system was treated as the MM region (Fig. S25†). The enamine intermediate was pulled toward 4-nitrobenzaldehyde arranged on the CNF model by an external force of 1000 kcal mol−1 Å−2 using a steered molecular dynamics (SMD) method.48 The potential of mean force (PMF) of the reaction process was estimated from the exponential Boltzmann-weighted ensemble average of nonequilibrium works obtained from 1000 SMD runs under ambient conditions (300 K and 1 bar) for 100 ps.
The CNF model was described by the GLYCAM06 force field.49 4-Nitrobenzaldehyde, the enamine intermediate, and DMF solvent were modeled using the GAFF force field.50 The QM/MM simulation was conducted using the SCC-DFTB-PA method with mio-1-1 parameters (Fig. S26†).51,52 The particle mesh Ewald algorithm was adopted for long-range interactions, and the cutoff for nonbonding interactions in the coordinate space was fixed at 12 Å. All MD and DFTB/MM-MD calculations were conducted using the SANDER and PMEMD. CUDA modules of the Amber 18 package with the NVIDIA Kepler GPU system.53–55 MD trajectory analysis was performed using the CPPTRAJ module of AmberTools 19.56
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by an Advanced Low Carbon Technology Research and Development Program (Grant No. JPMJAL1505 to T. K.) from Japan Science and Technology Agency, and a Grant-in-Aid (KAKENHI) for Challenging Exploratory Research (Grant No.: JP18K19233 to T. K.) from Japan Society for the Promotion of Science. The theoretical calculations were partly performed using Research Center for Computational Science, Okazaki Research Facilities, National Institutes of Natural Sciences (NINS), Japan. We thank Simon Partridge, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.Notes and references
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS.
- C. Tokoh, K. Takabe, J. Sugiyama and M. Fujita, Cellulose, 2002, 9, 65–74 CrossRef CAS.
- J. Sugiyama, R. Vuong and H. Chanzy, Macromolecules, 1991, 24, 4168–4175 CrossRef CAS.
- I. Usov, G. Nyström, J. Adamcik, S. Handschin, C. Schütz, A. Fall, L. Bergström and R. Mezzenga, Nat. Commun., 2015, 6, 7564 CrossRef CAS.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS.
- B. Wicklein, A. Kocjan, G. Salazar-Alvarez, F. Carosio, G. Camino, M. Antonietti and L. Bergström, Nat. Nanotechnol., 2015, 10, 277–283 CrossRef CAS.
- I. Capron, O. J. Rojas and R. Bordes, Curr. Opin. Colloid Interface Sci., 2017, 29, 83–95 CrossRef CAS.
- S. Fujisawa, E. Togawa and K. Kuroda, Sci. Technol. Adv. Mater., 2017, 18, 959–971 CrossRef CAS.
- M. Kaushik and A. Moores, Green Chem., 2016, 18, 622–637 RSC.
- M. Matsumoto and T. Kitaoka, Adv. Mater., 2016, 28, 1765–1769 CrossRef CAS.
- H. Koga, E. Tokunaga, M. Hidaka, Y. Umemura, T. Saito, A. Isogai and T. Kitaoka, Chem. Commun., 2010, 46, 8567–8569 RSC.
- Y. Tamura, K. Kanomata and T. Kitaoka, Sci. Rep., 2018, 8, 5021 CrossRef.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- K. Kanomata, N. Tatebayashi, X. Habaki and T. Kitaoka, Sci. Rep., 2018, 8, 4098 CrossRef.
- N. Ranaivoarimanana, K. Kanomata and T. Kitaoka, Molecules, 2019, 24, 1231 CrossRef.
- T. Serizawa, T. Sawada and M. Wada, Chem. Commun., 2013, 49, 8827–8829 RSC.
- M. Kaushik, K. Basu, C. Benoit, C. M. Cirtiu, H. Vali and A. Moores, J. Am. Chem. Soc., 2015, 137, 6124–6127 CrossRef CAS.
- S. J. Hanley, J. F. Revol, L. Godbout and D. G. Gray, Cellulose, 1997, 4, 209–220 CrossRef CAS.
