
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn all solid-state Li ion battery composed of low molecular weight crystalline electrolyte†
Prerna Joshi ab,
Raman Vedarajan*ac,
Anjaiah Sheelamd,
Kothandaraman Ramanujamd,
Bernard Malamane and
Noriyoshi Matsumi*af
ab,
Raman Vedarajan*ac,
Anjaiah Sheelamd,
Kothandaraman Ramanujamd,
Bernard Malamane and
Noriyoshi Matsumi*af
aSchool of Materials Science, Japan Advanced Institute of Science and Technology, Nomi, Ishikawa, Japan. E-mail: matsumi@jaist.ac.jp
bSurface Science Laboratory, Toyota Technological Institute, Nagoya, Japan
cInternational Advanced Research Centre for Powder Metallurgy and New Materials, Center for Fuel Cell Technology, Indian Institute of Technology (Madras)-Research Park, Chennai, India
dDepartment of Chemistry, Indian Institute of Technology (Madras), Chennai, India
eInstitut Jean Lamour, UMR 7198 – Université de Lorraine, Nancy Cedex, France
fElements Strategy Initiative for Catalysts & Batteries (ESICB), Kyoto University, Nishikyo-ku, Kyoto 615-8245, Japan
First published on 28th February 2020
Abstract
Conduction mechanisms in solid polymer electrolytes of Li ion batteries have always been a concern due to their theoretical limitation in conductivity value. In an attempt to increase the ionic conductivity of solid state electrolytes, used in lithium ion secondary batteries (LiBs), we studied the synthesis and conductive properties of a low molecular weight cyclic organoboron crystalline electrolyte. This electrolyte was expected to show better electrochemical properties than solid polymer electrolytes. The electrolyte was doped with LiTFSI salt via two different methods viz. (1) facile grinding of the crystalline sample with lithium salt under a nitrogen atmosphere and (2) a conventional method of solvent dissolution and evaporation under vacuum. The electrochemical properties were studied under specific composition of Li salt. The presence of crystallinity in the electrolyte can be considered as an important factor behind the high ionic conductivity of an all solid electrolyte of this type. Charge–discharge properties of the cell using the electrolyte were investigated in anodic half-cell configuration.
Introduction
Organic carbonate solvents used as the conventional liquid electrolytes1 have been employed in commercial lithium ion batteries (LiBs) due to their high ionic conductivities (1.7–11.1 S cm−1) at ambient temperature,2 low viscosities and impressive ability to form a stable and conductive solid electrolyte interface (SEI).2–5 However, demerits such as flammability of solvents (flash point < than 39 °C), reaction of Li-salts with the other materials in the electrolyte and impurities such as water, instability at high temperatures and failure of the solvent mixtures to work at both low and high temperatures demand an alternative with enhanced safety and a simple insertion procedure of the electrolyte during the manufacture of LiBs. This triggered the use of solid polymer electrolytes (SPEs) in LiBs6,7 which provide advantages such as simple design, resistance to shock and vibration, easy film formation, better processability, resistance to pressure and temperature variations, cost-effectiveness and lighter weight. However, current SPEs suffer from limitations ranging from poor ionic conductivity to low lithium ion transference number etc8,9.In this context, various types of SPEs broadly classified as gel-type polymers,10,11 solvent free polymers,10 inorganic crystalline compounds12 and inorganic glasses were discovered and investigated for their use in lithium ion batteries. These polymer electrolytes in general showed higher ionic conductivity and ease of processability when compared to the crystalline electrolytes and hence, are being studied extensively for the use in LiBs. These polymer electrolytes undergo segmental motion of polymer chains as a mode of ion conduction.13 Segmental motion of polymer chains results into simultaneous making and breaking of cation–oxygen interactions in polymers like polyethylene glycol (PEG), polyethylene oxide (PEO) etc. that provides free space for ions to diffuse under the influence of electric field and promote ionic motion and hence, enhance Li+ ion conduction.14 However, ion conduction in polymers require local relaxation and segmental motion that are greatly dependent on the glass transition temperature.15,16 Temperature restriction in cation mobility leads to a theoretical limitation in ionic conductivity based on the segmental motion that has been predicted to be 10−3 to 10−5 S cm−1.
Therefore, there has been a need to seek for an altogether new ion conductive mechanism17–19 that is different from ordinary polymer electrolytes. This has led to the development of several novel and interesting materials.20,21 Liquid crystals22–24 based on long alkyl chain ionic liquids, coordination polymers25 with long nano channels,26 single cation conductive inorganic glassy material,27 inorganic crystalline electrolytes12 such as lithium ion super conductors, anion doped π-conjugated systems28 without any radical carrier species and lithium alkoxide29,30 based metal organic frameworks (MOFs)31–34 are some of the novel materials which have exhibited enhanced ionic conductivity at ambient temperatures. All these examples with different ion conduction mechanism inspired us to design a non-polymeric electrolyte with an alternate ion conduction mechanism.35 The electrolyte aimed to design here is a low molecular weight crystalline organoboron electrolyte.
Research on incorporation of boron in electrolytes started in 1995 by Barthel et al.,36 with the design of a new lithium salt containing boron chelate complexes of aromatic, aliphatic diols or carboxylic acids as anions. Since then, several works on boron based complexes were reported such as boric esters of glycol (BEG solvents),37 polymer networks with boroxine rings,38–41 phenylboronic polymer electrolyte,42 boric ester type electrolytes,43 organoboron π-conjugated systems,28 organoboron electrolytes with 9-borabicyclo[3.3.1]nonane (9-BBN).44,45 Further, salts based on B were also introduced, among which lithium bis(oxalato)borate (LiBOB)46 gained widespread attention and is studied extensively for its properties. However, most of the work was solely concentrated on polymeric electrolytes and their modification to improve the ion conducting properties. Study on organic non-polymeric electrolytes was still untouched.
In the present work, needle crystalline organoboron compound was used as a scaffold for ion conductive path through boron–anion interaction. This was briefly reported by us previously, however, being an urgent publication,35 it involved the introduction to nano-channel based ion conduction mechanism. The mechanism is based on the boron–anion interaction between the organoboron compound and the Li salt anion. The intermolecular arrangement among the cyclic organoboron compound is expected to form nano-channels with B moiety responsible for the trapping of Li salt anions due to its electropositive nature. This will lead to freely available Li+ ions, that can move freely without any hindrance or drag from the anions. Further, the nano-channel structure will provide a directional route to the Li+ ions to travel between the electrodes. Oriented and defined ion conductive path will enhance possibility of long-distance migration of Li+ ions, which is, generally, low in the case of random walk of Li+ ions. This paper deals with the synthesis, chemical, thermal and electrochemical properties of the low molecular weight solid organoboron electrolyte with an aim to study its ion conduction mechanism, thorough characterization of its crystal structure and interfacial properties. Further, cell performance will also be presented for the first time in detail.
Materials and methods
Dehydrated ethylene glycol was purchased from WAKO Co. Ltd. and used as received. Hexane, for washing the product, was purchased from WAKO Co. Ltd. and was used as received. Lithium bis(triflouromethanesulfonyl)imide, used for doping with the crystalline compound, was procured from Kanto Chemicals.NMR spectroscopic analysis was done by using Bruker model Avance III 400. Fourier Transform-Infrared (FT-IR) spectroscopic analysis was carried out using JASCO FT/IR-4100. Raman spectroscopic analysis was done using Raman scattering equipment of Horiba, Jobin-Yvon make; model T64000. Transmission Electron Micrographs (TEM) were obtained using Transmission Electron Microscope, Hitachi H-7650. Single Crystal X-ray Diffraction (SC-XRD) was collected using X8 Kappa APEX-II (Bruker), fitted with Mo Kα X-ray source, 0.7107 Å at Department of Chemistry, Indian Institute of Technology Madras, India and was processed using VESTA 3.4.4.
Ionic conductivity was measured with a complex-impedance gain-phase analyzer Solartron model 1260, under the frequency range from 0.1 Hz to 1 MHz using an AC amplitude of 10 mV over a temperature range of 30–60 °C. The sample was sandwiched between two gold-coated blocking electrodes. Prior to the experiment all the samples were thoroughly dried under reduced pressure at room temperature. The temperature dependence of ionic conductivity was studied over a range of 30–60 °C at an interval of 3 °C between two consecutive temperatures. Further, all the ionic conductivities mentioned further, are reported at 51 °C. Li+ ion transference number measurements were done by Vincent–Bruce–Evans method.47 DC current measurements were done on a potentiostat/galvanostat of Princeton Applied Research; model Versastat-3 with an applied constant potential of 30 mV. AC impedance analysis for measuring the charge transfer resistance before and after DC polarization was carried out with the same instrument with an AC amplitude of 10 mV.
Potential window measurement was done using potentiostat/galvanostat (Princeton Applied Research model Versastat-3) in a 3E beaker type cell in the glove box under Argon atmosphere. Electrolyte used was 0.01 M 1 in 0.1 M LiTFSI in 15 mL EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DC = 1:1 as the electrolyte. The potential window of the cell was analyzed by cyclic voltammetric analysis between −3.5 V to +3.5 V using Pt electrode (wire) as the counter electrode, Ag/AgNO3 as the reference electrode and Pt foil of 1 × 1 cm2 as the working electrode at a scan rate of 5 mV s−1.
:DC = 1:1 as the electrolyte. The potential window of the cell was analyzed by cyclic voltammetric analysis between −3.5 V to +3.5 V using Pt electrode (wire) as the counter electrode, Ag/AgNO3 as the reference electrode and Pt foil of 1 × 1 cm2 as the working electrode at a scan rate of 5 mV s−1.
For charge–discharge studies, graphite based anodic half-cells were prepared using CR2025 type coin cells with graphite as the working electrode (diameter = 15 mm, PIOTREK), lithium metal as the counter electrode (diameter = 15 mm, Honjo metals, Japan) and a ring shaped polypropylene based membrane (Celgard®) as separator (outer diameter = 16 mm, inner diameter = 12 mm). 30 μL of EC:DEC was added to wet the surface of the electrode. The prepared graphite based anodic half-cells were charged and discharged at room temperature (22 ± 1 °C) in a galvanostatic (constant current) mode with restriction in potential using compact charge and discharge system of EC Frontier; ECAD-1000. The cut off potential limits were chosen to be 2.3 V and 0.03 V for 0.5C; 1.8 V and 0.03 V for 2C.
Experimental
Preparation of cyclic crystalline product.35 To mesitylborane43,48 (1.42 g, 10.7 mmol), equimolar amount of dehydrated ethylene glycol (0.598 mL, 10.7 mmol) was added at 0 °C and the reaction mixture was kept for stirring at room temperature for 5 hours under N2 atmosphere (Scheme 1). The resulting mixture was washed with hexane several times and was dried under vacuum for 3 hours to obtain crystalline white powder as the product (1.93 g, 9.99 mmol, 93%). The final product was recrystallized using hexane to obtain needle shaped crystals. Fig. 1 shows the image of single crystal of 1. The synthesized low molecular weight cyclic organoboron electrolyte, 1 was characterized by 11B-NMR in DMSO-d6 at 400 MHz showing a peak at 31.3 ppm referring to incorporation of boron as a boric ester (Fig. 2a). A single peak in 11B-NMR confirmed the presence of a single pure environment of boron. Further, 1H-NMR confirmed the complete structure of 1 (Fig. 2b). The absence of broad peaks and presence of sharp peaks along with appropriate proton integrals were also in accord with the formation of cyclic structure of 1. [11B-NMR (128 MHz, DMSO-d6)]: 31.3 ppm; [1H-NMR (400 MHz, DMSO-d6): 2.27–2.08 (–CH3 protons of mesityl group), 4.30 (–CH2– groups of ethylene glycol), 6.79–6.72 (phenyl ring protons)].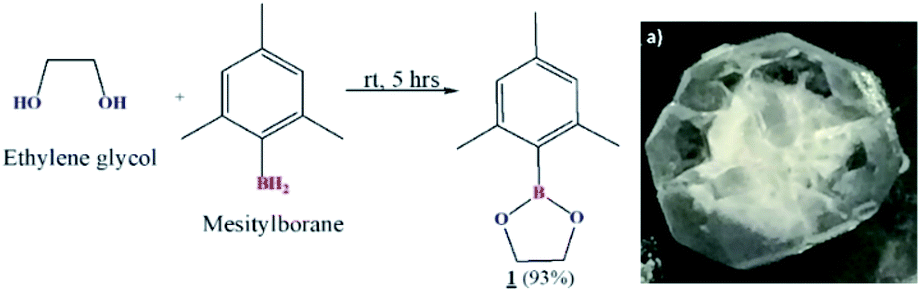 | ||
| Fig. 1 Reaction Scheme 1 and (a) single crystal image of 1. | ||
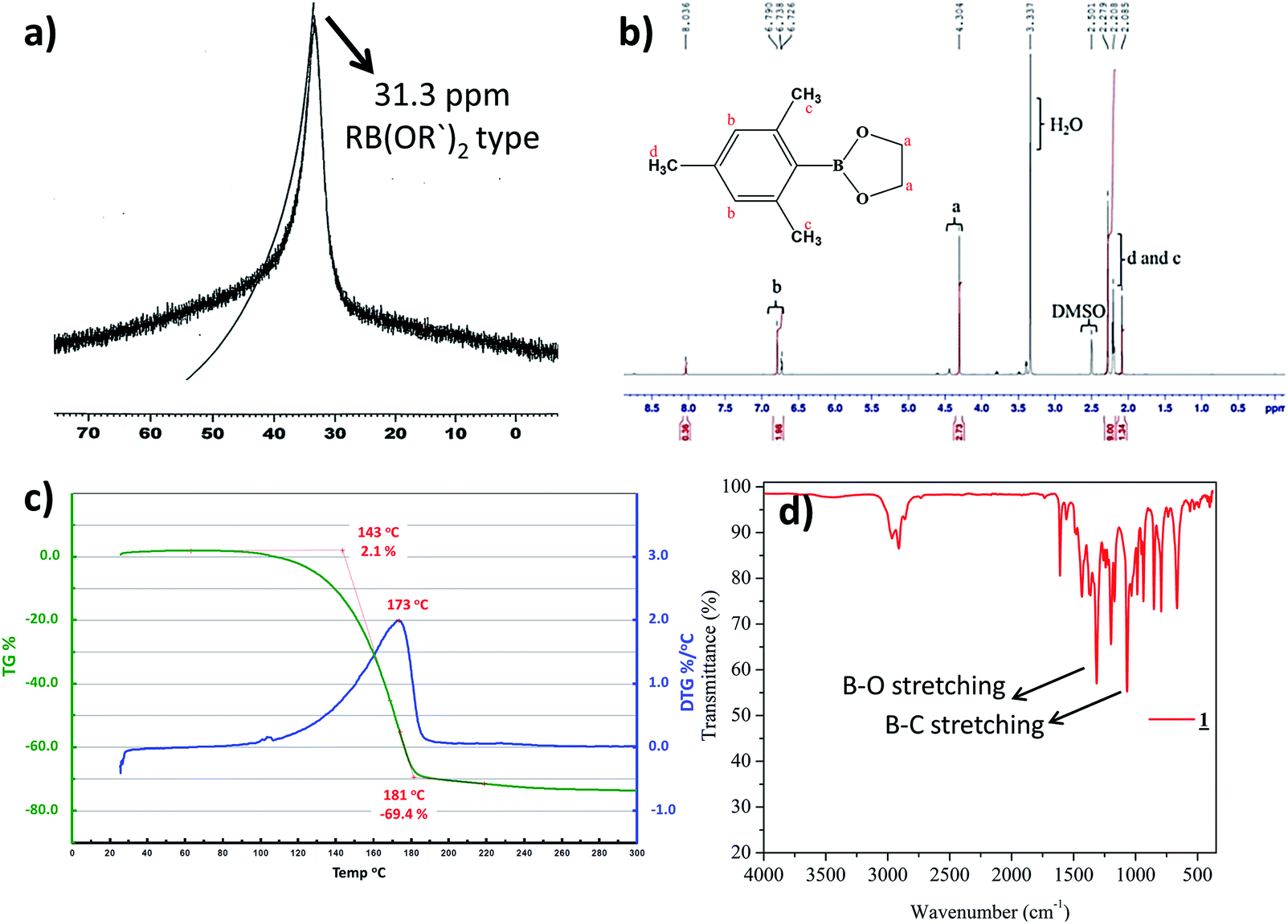 | ||
| Fig. 2 (a) 11B-NMR of 1 in DMSO-d6, (b) 1H-NMR of 1 in DMSO-d6 (c) TGA profile of 1 (d) IR Spectrum of 1. | ||
Results and discussion
TGA was measured under argon atmosphere between 30 °C to 300 °C at the rate of 10 °C min−1. TGA analysis of the cyclic organoboron electrolyte showed that the compound was thermally stable up to 100 °C (Fig. 2c). The FT-IR analysis (Fig. 2d) confirmed the presence of B–O bond and B–C bond which supported the NMR data. The observed FT-IR peaks are as follows:49,50 (3000–2950 cm−1, aromatic CH stretching conjugated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C; 1620–1580 cm−1, aromatic CH2CH2 stretch; 1450–1300 cm−1, B–O bond stretching; 1050–950 cm−1, B–C stretching).
C; 1620–1580 cm−1, aromatic CH2CH2 stretch; 1450–1300 cm−1, B–O bond stretching; 1050–950 cm−1, B–C stretching).
Fig. 3 shows the transmission electron micrographs of the crystalline material 1. A definite geometry and an unresolved lattice fringes like pattern apparently evinces the formation of organic crystals. For electrochemical analysis, 1 was doped with Li salt, lithium bis (triflouromethanesulfonyl)imide, LiTFSI,51 which changed the physical aspect of 1 from transparent crystal to an opaque powder. Addition of Li salt was carried out by two different methods. In the first case, the conventional method of inserting the salt using a solvent (THF)6 was employed. In the second case, a rather simple grinding of 1 with LiTFSI was adapted under N2 atmosphere.
 | ||
| Fig. 3 Transmission electron micrographs of 1. | ||
Ionic conductivity studies
In our previously reported work,35 the Arrhenius plots of samples prepared by conventional method showed a monotonous increase in ionic conductivity with increase in temperature under relatively low concentrations of lithium salt (Arrhenius behaviour). On the other hand, the samples prepared by grinding method obeyed different behaviour exhibiting lower activation energy of ion transport. This implied that different ion conduction mechanisms prevailed in the two samples prepared by the two different methods. In the first case, the maximum ionic conductivity was observed to be 2.7 × 10−4 S cm−1 while the maximum ionic conductivity observed by the facile grinding method reached 7.1 × 10−4 S cm−1. The conductivities shown by the samples prepared by grinding method were significantly higher at the optimal concentration of lithium salt (1:LiTFSI = 1:2). This can be due to the retainment of crystallinity of a Lewis acidic ordered structure by grinding which otherwise might be lost in the case of conventional samples due to solvation. The temperature dependence of ionic conductivity was further studied by Vogel–Fulcher–Tammann (VFT72–75) analysis (cited from ref. 35).
VFT plots for the samples prepared by both the methods showed lower values of ionic conductivity at lower concentration of lithium salt which were attributed to the minimal availability of ions for conduction. The lowering of ionic conductivity at higher concentration of salt can be due to an increased activation energy required for ion transportation. That is, excess of LiTFSI led to insufficient ordering through boron–anion interaction. High conductivity seen at an optimal concentration of lithium salt in the sample prepared by grinding can be hypothesized to be due to the formation of nanochannels15,35,52–54 by an arrangement of anions scaffolded by the undisturbed crystalline nature of the boron compound through boron–anion interaction,44,55–59 allowing easier and faster transportation of ions by ion hopping mechanism.10 This becomes evident from the fact that ionic conductivity for a concentration of 1:LiTFSI = 1:2 prepared by conventional method shows a very low value as compared to the sample prepared by grinding of same composition. This can be due to the absence of any such nanochannel formation in the conventional method (cited from ref. 35).
A closer look at the VFT parameters of 3 samples prepared by grinding method, showed very low activation energy of ion transport (B). This led to markedly higher ionic conductivity. On the other hand, the samples prepared by conventional method showed relatively high activation energy and low ionic conductivity under the same composition. As a specific instance, comparison between grinding and conventional method for a composition of 1:LiTFSI = 1:1 should be noted. Although the carrier ion number in the sample prepared by grinding is only 1.72, its ionic conductivity is 3.5 times higher than that of the sample prepared by conventional method of same composition in which carrier ion number was 59.2. Hence, in the case of grinded samples, low activation energy of ion transport resulted into high ionic conductivity. This evinces our hypothesis of formation of special channelized pathways resulting into low activation energy allowing facile transportation of Li+ ions (cited from ref. 35).
X-ray diffraction analysis
XRD analysis was done for the sample with highest ionic conductivity (1:LiTFSI = 1:2, prepared by grinding method). The data obtained is shown in Fig. 4. This data was compared with the pure crystalline sample, 1 and with the sample prepared by conventional method in same molar ratio. XRD patterns obtained for the three samples gave a clear idea about the crystalline structure of 1. In the case of conventional sample that involves the use of THF, the crystallinity of the sample was lost. Pure sample 1 and the sample prepared by grinding method showed similar peaks at the same 2θ values. The high ionic conductivity of the grinded sample 1:LiTFSI = 1:2 (grinding) can be associated with this intactness of the 100% peak and similarity in other peaks can be attributed to the retainment of crystallinity leading to formation of channels for ion conduction. However, in the case of solvent treated sample, the 100% peak shifted to 2θ 14.8 indicating the change in structure.
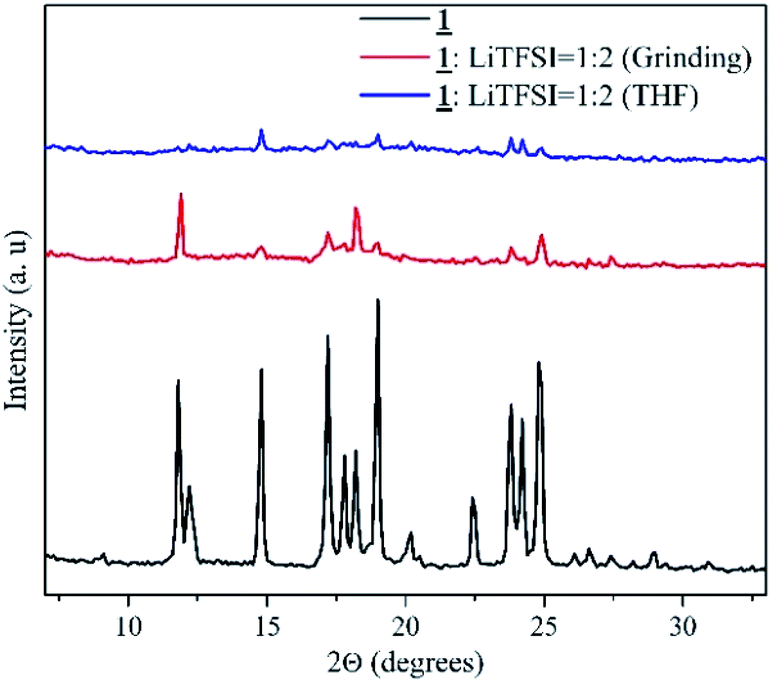 | ||
| Fig. 4 XRD pattern of 1 and sample of 1 doped with Li salt by two methods in 1:2 molar ratio. | ||
SC-XRD analysis of the crystalline material also supports the nano-channel formation for the ion conduction mechanism. Fig. 5 shows the lattice structure of the crystalline material having carbon, oxygen and boron arranged in a zigzag channel manner. The structure of molecule C9BO2C2 (1) was analysed to be orthorhombic system with space group Cmc2 (non-centrosymmetric) with the cell parameters a = 29.865 (2) Å, b = 10.2827 (4) Å and c = 10.8622 (7) Å. Here, in this pattern the total number of constituent atoms present do not correspond to the actual formula unit structure. This can be very likely due to the boron carbon (B–C) single bond rotation which is free to rotate. Within the temperature range of ionic conductivity measurement, the rotation is expected to be very fast. If the rotation is very fast, in that case, the atoms appear at different position in space according to the rotational conformation, instead of one unit and hence, determination of electron density becomes somewhat difficult. Despite the fact, it is still possible to see the formation of nano-channels responsible for the ion conduction. The dimensions of the cuboidal box are estimated to be 10.86 Å (X and Y) and 29.8 Å (Z).
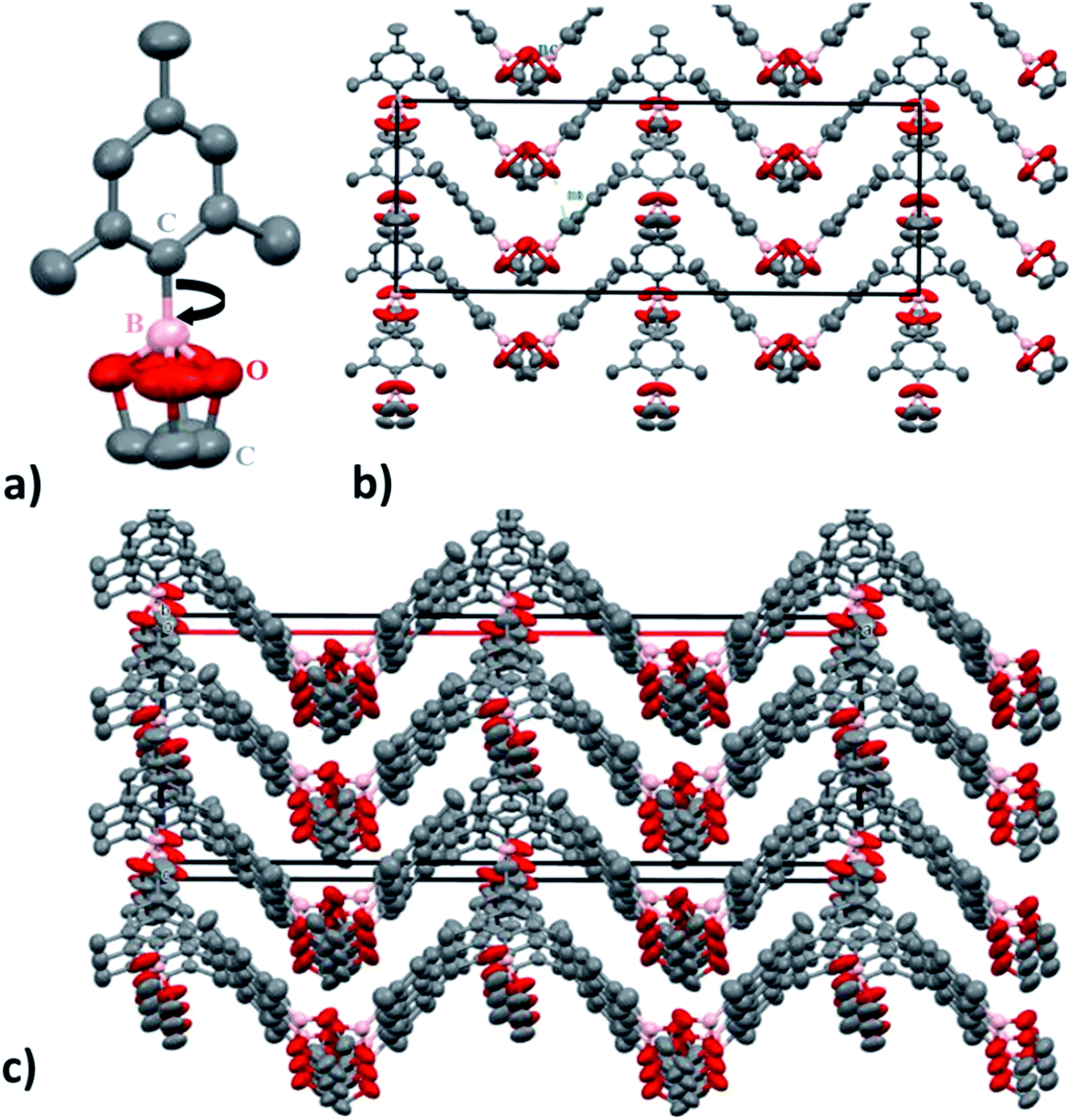 | ||
| Fig. 5 (a) C9BO2C2 molecule (b) (010) projection of the C9BO2C2 (1) structure. For a better understanding of the real size of the nanochannels (nc), the atoms are represented with their thermal anisotropic ellipsoids. (c) 3D view of the C9BO2C2 (1) structure showing the nanochannels (nc) running along the b parameter. | ||
From this, channel width is estimated to be 5.428 (5) Å. Since, ionic radius of Li+ is 90 pm (0.9 Å), it is very easy for the Li ions to migrate and transfer through the channel promoting ion conduction. Also, even if solvation of Li+ occurs due to the presence of EC as a wetting additive and the TFSI anions as well, the ionic radius for the ion would change to ∼2.1 Å,60 which is still 2.5 times smaller than the nanochannel width. Hence, nano-channel formation is expected to not hinder the efficient and directional movement of Li ions between the electrodes thereby, increasing the ionic conductivity by providing defined ion conductive pathway enabling long distance migration under higher probability than random walk fashion.
Li ion transference number
The cell employed for measuring the lithium ion transference number was a symmetrical cell consisting of a Li/electrolyte/Li configuration. The transference number was measured for the sample for the molar ratio 1:LiTFSI = 2:1 under argon atmosphere. For an all solid state electrolyte of this type, the Li ion transference number was found out to be 0.28. The transference number was also measured using the crystalline sample as a co-electrolyte to study the anion-trapping effect of boron (Fig. 6). In this analysis, following the similar procedure for transference number measurement, 1 was added in various molar ratio from 0.25 to 1.25 moles to 15 mL solution of 1.0 M LiTFSI in EC:DEC = 1:1 (v/v) where 1.0 M LiTFSI in EC:DEC = 1:1 (v/v) acted as the electrolyte and 1 in various molar ratio from 0.25 to 1.25 moles acted as the co-electrolyte. Interestingly, as the amount of boron increased in the electrolytic solution, the transference number increased to a maximum value of 0.92. However, after reaching the saturation, the electrolyte behaved similar to a solid state electrolyte and the Li+ ion transference number decreased to a similar value (0.22) like the solid state electrolyte mentioned earlier (0.28).
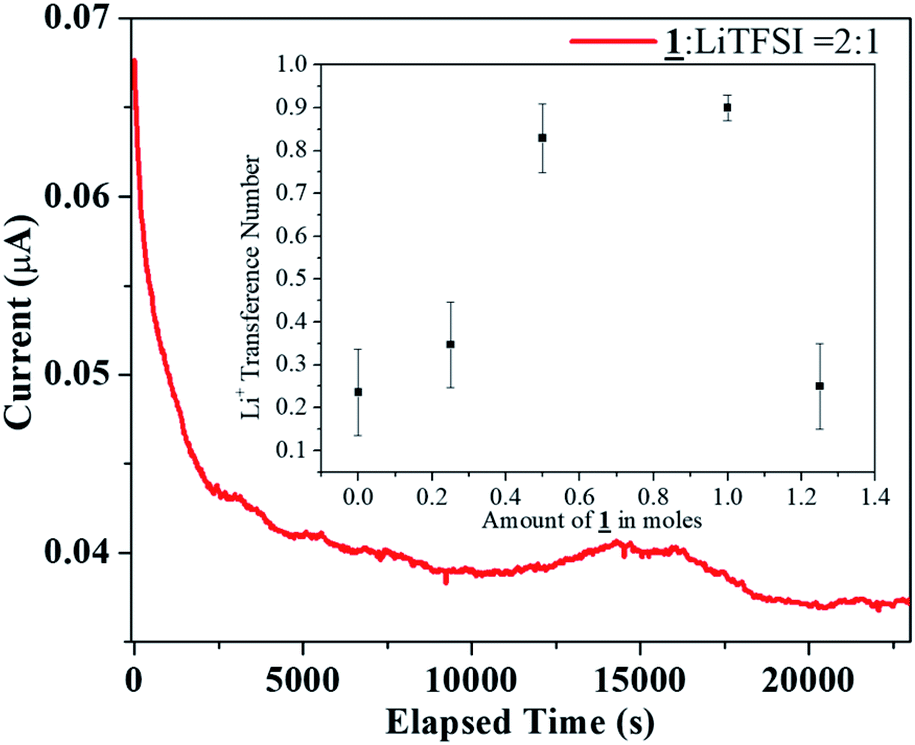 | ||
| Fig. 6 DC Polarization data for the sample 1:LiTFSI = 2:1 (inset: Li transference number of 1 as co-electrolyte with 1.0 M LiTFSI in EC:DEC = 1:1). | ||
The Li ion transference number is the fraction of the total ionic conductivity because of the Li ions. As a mean to increase the Li ion transference number, alternative methods such as ceramic based single ion conductors,76,77 dry polymer electrolytes (that involves addition of a small-molecule salt to a polymer),78 additives containing polymer electrolytes membranes79 and liquid electrolytes with polymeric anions have been studied extensively.80 These studies showed that in the case of dry polymer electrolytes usually the tLi+ is between 0.3–0.4 due to the strong solvation of the lithium relative to the bulky anion by the polymer backbone.78 Also, use of strong Lewis acid polymers81 were suggested to enhance the solvation of anion as a method to increase tLi+. A few research reported affixing anions to the polymer backbone making them immobile as the most common approach to increase the tLi+.82 The similar approach of immobilizing the anions using the Lewis acidic boron,83,84 allowing boron–anion interaction was used here in the current research.
In the case of polymer electrolytes, with the increase in the additive content, the tLi+ is reported to first increase and then decrease. This behaviour is attributed to the enhanced degree of Lewis acid–base interaction between the polymer and the additives.85 Further, Appetecchi et al. reported that because of the unique interaction of polymers such as poly (acrylonitrile), poly (methyl methacrylate) in EC and propylene carbonate (PC) with anion in LiTFSI, tLi+ as high as 0.8 can be achieved.86 A single ion conducting poly(arylene ether) based electrolyte was reported to show a tLi+ of 0.93 due to facilitated electrostatic interaction between oxygen atoms and lithium ions.87
The literature review provides an insight towards the obtained trend in the tLi+ when organoboron compound is added as a co-electrolyte. With lower concentration of organoboron compound, the immobilization of TFSI− anions by the vacant p-orbital of boron is less effective. Also, the ion conduction would occur due to solvation of both Li+ and TFSI− ions, resulting in low tLi+. With an increase in the boron amount, enhanced anion trapping due to boron–anion interaction would result in availability of only Li+ ions to contribute to the total transference number. Hence, owing to the selective cation conduction, the Li+ ion transference number increased to a value of 0.92 in the 1/LiTFSI range of 0 to 1.0. However, the trapping of the anion at high concentration (1.25 moles of 1 as co-electrolyte) was not as effective as in lower concentrations thereby resulting in a tLi+ value of 0.22, as it behaved similar to the solid electrolyte after reaching saturation (Fig. 6 inset).
Electrochemical stability
Potential window measurement was done using 0.01 M 1 in 0.1 M LiTFSI in 15 mL EC:DC = 1:1 as the electrolyte by cyclic voltammetric analysis between −3.5 V to +3.5 V. From −3.3 V to +2.5 V there occurred no electrochemical reaction. The potential window of the sample was found to be 5.3 V (Fig. 7).
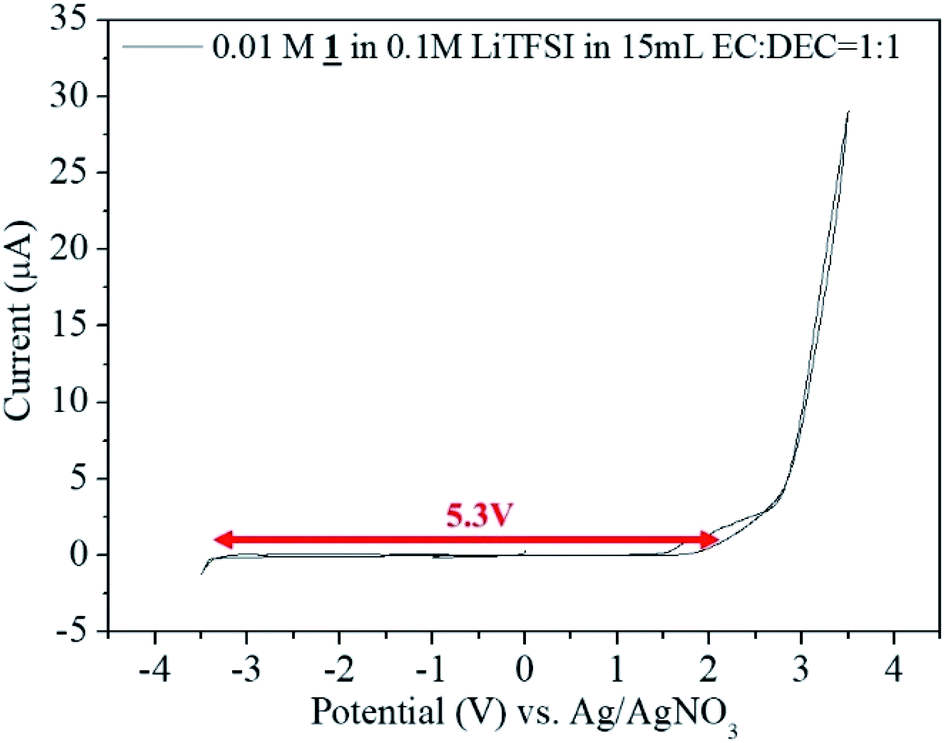 | ||
| Fig. 7 Cyclic Voltammogram for the sample 1 in 0.1 M LiTFSI in 15 mL EC:DEC = 1:1 vs. Ag/AgNO3. | ||
Charge–discharge studies
For charge–discharge analysis of sample 1, powdered electrolyte prepared by the grinding method in molar ratio 1: LiTFSI = 2:1 (sample with highest ionic conductivity) was chosen. The choice of electrolyte was done on the basis of the observed ionic conductivity at 51 °C. Along with the electrolyte used, optimum amount of EC:DEC = 1:1 (30 μL) was added to wet the surface of the electrode while fabricating the cell in order to ensure a steady SEI3,4 formation. After assembling, they were kept for stabilization. The prepared anodic coin half-cells were charged and discharged at 0.5C (0.154 mA) and 2C (0.616 mA). The interfacial characteristics were monitored by measuring the charge-transfer resistance before and after charge–discharge analysis via EIS technique.61,62
The anodic half-cell fabricated using graphite and Li metal as anode and cathode, respectively and electrolyte 1 is termed as cell 1. Cell 1 was fabricated using electrolyte 1 and its charge discharge was first carried out at a lower current rate i.e., 0.5C (0.154 mA) and was increased to 2C (0.616 mA) after 10 cycles. The result of charge discharge at 0.5C for 10 cycles is shown in Fig. 8a and following cycles at 2C is shown in Fig. 8b. The cell was able to deliver a charge–discharge capacity of 120–150 mA h g−1 during 15 cycles at 2C, however a reduction in the capacity was observed after 10th cycle. The capacity decreased to 110 mA h g−1 till 10th cycle.
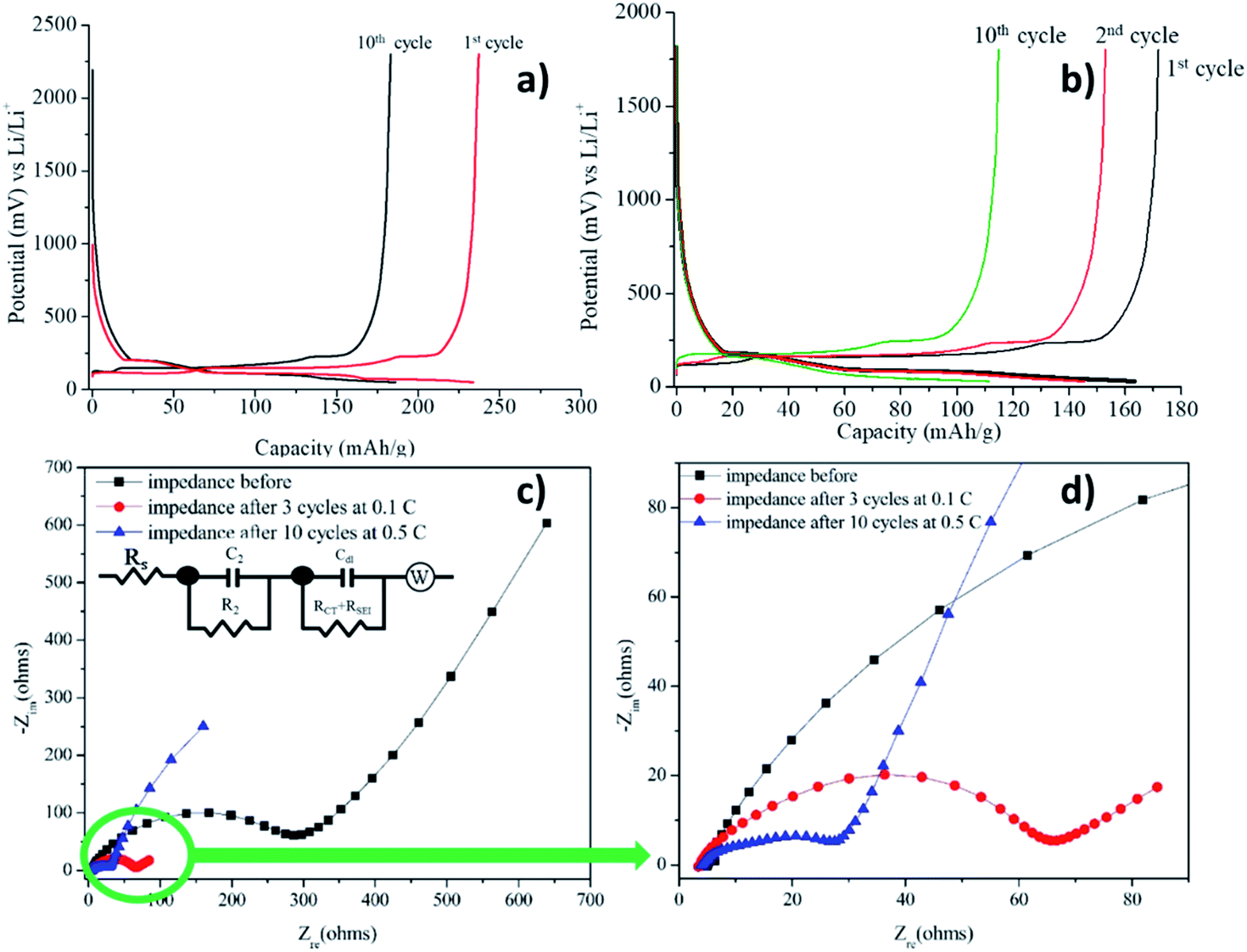 | ||
| Fig. 8 Charge discharge profiles of 1: LiTFSI = 1:2 with 30 μL of EC:DEC = 1:1 at (a) 0.5C (b) 2C. Inset shows the coulombic efficiency profile of anodic half-cells at corresponding current rates. (c) Impedance analysis before and after charge–discharge with the equivalent circuit, EqC1, used for fitting the charge–discharge data for cell 1, (d) zoom in version of (c). | ||
As it can be clearly observed, the cell was able to charge up to ∼250 mA h g−1 and discharge completely. A plateau observed at 0.2 V while charging marked the intercalation of the Li+ ions into graphite. The charge–discharge profiles showed only a slight decrease in the capacity from 1st to 10th cycle. This can be attributed to the formation of the stable SEI during cycling which prevented the drastic decrease in the capacity after 10 cycles. A similar kind of plateau was observed in the discharging profiles as well. Coulombic efficiency of the cell was observed to be ∼100% for all the cycles.
Interfacial characteristics
Impedance analysis before charge–discharge of cell 1 was done to determine the interfacial resistance (Fig. 8c and d) and was evaluated by using an equivalent circuit, EqC1, provided in the inset in Fig. 8c. In general, the anodic SEI layer is formed by the consumption of Li ions, salts and solvents during charge–discharge cycles, mainly in the first cycle and gradually in the frequent number of cycles.2 A conductive SEI layer provides high lithium selectivity and permeability with tolerance to expansion and contraction stresses.63 Table 1 shows the values of all the resistances present in the system evaluated by the equivalent circuit, EqC1. The equivalent circuit consists of three resistances. The high frequency resistance Rs, refers to the solution resistance, followed by R2 which is designated to the migration of Li ion to the surface. The low frequency tail refers to the combination of charge transfer resistance (Rct) + resistance of the SEI layer (RSEI).64| Imp. data | Solution resistance, Rs/R1 (Ω) | Li migration through surface, R2 (Ω) | Interfacial resistance, Rct + RSEI (Ω) |
|---|---|---|---|
| Impedance before | 5.193 | 18.34 | 237.2 |
| Impedance after 10 cycles | 3.567 | 630.5 | 26.09 |
A review of literature shows that a variety of Nyquist plots are reported for all-solid state batteries depending on the operating conditions such as electrodes used, electrolyte materials, temperature of operation etc. Depending on the electrode structure and cell configuration, different spectra were fitted by different equivalent circuits. Zhang et al. reported potential dependent interfacial processes on a composite cathode for an all-solid state Li battery. They reported two semicircles located in the mid frequency region (from 1 kHz to 100 Hz) and low-frequency region (∼1 Hz) assigned to resistance of the solid electrolyte and the interface between the negative electrode and the solid electrolyte.65 Shin et al. reported a LiTFSI based electrolyte for an all solid Li–S battery and reported a combination of bulk solution resistance, a small grain boundary resistance, interfacial resistance denoted by Rss corresponding to solid–solid interfaces at cathode and anode and a solid–liquid interfacial resistance.66 Further, Braun et al. reported the physical cell modelling for ceramic based all solid-state cells based on the Newman's approach67 in which impedance behaviour of a fully charged LiCoO2–Li10GeP2S12/Li10GeP2S12/Li4Ti5O12–Li10GeP2S12 cell was calculated. The obtained Nyquist diagram was defined into three segments: high frequency intersection referring to the total ohmic resistance, the middle frequency part (1 MHz to 1 Hz) referring to total charge-transfer resistance due to active material/electrolyte interfaces and a low frequency tail (<1 Hz) corresponding to the solid-state diffusion of Li ion in active materials.68
For the current organoboron electrolyte, the charge transfer resistance, Rct, was obtained as 237.2 Ω before the charge–discharge was started. However, after 10 cycles, interfacial resistance for the cell reduced to 26.09 Ω indicating the formation of a conducting interface, termed as the solid electrolyte interface (SEI)66,69,70 and occurrence of an effective charge-transfer process due to the formation of a stable and highly conductive SEI layer. A reduction in Rct with number of cycles along with no reduction in capacity evinces the formation of a stable SEI.64,71 Formation of such an SEI layer is advantageous in improving the performance of the cell.
Conclusions
In this work, the detailed synthesis and electrochemical properties of a low molecular weight cyclic organoboron crystalline electrolyte synthesized using ethylene glycol and mesityl borane was discussed. The electrolyte was obtained as needle shaped crystal in high yields. The crystalline nature of the electrolyte has an important characteristic feature and showed interesting effect on the ionic conductivity after externally doping with the lithium salt via two different methods namely conventional method and grinding method. The electrochemical analysis of the sample obtained by grinding method exhibited high ionic conductivity of 7.1 × 10−4 S cm−1 at an optimized concentration of lithium salt (1:LiTFSI = 1:2). The presence of crystallinity in Lewis acidic 1 can be considered as an important factor behind the high ionic conductivity of the electrolyte prepared by grinding due to more regulated ion conduction path in the matrices. The electrolyte was also tested for their charge-discharge characteristics and its interfacial characteristics by studying impedance before and after charge–discharge when the cell was completely discharged for Li ion secondary batteries. The crystalline sample, 1 when mixed with LiTFSI in a molar ratio of 1:2 by grinding method showed an impressive charge–discharge behaviour with a charging capacity in the range of 150–200 mA h g−1 and was studied with different current rates. A stable and a conductive interface was formed and as a result the interfacial resistance decreased to a lower value of 26.09 Ω from 237.2 Ω after 10 cycles of charge–discharge. All the above results confirm a real-time application of such an all solid state organoboron based low molecular weight electrolyte in lithium ion batteries.
Conflicts of interest
There are no conflicts to declare.References
- D. Aurbach, Y. Talyosef, B. Markovsky, E. Markevich, E. Zinigrad, L. Asraf, J. S. Gnanaraj and H. J. Kim, Electrochim. Acta, 2004, 50, 247–254 CrossRef CAS.
- A. M. Andersson and K. Edström, J. Electrochem. Soc., 2001, 148, A1100–A1109 CrossRef CAS.
- V. A. Agubra and J. W. Fergus, J. Power Sources, 2014, 268, 153–162 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 1–26 CrossRef CAS.
- A. M. Stephan, K. S. Nahm, M. Anbu Kulandainathan, G. Ravi and J. Wilson, Eur. Polym. J., 2006, 42, 1728–1734 CrossRef CAS.
- M. Wakihara, Y. Kadoma, N. Kumagai, H. Mita, R. Araki, K. Ozawa and Y. Ozawa, J. Solid State Electrochem., 2012, 16, 847–855 CrossRef CAS.
- Y. S. Kim, Y. G. Cho, D. Odkhuu, N. Park and H. K. Song, Sci. Rep., 2013, 3, 1917 CrossRef PubMed.
- W. H. Meyer, Adv. Mater., 1998, 10, 439 CrossRef CAS.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904–7929 CrossRef CAS.
- A. M. Stephan, Eur. Polym. J., 2006, 42, 21–42 CrossRef.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- Q. Li, E. Wood and H. Ardebili, Appl. Phys. Lett., 2013, 102(1–5), 243903 CrossRef.
- G. Mao, R. F. Perea, W. S. Howells, D. L. Price and M. L. Saboungi, Nature, 2000, 405, 163–165 CrossRef CAS PubMed.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520–523 CrossRef CAS PubMed.
- A. M. Christie, S. J. Lilley, E. Staunton, Y. G. Andreev and P. G. Bruce, Nature, 2005, 433, 50–53 CrossRef CAS PubMed.
- W. A. Henderson, D. M. Seo, Q. Zhou, P. D. Boyle, J. H. Shin, H. C. De Long, P. C. Trulove and S. Passerini, Adv. Energy Mater., 2012, 2, 1343–1350 CrossRef CAS.
- L. Ramõn-Gimenez, R. Storz, J. Haberl, H. Finkelmann and A. Hoffmann, Macromol. Rapid Commun., 2012, 33, 386–391 CrossRef PubMed.
- A. Sato, T. Okumura, S. Nishimura, H. Yamamoto and N. Ueyama, J. Power Sources, 2005, 146, 423–426 CrossRef CAS.
- T. Yong, J. Wang, Y. Mai, X. Zhao, H. Luo and L. Zhang, J. Power Sources, 2014, 254, 29–32 CrossRef CAS.
- M. Ranger and M. Leclerc, Macromolecules, 1999, 32, 3306–3313 CrossRef CAS.
- K. Hoshino, M. Yoshio, T. Mukai, K. Kishimoto, H. Ohno and T. Kato, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3486–3492 CrossRef CAS.
- C. T. Imrie, M. D. Ingram and G. S. McHattie, J. Phys. Chem. B, 1999, 11, 832–834 CAS.
- T. Kato, Y. Kubota and T. Uryu, Polym. Bull., 1997, 38, 551–554 CrossRef CAS.
- Y. Kubota, M. Takata, T. C. Kobayashi and S. Kitagawa, Coord. Chem. Rev., 2007, 251, 2510–2521 CrossRef CAS.
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS.
- M. J. G. Jak, F. G. B. Ooms, E. M. Kelder, W. J. Legerstee, J. Schoonman and A. Weisenburger, J. Power Sources, 1999, 80, 83–89 CrossRef CAS.
- N. Matsumi, M. Nakashiba and H. Ohno, Polym. Bull., 2003, 50, 259–264 CrossRef CAS.
- R. Ameloot, M. Aubrey, B. M. Wiers, A. P. Gõmora-Figueroa, S. N. Patel, N. P. Balsara and J. R. Long, Chem. - Eur. J., 2013, 19, 5533–5536 CrossRef CAS PubMed.
- B. M. Wiers, M. L. Foo, N. P. Balsara and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14522–14525 CrossRef CAS PubMed.
- J. Shin, M. Kim, J. Cirera, S. Chen, G. J. Halder, T. A. Yersak, F. Paesani, S. M. Cohen and Y. S. Meng, J. Mater. Chem. A, 2015, 3, 4738–4744 RSC.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2005, 44, 4670–4679 CAS.
- C. Combelles, M. Ben Yahia, L. Pedesseau and M. L. Doublet, J. Phys. Chem. C, 2010, 114, 9518–9527 CrossRef CAS.
- P. Joshi, R. Vedarajan and N. Matsumi, Chem. Commun., 2015, 51, 15035–15038 RSC.
- J. Barthel, M. Wühr, R. Buestrich and H. J. Gores, J. Electrochem. Soc., 1995, 142, 2527–2531 CrossRef CAS.
- S. S. Zhang and C. A. Angell, J. Electrochem. Soc., 1996, 143, 4047–4053 CrossRef CAS.
- M. A. Mehta and T. Fujinami, Chem. Lett., 1997, 26, 915–916 CrossRef.
- M. A. Mehta and T. Fujinami, Solid State Ionics, 1998, 113–115, 187–192 CrossRef CAS.
- M. A. Mehta, T. Fujinami and T. Inoue, J. Power Sources, 1999, 81–82, 724–728 CrossRef CAS.
- M. A. Mehta, T. Fujinami, S. Inoue, K. Matsushita, T. Miwa and T. Inoue, Electrochim. Acta, 2000, 45, 1175–1180 CrossRef CAS.
- X. Sun and C. Austen Angell, Electrochim. Acta, 2001, 46, 1467–1473 CrossRef CAS.
- N. Matsumi, K. Sugai and H. Ohno, Macromolecules, 2002, 35, 5731–5733 CrossRef CAS.
- T. Mizumo, K. Sakamoto, N. Matsumi and H. Ohno, Electrochim. Acta, 2005, 50, 3928–3933 CrossRef CAS.
- T. Mizumo, K. Sakamoto, N. Matsumi and H. Ohno, Chem. Lett., 2004, 33, 396–397 CrossRef CAS.
- K. Xu, S. S. Zhang, U. Lee, J. L. Allen and T. R. Jow, J. Power Sources, 2005, 146, 79–85 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28(13), 2324–2328 CrossRef CAS.
- N. Matsumi and Y. Chujo, Polym. Bull., 1997, 38, 531–536 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Elsevier, 1991 Search PubMed.
- P. R. Griffiths, Vib. Spectrosc., 1992, 4, 121 CrossRef.
- V. Aravindan, J. Gnanaraj, S. Madhavi and H. K. Liu, Chem.–Eur. J., 2011, 17, 14326–14346 CrossRef CAS PubMed.
- S. Vorrey and D. Teeters, Electrochim. Acta, 2003, 48, 2137–2141 CrossRef CAS.
- T. Nakamura, T. Akutagawa, K. Honda, A. E. Underhill, A. T. Coomber and R. H. Friend, Nature, 1998, 394, 159–162 CrossRef CAS.
- S. B. R. S. Adnan and N. S. Mohamed, Int. J. Electrochem. Sci., 2012, 7, 9844–9858 CAS.
- N. Matsumi, K. Sugai, K. Sakamoto, T. Mizumo and H. Ohno, Macromolecules, 2005, 38, 4951–4954 CrossRef CAS.
- N. Matsumi, M. Nakashiba, T. Mizumo and H. Ohno, Macromolecules, 2005, 38, 2040–2042 CrossRef CAS.
- N. Matsumi, K. Sugai and H. Ohno, Macromolecules, 2002, 35, 5731–5733 CrossRef CAS.
- N. Matsumi, K. Sugai, M. Miyake and H. Ohno, Macromolecules, 2006, 39, 6924–6927 CrossRef CAS.
- D. Shanmukaraj, S. Grugeon, G. Gachot, S. Lauelle, D. Mathlron, J. M. Tarascon and M. Armand, J. Am. Chem. Soc., 2010, 132(9), 3055–3062 CrossRef CAS PubMed.
- Q. Li, S. Tan, L. Li, Y. Lu and Y. He, Sci. Adv., 2017, 3(7), e1701246 CrossRef PubMed.
- J. Jiang, Z. Lin, Q. Ju, Z. Ma, C. Zheng and Z. Wang, Energy Procedia, 2017, 105, 844–849 CrossRef CAS.
- U. Westerhoff, T. Kroker, K. Kurbach and M. Kurrat, J. Electrochem. Energy Convers. Storage, 2016, 8, 244–256 CrossRef.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- D. Aurbach, B. Markovsky, I. Weissman, E. Levi and Y. Ein-Eli, Electrochim. Acta, 1999, 45, 67–86 CrossRef CAS.
- W. Zhang, D. A. Weber, H. Weigand, T. Arlt, I. Manke, D. Schröder, R. Koerver, T. Leichtweiss, P. Hartmann, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 17835–17845 CrossRef CAS PubMed.
- M. Shin and A. A. Gewirth, Adv. Energy Mater., 2019, 9, 1900938 CrossRef.
- J. Newman and W. Tiedemann, AIChE J., 1975, 21, 25–41 CrossRef CAS.
- P. Braun, C. Uhlmann, M. Weiss, A. Weber and E. Ivers-Tiffée, J. Power Sources, 2018, 393, 119–127 CrossRef CAS.
- W. C. West, Z. D. Hood, S. P. Adhikari, C. Liang, A. Lachgar, M. Motoyama and Y. Iriyama, J. Power Sources, 2016, 312, 116–122 CrossRef CAS.
- P. Joshi, K. Iwai, S. G. Patnaik, R. Vedarajan and N. Matsumi, J. Electrochem. Soc., 2018, 165, A493–A500 CrossRef CAS.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- J. Rault, J. Non-Cryst. Solids, 2000, 271, 177–217 CrossRef CAS.
- H. Vogel, Phys. Z., 1921, 22, 645–646 CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339 CrossRef CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 4, 156 Search PubMed.
- H. Aono, E. Sugimoto, Y. Sadaoka, N. Imanaka and G. Adachi, J. Electrochem. Soc., 1989, 136(2), 590 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46(41), 7778–7781 CrossRef CAS PubMed.
- K. Timachova, H. Watanabe and N. P. Balsara, Macromolecules, 2015, 48(21), 7882–7888 CrossRef CAS.
- I. Villaluenga, S. Inceoglu, X. Jiang, X. C. Chen, M. Chintapalli, D. R. Wang, D. Devaux and N. P. Balsara, Macromolecules, 2017, 50(5), 1998–2005 CrossRef CAS.
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, ACS Energy Lett., 2017, 2, 2563–2575 CrossRef CAS.
- B. M. Savoie, M. A. Webb and T. F. Miller, J. Phys. Chem. Lett., 2017, 8(3), 641–646 CrossRef CAS PubMed.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. RodriguezMartinez, M. B. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- N. Matsumi, K. Sugai and H. Ohno, Macromolecules, 2003, 36(7), 2321–2326 CrossRef CAS.
- S. Liang, U. H. Choi, W. Liu, J. Runt and R. H. Colby, Chem. Mater., 2012, 24(12), 2316–2323 CrossRef CAS.
- K. E. Aifantis, S. A. Hackney and R. V. Kumar, High Energy Density Lithium Batteries: Materials, Engineering, Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2010, pp. 165–208 Search PubMed.
- G. B. Appetecchi, F. Croce and B. Scrosati, Electrochim. Acta, 1995, 40(8), 991–997 CrossRef CAS.
- Y. Chen, H. Ke, D. Zeng, Y. Zhang, Y. Sun and H. Cheng, J. Membr. Sci., 2017, 525, 349–358 CrossRef CAS.
Footnote |
| † CCDC 1964230. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09559d |
| This journal is © The Royal Society of Chemistry 2020 |