- I. Usov, G. Nyström, J. Adamcik, S. Handschin, C. Schütz, A. Fall, L. Bergström and R. Mezzenga, Nat. Commun., 2015, 6, 7564 CrossRef CAS.
- H. Goto, J. Jwa, K. Nakajima and A. Wang, J. Appl. Polym. Sci., 2014, 131, 41118 CrossRef.
- H. Yu and J. Deng, Macromolecules, 2016, 49, 7728–7736 CrossRef CAS.
- P. Krattiger, R. Kovasy, J. D. Revell, S. Ivan and H. Wennemers, Org. Lett., 2005, 7, 1101–1103 CrossRef CAS.
- Z. Tang, Z.-H. Yang, X.-H. Chen, L.-F. Cun, A.-Q. Mi, Y.-Z. Jiang and L.-Z. Gong, J. Am. Chem. Soc., 2005, 127, 9285–9289 CrossRef CAS.
- T. Kano, J. Takai, O. Tokuda and K. Maruoka, Angew. Chem., Int. Ed., 2005, 44, 3055–3057 CrossRef CAS.
- Y. S. Wu, Y. Chen, D. S. Deng and J. Cai, Synlett, 2005, 2005, 1627–1629 CrossRef.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas, J. Am. Chem. Soc., 2001, 123, 5260–5267 CrossRef CAS.
- N. Zotova, A. Franzke, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2007, 129, 15100–15101 CrossRef CAS.
- T. Kanemitsu, A. Umehara, M. Miyazaki, K. Nagata and T. Itoh, Eur. J. Org. Chem., 2011, 2011, 993–997 CrossRef.
- K. Mazeau, Carbohydr. Polym., 2011, 84, 524–532 CrossRef CAS.
- K. Mazeau and M. Wyszomirski, Cellulose, 2012, 19, 1495–1506 CrossRef CAS.
- M. H. Haindl, J. Hioe and R. M. Gschwind, J. Am. Chem. Soc., 2015, 137, 12835–12842 CrossRef CAS.
- M. Orlandi, M. Ceotto and M. Benaglia, Chem. Sci., 2016, 7, 5421–5427 RSC.
- T. He, K. Li, M. Y. Wu, M. B. Wu, N. Wang, L. Pu and X. Q. Yu, Tetrahedron, 2013, 69, 5136–5143 CrossRef CAS.
- C. Wu, X. Long, S. Li and X. Fu, Tetrahedron: Asymmetry, 2012, 23, 315–328 CrossRef CAS.
- J. Rosenthal and D. I. Schuster, J. Chem. Educ., 2003, 80, 679–690 CrossRef CAS.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- Y. Okita, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1696–1700 CrossRef CAS.
- S. Bahmanyar and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 11273–11283 CrossRef CAS.
- S. Bahmanyar and K. N. Houk, J. Am. Chem. Soc., 2001, 123, 12911–12912 CrossRef CAS.
- S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475–2479 CrossRef CAS.
- T. Saito, T. Uematsu, S. Kimura, T. Enomae and A. Isogai, Soft Matter, 2011, 7, 8804–8809 RSC.
- Y. Kobayashi, T. Saito and A. Isogai, Angew. Chem., Int. Ed., 2014, 53, 10394–10397 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS.
- S. Park, F. Khalili-Araghi, E. Tajkhorshid and K. Schulten, J. Chem. Phys., 2003, 119, 3559–3566 CrossRef CAS.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk and T. Frauenheim, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- G. D. M. Seabra, R. C. Walker, M. Elstner, D. A. Case and A. E. Roitberg, J. Phys. Chem. A, 2007, 111, 5655–5664 CrossRef.
- R. Salomon-Ferrer, A. W. Götz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS.
- S. Le Grand, A. W. Götz and R. C. Walker, Comput. Phys. Commun., 2013, 184, 374–380 CrossRef CAS.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, Y. Huang, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, Univ. California, San Fr., 2018 Search PubMed.
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07412h |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |