DOI:
10.1039/D0QO00387E
(Review Article)
Org. Chem. Front., 2020,
7, 1743-1778
Recent developments in 1,6-addition reactions of para-quinone methides (p-QMs)
Received
30th March 2020
, Accepted 28th May 2020
First published on 28th May 2020
Abstract
In recent years, para-quinone methides (p-QMs) have emerged as attractive and versatile synthons in organic synthesis owing to their high reactivity. Consequently, p-QM chemistry has attracted increasing attention and remarkable advances have been achieved. Among the numerous transformations involving p-QMs, catalytic reactions play a pivotal role, and a variety of catalytic systems mediated by Lewis acids, Brønsted acids, bases, transition metals, N-heterocyclic carbenes, and other catalysts have been established for performing 1,6-conjugate addition reactions. Various molecular scaffolds have been constructed using p-QMs to obtain the core structures of numerous natural and synthetic substances of chemical and biomedical relevance. In this review, we provide a comprehensive overview of recent progress in this rapidly growing field by summarizing the 1,6-conjugate addition and annulation reactions of p-QMs with consideration of their mechanisms and applications.
 Jia-Yin Wang | Jia-Yin Wang was born in 1993 in Jiangsu Province (China). He received his BS in 2016 at Xuzhou Institute of Technology. He received his MS in 2019 at Jiangsu Normal University under the supervision of Prof. Shu-Jiang Tu and Prof. Bo Jiang. Currently, he is pursuing his PhD at Nanjing University under the supervision of Prof. Guigen Li and Prof. Bo Jiang (JSNU). His research interests include radical triggered bicyclization and asymmetric transformation. |
 Wen-Juan Hao | Wen-Juan Hao was born in 1983 in Jiangsu province, China. She received her B.S. in 2006 and M.S. in 2009 at Jiangsu Normal University under the supervision of Prof. Shu-Jiang Tu, Ph.D. in 2014 at Soochow University with Prof. Shu-Jun Ji and then joined the School of Chemistry and Materials Science at Jiangsu Normal University as a lecturer and is currently promoted to Associate Professor. She was as a Visiting Scholar at Nanyang Technological University from July 2016 to January 2017 in Singapore with Prof. Choon Hong Tan. Her research interest includes synergistic catalysis, heterocyclic syntheses, and radical domino reactions. |
 Shu-Jiang Tu | Shu-Jiang Tu was born in 1957 in Jiangsu (China) and received his B.S. in 1983 at Jiangsu Normal University. He was appointed as Assistant Professor at the Xuzhou Normal University in 1999 and was promoted to Full Professor in 2003. His current interests are the development of new synthetic methods, green chemistry, and microwave multicomponent syntheses. |
 Bo Jiang | Bo Jiang was born in 1981 in Jiangsu (China). He received his B.S. in 2004 and M.S. in 2007 at Jiangsu Normal University under the supervision of Prof. Shu-Jiang Tu, Ph.D. in 2010 at Soochow University with Prof. Shu-Jiang Tu and Prof. Guigen Li. He joined Jiangsu Normal University as a lecturer and was promoted to Full Professor in 2019. He was as an Associate Research Professor at Texas Tech University from Aug. 2015 to Feb. 2017 in USA with Prof. Guigen Li. His research interest includes asymmetric catalysis, radical transformations, and the innovation of synthetic methods. |
1. Introduction
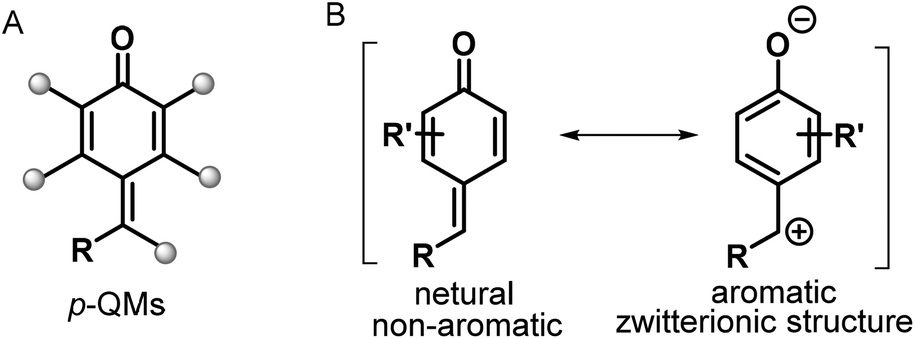
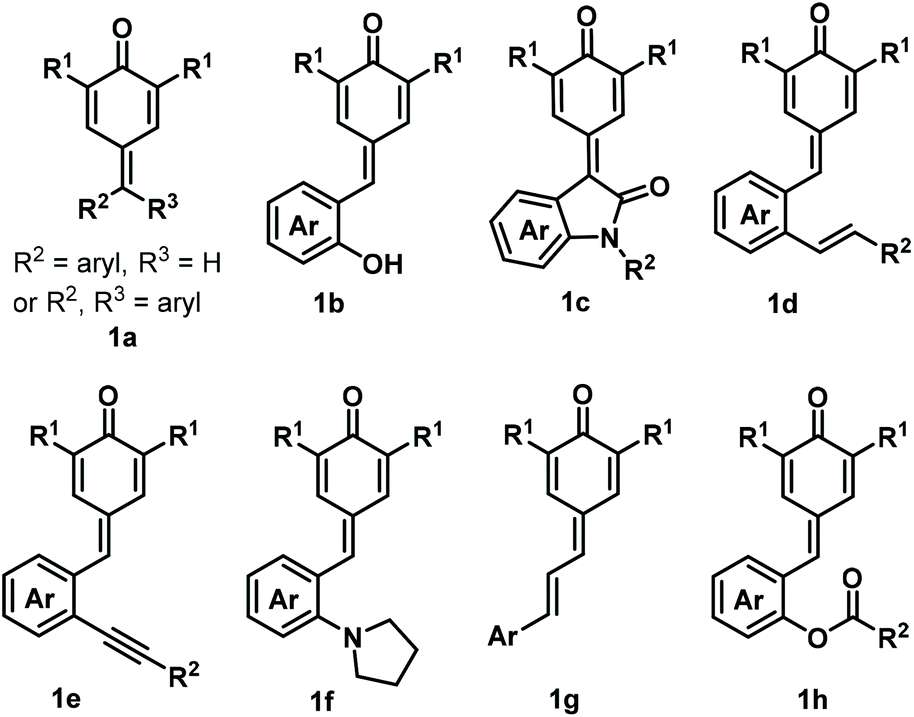
para-Quinone methides (p-QMs; Fig. 1, type A) are ubiquitous structural motifs present in a wide variety of biologically active natural products.1p-QMs also play numerous roles in biological processes, such as lignin biosynthesis,2 adrenergic receptors,3 enzyme inhibition,4 and DNA alkylation5 and cross-linking.6 Owing to their unique structural assembly comprising reactive carbonyl and olefinic moieties, p-QMs undergo resonance between neutral and zwitterionic structures (Fig. 1, type B) and display remarkable chemical reactivity as versatile acceptors for 1,6-addition reactions, including Michael addition and radical addition. As Michael acceptors, p-QMs have been successfully subjected to 1,6-addition or annulation reactions with carbon-, phosphorus-, and nitrogen-centered nucleophiles under various catalytic systems. Interestingly, p-QMs have also been exploited as highly reactive radical acceptors for capturing various carbon- and nitrogen-centered radicals via radical addition/cyclization or radical cross-coupling. These transformations provide numerous elegant and practical methods for the formation of a variety of carbon–carbon and carbon–heteroatom bonds, permitting the synthesis of important molecules that are difficult to obtain by other approaches. Since two pioneering works describing the synthesis of diarylmethines from p-QMs via 1,6-conjugate addition were independently reported by Fan7a and Jørgensen,7b several reaction modes of p-QMs have been thoroughly explored,8 including [2 + 1], [4 + 1], [3 + 2], [4 + 2], [4 + 3], and double annulations involving various p-QM substrates containing different functional groups and substitution patterns (Fig. 2, 1a–1h), resulting in new three-, five-, and six-membered and polycyclic systems. In the past years, Li, Bernardi, Enders, and Tortosa independently highlighted achievements in the asymmetric transformations of p-QMs.9 Undoubtedly, implementing catalytic approaches involving of p-QMs is one large research field, which includes both chiral and achiral aspects. Owing to the increasing attention that p-QM chemistry has received in recent years, especially with respect to non-asymmetric transformations, there is a strong demand to summarize these recent advances. Therefore, the purpose of this review is to provide our perspective on the strategic design, reaction discovery, and fundamental characteristics of the 1,6-conjugate addition and annulation reactions of p-QMs with an emphasis on achiral transformations in consideration with recent asymmetric advances.
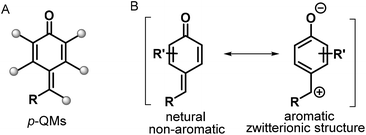 |
| | Fig. 1
p-QMs and their resonance structures. | |
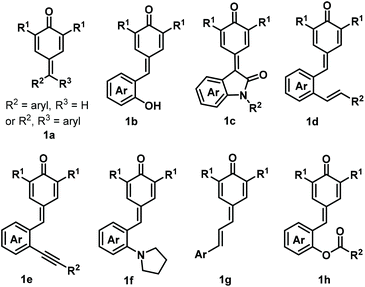 |
| | Fig. 2
p-QMs and their derivatives. | |
2. Chemistry of p-QMs
2.1. 1,6-Addition of nucleophiles to p-QMs
2.1.1. Catalyst-free 1,6-addition.
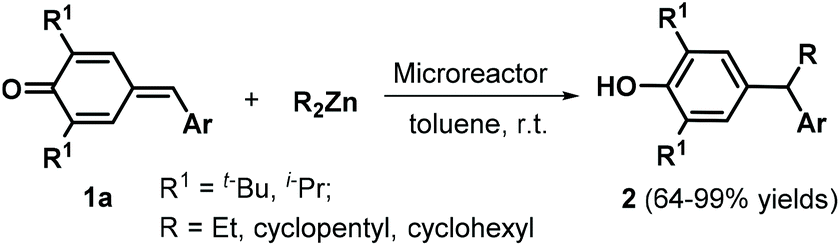
Environmentally benign and sustainable synthetic approaches to target compounds have attracted increasing attention in recent years, with considerable research effort devoted to the development of reaction conditions for green and sustainable chemistry, such as metal-free catalysis,10 catalyst-free synthesis, and the utilization of green solvents.11 In this context, Anand and co-workers reported a straightforward strategy for the 1,6-conjugate addition of dialkylzinc reagents to p-QMs 1a under continuous-flow conditions in a microreactor (Scheme 1).12 Upon injection of a toluene solution of p-QMs 1a and 2.0 equivalents of dialkylzinc reagent at a flow rate of 5 μL min−1 (residence time in the microreactor = 10 min), the reactants were smoothly transformed into unsymmetrical alkyl diarylmethanes 2 at room temperature. This protocol was generally applicable to a wide range of p-QMs 1a and dialkylzinc reagents without any catalyst or promoter.
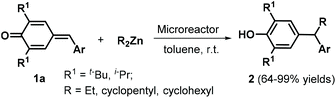 |
| | Scheme 1 1,6-Addition of dialkylzinc reagents to p-QMs. | |
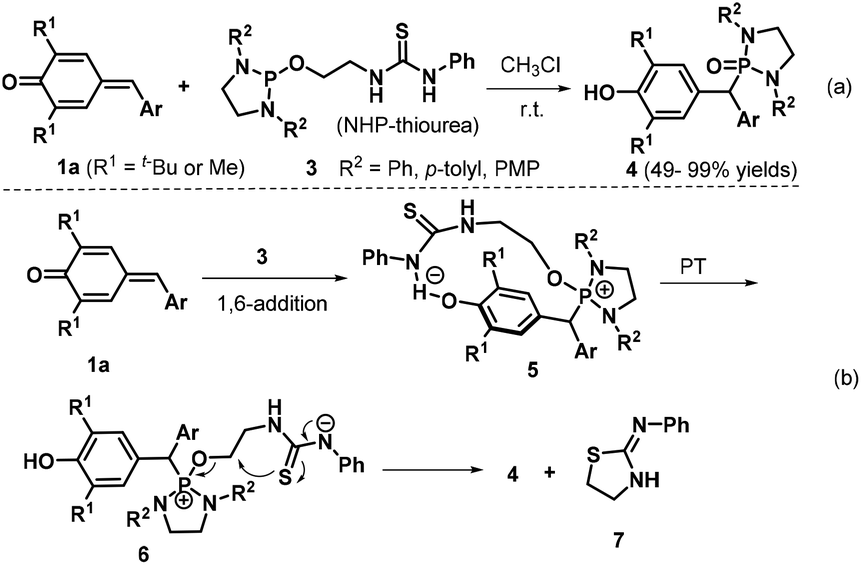
In 2017, Kang et al. synthesized diaryl diazaphosphonates 4via 1,6-hydrophosphonylation of p-QMs 1a with N-heterocyclic phosphine (NHP)-thioureas 3 under catalyst- and additive-free conditions.13 This transformation proceeded smoothly in CHCl3 at room temperature over 16 h to afford 20 examples of the desired product (Scheme 2a). When the 2,6-diisopropylphenyl-substituted NHP-thiourea 3 was employed under the optimized reaction conditions, the reaction did not proceed. On the basis of control experiments, the authors proposed a plausible reaction pathway (Scheme 2b). The 1,6-conjugate addition between the p-QM 1a and the bifunctional NHP-thiourea 3, which is activated by hydrogen bond formation with the thiourea Brønsted acid, generates the diazaphosphonium intermediate 5. Subsequent proton transfer (PT) affords the anionic thiourea intermediate 6, which undergoes intramolecular nucleophilic substitution to furnish the final product 4 alongside thiazolidine 7.
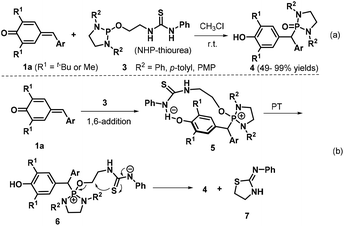 |
| | Scheme 2 Catalyst-free synthesis of diaryl diazaphosphonates. | |
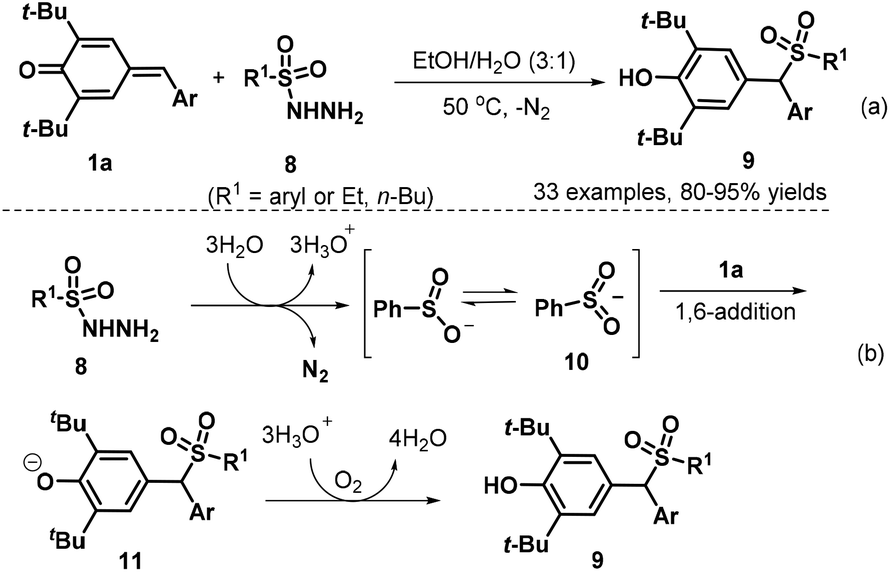
Subsequently, the Liu group developed a 1,6-conjugate addition/sulfonylation reaction of p-QMs 1a with sulfonyl hydrazides 8 to obtain diarylmethyl sulfones 9. The reaction proceeded efficiently at 50 °C under catalyst-free conditions using EtOH–H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as the reaction medium, thus providing a highly chemo- and regioselective synthesis of 33 examples of new diarylmethyl sulfones 9 in 80–95% yields (Scheme 3a). Various types of sulfonyl hydrazides 8 bearing either aryl or alkyl groups were well tolerated in this conjugate addition reaction, and the authors proposed a plausible mechanism for this catalyst-free transformation. In the first step, the sulfinyl anion is formed from sulfonyl hydrazide 8 in the presence of water with the release of N2. The resonance structure of the sulfinyl anion, sulfur-centered anion 10, undergoes 1,6-conjugate addition to the p-QM 1a to afford intermediate 11. The desired product 9 is formed upon proton transfer from hydronium ions to the intermediate 11 (Scheme 3b).14 The similar unsymmetrical gem-diarylmethyl sulfones 9 could also be synthesized through HOAc-promoted 1,6-conjugate sulfonylation of p-QMs 1a with arylsulfinate sodium reagents.15
:1 v/v) as the reaction medium, thus providing a highly chemo- and regioselective synthesis of 33 examples of new diarylmethyl sulfones 9 in 80–95% yields (Scheme 3a). Various types of sulfonyl hydrazides 8 bearing either aryl or alkyl groups were well tolerated in this conjugate addition reaction, and the authors proposed a plausible mechanism for this catalyst-free transformation. In the first step, the sulfinyl anion is formed from sulfonyl hydrazide 8 in the presence of water with the release of N2. The resonance structure of the sulfinyl anion, sulfur-centered anion 10, undergoes 1,6-conjugate addition to the p-QM 1a to afford intermediate 11. The desired product 9 is formed upon proton transfer from hydronium ions to the intermediate 11 (Scheme 3b).14 The similar unsymmetrical gem-diarylmethyl sulfones 9 could also be synthesized through HOAc-promoted 1,6-conjugate sulfonylation of p-QMs 1a with arylsulfinate sodium reagents.15
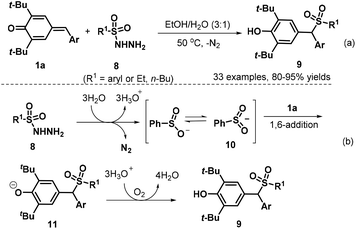 |
| | Scheme 3 1,6-Addition of sulfonyl hydrazides to p-QMs. | |
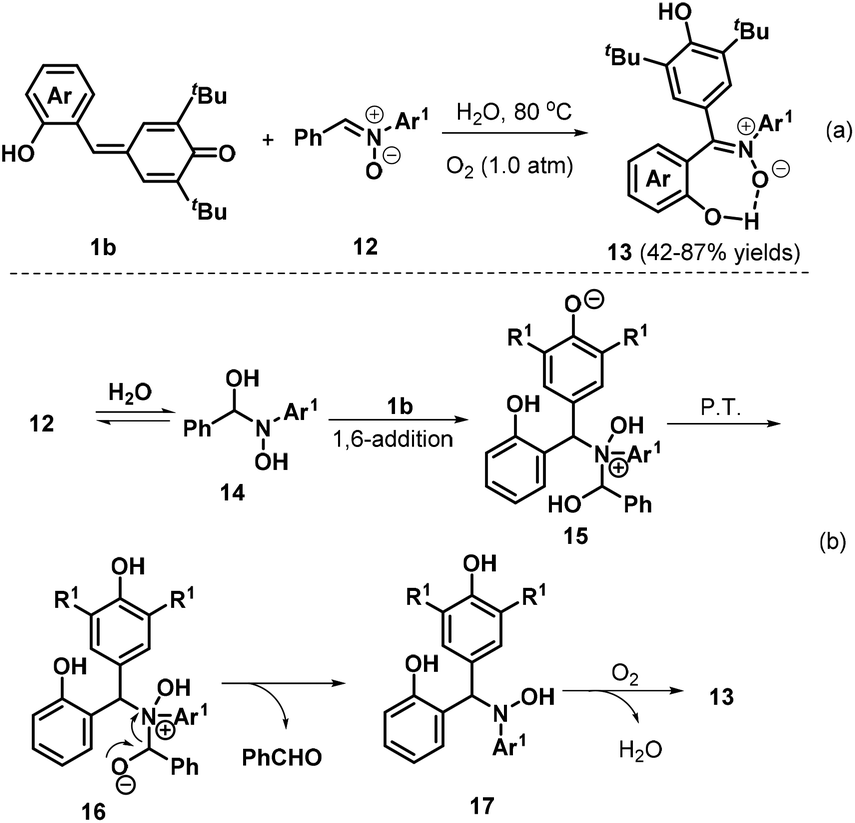
Very recently, Tu, Jiang, and co-workers described a new and environmentally benign protocol for effectively synthesizing triarylated (Z)-nitrones 13 with high stereoselectivity and generally excellent yields via a metal- and catalyst-free H2O-mediated 1,3-dipolar transfer reaction of o-hydroxyphenyl-substituted p-QM 1b and diarylated nitrones 12 (Scheme 4a).16 Interestingly, in this reaction, H2O played a dual role as both a mediator and a green solvent, with oxygen serving as a green oxidant. The reaction was tolerant to both electron-donating and electron-withdrawing substituents at various positions of the nitrone phenyl ring. The authors proposed a reasonable mechanism for this reaction, as depicted in Scheme 4b. In the first step, nucleophilic addition of H2O to nitrone 12 generates an unstable α-amino alcohol intermediate 14 in a reversible process. Fast 1,6-addition of the nitrogen atom of 14 to p-QM 1b affords intermediate 15, followed by proton transfer and the release of benzaldehyde to furnish intermediate 17. Finally, intermediate 17 is converted to product 13via oxidation in the presence of O2.
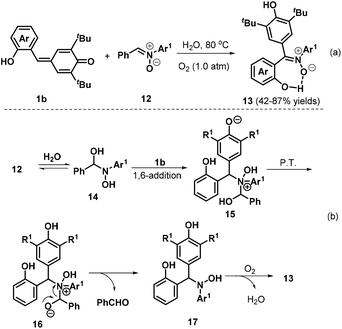 |
| | Scheme 4 H2O-Mediated reaction of p-QMs with nitrones. | |
2.1.2. Base-promoted 1,6-addition.
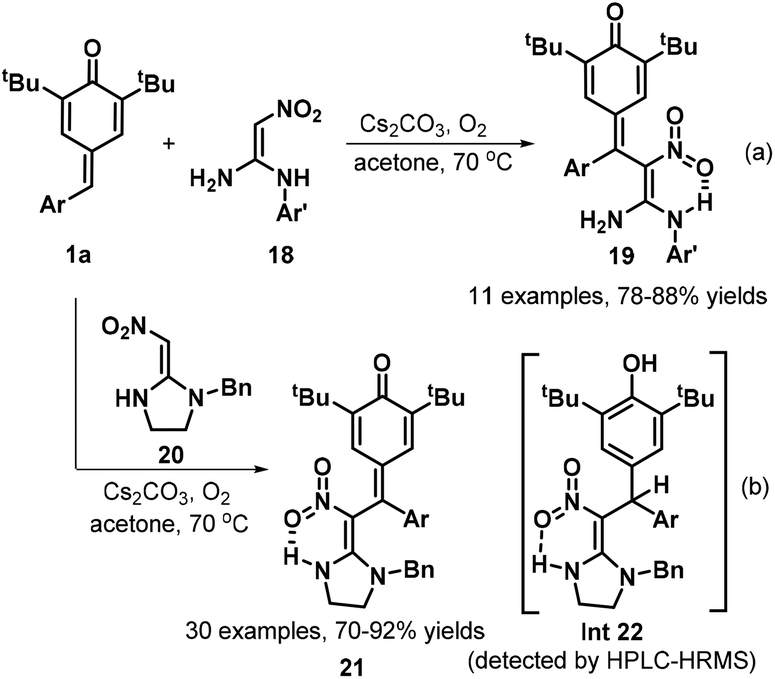
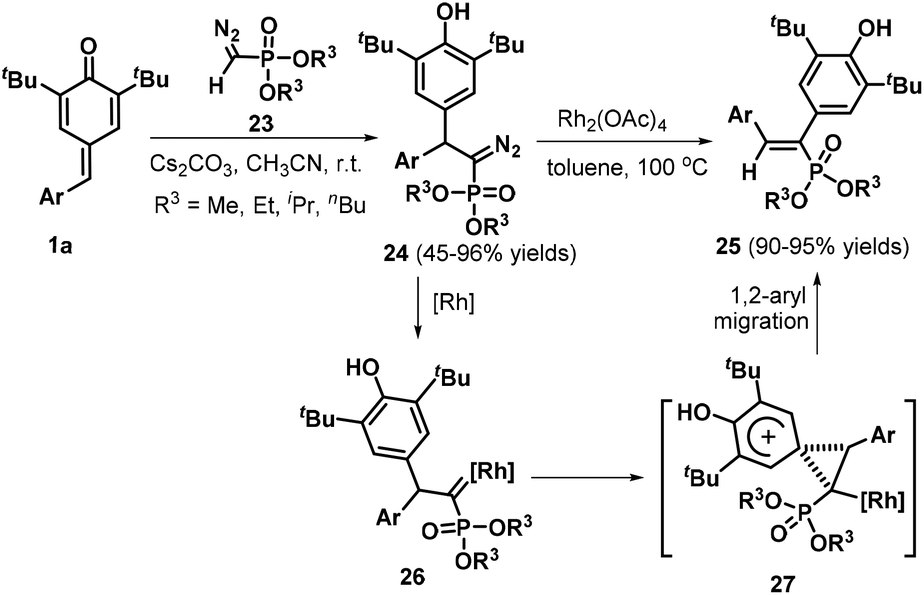
Owing to the intrinsic electrophilic nature and aromatization driving force of the cyclohexadiene moiety, p-QMs act as strongly electrophilic acceptors in 1,6-conjugate addition reactions, especially in the presence of certain bases.17 Guided by this principle, Yan et al. developed a concise and practical strategy for the synthesis of a variety of QM-containing β-nitroenamine derivatives 19via a Cs2CO3-promoted 1,6-conjugate addition of N-benzyl β-nitroenamines 18 with p-QMs 1a in acetone under an air atmosphere (Scheme 5a).18 Evaluation of the reaction scope using 1,1-enediamine 20 as the enamine component revealed that the reaction worked well, providing the corresponding cyclic β-nitroenamines 21 containing a QM moiety. Control experiments demonstrated that the reaction under the standard conditions afforded intermediate 22, which was detected by HPLC-HRMS (Scheme 5b). On the basis of the experimental results, the authors proposed a plausible mechanism comprising base-promoted 1,6-addition, imine–enamine tautomerization, and oxidation by atmospheric oxygen. In another impressive work, Mohanan and co-workers documented the 1,6-conjugate addition of Seyferth–Gilbert reagent (SGR) 23 to p-QMs 1a (Scheme 6). In the presence of Cs2CO3, SGR 23 reacted with p-QMs 1a in CH3CN, furnishing diarylmethylated diazomethylphosphonates 24 in yields ranging from 45% to 96%. This reaction featured a broad substrate scope and high functional group tolerance.19 To broaden the synthetic utility of these products, the authors also subjected the conjugate adducts 24 to treatment with rhodium acetate, which provided the corresponding 1,2-diaryl alkenylphosphonates 25 in good yields via 1,2-aryl migration of the rhodium carbenoid intermediate 27.
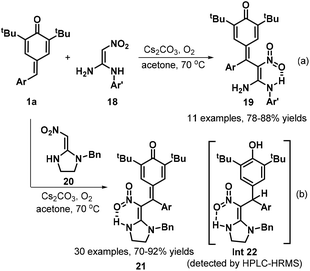 |
| | Scheme 5 Synthesis of β-nitroenamine derivatives. | |
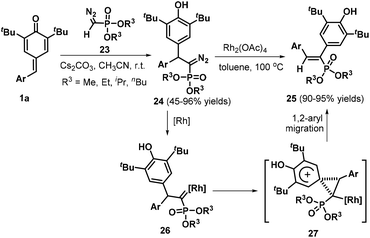 |
| | Scheme 6 1,6-Addition of SGR to p-QMs. | |
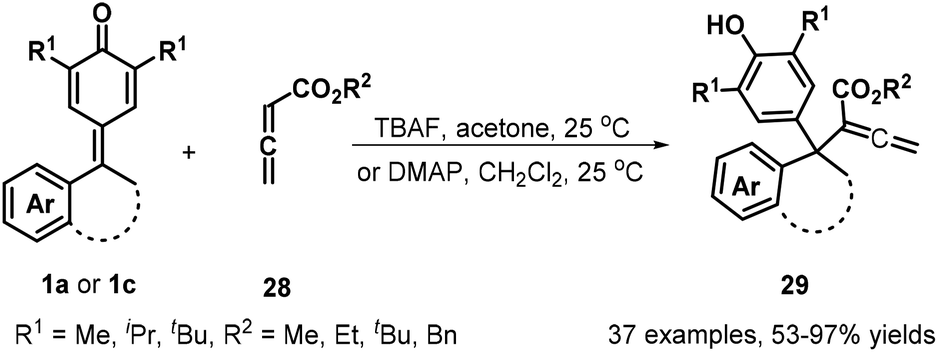
The same group subsequently demonstrated that the use of allenic esters 28 in combination with tetrabutylammonium fluoride (TBAF) activation enabled 1,6-conjugate addition for the synthesis of 37 structurally relevant α-diarylmethylated allenic esters 29 in 53–97% yields (Scheme 7). Furthermore, this transformation could be extended to isatin-derived p-QMs 1c, delivering allenic esters containing 3,3-disubstituted oxindoles 29.20a At the same time, Chandra et al. described a Rauhut–Currier reaction between 1a and 28 for forming similar allenoates 29 in high to excellent yields by using DMAP as a nucleophilic catalyst (Scheme 7).20b
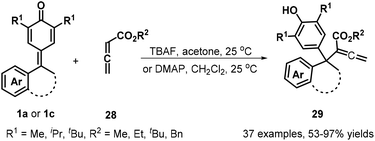 |
| | Scheme 7 Synthesis of α-diarylmethylated allenic esters. | |
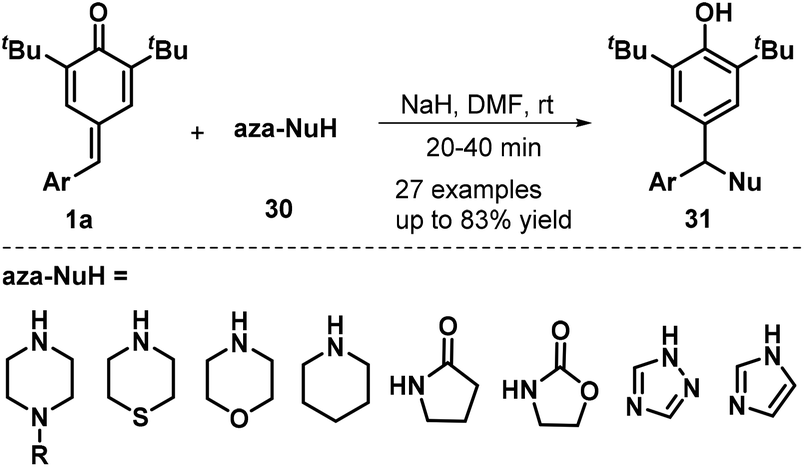
Heterocyclic amines and amides were also found to serve as N-nucleophiles 30 for the synthesis of nitrogen-containing trisubstituted methanes 31 in moderate to good yields via NaH-mediated 1,6-aza-Michael addition to p-QMs 1a (Scheme 8). A wide range of N-nucleophiles 30, including N-substituted piperazines, thiomorpholine, morpholine, piperidine, pyrrolidin-2-one, oxazolidin-2-one, 1H-1,2,4-triazole, and 1H-imidazole, were compatible with this base-promoted reaction.21
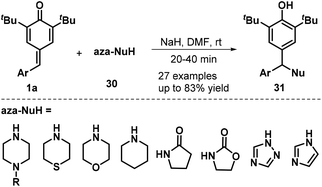 |
| | Scheme 8 1,6-Aza-Michael addition to p-QMs. | |
2.1.3. Lewis acid-promoted 1,6-addition.
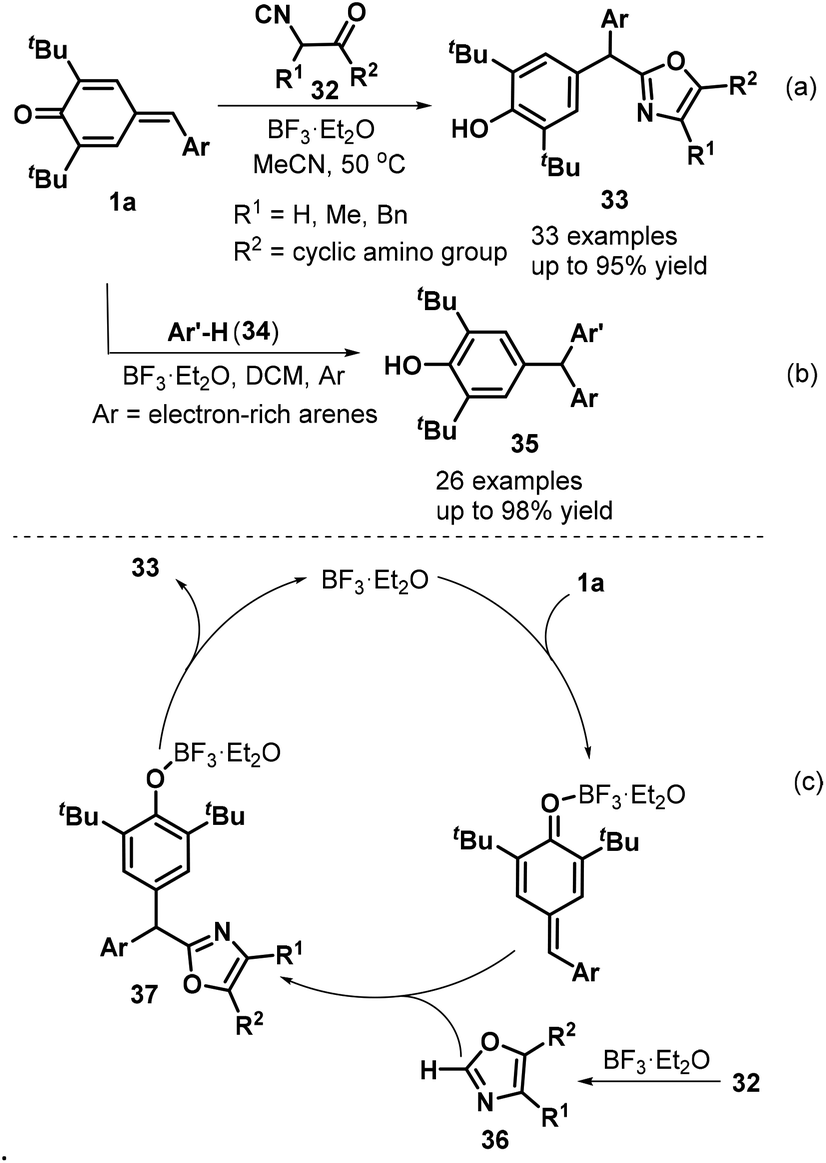
Lewis acid-promoted 1,6-addition of various nucleophilic and electrophilic reagents to p-QMs 1 has become an important research area that provides an efficient strategy for the construction of polysubstituted arylmethanes. Interestingly, Lin, Yao, and co-workers described an elegant BF3·Et2O-catalyzed 1,6-conjugate addition involving p-QMs 1a and α-isocyanoacetamides 32 that successfully provided access to 33 examples of oxazole-substituted arylmethanes 33 in 46–95% yields. However, α-isocyanoacetates were ineffective reaction partners in this transformation (Scheme 9a). During the reaction process, BF3·Et2O catalyzed the cyclization of the α-isocyanoacetamide to generate an oxazole 36, which underwent 1,6-conjugate addition to the p-QMs 1a to give the target products 33 (Scheme 9c). The authors also used electron-rich aromatic compounds 34 to expand the synthetic utility. This reaction proceeded readily under mild conditions to afford diverse unsymmetrical triarylmethanes 35 in 41–98% yields (Scheme 9b).22
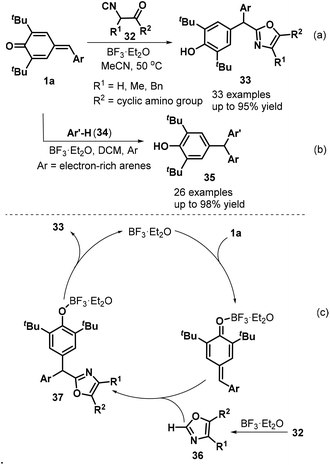 |
| | Scheme 9 BF3·Et2O-Catalyzed synthesis of arylmethanes. | |
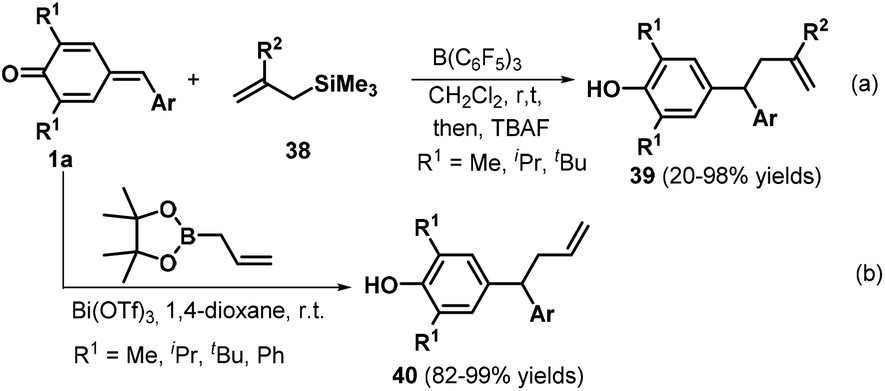
Subsequently, the Anand group reported an interesting transformation of allyltrimethylsilanes. These researchers discovered that the B(C6F5)3-catalyzed reaction of p-QMs 1a and allyltrimethylsilanes 38 led to the rapid formation of the expected unsymmetrical allyl diarylmethanes 39 with moderate to excellent yields via 1,6-conjugate addition (Scheme 10a).23 Various aryl-substituted allyltrimethylsilanes worked well in this reaction, whereas a quinoline-based analogue exhibited lower reactivity with a yield of only 20%. Unfortunately, the p-QM derived from thiophene-2-carboxaldehyde resulted in the formation of a complex mixture under the reaction conditions. In this protocol, B(C6F5)3 is employed as a Lewis acid catalyst to mediate the 1,6-conjugate allylation of p-QMs 1a. Following this study, Li and co-workers reported the bismuth-catalyzed 1,6-allylation addition of p-QMs 1a with 2-allyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, affording 25 examples of the allylation product 40. Optimization of the conditions revealed that the reaction proceeded smoothly in the presence of 0.5–5 mol% Bi(OTf)3 in 1,4-dioxane at room temperature (Scheme 10b).24
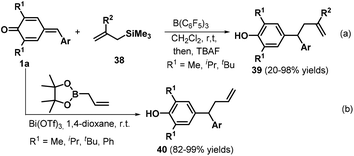 |
| | Scheme 10 1,6-Addition to form allyl diarylmethanes. | |
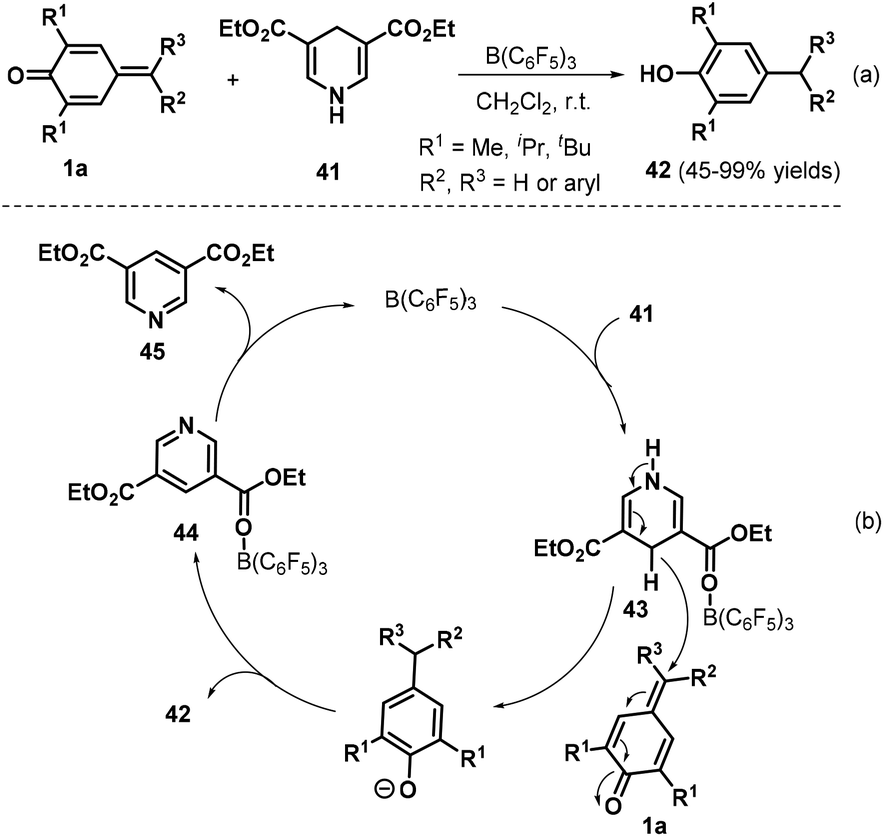
Exchanging allyltrimethylsilanes for Hantzsch ester 41, Anand and co-workers presented a B(C6F5)3-catalyzed reduction of p-QMs 1a, leading to 31 examples of unsymmetrical diaryl- and triarylmethanes 42 in 45–99% yields (Scheme 11a).25 Notably, this protocol was applied to the synthesis of beclobrate, a potent cholesterol- and triglyceride-lowering drug that is used for the treatment of hyperlipidemia. Mechanistically, B(C6F5)3 activates the Hantzsch ester 41 to afford the corresponding Lewis acid–base complex 43, which mediates hydride transfer to p-QM 1a, and subsequent protonation yields the desired product 42 along with complex 44. The complex 44 releases the pyridine byproduct 45 to regenerate the catalyst (Scheme 11b).
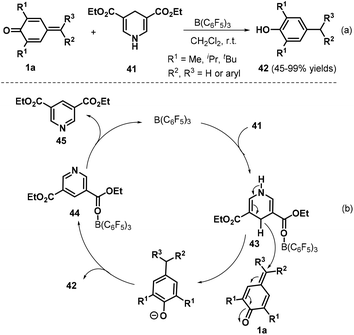 |
| | Scheme 11 B(C6F5)3-Catalyzed reduction of p-QMs. | |
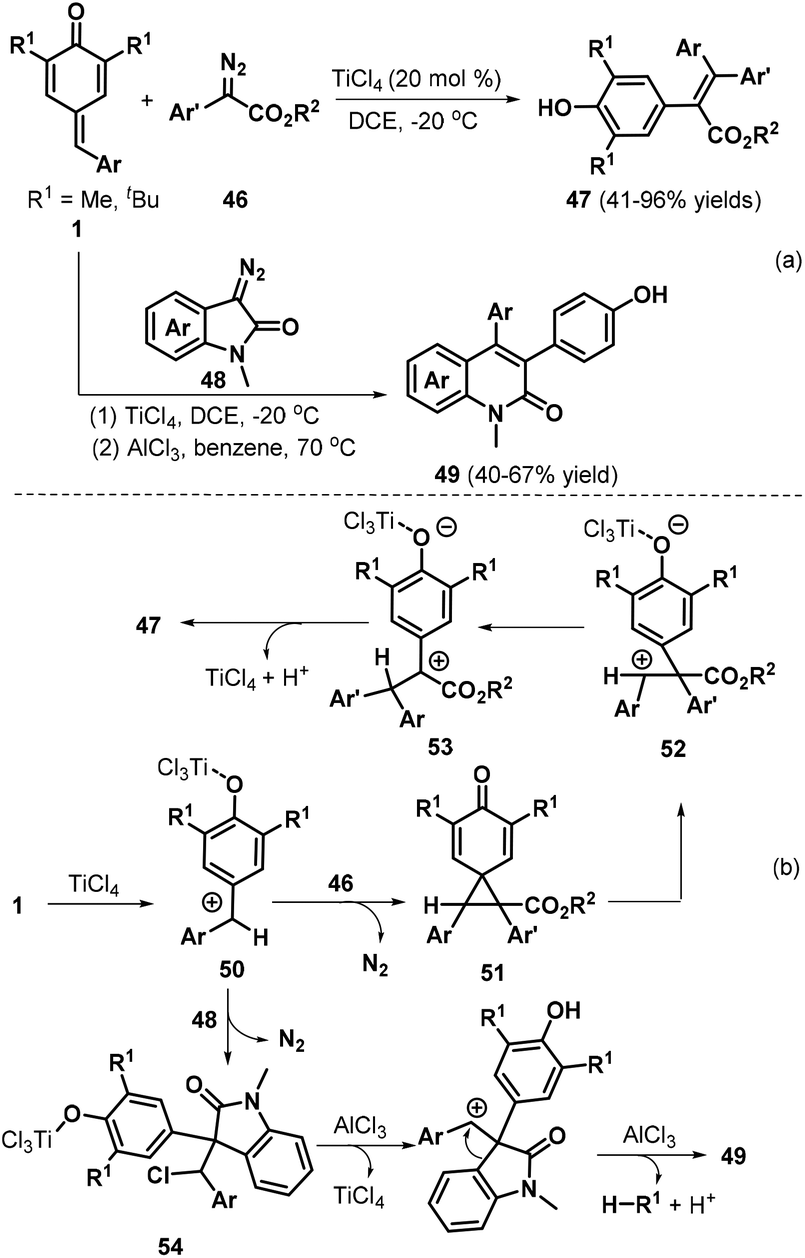
Cui and co-workers reported a remarkable insertion reaction between diazo compounds and p-QMs 1a for the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds.26 The TiCl4-catalyzed reaction of p-QMs 1a with α-aryl diazoesters 46 at low temperature enabled aryl group migration to afford a wide array of functionalized tetrasubstituted alkenes 47 (Scheme 12a). Next, the authors reacted p-QMs 1a with diazooxindole 48. Careful screening of the reaction conditions revealed that the two-step reaction resulted in quinolinone derivatives 49, in which 1.0 equivalents of TiCl4 were required as the promoter in the first step and AlCl3 promoted the aryl migration and de-tert-butylation in the second step. Furthermore, the authors combined these two steps in a one-pot reaction for quinolinone synthesis. Moreover, the authors also presented possible mechanisms for the formation of these products (Scheme 12b). The p-QM is first activated by TiCl4 to generate cationic intermediate 50, which is trapped by α-aryl diazoester 46 to afford cyclopropanation intermediate 51 with the release of nitrogen. Subsequent cyclopropane fragmentation delivers cationic intermediate 52 with a TiCl4-activated quinone moiety, which undergoes rearrangement to form cationic intermediate 53. Next, intermediate 53 is transformed into tetrasubstituted alkene 47via hydrogen elimination with the regeneration of TiCl4. In contrast, when intermediate 50 is generated in situ from the diazooxindole, direct chloride addition to the benzyl cation affords intermediate 54, which undergoes successive AlCl3-catalyzed dechlorination, ring expansion, and de-tert-butylation to yield quinolinone 49.
C double bonds.26 The TiCl4-catalyzed reaction of p-QMs 1a with α-aryl diazoesters 46 at low temperature enabled aryl group migration to afford a wide array of functionalized tetrasubstituted alkenes 47 (Scheme 12a). Next, the authors reacted p-QMs 1a with diazooxindole 48. Careful screening of the reaction conditions revealed that the two-step reaction resulted in quinolinone derivatives 49, in which 1.0 equivalents of TiCl4 were required as the promoter in the first step and AlCl3 promoted the aryl migration and de-tert-butylation in the second step. Furthermore, the authors combined these two steps in a one-pot reaction for quinolinone synthesis. Moreover, the authors also presented possible mechanisms for the formation of these products (Scheme 12b). The p-QM is first activated by TiCl4 to generate cationic intermediate 50, which is trapped by α-aryl diazoester 46 to afford cyclopropanation intermediate 51 with the release of nitrogen. Subsequent cyclopropane fragmentation delivers cationic intermediate 52 with a TiCl4-activated quinone moiety, which undergoes rearrangement to form cationic intermediate 53. Next, intermediate 53 is transformed into tetrasubstituted alkene 47via hydrogen elimination with the regeneration of TiCl4. In contrast, when intermediate 50 is generated in situ from the diazooxindole, direct chloride addition to the benzyl cation affords intermediate 54, which undergoes successive AlCl3-catalyzed dechlorination, ring expansion, and de-tert-butylation to yield quinolinone 49.
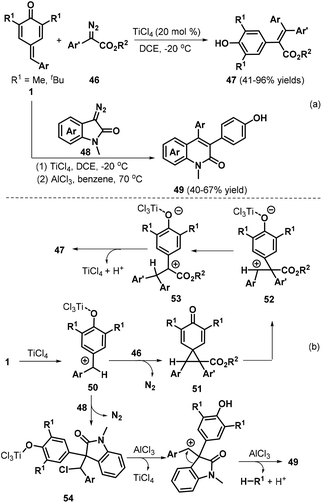 |
| | Scheme 12 TiCl4-Catalyzed reaction of p-QMs. | |
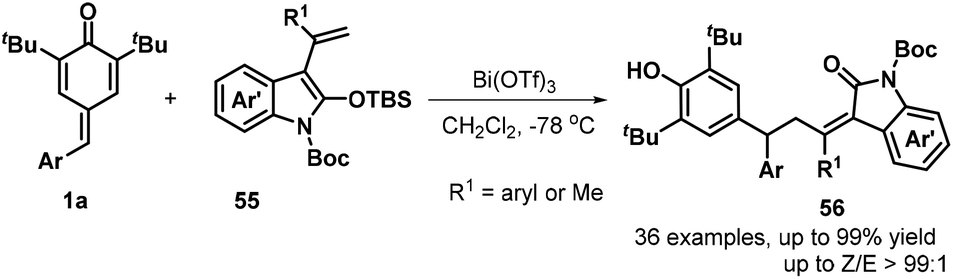
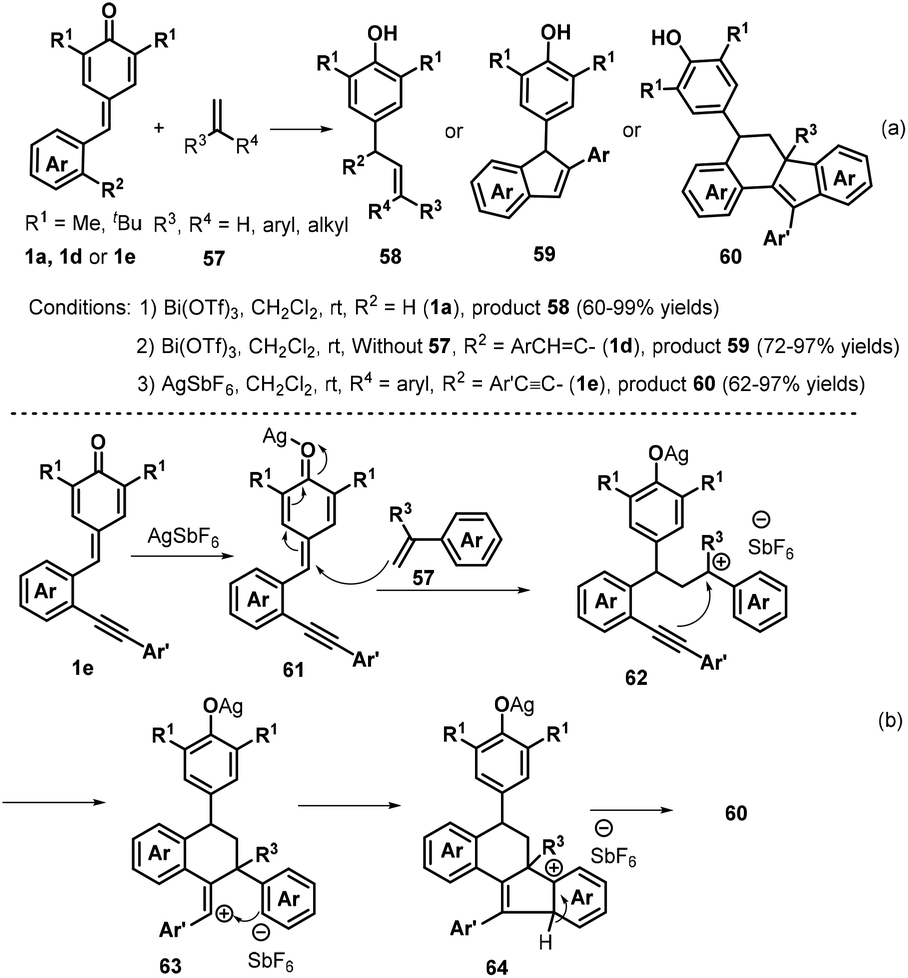
In subsequent contributions, a bismuth-catalyzed vinylogous nucleophilic 1,6-conjugate addition reaction of p-QMs 1a and 3-propenyl-2-silyloxyindoles 55 generated substituted α-alkylidene-δ-diaryl-2-oxindoles 56 (Scheme 13).27 Optimization of the reaction conditions revealed that the use of 10 mol% Bi(OTf)3 in dichloromethane (DCM) at −78 °C for 12 h resulted in the best product yield. A wide range of α-alkylidene-δ-diaryl-2-oxindoles 56 were obtained in 55–99% yields and 94:6–98:2 Z/E ratio under the optimized reaction conditions. Subsequently, Anand and co-workers reported a similar bismuth-catalyzed intermolecular 1,6-hydroolefination of p-QMs 1a with olefins, which provided access to 21 examples of vinyl diarylmethanes 58 in up to 99% yield (Scheme 14a). The bismuth-catalyzed reaction also tolerated the intramolecular version to afford six examples of arylated indenes 59 in up to 97% yield. Furthermore, in the presence of AgSbF6 as catalyst, 2-alkenylated p-QMs 1e and 1,1-diphenylethylene 57 underwent 1,6-conjugate electrophilic addition and alkene–alkyne cyclization to furnish a series of substituted dihydrobenzo[a]fluorene derivatives 60 in good yields and excellent diastereoselectivity.28 This transformation was also exploited for the total synthesis of the resveratrol-derived natural product (±)-isopaucifloral F. The authors proposed a plausible mechanism for products 60 (Scheme 14b). The silver salt initially activates the carbonyl group of the p-QM 1e and 1,6-addition of olefin 57 then generates reactive carbocation intermediate 62, followed by electrophilic cyclization to form 63, Friedel–Crafts-type cyclization to provide aryl carbocation intermediate 64, and finally aromatization to generate the final product 60.
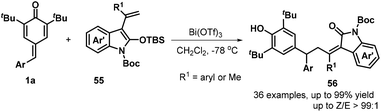 |
| | Scheme 13 Bismuth-catalyzed reaction of p-QMs with oxyindoles. | |
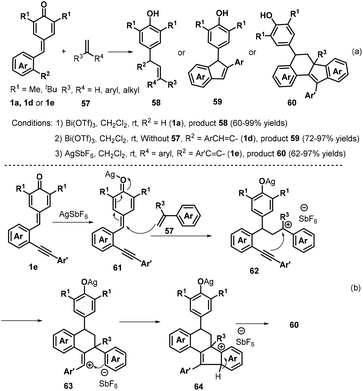 |
| | Scheme 14 1,6-Hydroolefination of p-QMs with olefins. | |
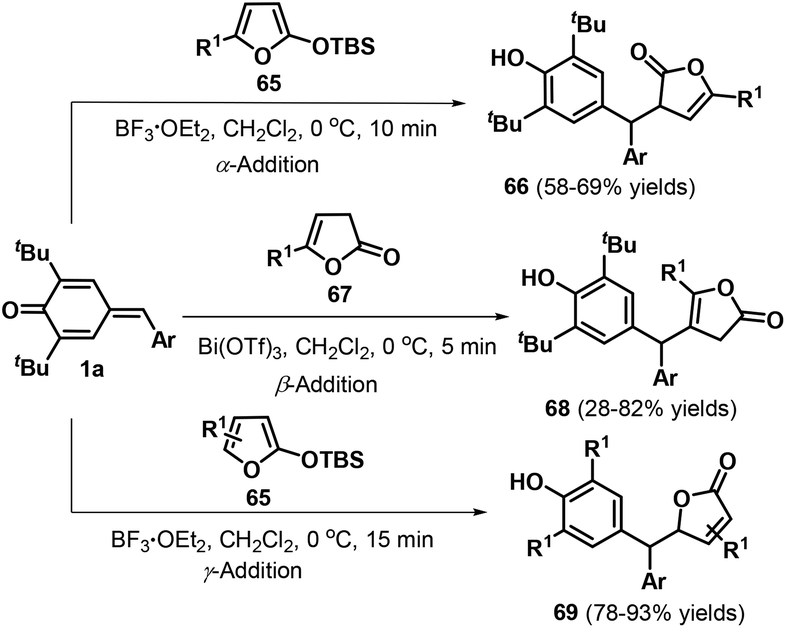
Kumar and co-workers reported another impressive example of the selective 1,6-conjugate addition of p-QMs 1a by switching the nucleophilic reactivity of the α, β, and γ positions of butenolides 65 and lactone 67 (Scheme 15).29 For example, α-addition could be achieved by reacting p-QMs 1a and butenolides 65 (R1 ≠ H) in the presence of BF3·Et2O, whereas the use of lactone 67 in the presence of Bi(OTf)3 afforded the β-adducts 68. Butenolides 65 lacking a substituent in the C5 position underwent γ-addition mediated by BF3·Et2O to yield 5-substituted furan-2(5H)-ones 69. All of these reactions rapidly reached completion in approximately 15 min and featured wide substrate scope and good functional group tolerance under mild conditions.
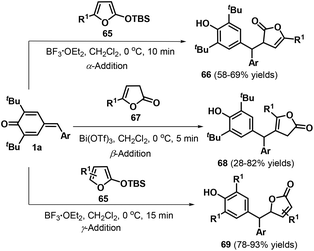 |
| | Scheme 15 Selective 1,6-addition of p-QMs and butenolides. | |
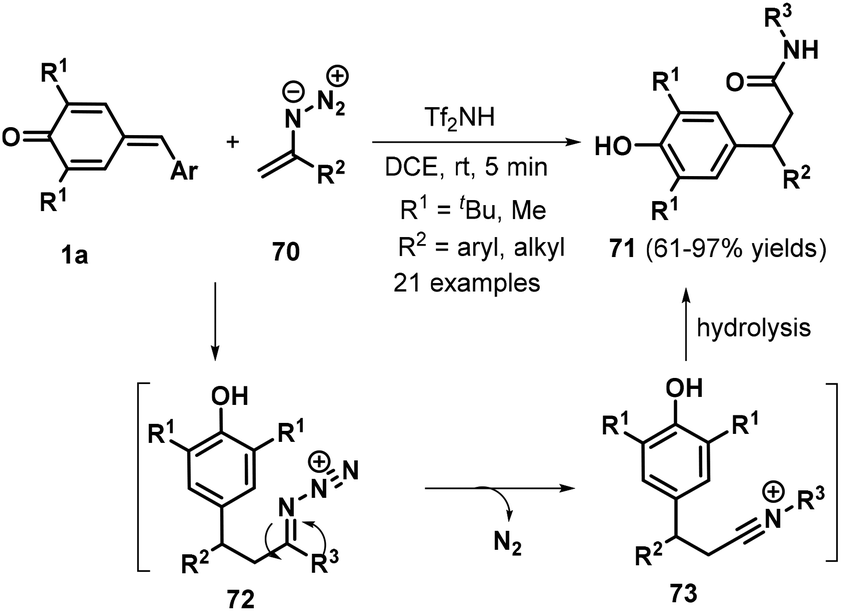
In 2017, the same researchers reported a Tf2NH-catalyzed 1,6-conjugate addition/Schmidt-type rearrangement of p-QMs 1a with vinyl azides 70, providing access to 21 examples of β-bisarylated amides 71 in 61–97% yields (Scheme 16).30 Vinyl azides bearing either variously substituted aryl groups or alkyl functionalities both reacted smoothly to deliver the desired products in generally good yields. The authors also proposed a plausible mechanism for this reaction (Scheme 16). 1,6-Conjugate addition of vinyl azide 70 to proton-activated p-QM 1a generates intermediate 72, which undergoes Schmidt rearrangement to form the nitrilium ion intermediate 73. Subsequent hydrolysis affords the target product 71.
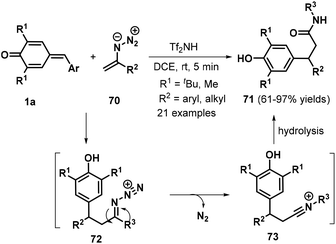 |
| | Scheme 16 Tf2NH-Catalyzed rearrangement of p-QMs. | |
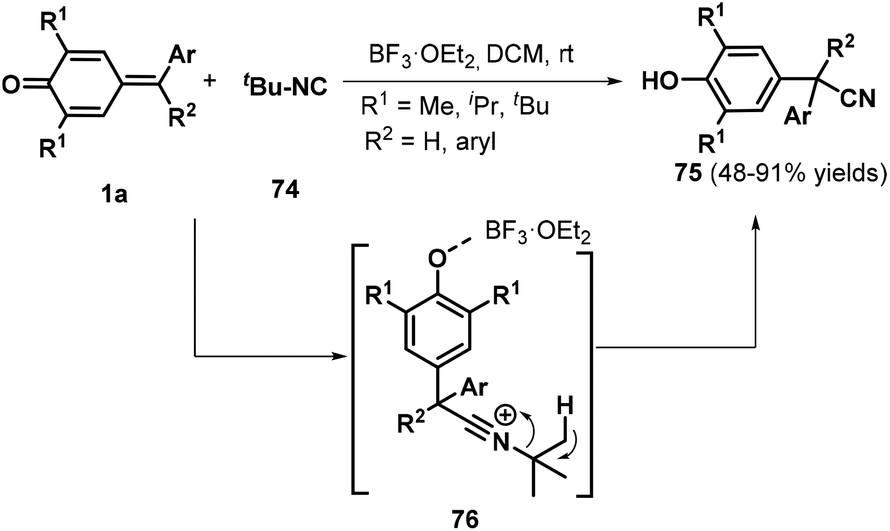
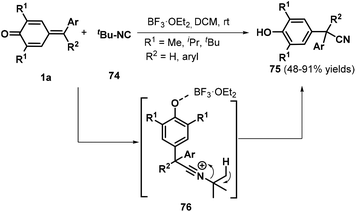
Shortly after this disclosure, Muthukrishnan et al. used tert-butyl isocyanide 74 as a cyanide source to accomplish the cyanation of p-QMs 1a, permitting the synthesis of α-diaryl and α-triaryl nitriles 75 in good to excellent yields under mild reaction conditions (Scheme 17).31 Notably, an alkyne-tethered p-QM was also compatible with the optimized reaction conditions. However, pyridyl-containing p-QMs did not react under the standard conditions. The reaction proceeded via the 1,6-addition of tert-butyl isocyanide 74 to BF3·Et2O-activated p-QM 1a, subsequent formation of key zwitterionic nitrilium ion intermediate 76, and the liberation of isobutylene.
 |
| | Scheme 17 1,6-Addition of tert-butyl isocyanide to p-QMs. | |
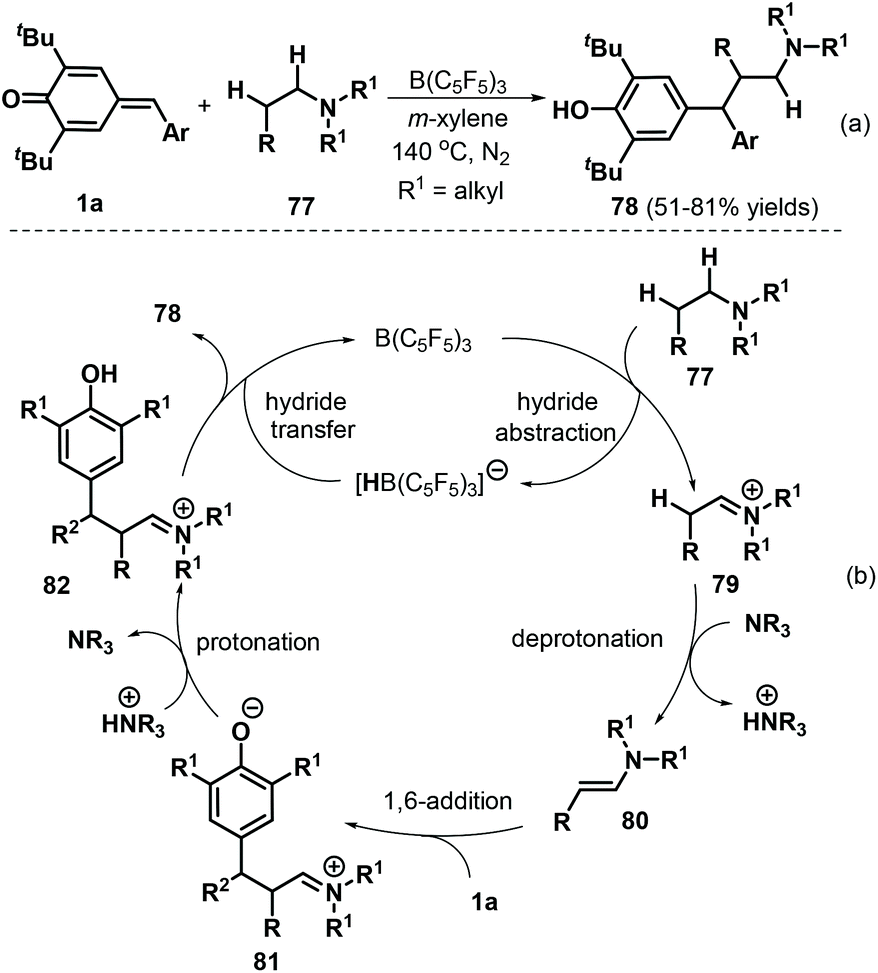
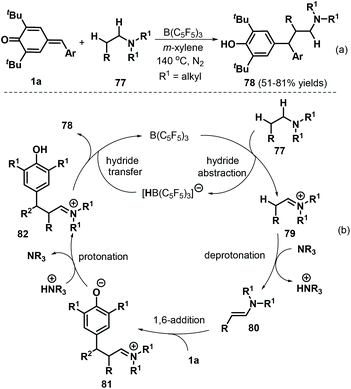
More recently, Ma and co-workers described an elegant intermolecular redox-neutral β-alkylation of acyclic tertiary amines 77 with p-QMs 1a under B(C6F5)3 catalysis, which delivered a diverse array of functionalized tertiary amines 78 in good yields of up to 81% (Scheme 18a). This procedure utilized commercially available substrates and catalyst and promoted direct functionalization of the C(sp3)–H bond at the β-position of acyclic tertiary amines 77via 1,6-conjugate addition to p-QMs 1a.32 Acyclic amines, including triethylamine, N,N-diethylbutylamine, and tripropylamine, smoothly underwent reaction under these conditions. In contrast, cyclic tertiary amines such as N-benzylpiperidine and N-mesitylpiperidine displayed lower conversions. This reaction starts with hydride abstraction from tertiary amine 77 by B(C6F5)3 to form iminium ion intermediate 79 and [HB(C6F5)3]− (Scheme 18b). Deprotonation of intermediate 79 then generates enamine intermediate 80, which reacts with p-QM 1avia 1,6-conjugate addition to produce functionalized iminium intermediate 81. Subsequent protonation of 81 generates strongly electrophilic iminium intermediate 82, which undergoes hydride transfer from [HB(C6F5)3]− to generate product 78 and complete the catalytic cycle.
 |
| | Scheme 18 1,6-Addition of acyclic tertiary amines to p-QMs. | |
2.1.4. Metal-catalyzed 1,6-addition.
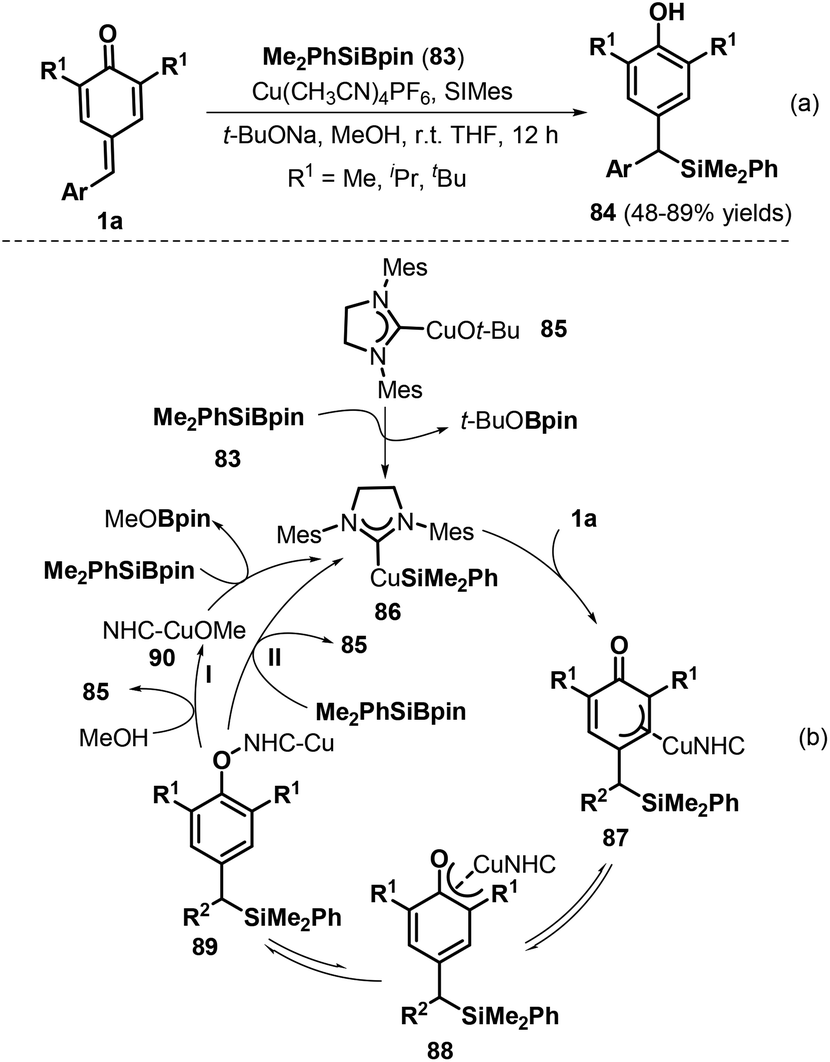
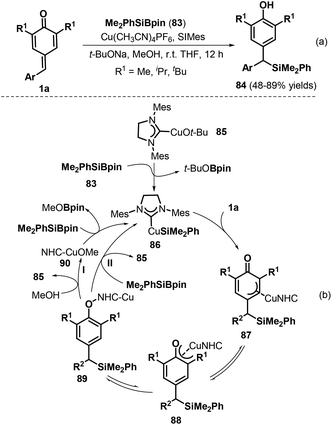
The construction of carbon–silicon bonds is an important research topic in synthetic organic chemistry owing to the fact that silicon-containing molecules are crucial precursors for producing bioactive natural products via various transformations.33 Specifically, copper(I)-catalyzed silylation reactions have emerged as a powerful strategy for carbon–silicon bond formation. In 2015, Tortosa and co-workers pioneered a copper(I)-catalyzed silylation of p-QMs 1a using silaborane reagent 83 under mild reaction conditions to obtain a series of dibenzylic silanes 84 with high yields (Scheme 19a).34 This transformation represented the first example of 1,6-conjugate addition to p-QMs 1a for carbon–silicon bond formation. In this process, the silyl–copper(I)-N-heterocyclic carbene (NHC) species 86, generated by the reaction of copper alkoxide 85 and the silaborane reagent, inserts into the exocyclic double bond of the p-QM 1a through cleavage of the copper–silicon bond to produce π-allylcopper intermediate 87, which isomerizes to copper phenoxide intermediate 88 (Scheme 19b). Subsequent transformation may occur via two reaction pathways. In pathway I, protonolysis provides product 84 in the presence of MeOH with release of NHC–CuOMe 90 to restart the catalytic cycle. In pathway II, intermediate 89 reacts directly with the silaborane to afford product 84 and silyl–copper species 86 with regeneration of the catalyst.
 |
| | Scheme 19 Copper(I)-catalyzed silylation toward dibenzylic silanes. | |
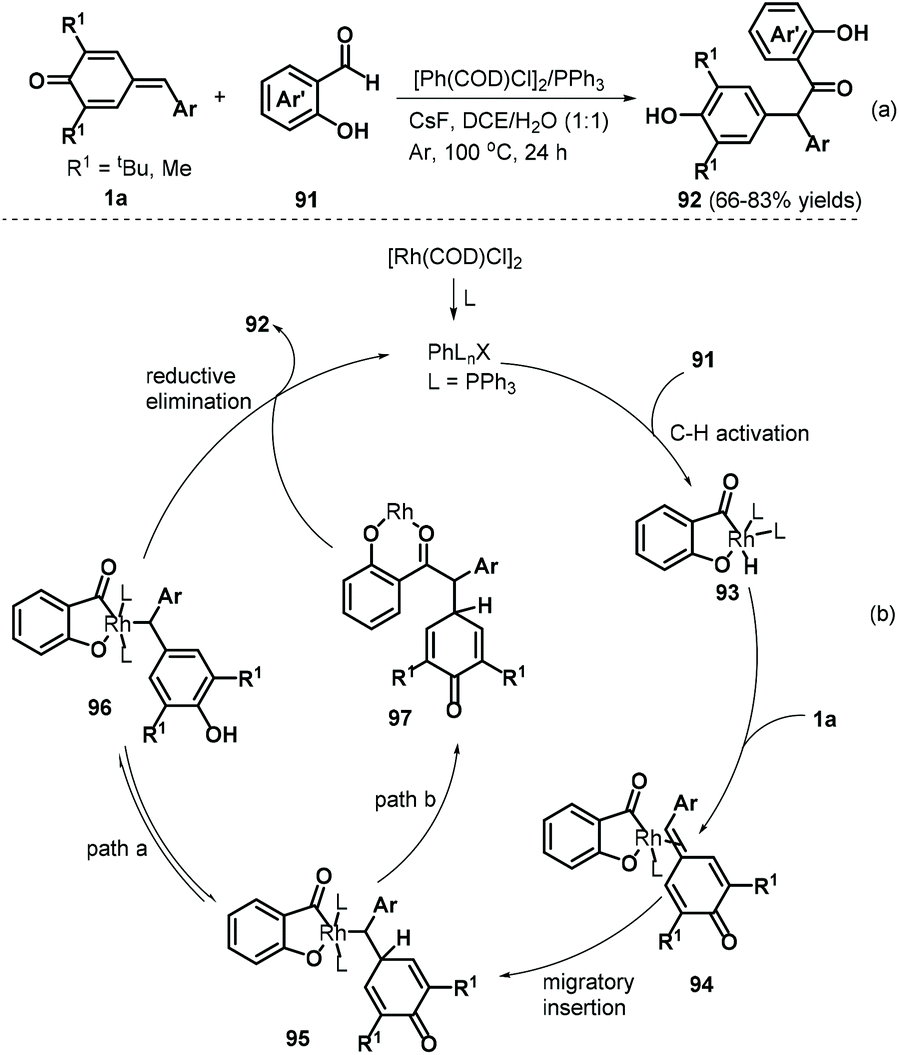
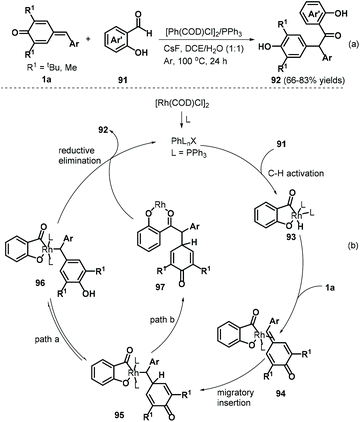
A follow-up work by the Lin group described an interesting rhodium-catalyzed C–H activation for 1,6-addition to p-QMs 1a.35 The authors reacted salicylaldehydes 91 and p-QMs 1a in a catalytic system comprising [Rh(COD)Cl]2 (2.5 mol%), PPh3 (5 mol%), and the additive CsF (1.0 equivalents) under an argon atmosphere, resulting in a series of α,α-diaryl-2-hydroxyacetophenones 92 (Scheme 20a). A series of salicylaldehydes bearing electron-donating and electron-withdrawing groups at the C5, C4, and C3 positions all afforded the corresponding products in 66–83% yields. However, both C6-methoxy-substituted salicylaldehyde and 3-(methylthio)butanal failed to participate in the reaction. This method displayed good yields, high chemoselectivity, good functional group tolerance, and gram-scale capacity. Mechanistically, this reaction involves hydroxy-assisted C–H activation of the aldehyde group by the rhodium catalyst to form rhodium species 93, which chelates the exocyclic double bond of p-QM 1a to afford metal–organic intermediate 94 (Scheme 20b). Subsequent migratory hydride insertion into the exo-methylene moiety delivers intermediate 95, which can undergo two reaction pathways. In pathway a, intermediate 96 is formed by isomerization of intermediate 95, leading to final product 92via reductive elimination with regeneration of the active rhodium complex for the subsequent catalytic cycle. In pathway b, intermediate 95 undergoes reductive elimination to generate intermediate 97, which then isomerizes to form product 92 with regeneration of the rhodium catalyst.
 |
| | Scheme 20 Rhodium-catalyzed C–H activation for 1,6-addition to p-QMs. | |
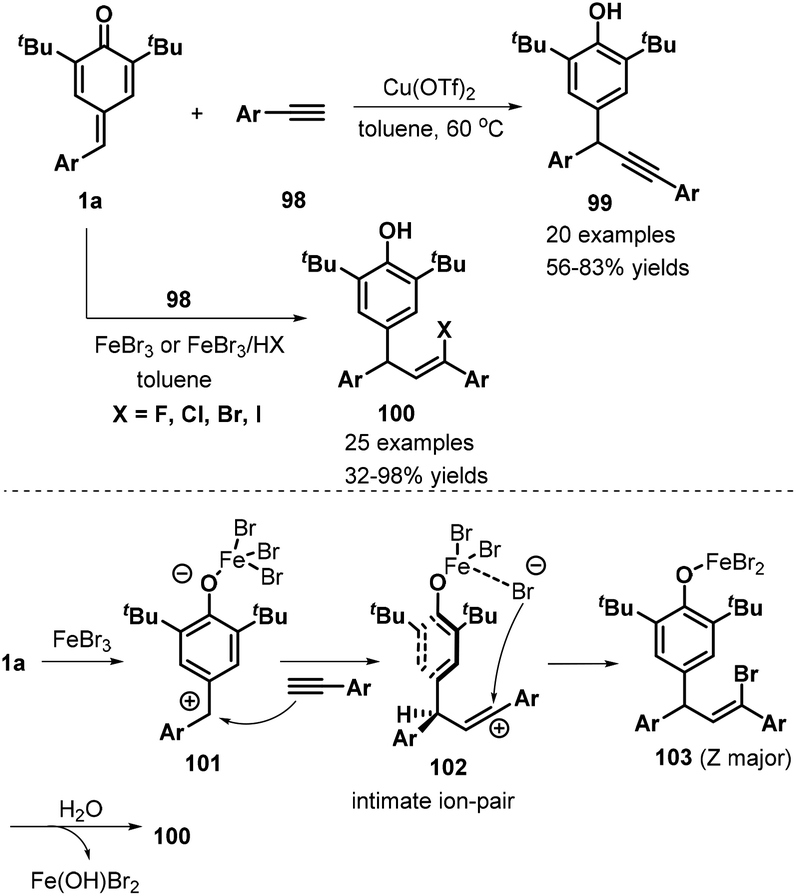
Very recently, Liu, Zhang, and co-workers developed a remarkable catalyst-controlled chemodivergent 1,6-addition of p-QMs 1a and terminal alkynes 98, which afforded a variety of structurally diverse alkynyl- or vinyl-substituted diarylmethanes 99 or 100 with good yields (Scheme 21).36 Heteroaryl-, ferrocenyl-, and enyne-derived terminal alkynes all served as suitable substrates in this transformation. The authors also synthesized a TMS-substituted p-QM, although the subsequent reaction did not proceed under the standard conditions. In this transformation, direct 1,6-addition of the p-QMs and terminal alkynes occurred under copper catalysis, whereas iron catalysis mediated a stereoselective three-component reaction between the p-QM, alkyne, and the halogen present in the iron salt or acid HX (fluoride, chloride, bromide, or iodide). Notably, the haloolefin products were obtained with Z/E ratios ranging from 1:1 to >20:1 via the iron-catalyzed 1,6-electrophilic addition and halogen addition.
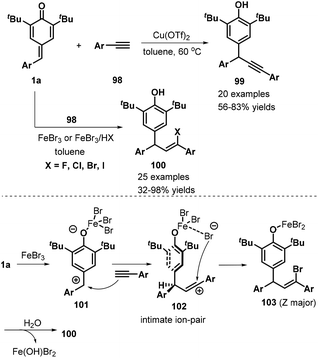 |
| | Scheme 21 Synthesis of alkynyl- and vinyl-substituted diarylmethanes. | |
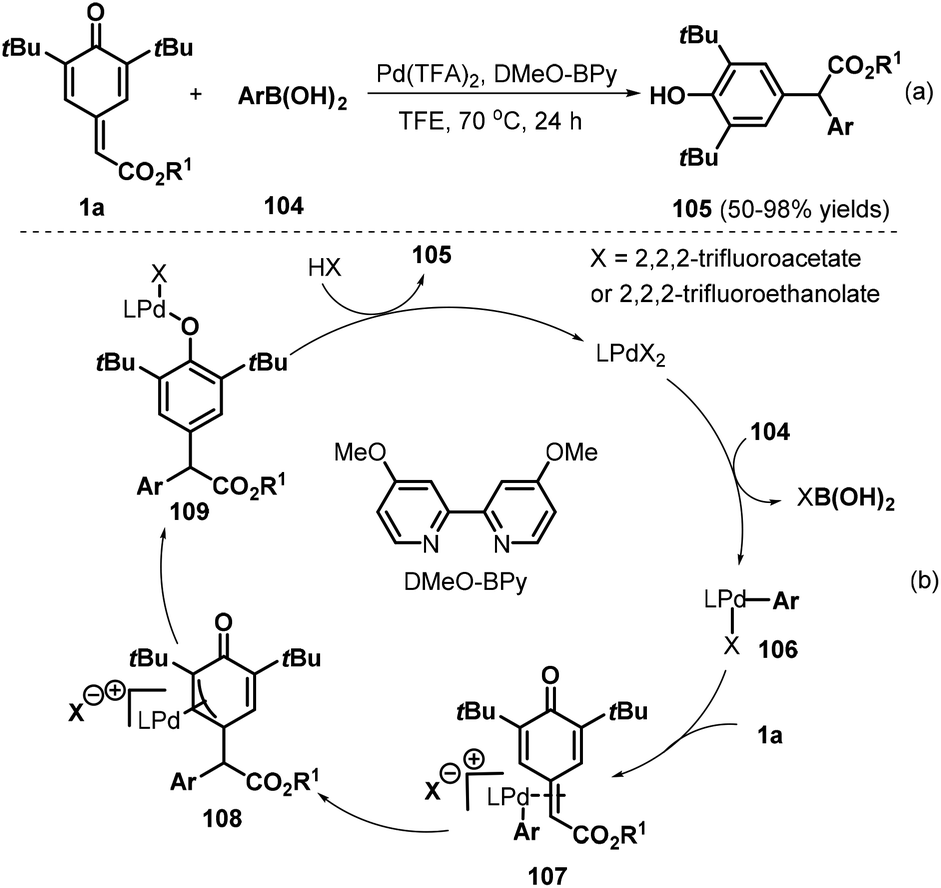
Besides copper and rhodium catalysts, palladium(II) catalysts have also proved suitable for 1,6-addition to p-QMs. The Zhang group developed an interesting palladium(II)-catalyzed 1,6-conjugate addition of arylboronic acids 104 to p-QMs 1a for the preparation of diarylacetates 105 by using 4,4′-dimethoxy-2,2′-bipyridine (DMeO-BPy) as the ligand and trifluoroethanol (TFE) as the reaction medium (Scheme 22a).37 Various ester-substituted p-QMs and arylboronic acids reacted smoothly under the optimal reaction conditions. However, arylboronic acids bearing an ester group at the para position exhibited lower reactivity. The reaction pathway involves transmetalation, ligand exchange, phenyl insertion into the double bond, tautomerization, and protonation (Scheme 22b).
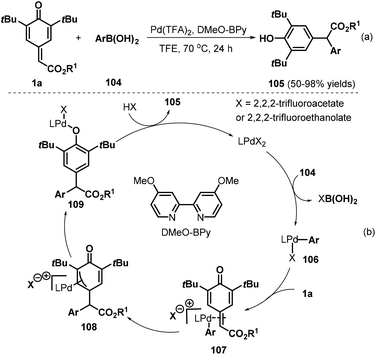 |
| | Scheme 22 Synthesis of diarylacetates. | |
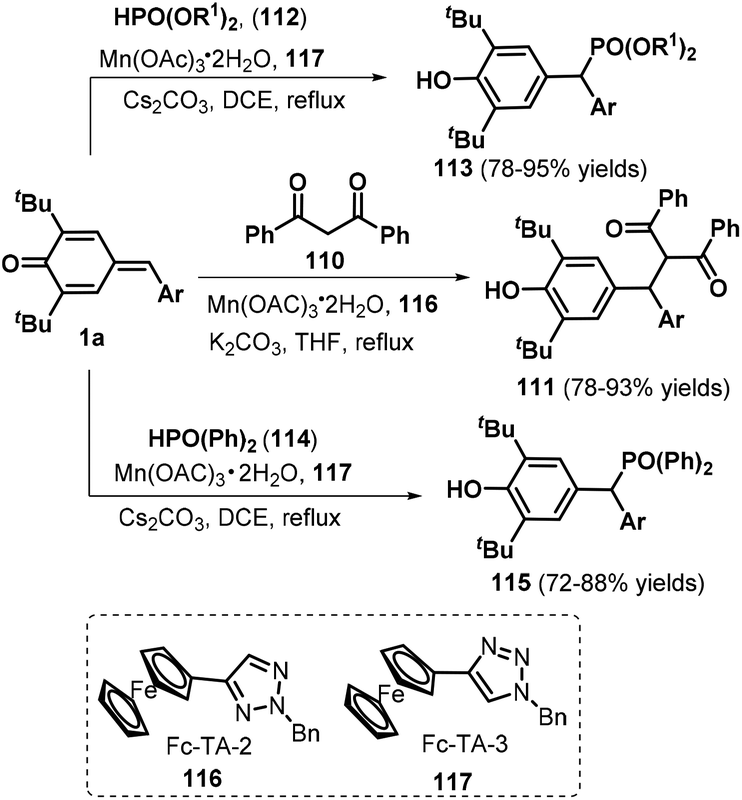
Recently, Wang and co-workers employed Mn(OAc)3˙ as the catalyst to realize 1,6-nucleophilic addition between p-QMs 1a and carbon or phosphorus nucleophiles, such as 1,3-diphenylpropane-1,3-dione 110, diethylphosphite 112, and diphenylphosphine oxide 114, in the presence of ferrocenyl triazole (Fc-TA) ligands 116 and 117 (Scheme 23).38 This protocol provides an efficient method for obtaining functionalized methines and their analogues in good to high yields. An improved variation starting from p-QMs 1a and phosphorus nucleophiles was subsequently documented using different bases including Cs2CO339 and DBU40 or aqueous solution.41 Furthermore, Lu, Jiang, and co-workers presented a one-pot, two-step synthesis of functionalized ketones via the 1,6-addition of β-ketoacids to p-QMs 1a and subsequent Et3N-assisted decarboxylation.42
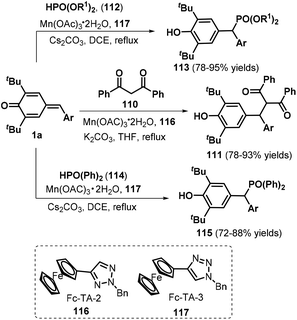 |
| | Scheme 23 Manganese-catalyzed 1,6-addition toward functionalized methines. | |
2.1.5. NHC-catalyzed 1,6-addition.
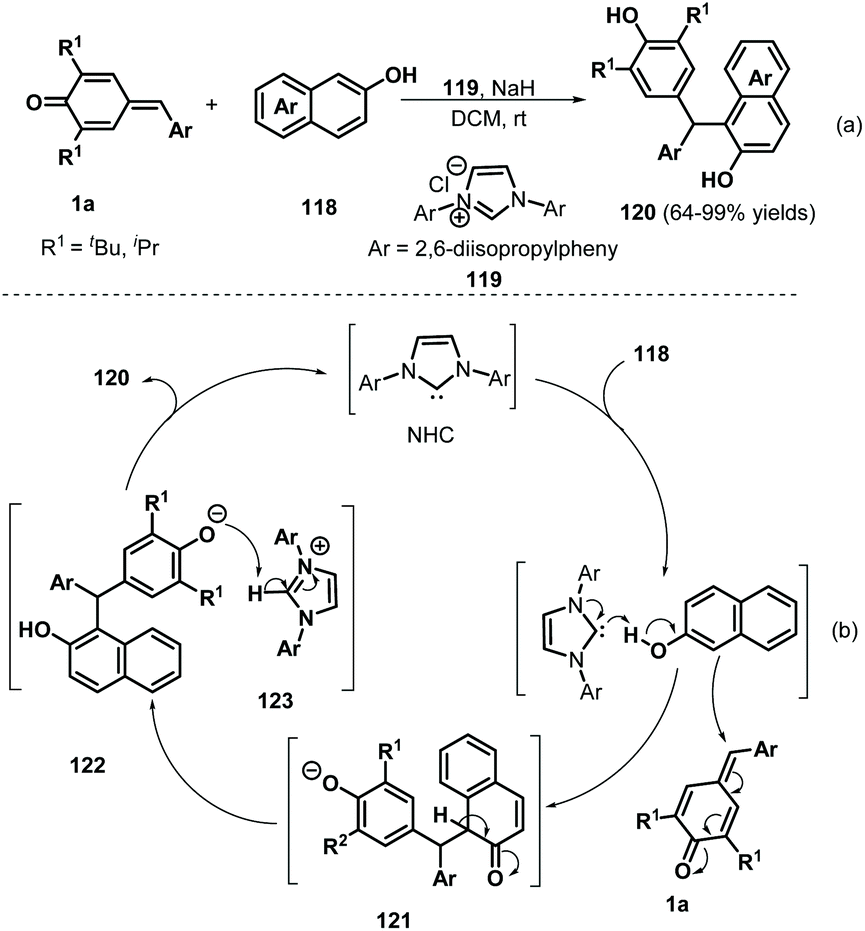
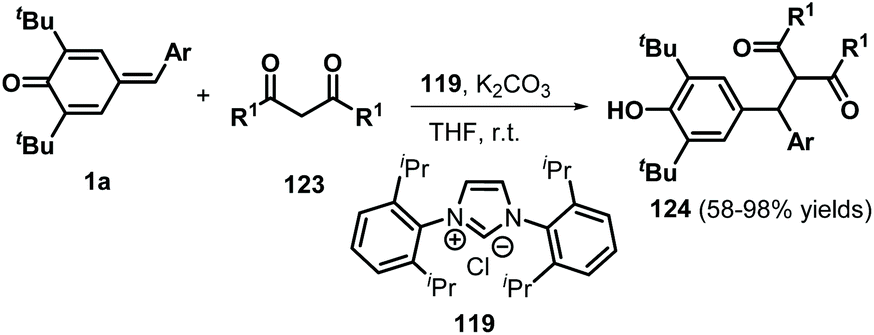
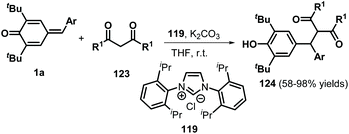
Over the past decades, NHC catalysis has emerged as a versatile tool for carbon–carbon and carbon–heteroatom bond formation.43 In 2016, Anand and co-workers reported an NHC-catalyzed 1,6-conjugate addition of 2-naphthols 118 to p-QMs 1a to afford a series of functionalized unsymmetrical triarylmethanes 120 in an atom-economical manner (Scheme 24a).44 This reaction proceeded readily for various 2-naphthol derivatives to generate the expected triarylmethanes in high yields. However, simple phenol was not converted into the corresponding product under the standard conditions. In this transformation, the NHC 119 acts as a Brønsted base to abstract the phenolic proton of the 2-naphthol to form a 2-naphthoxide anion, which undergoes 1,6-addition of p-QM 1a to generate intermediate 121 (Scheme 24b). Aromatization of 121 followed by proton transfer from 123 to 122 results in the formation of the final product with liberation of the NHC. Guin et al. demonstrated a similar catalytic strategy for the synthesis of functionalized trisubstituted methanes 124 in 58–98% yields using 119 as a Brønsted base catalyst (Scheme 25).45
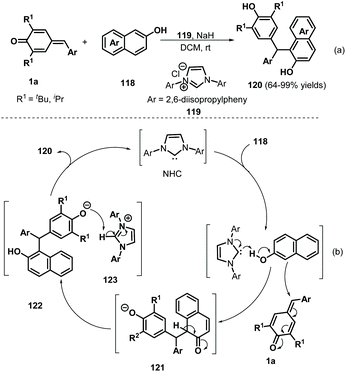 |
| | Scheme 24 NHC-Catalyzed 1,6-addition of 2-naphthols to p-QMs. | |
 |
| | Scheme 25 NHC-Catalyzed synthesis of trisubstituted methanes. | |
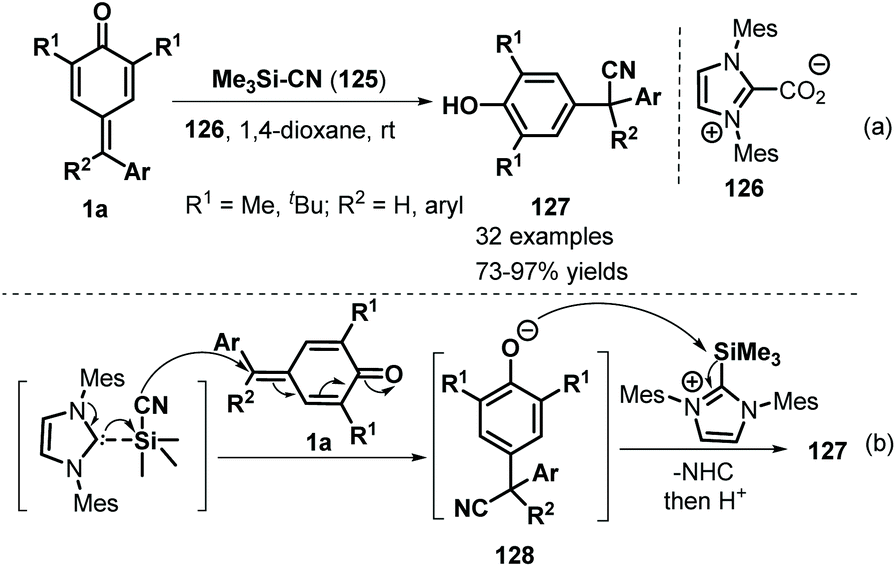
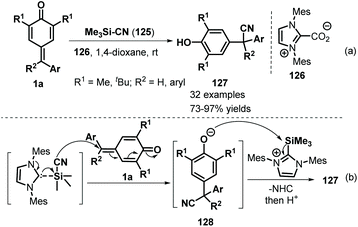
Inspired by the extensive studies on NHC-catalyzed 1,6-addition reactions of p-QMs, Anand and co-workers described another interesting NHC-catalyzed reaction involving silicon activation for vinylogous conjugate cyanation of p-QMs 1a (Scheme 26). The NHC-catalyzed reaction of 1a proceeded smoothly using Me3SiCN 125 as a cyanide source, providing general and straightforward access to a series of α-diaryl- and α-triarylnitriles 127 in good to excellent yields.46 Notably, this protocol was the first example of NHC-catalyzed 1,6-conjugate addition of Me3SiCN to a dienone system for cyanation.
 |
| | Scheme 26 Synthesis of α-diaryl- and α-triarylnitriles. | |
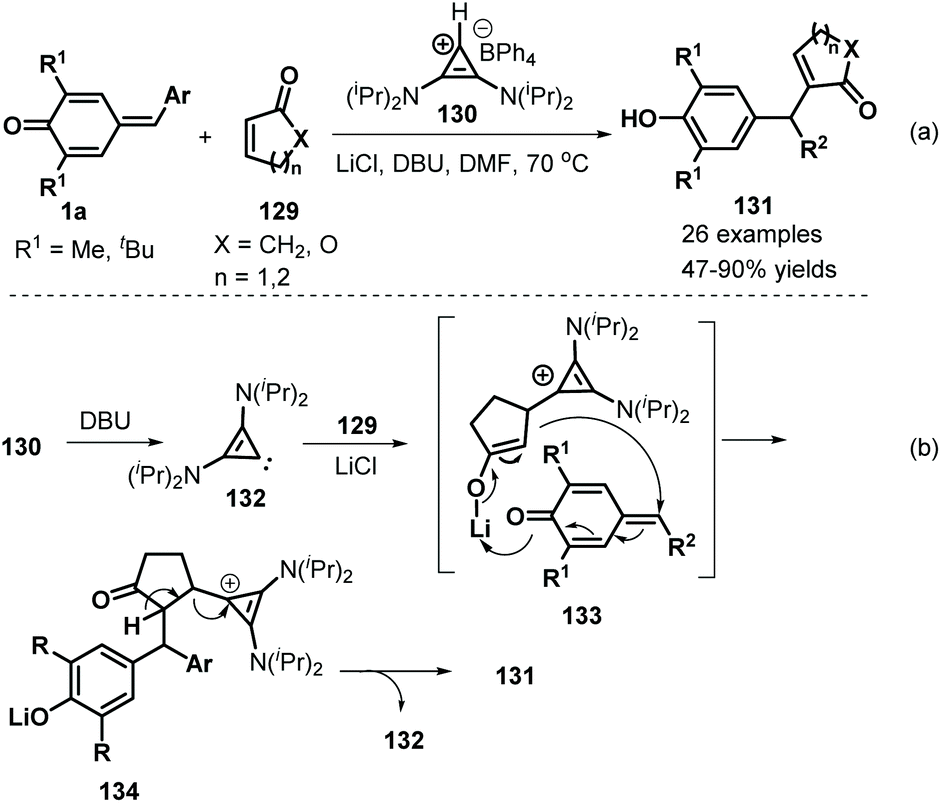
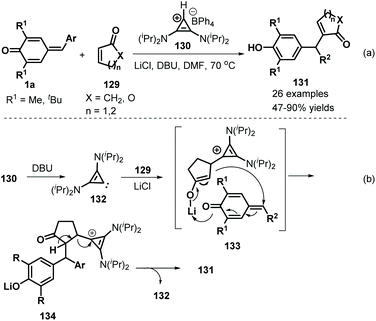
Anand and co-workers employed bis(amino)cyclopropenylidene 130 as an effective NHC catalyst to mediate the 1,6-addition of p-QMs 1a and α,β-unsaturated carbonyls 129. This reaction proceeded readily to furnish 26 examples of vinyl diarylmethanes 131 in moderate to good yields (Scheme 27a).47 Various α,β-unsaturated carbonyls, including 2-cyclopentenone, 2-cyclohexen-1-one and 5,6-dihydro-2H-pyran-2-one, reacted smoothly. Unfortunately, 2-(5H)-furanone, α,β-unsaturated lactams, and acyclic enones such as vinyl ketones, acrylates, and acrylamides were all found to be unreactive. A plausible mechanism for the formation of these products may include in situ generation of carbene intermediate 132, Michael addition in the presence of lithium salts, and 1,6-addition alongside regeneration of the carbene intermediate 133 (Scheme 27b).
 |
| | Scheme 27 1,6-Addition of p-QMs and α,β-unsaturated carbonyls. | |
2.1.6. Radical-enabled 1,6-addition.
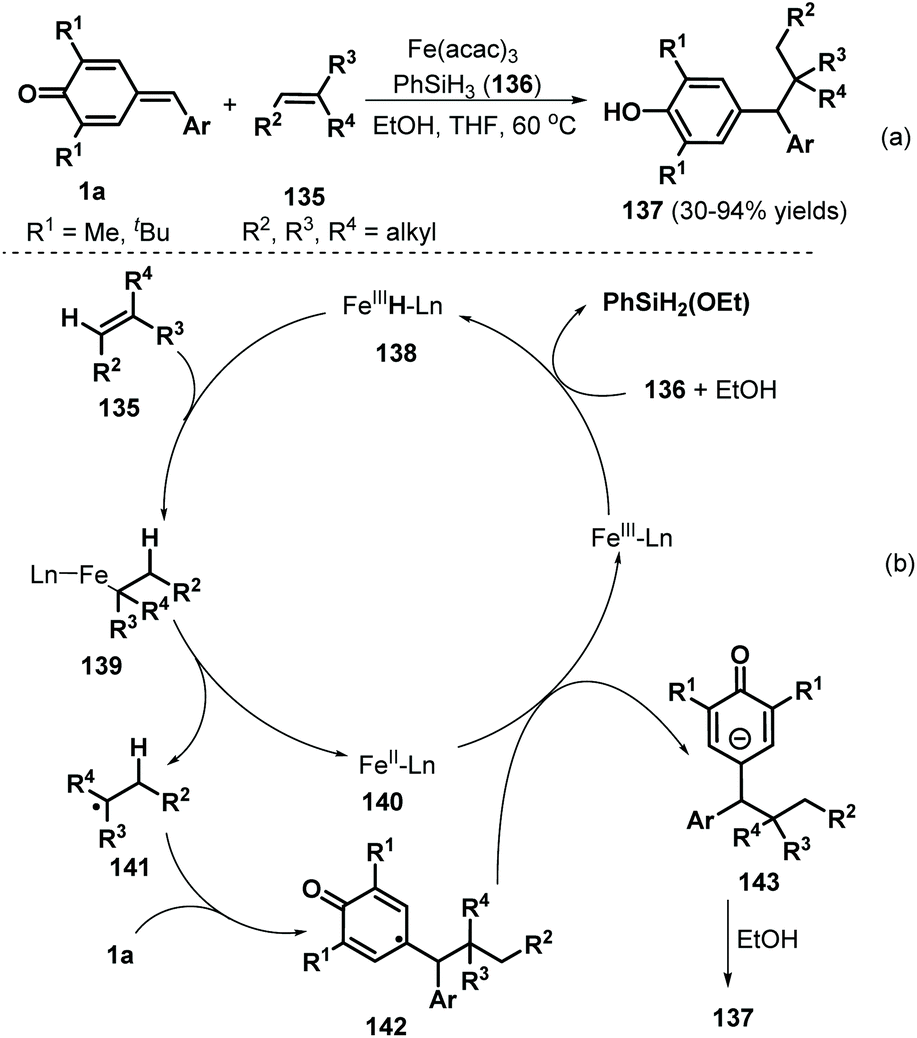
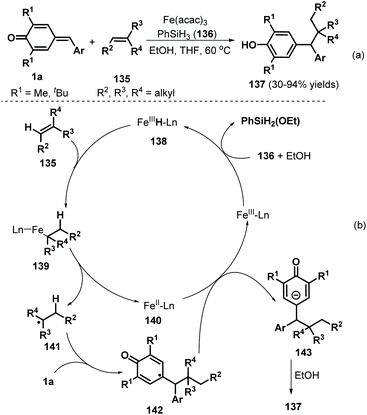
Cascade radical reactions permit the formation of multiple carbon–carbon and carbon–heteroatom bonds and have become a valuable synthetic strategy for accessing densely functionalized structures in a highly controlled manner with good functional group tolerance. Undoubtedly, the design and preparation of versatile radical acceptors is the key to developing radical chemistry approaches for the construction of skeletally diverse molecules. Compounds possessing unsaturated structures such as alkene and alkyne moieties have proven to be versatile radical acceptors. p-QMs have often been employed to capture various radical species via 1,6-radical addition to construct diarylated methanes. In 2016, the Cui group reported a catalytic radical-triggered 1,6-addition involving p-QMs 1a and olefins 135 (Scheme 28a). This radical-induced reaction was conducted in a mixed solvent system containing THF and EtOH at 60 °C using Fe(acac)3 and PhSiH3136, generating the corresponding phenols 137 in 30–94% yields.48 The hydroalkylation process was readily applicable to terminal olefins bearing hydroxy, amide, ester, and PMP moieties and tolerated not only cyclic olefins such as dihydropyran but also internal alkenes containing ester, ketone, and hydroxy groups and (S)-(−)-β-citronellol. On the basis of several control experiments, the authors proposed that the iron(III) catalyst is first converted to iron hydride species 138 in the presence of PhSiH3 and EtOH, followed by the addition of olefin 136 and dissociation of intermediate 139 to generate iron(II) species 140 and alkyl radical 141 (Scheme 28b). The latter component then undergoes radical addition of p-QM 1a to form intermediate 142, which is reduced by the iron(II) species 140 to furnish the phenol product 137 in the presence of EtOH with regeneration of the iron(III) catalyst.
 |
| | Scheme 28 Radical 1,6-addition of p-QMs and olefins. | |
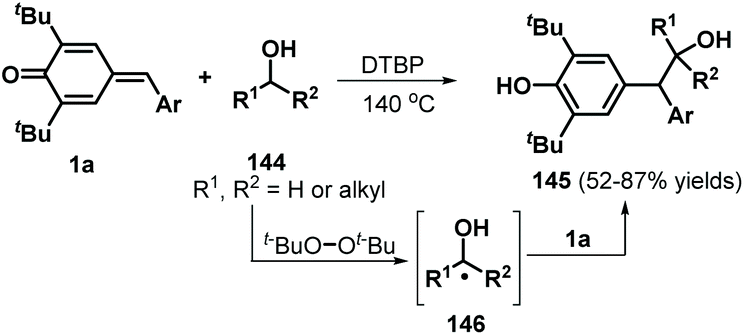
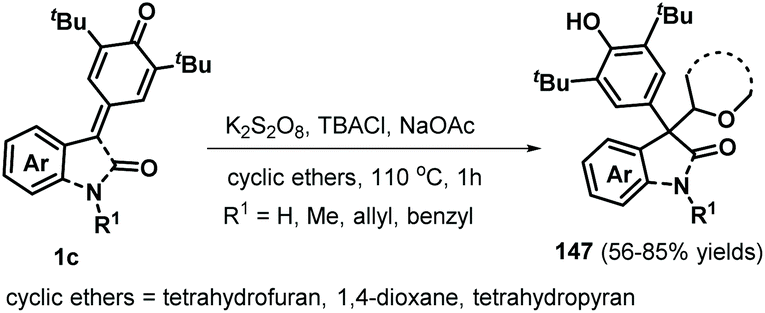
Following this successful work, the same group reported another α-oxy radical-induced 1,6-addition of p-QMs 1a to obtain phenols 145 under metal-free conditions using di-tert-butyl peroxide (DTBP) as a radical initiator (Scheme 29).49 Various primary alcohols, such as methanol, ethanol, and n-propanol, reacted well and underwent α-oxy addition to p-QMs 1a. Secondary alcohols such as isobutanol and cyclopentanol also furnished the corresponding functionalized alcohols in moderate yields. In the presence of DTBP, alcohol 144 is transformed to α-oxy radical 146, which is trapped by the p-QM to afford products 145 through hydrogen atom transfer from alcohol 144. In a subsequent work, cyclic ethers were converted into carbon-centered radicals under oxidative conditions (K2S2O8/tetrabutylammonium chloride (TBACl)), which were then captured by p-QMs 1c to afford cyclic ether-substituted diarylmethanes and triarylmethanes 147 with good yields (Scheme 30). Tetrahydrofuran, 1,4-dioxane, and tetrahydropyran as representative cyclic ethers also reacted well. However, 1,3,5-trioxane and 1,2-dimethoxyethane were incompatible.50
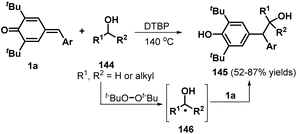 |
| | Scheme 29 Radical 1,6-addition of p-QMs toward functionalized alcohols. | |
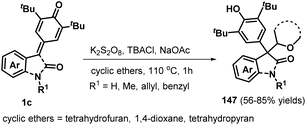 |
| | Scheme 30 Radical 1,6-addition of cyclic ethers to p-QMs. | |
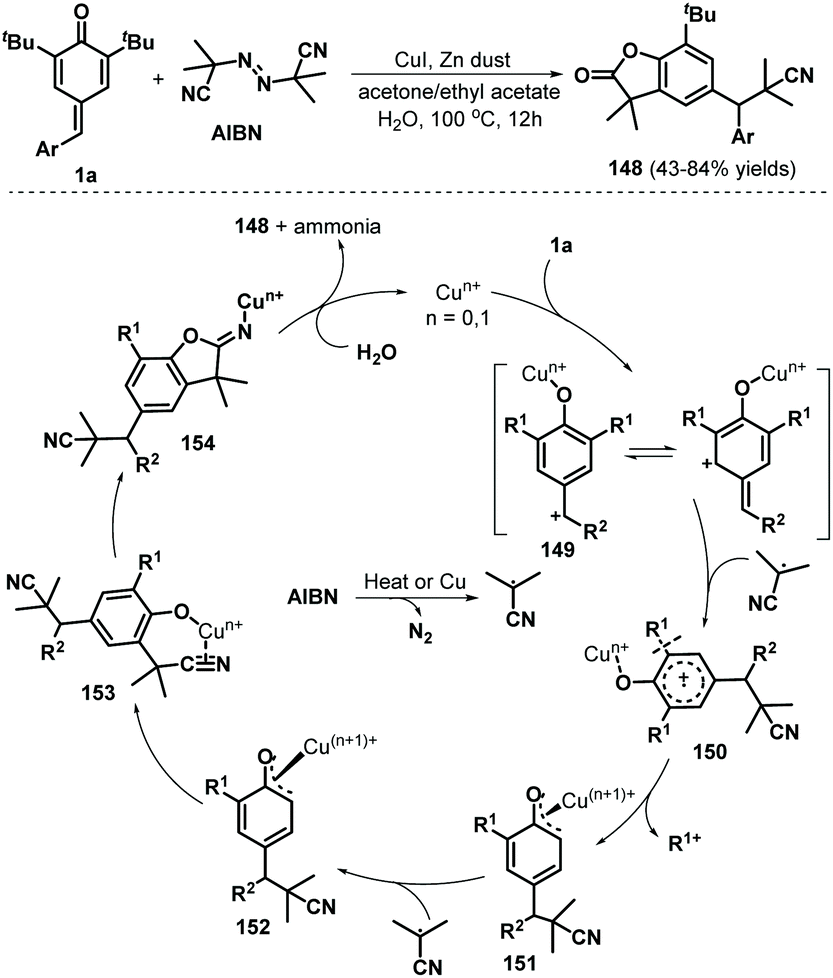
Tang, Chen, and co-workers reported the use of a radical cascade reaction of p-QMs 1a in the presence of azodiisobutyronitrile (AIBN) and water to access benzofuran-2(3H)-ones in 43–84% yields (Scheme 31). Various p-QMs bearing electron-rich, electron-neutral, or electron-deficient aromatic moieties, some of which contained relatively bulky substituents, reacted well with AIBN in the presence of CuI (20 mol%) to afford cyano-containing benzofuran-2(3H)-ones 148. Unfortunately, the desired products were not obtained after replacement of the t-Bu substituents with methyl or isopropyl groups. This cascade reaction is believed to proceed via 1,6-conjugate addition/aromatization, α-cyanoalkylation with cleavage of the unstrained and non-polar C(aryl)–C(t-Bu) bond, and downstream cyano-insertion/cyclization/hydrolysis.51
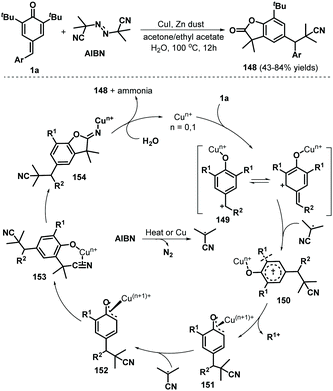 |
| | Scheme 31 Radical reaction of p-QMs with AIBN. | |
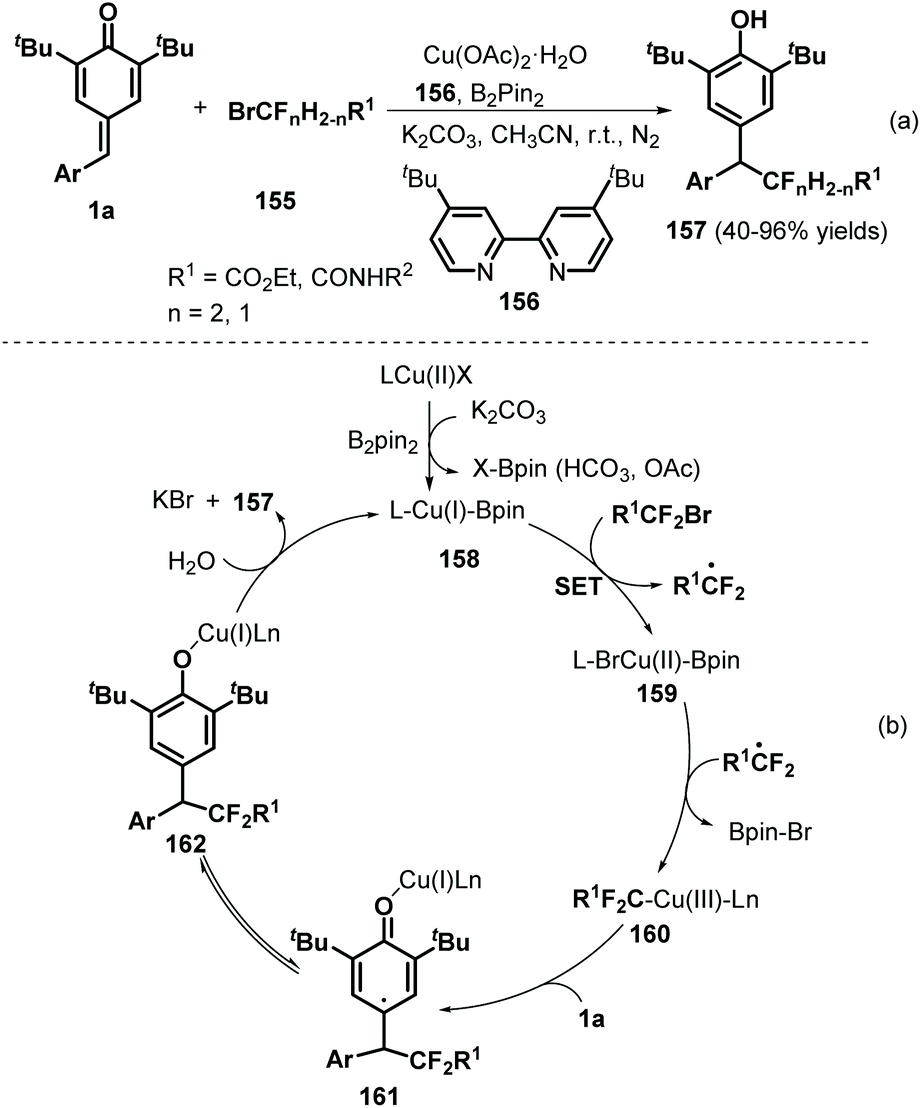
Using Cu/B2Pin2 as a catalytic system, Song and co-workers documented an elegant 1,6-hydrodifluoroacetylation of p-QMs 1a with difluoroalkyl bromides 155 (Scheme 32a).52 This reaction featured a broad substrate scope and B2Pin2 as the reductant, resulting in the formation of a C(sp3)–CF2 bond. Both difluoromethylated and monofluoromethylated bromides and a variety of p-QMs were compatible with this reaction. During the reaction, the copper salt is considered to be reduced by B2Pin2 to afford Cu(I)–BPin species 158 (Scheme 32b). Then, oxidative addition of 158 into the difluoroalkyl bromide via single-electron transfer (SET) generates Cu(II)–BPin species 159 and a difluoroalkyl radical, which react to form Cu(III) species 160 with the elimination of Br–BPin. Subsequently, intermediate 160 is further trapped by p-QM 1a to generate carbon-centered radical 161, which yields the desired product 157 after isomerization and proton transfer with regeneration of the Cu(I) species for the next catalytic cycle.
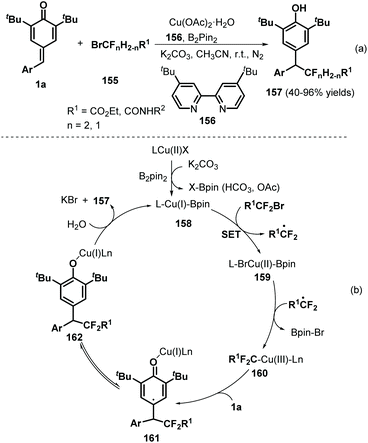 |
| | Scheme 32 1,6-Hydrodifluoroacetylation of p-QMs. | |
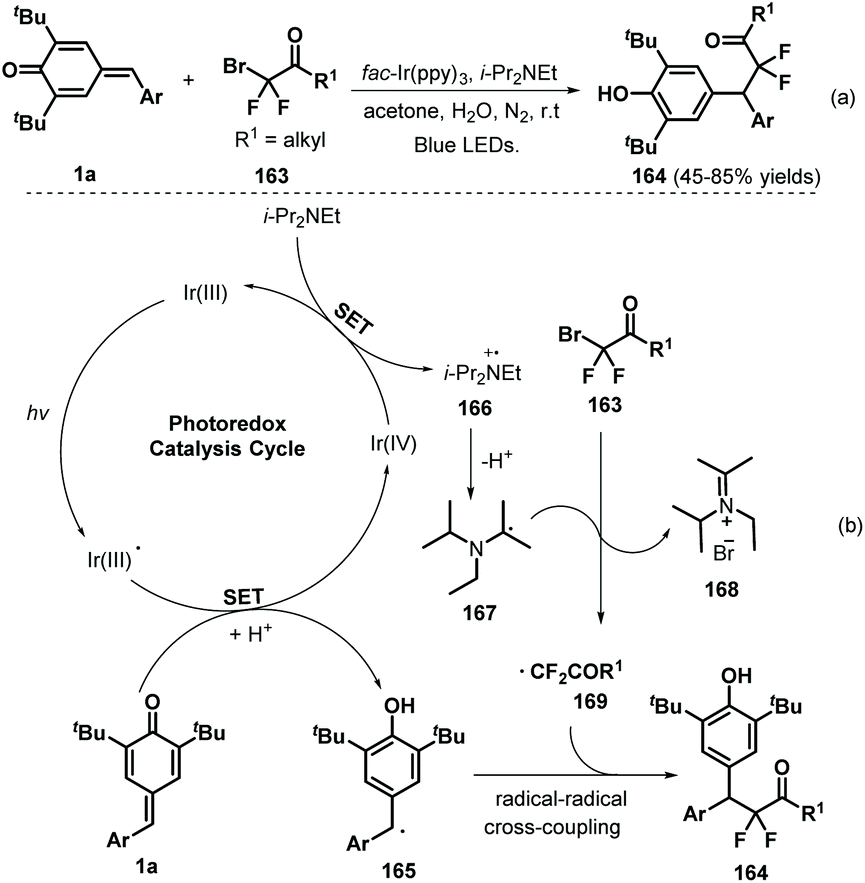
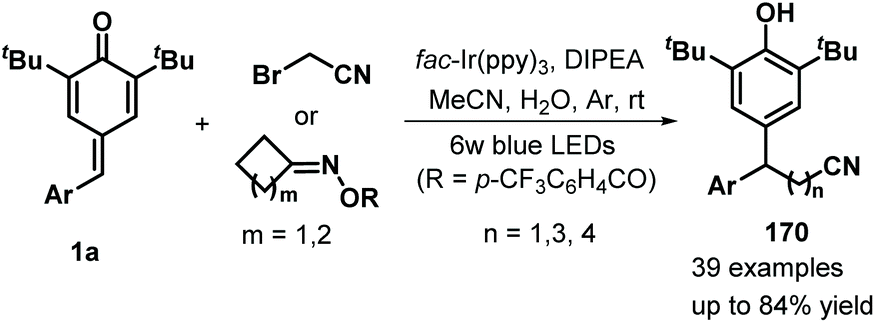
Later, Xu and co-workers applied visible-light photocatalysis to achieve 1,6-hydrodifluoroacetylation between p-QMs 1a and difluoroalkyl bromides 163, leading to 22 examples of difluoroalkylated diarylmethanes 164 in 45–85% yields via radical cross-coupling (Scheme 33a).53 Difluoroalkylating reagents containing various functional groups, including carbonyl, ester, acylamino, and heteroaryl moieties, reacted well to afford the expected products in moderate to good yields. The reaction pathway consists of SET, deprotonation, and radical–radical cross-coupling (Scheme 33b). In another example of visible-light photocatalysis, Li and co-workers exploited bromoacetonitrile as an α-cyanomethyl radical precursor for the cyanomethylation of p-QMs 1a (Scheme 34). The use of cyclobutanone oximes as γ-cyanoalkyl radical donors permitted the successful synthesis of 3-cyanopropyl diarylmethanes 170 with good yields. Unfortunately, a p-QM with a methyl moiety did not react, which may be attributable to the stability of the radical intermediate. The catalytic process is similar to that described for the above transformation.54
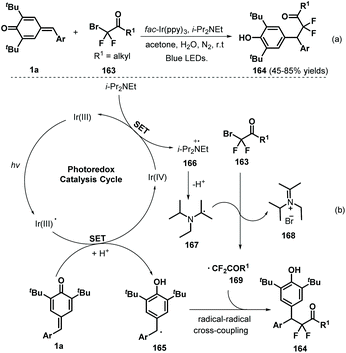 |
| | Scheme 33 Photocatalytic 1,6-hydrodifluoroacetylation of p-QMs. | |
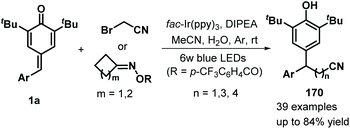 |
| | Scheme 34 Synthesis of cyanated diarylmethanes. | |
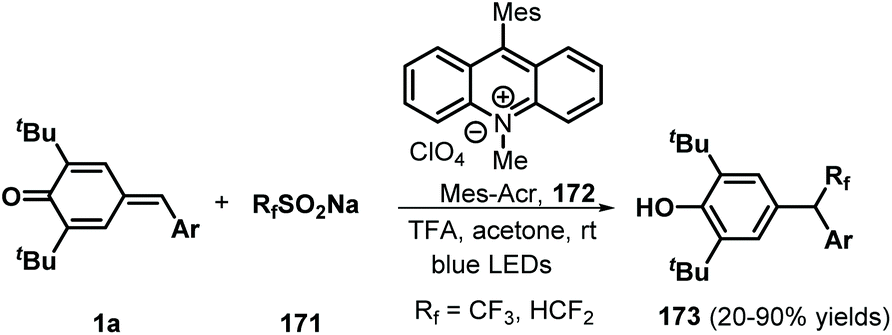
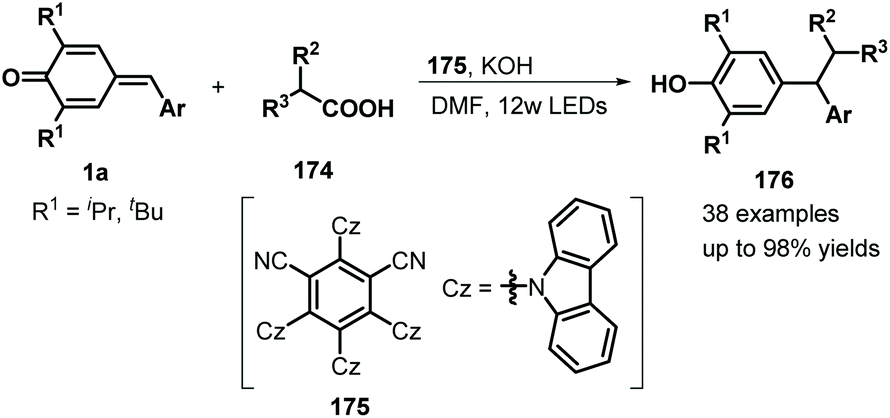
Similarly, difluoromethylated diarylmethanes 173 were obtained in high yields via visible-light-initiated photoredox catalysis of p-QMs 1a and CF3SO2Na/HCF2SO2Na 171 (Scheme 35).55 It should be noted that the strong inductive effect of fluorine atoms may have played a critical role in the reactivity of the sodium sulfinates. A similar photocatalytic reaction for the trifluoromethylation of p-QMs 1a was described by Sureshkumar and co-workers using 2,4,6-tri-p-tolylpyrylium tetrafluoroborate (T(p-CH3)PPT) as the photocatalyst.56 As a continuation of the above study, Lu, Weng, and co-workers developed a new photocatalytic decarboxylative 1,6-conjugate addition between carboxylic acids 174 and p-QMs 1a to obtain 1,1,2-triarylethanes and 2,2-diarylethylamines 176 in good to excellent yields under metal-free conditions (Scheme 36). A range of structurally diverse (hetero)arylacetic, secondary fatty, and amino acids were suitable for this addition reaction. During this reaction, 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) 175 served as a photocatalyst, accelerating decarboxylation of the carboxylic acid via SET.57
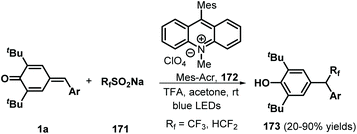 |
| | Scheme 35 Synthesis of difluoromethylated diarylmethanes. | |
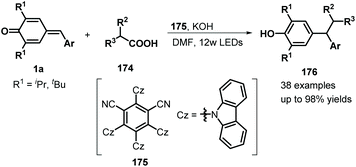 |
| | Scheme 36 Photocatalytic decarboxylation of carboxylic acids. | |
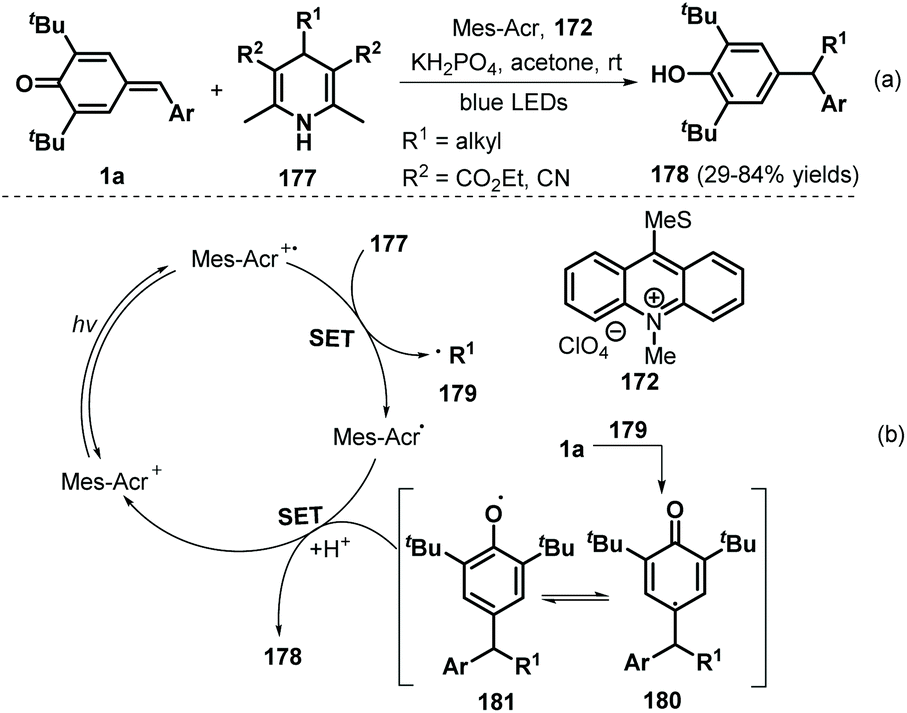
In 2018, the Liu group demonstrated the visible-light-initiated alkylation of p-QMs 1a using 4-substituted Hantzsch esters/nitriles 177 as the alkyl radical precursor and KH2PO4 as an additive, which afforded a wide range of functionalized phenols 178 in reasonable yields (Scheme 37a).58 Hantzsch esters/nitriles bearing various alkyl groups were compatible with this reaction, and the mechanism is very similar to that described in previous works,56,57 consisting of SET and radical addition (Scheme 37b).
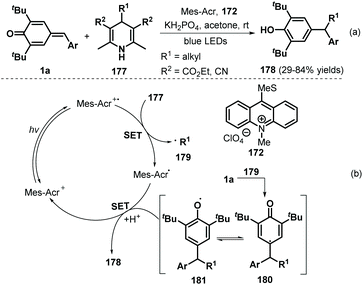 |
| | Scheme 37 Visible-light-initiated alkylation of p-QMs. | |
2.1.7. Catalytic asymmetric 1,6-addition.
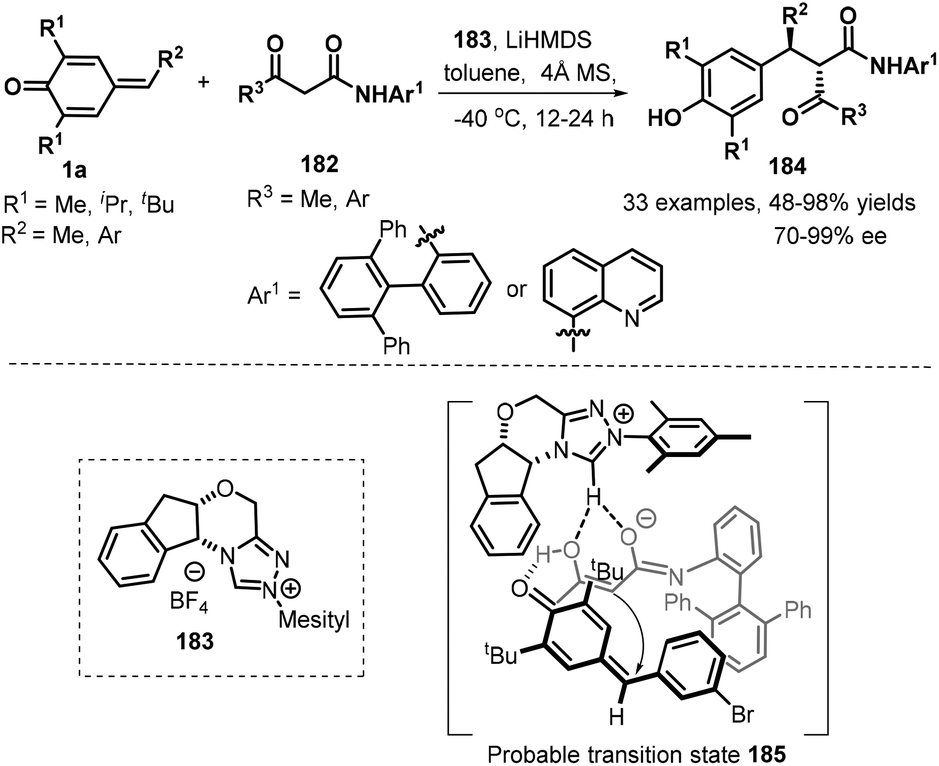
Catalytic asymmetric 1,6-addition reactions in recent years have proven to be a powerful tool for the generation of chiral molecules with important biological and medical interests in modern synthetic chemistry. As a versatile and readily available class of synthetic intermediates, p-QMs have been extensively used to construct chiral diarylmethane or triarylmethane frameworks via asymmetric 1,6-conjugate addition. Consequently, efforts have been devoted to developing organocatalytic asymmetric nucleophilic addition between p-QMs and different nucleophiles. After recent comprehensive review highlighted by Li and co-workers,9a many elegant asymmetric transformations of p-QMs have been documented. Guin and co-worked developed a novel highly diastereo- and enantioselective 1,6-addition of 1,3-ketoamides 182 to p-QMs 1a using chiral N-heterocyclic carbenes 183 as Brønsted base catalysts, furnishing the desired products 184 with 48–98% yields and high stereoselectivity (54:46–99:1 dr and 70–99% ee). A series of p-QMs 1a with different substituents at both aromatic ring and quinone moiety were easily transformed into the corresponding products. Notably, this reaction could be extended to the alkylated p-QM as well, providing the expected product with 66% yield and 96% ee, albeit with an approximate 1:1 dr. Similarly, α-aroylacetamides with a methyl or an aryl group relative to carbonyl moiety and an aryl or a quinolin-8-yl functionality linked by amide unit were found to be applicable to this asymmetric reaction, furnishing the desired products in good yields and enantioselectivity. Some of these products could be applied to synthesize highly enantioenriched β and γ-lactam derivatives. On the basis of 1H NMR spectroscopy studies, it is believed that the reaction proceeds through a chiral ion-pair intermediate consisting of the enolate and the azolium ion (Scheme 38).59
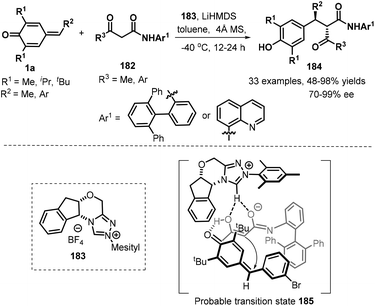 |
| | Scheme 38 NHC-Catalyzed asymmetric 1,6-addition of p-QMs. | |
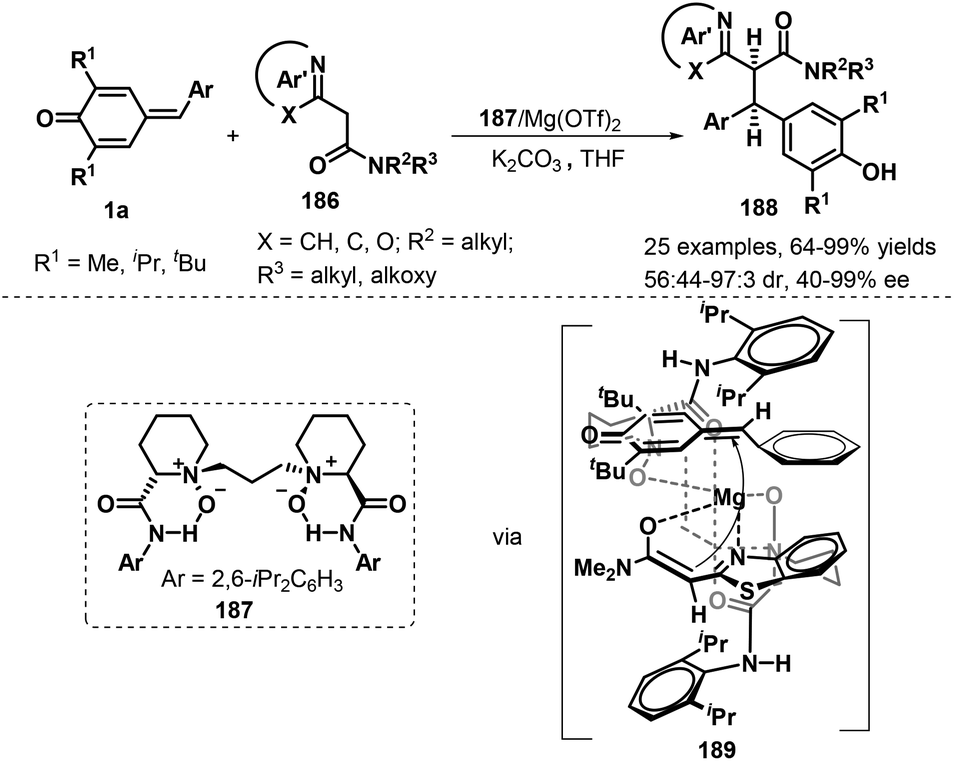
Another interesting catalytic asymmetric approach involving 1,6-addition to p-QMs 1a was demonstrated by Feng's group. During the reaction process, chiral Mg(II)/N,N′-dioxide 187 complex could effectively promote the diastereo- and enantioselective 1,6-addition of azaarylacetamides 177 to p-QMs 1a, and a series of chiral azaarene derivatives 186 were provided in 64–99% yields, 56:44→19:1 dr, and 87–99% ee. Various azaarylacetamides including benzothiazole, benzoxazole, pyridine, and pyrazine all reacted with p-QMs 1a smoothly under the mild conditions. In addition, it is proposed that tetradentate N,N′-dioxide ligand and azaarylacetamides coordinates with the central metal Mg(II) to form a slightly distorted octahedral structural transition state. Because of the steric hinderance, the Si-face of the enolate of azaarylacetamides prefers to attack the Si-face of 1a to afford chiral product 188 (Scheme 39).60 This inference would be consistent with the experimental observations that decreasing the steric hindrance of p-QMs by replacing the tert-butyl group with an isopropyl or methyl resulted in reduced diastereoselectivity.
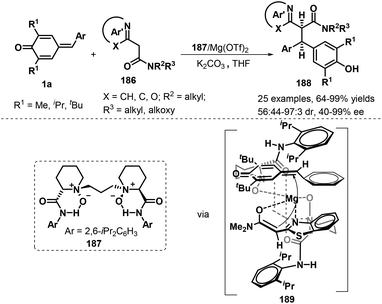 |
| | Scheme 39 1,6-Addition of 2-azaarylacetamides to p-QMs. | |
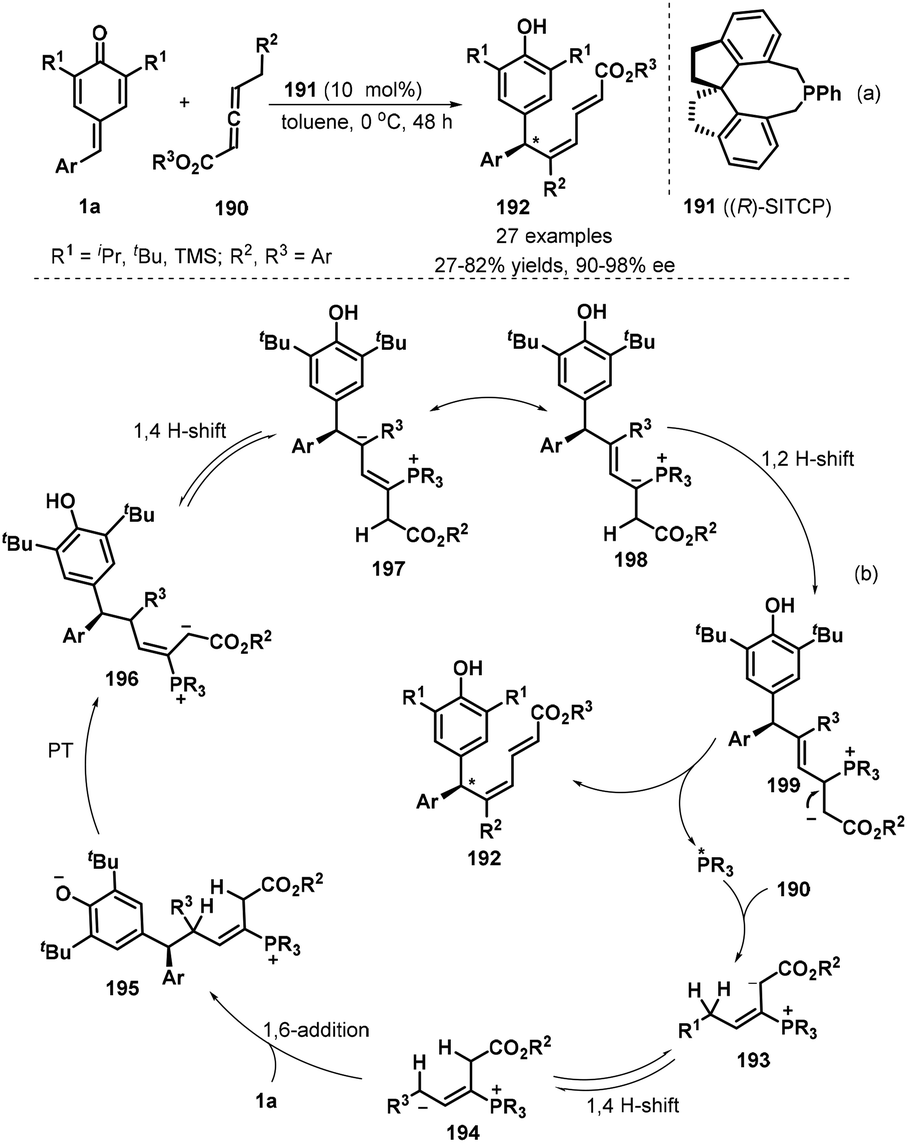
Xu and co-workers developed the first chiral phosphine-catalyzed isomerization/1,6-conjugate addition of substituted allenoates 190 to p-QMs 1a. The reaction worked well in toluene in the presence of 10 mol% of (R)-SITCP 191 as a chiral phosphine catalyst at 0 °C without any additives in a one-pot, generating 27 chiral dienylated bisarylmethides 192 in 27–82% yields and 90–98% ee (Scheme 40a).61 Various substituents such as t-butyl, i-propyl and TMS on the p-QM 1a unit were compatible. However, the introduction of methyl group into p-QM framework led to severe decomposition with no desired product isolated. Besides aryl-substituted p-QMs, pyridine- and indole-containing counterparts also displayed good tolerance by obtaining in both good yield and high enantioselectivity. The proposed mechanism starts with nucleophilic addition of chiral phosphine *PR3 to allenoates, which undergoes 1,4-H shift, 1,6-addition, proton transfer (PT) and 1,2-H shift to achieve the asymmetric dienylation reaction p-QMs (Scheme 40b).
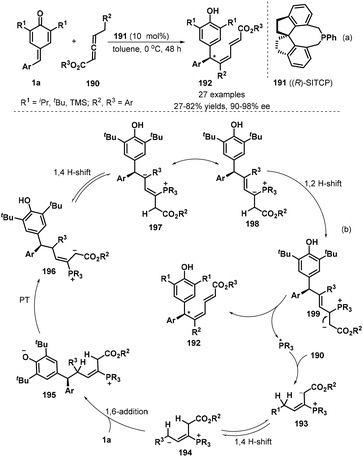 |
| | Scheme 40 Enantioselective dienylation of p-QMs. | |
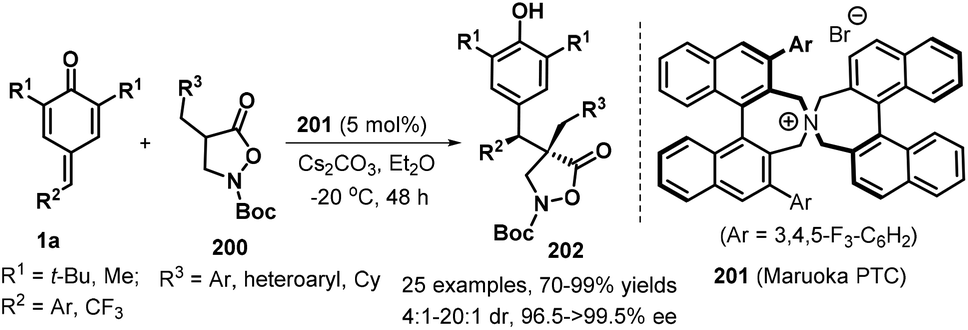
Binaphthyl-based spiro ammonium salt proved to be a high-effieicent chiral phase-transfer catalyst (Maruoka PTC 201), which could promote asymmetric 1,6-conjugate addition of isoxazolidin-5-ones 200 to p-QMs 1a. A variety of differently substituted isoxazolidin-5-ones 200 and p-QMs 1a were performed similarly in the reaction, giving access to 25 examples of functionalized isoxazolidin-5-one derivatives 202 in good yields (70–99% yields) and with excellent enantioselectivities (96.5→99.5% ee) and good diastereoselectivities (4:1 to 20:1 dr, Scheme 41).62N-Cbz-protected isoxazolidin-5-one enabled a similarly highly enantioselective direction to access the corrosponding product, despite with lower yield of 35%. The authors also demnstarted late-stage appliation of these products for the preapration of N-protected β-amino acid and dipeptide.
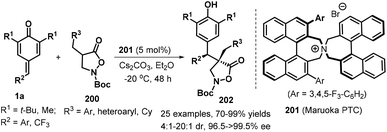 |
| | Scheme 41 Asymmetric 1,6-addition of isoxazolidin-5-ones to p-QMs. | |
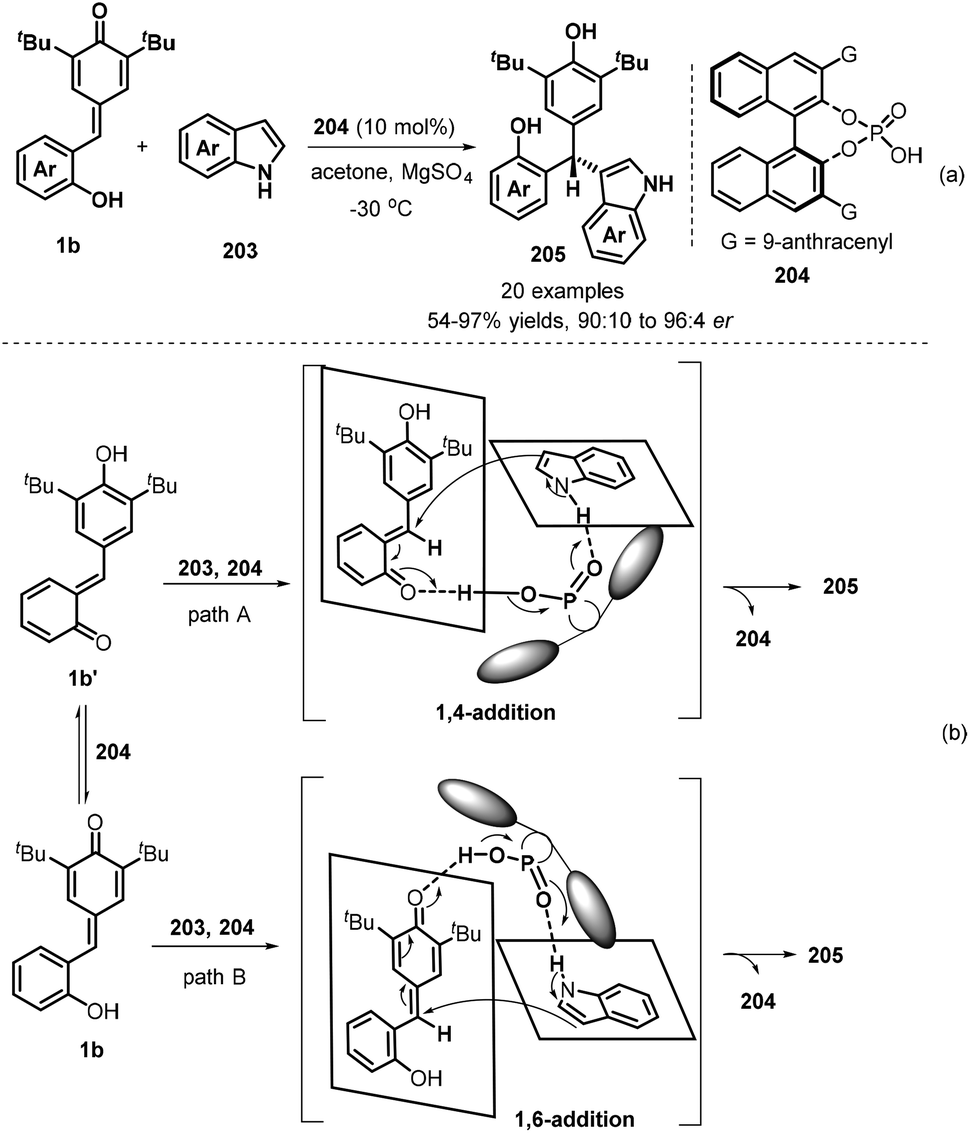
Hydrogen bonding activation of chiral phosphoric acids has applied in asymmetric 1,6-conjugate addition of p-QMs, which also exhibits high enantioselectivity. Since the pioneering work of chiral phosphoric acids-catalysed 1,6-addition of p-QMs reported by Sun's group,63 many asymmetric versions were developed by using different nucleophilies. In 2019, Shi and co-workers established a catalytic asymmetric conjugate 1,6-addition of indoles 203 to p-QMs 1b in the presence of chiral phosphoric acid 204 and using acetone as solvent in −30 °C for 12 hours, which afforded chiral indole-containing triarylmethanes 205 in generally high yields (54–98%) and good enantioselectivities (90:10–96:4 er, Scheme 42a). By conducting control experiments, the authors believed that the reaction had a great possibility to undergo 1,4-conjugate addition of indoles to in situ-generated o-QMs 1b′ from 1b in the presence of chiral phosphoric acid, although its 1,6-conjugate addition pathway may occur (Scheme 42b).64
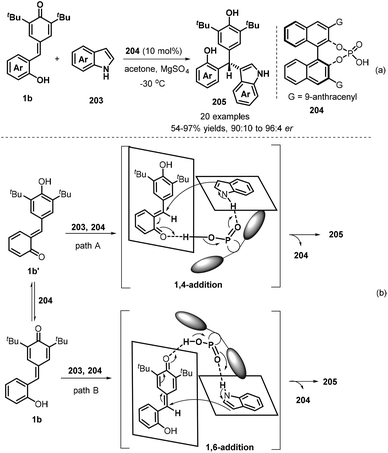 |
| | Scheme 42 Asymmetric addition of indoles to p-QMs. | |
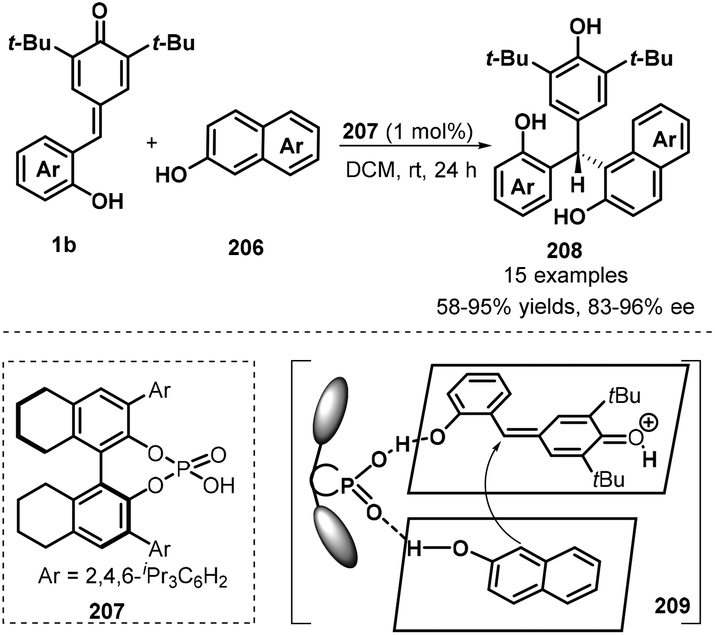
Another example of the application of chiral phosphoric acid 207 as a Brønsted catalyst in asymmetric 1,6-conjugate addition of p-QMs 1b was reported by Li and co-workers (Scheme 43).65 In this work, 2-naphthols 206 were selected as C-nucleophiles, enabling the enantioselective addition to p-QMs 1b to construct chiral naphthol-containing triarylmethane skeleton 208. This strategy not only was applicable to various p-QMs 1b bearing different types of substituents but also could carry out with various substituents including electron-withdrawing (Br, CN, CO2Me, CO2Et) and electron-donating groups (MeO, EtO) at different positions of the aromatic ring of the 2-naphthols 206, giving generally good yields (58–95%) and high enantioselectivities (83–96%). The proposed mechanism is analogue to Shi's protocol, in which 1,4-conjugate addition of 2-naphthols 206 to in situ-generated o-QMs 1b′ from 1b proceeds in the presence of chiral phosphoric acid.
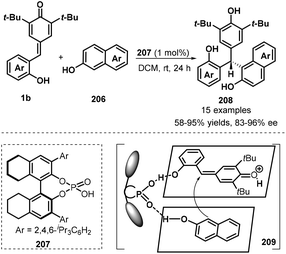 |
| | Scheme 43 Enantioselective addition of 2-naphthols to p-QMs. | |
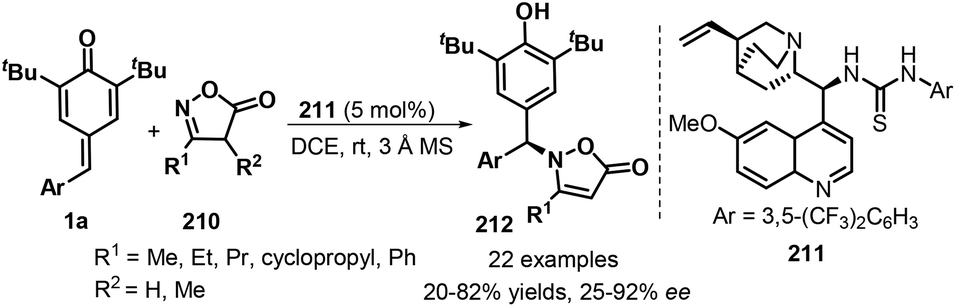
An impressive protocol involving the asymmetric 1,6-aza-Michael addition to p-QMs 1a was described by Blay and co-workers in 2020. Here, quinine-derived thiourea 211 as a bifunctional catalyst was adopted in the presence of 3 Å molecular sieve (MS) to promote the enantioselective 1,6-aza-addition reaction between p-QMs 1a and isoxazolinones 210 as N-nucleophiles, leading to chiral isoxazolinone-containing diarylmethanes 212 with moderate to good yields (20–82%) and enantioselectivity (25–92% ee). Notably, for a same substituent in p-QMs 1a, ortho- or para-substituted rings showed higher reactivity profiles than meta-substituted ones (Scheme 44).66
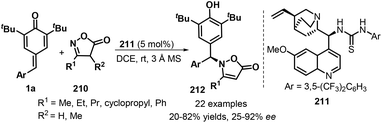 |
| | Scheme 44 Asymmetric 1,6-aza-Michael addition to p-QMs 1a. | |
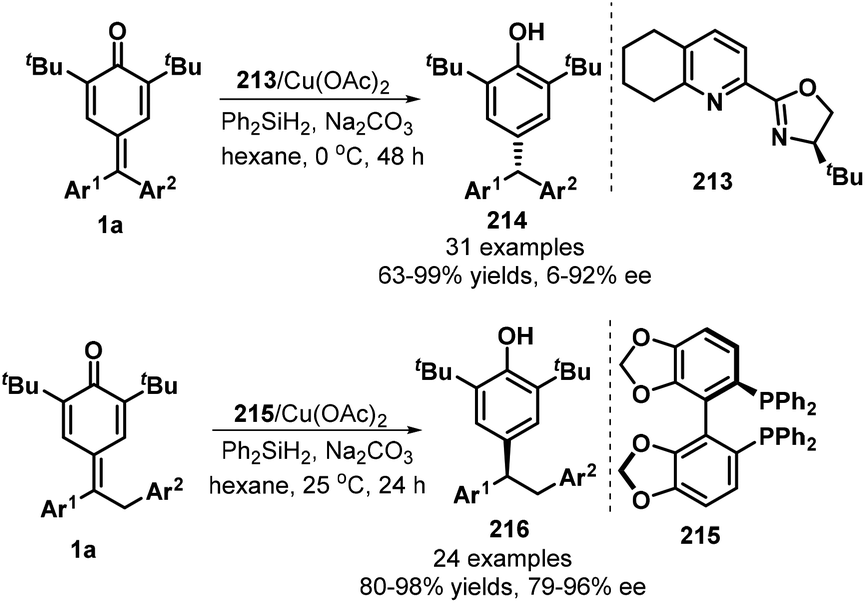
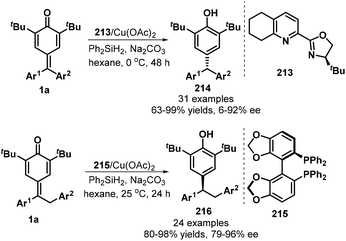
Chiral copper complexes could be applied in the asymmetric 1,6-conjugate reduction of p-QMs, as demonstrated by Fan's group in 2019 (Scheme 45).67 By using tetrahydroQuinox 213 as ligand and Ph2SiH2 as hydrogen source, CuH-catalyzed reaction of diarylated p-QMs 1a in the presence of Na2CO3 in hexane at 0 °C provided chiral triarylmethanes 214 in good yields (63–99%) with high enantioselectivities (up to 96% ee). When benzyl-substituted p-QMs were exploited as 1,6-addition acceptors, the combination of Cu(OAc)2 with 4,4′-bi-1,3-benzodioxole-5,5′-diylbis(diphenylphosphane) (SEGPHOS, 215) ligand drove this transformation into 24 examples of 1,1,2-triarylethanes 216 in 80–98% yields with 79–96% ee. Generally, meta- and para-substituted p-QMs were reduced to the triarylmethanes with low enantioselectivities.
 |
| | Scheme 45 Asymmetric 1,6-conjugate reduction of p-QMs. | |
2.2. 1,6-Addition/annulation reactions of p-QMs
2.2.1. 1,6-Addition/intramolecular annulation of p-QMs.
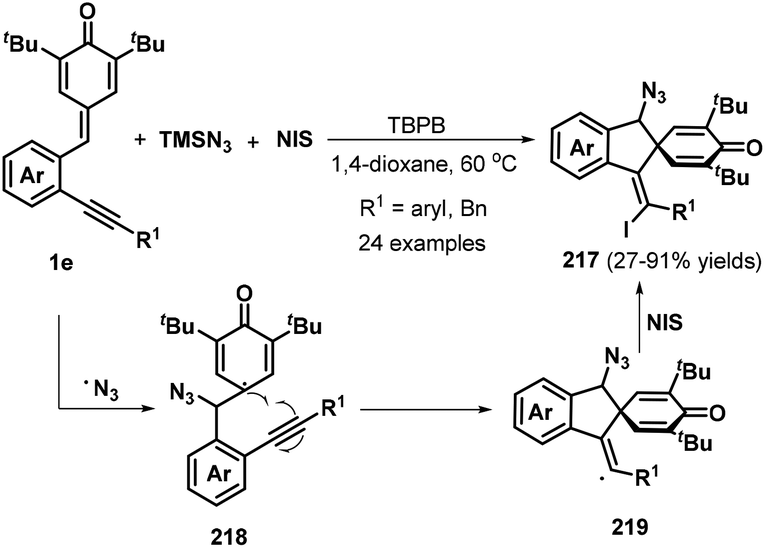
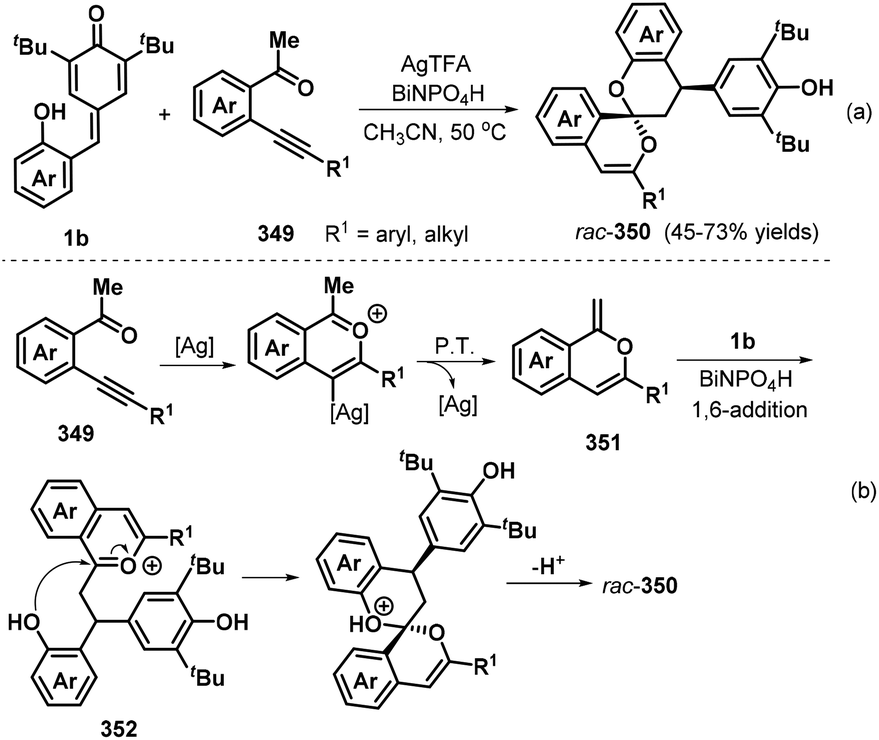
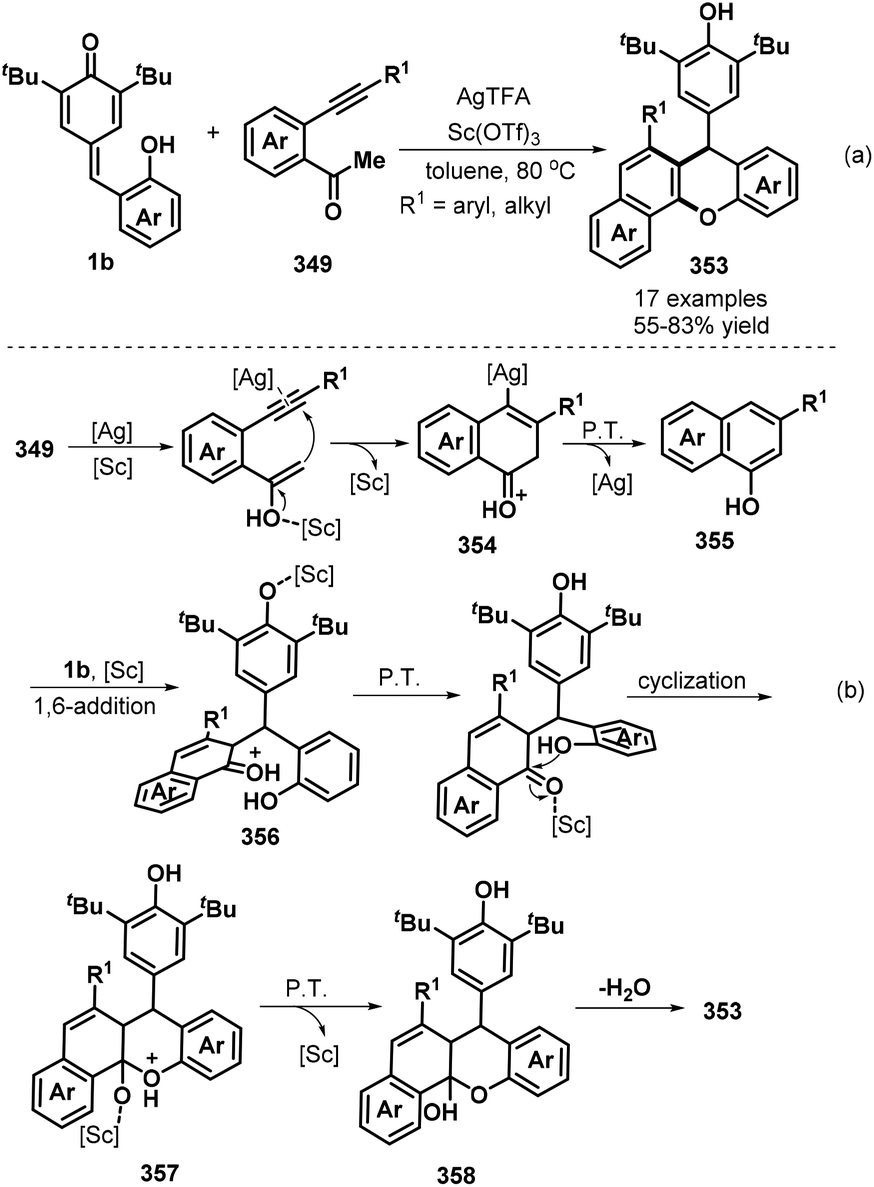
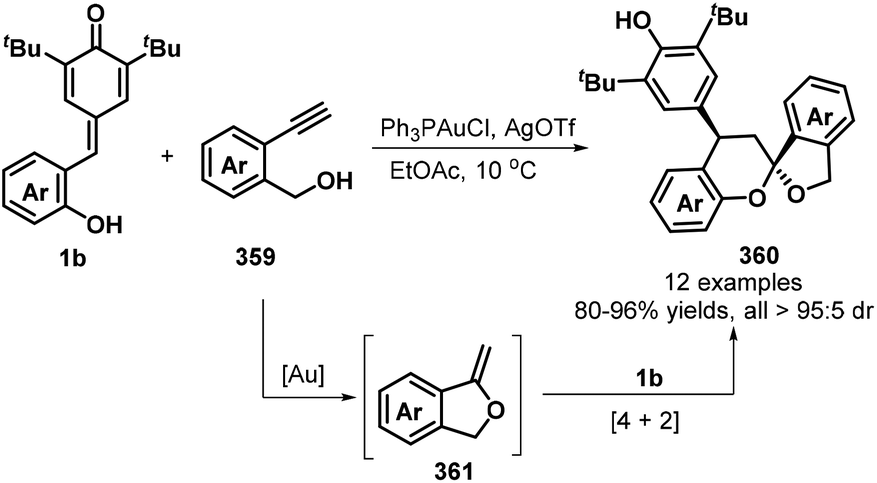
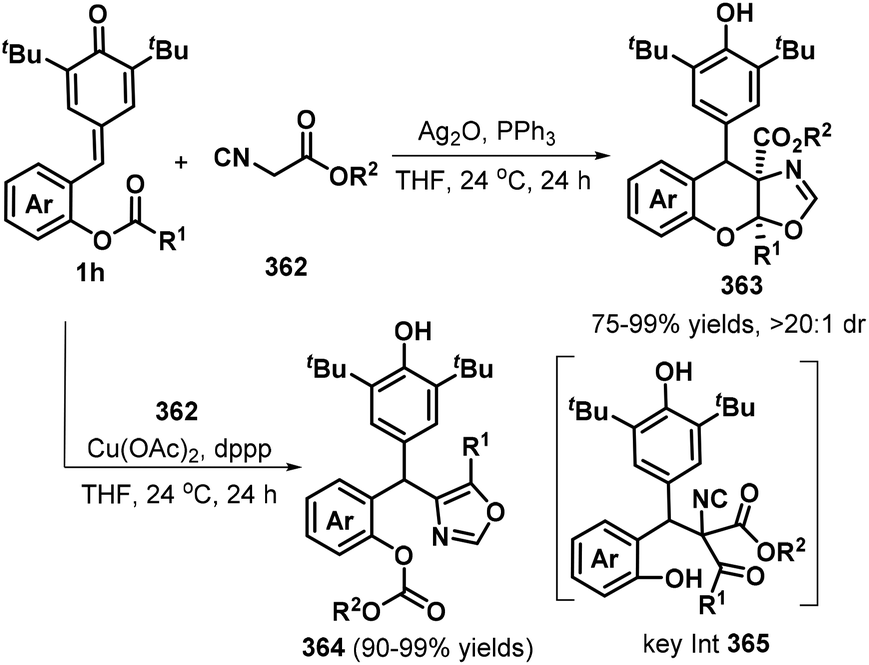
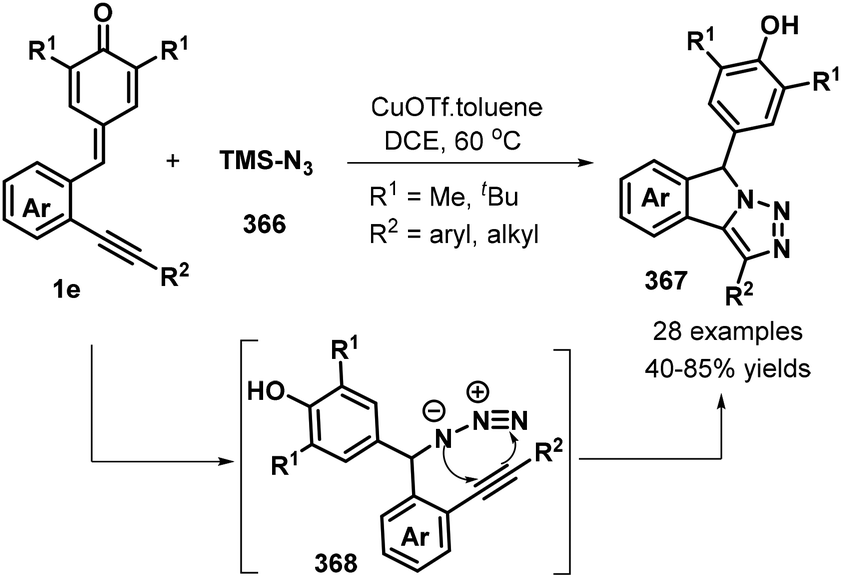
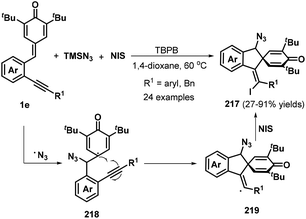
o-Alkynylphenyl-substituted p-QMs containing both alkene and alkyne moieties have emerged as versatile 1,5-enyne equivalents with high reactivity toward 1,6-addition/intramolecular annulation reactions. Owing to the differences in the electronic properties of the alkene and alkyne moieties and the aromatization driving force of the cyclohexadiene ring, radical species and nucleophiles preferentially attack the alkene unit via 1,6-addition followed by intramolecular cyclization across the alkyne moiety to form useful spiro-cyclohexadiene targets with excellent regioselectivity. In a seminal report, Yao, Lin, and co-workers documented an elegant 1,6-addition/intramolecular annulation for the direct conversion of o-alkynylphenyl-substituted p-QMs 1e to functionalized spiro[4.5]deca-6,9-dien-8-ones 217 (Scheme 46). In this process, TMSN3 first reacts with tert-butyl peroxybenzoate (TBPB) to generate the N3 radical, which then undergoes radical addition to the o-alkynylphenyl-substituted p-QM. Subsequent 5-exo-dig cyclization yields vinyl radical 219, which is trapped by N-iodosuccinimide (NIS) to furnish the corresponding product 217 in good to excellent yields.68 This three-component protocol permits the direct construction of three new σ bonds (C–N, C–C, and C–I) in a one-pot process with complete chemo- and regioselectivity.
 |
| | Scheme 46 Three-component reaction of p-QMs with TMSN3 and NIS. | |
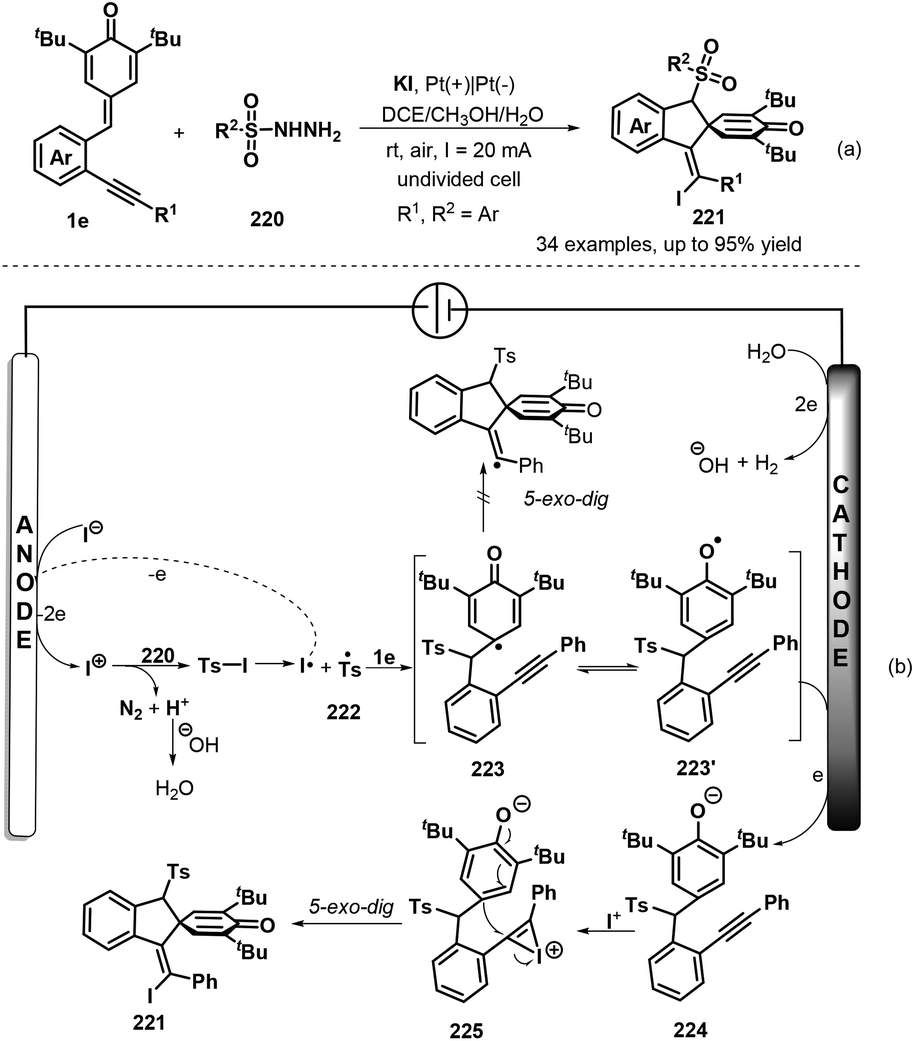
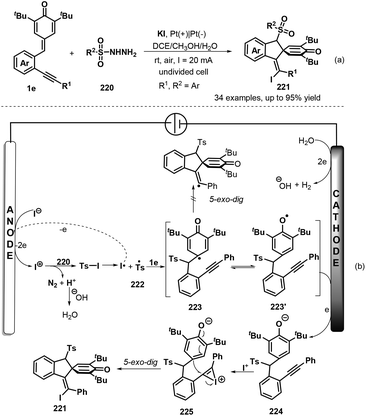
To develop practical and green methods to construct these skeletons, recently, Jiang, Tu and co-workers established an electrochemical three-component annulation-iodosulfonylation of p-QMs 1e by using available arylsulfonyl hydrazides 220 and potassium iodide under environmentally benign conditions, offering a sustainable and efficient access to construct 34 examples of spirocyclohexadienone-containing (E)-indenes 221 in 30%–95% yields (Scheme 47a).69 However, substrate with a n-butyl or a cyclopropyl group on the alkynyl moiety of p-QMs was not adaptable for this reaction Notably, potassium iodide plays triple roles of an electrolyte and a redox catalyst as well as an iodination reagent. Based on the control experiments, the authors proposed a plausible mechanism depicted in Scheme 47b. Initially, the anodic oxidation of iodide ion generates iodonium I+ with losing two electrons, which reacts with p-tolylsulfonyl hydrazide 220 to give arylsulfonyl iodide intermediate, followed by homolysis to yield arylsulfonyl radical 222 and iodine radical. The latter was converted into I+ species by electric current whereas sulfonyl radical 222 adds to activated carbon–carbon double bond of p-QM 1e through 1,6-addition, generating cyclohexadienone radical 223, which undergoes aromatic zwitterionic resonances to access phenol radical 223′. Then, the cathodic reduction of intermediate 223′ occurs to produce phenol anion 224. Subsequently, iodonium I+ is intercepted by carbon–carbon triple bond of 224 to yield highly reactive intermediate 225, which is converted into the final product 221 through intramolecular ipso-cyclization. The concomitant cathodic half-reaction involves the reduction of two protons to H2.
 |
| | Scheme 47 Electrochemical three-component annulation-iodosulfonylation of p-QMs. | |
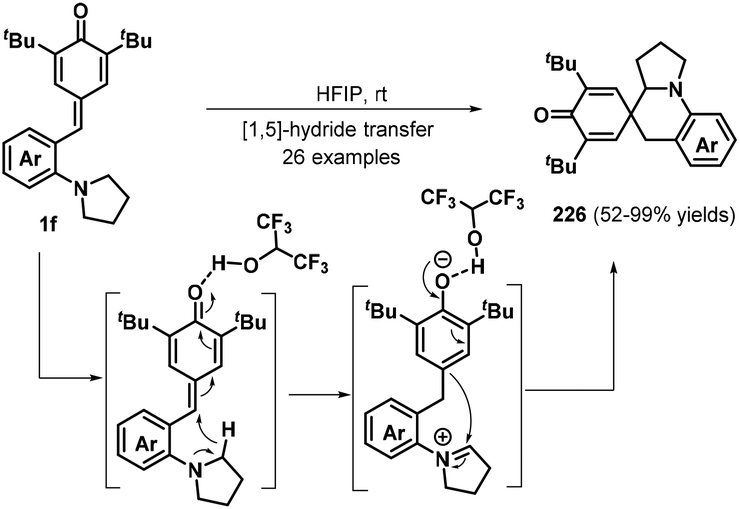
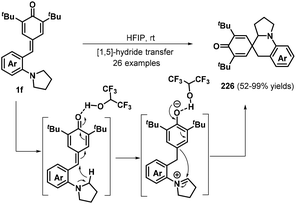
As a different approach for realizing the 1,6-addition/intramolecular annulation of p-QMs, the Xiao group prepared o-pyrrolidin-1-ylphenyl-substituted p-QMs 1f and transformed these compounds into 2-azaspiro[5.5]undeca-7,10-dien-9-ones 226via hydrogen-bonding-activated [1,5]-hydride transfer70 and 1,2-addition in hexafluoroisopropanol (HFIP) (Schemes 48). Furthermore, an o-dimethylaminophenyl analogue also proved suitable for this transformation. Nevertheless, substrates containing electron-donating substituents such as methyl and methoxy groups afforded complex mixtures that were difficult to purify.70a
 |
| | Scheme 48 Intramolecular annulation of p-QMs. | |
 |
| | Scheme 49 [2 + 1] annulation of p-QMs with α-bromomalonates. | |
2.2.2. 1,6-Addition/[2 + 1] annulation of p-QMs.
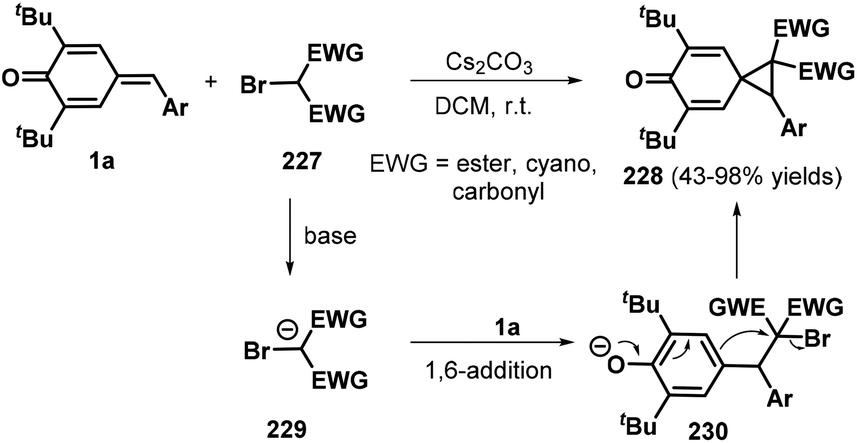
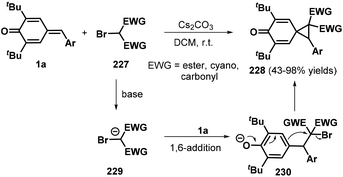
Annulation reactions remain a hot topic in modern organic synthesis as they provide a facile method for synthesizing large functionalized cyclic molecular libraries with precisely defined stereochemical arrangements from simple precursors with high step economy, efficiency, and scalability. Specifically, [2 + 1] annulation has been recognized as an important and step-economical synthetic strategy for the assembly of three-membered rings with an extreme convergence. Owing to their ring tension, such targets could exhibit unique reactivity profiles that enable their direct ring-opening/expansion to construct valuable molecular architectures that are not accessible via other methods. Thus, the development of [2 + 1] annulation for small-ring synthesis is of substantial interest.71 To this end, the Lin and Yao group explored a 1,6-conjugate addition/[2 + 1] annulation strategy for the formation of spiro[2.5]octa-4,7-dien-6-ones 228 from p-QMs 1a and α-bromomalonates 227 under Cs2CO3-promoted conditions (Scheme 49). All of the desired products contained two consecutive quaternary centers and were obtained in high yields of up to 98%. Various electron-withdrawing groups, such as allyl ester, cyano, and carbonyl moieties, were well tolerated in this reaction, affording the desired products in good yields. The authors proposed a mechanism involving 1,6-conjugate addition and intramolecular nucleophilic substitution.72 Exchanging the α-bromomalonates for benzyl chlorides successfully afforded spiro[2.5]octa-4,7-dien-6-ones 228via Cs2CO3-mediated tandem 1,6-addition/[2 + 1] annulation.73
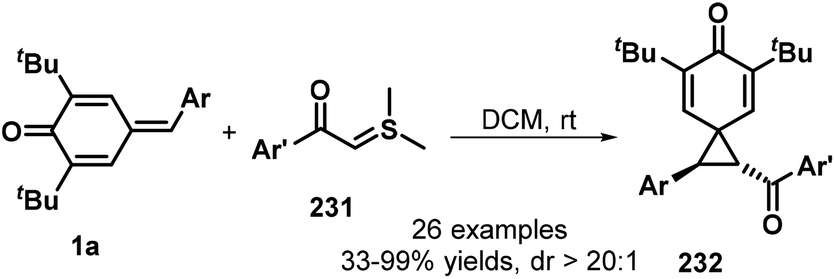
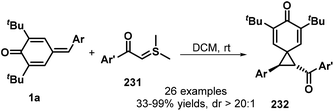
In a similar manner, the same group then exploited sulfur ylides 231 as the C1 synthon in this 1,6-conjugate addition/[2 + 1] annulation (Scheme 50). This reaction proceeded smoothly in DCM at ambient temperature without any catalyst or base, affording the functionalized spiro[2.5]octa-4,7-dien-6-ones 232 in 33–99% yields with >20:1 dr. This transformation also exhibited good functional group tolerance and scalability.74 A similar reaction between p-QMs 1a and sulfonium salts in the presence of DBU was reported by Fan and co-workers.75
 |
| | Scheme 50 [2 + 1] annulation of p-QMs with sulfur ylides. | |
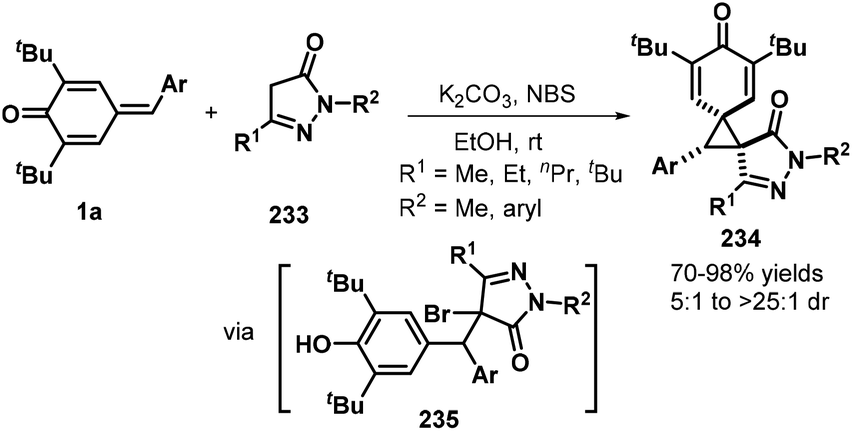
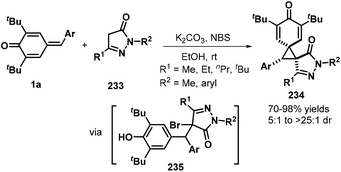
Das and co-workers reported an N-bromosuccinimide (NBS)-mediated reaction between p-QMs 1a and pyrazolones 233, which afforded a series of new bis-spiro[cyclohexadienone–cyclopropane–pyrazolone] compounds 234 in good yields and diastereoselectivity. Pyrazolones 233 containing various aryl and alkyl nitrogen-protecting groups and C5 substituents were well tolerated, furnishing the corresponding bispirocyclic products in 70–98% yields and dr values of 5:1 to >25:1. However, substrates containing either phenyl or trifluoromethyl groups at the pyrazolone C5 position did not undergo spirocyclization. A plausible mechanism consists of 1,6-conjugate addition, bromination, and de-aromatic nucleophilic substitution (Scheme 51).76
 |
| | Scheme 51 NBS-Mediated reaction of p-QMs and pyrazolones. | |
2.2.3. 1,6-Addition/[4 + 1] annulation of p-QMs.
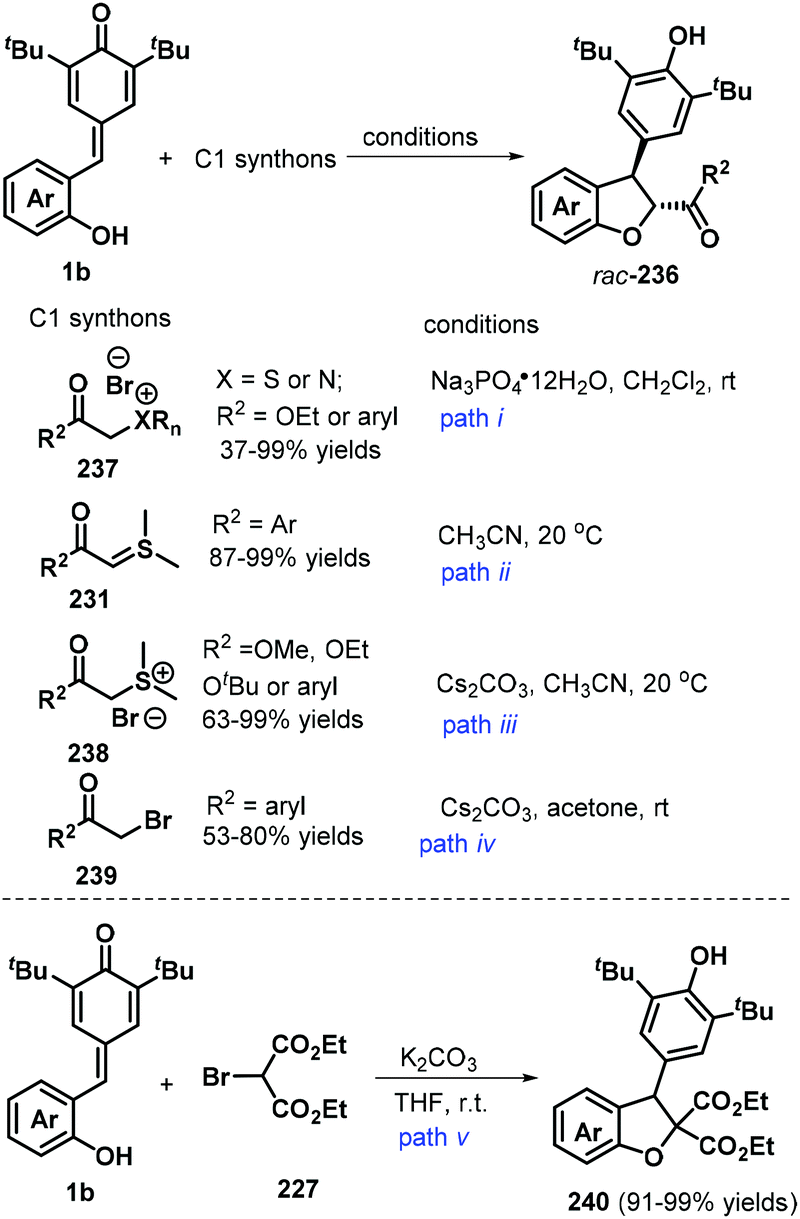
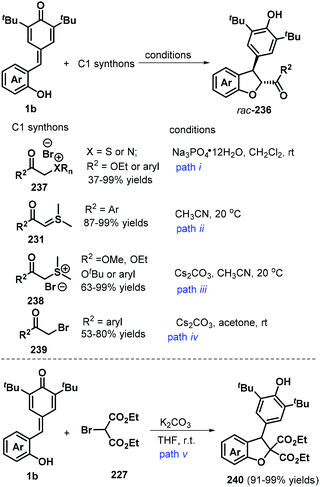
2,3-Dihydrobenzofurans are among the most important and versatile oxygen-containing heterocycles and frequently occur in many natural and synthetic bioactive compounds, such as those exhibiting remarkable anticancer or antibacterial activities.77 The 1,6-addition/[4 + 1] annulation of o-hydroxyphenyl-substituted p-QMs using one-carbon synthons offers a convenient and mild strategy for the preparation of 2,3-dihydrobenzofuran derivatives. For instance, the Huang and Yao group reported an impressive method for synthesizing trans-2,3-dihydrobenzofurans 236via the 1,6-addition/[4 + 1] annulation of o-hydroxyphenyl-substituted p-QM 1b using sulfonium or ammonium bromides 238 (Scheme 52, path i).78 This transformation afforded a series of products in 34–99% yields with high diastereoselectivity (>20:1 dr). This domino-type process featured good functional group tolerance and scalability, mild reaction conditions, high yields of up to 99%, and high stereoselectivity. However, in this report, a sulfonium salt containing an alkyl ester group provided the corresponding product in only low yield (37%).78 The reaction proceeds via 1,6-conjugate addition, proton transfer, and intramolecular SN2 nucleophilic substitution. In subsequent works, sulfur ylides 231 (Scheme 52, path ii),79 sulfonium salts 238 (Scheme 52, path iii),80 α-haloketones 239 (Scheme 52, path iv),81 and α-bromomalonates 227 (Scheme 52, path v)82 were applied as C1 synthons and independently reacted with o-hydroxyphenyl-substituted p-QM 1b in similar 1,6-addition/[4 + 1] annulation processes, affording 2,3-dihydrobenzofurans 236 and 240.
 |
| | Scheme 52 [4 + 1] annulation toward 2,3-dihydrobenzofurans. | |
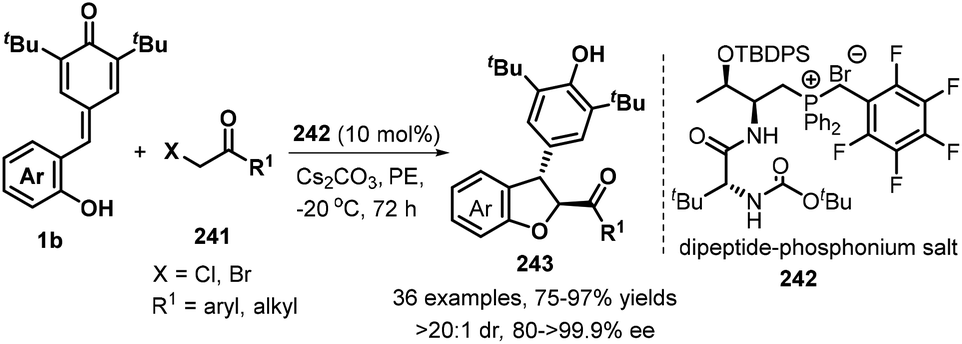
As demonstrated in the above examples, the synthesis of achiral 2,3-dihydrobenzofurans from p-QMs has been well-developed. In contrast, catalytic asymmetric [4 + 1] annulation reaction of p-QMs toward optically pure 2,3-dihydrobenzofurans still constitutes a daunting challenge. Until 2019, Wang's group reported the first catalytic enantioselective 1,6-addition/[4 + 1] annulation of p-QMs 1b and α-halogenated ketones 241 by employing O-TBDPS-protected L-Thr-D-tert-Leu-derived phosphonium salt 242 as a bifunctional catalyst. They discovered that the bifunctional catalyst promotes this transformation in petroleum ether (PE) in the presence of Cs2CO3, furnishing functionalized trans-2,3-dihydrobenzofurans 243 exclusively with high enantioselectivities. α-Halogenated ketones 241 having electron-withdrawing and -donating aromatic groups were tolerated, as well as cyclic ketones such as 3,4-dihydronaphthalen-1(2H)-one and benzofuran-3(2H)-one and bulky alkyl group such as t-butyl group. Mechanistic studies supported the annulation cascade consisting of intermolecular nucleophilic substitution and subsequent intramolecular 1,6-addition. Furthermore, the authors also elaborated a triad of hydrogen-bonding interactions in the L-Thr-derived dipeptide system for the key to the high enantioselectivity of this catalytic reaction by conducting DFT calculations (Scheme 53).83
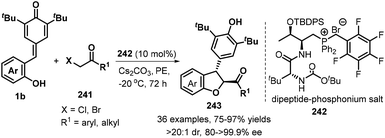 |
| | Scheme 53 [4 + 1] annulation reaction between p-QMs and α-halogenated ketones. | |
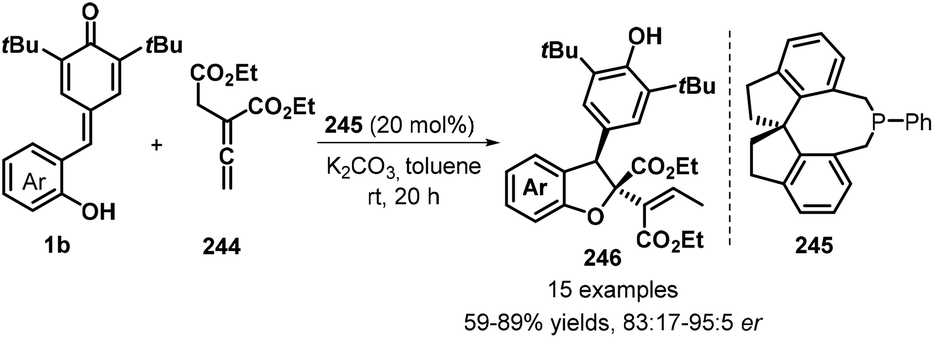
A relatively new development in this area is the use of allenoates as C1 synthons for [4 + 1] annulation. Waser group's research revealed the use of commercially available (R)-SITCP 245 as a chiral spiro phosphine catalyst promotes the reaction of 1b with allenoates 244 to give enantioenriched dihydrobenzofurans 246 as single diastereomers in yields up to 90% and with up to 95:5 er (Scheme 54).84 This methodology could tolerate benzyl and ethyl esters. The extension of this procedure to tert-butyl ester was found to be suitable, but with somewhat lower enantiomeric ratios.
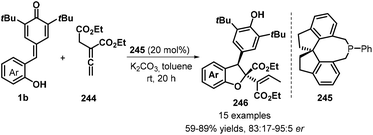 |
| | Scheme 54 Enantioselective catalytic [4 + 1]-cyclization of p-QMs with allenoates. | |
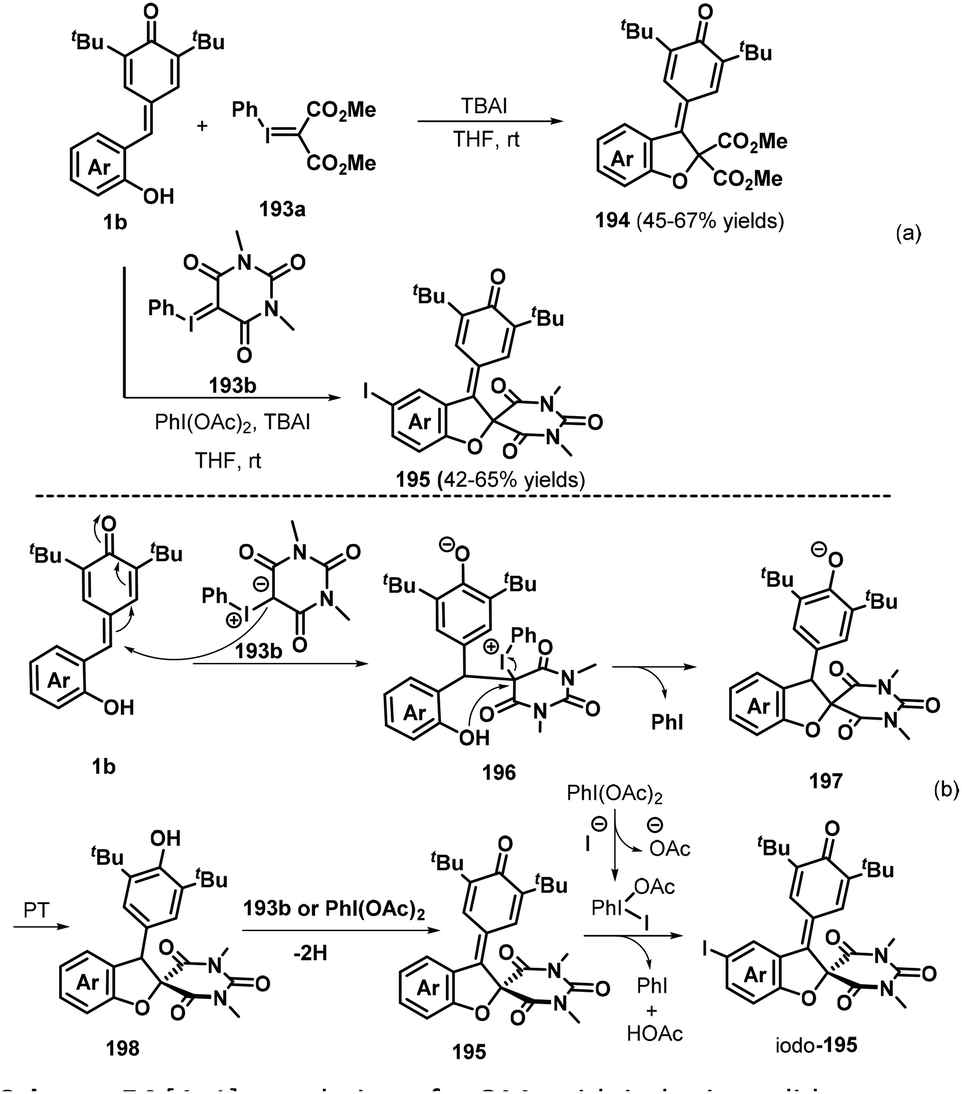
By using acyclic and cyclic phenyl iodonium ylides 247a and 247b as the C1 component, Tu, Jiang, and co-workers recently presented a new dehydrogenative [4 + 1] annulation of o-hydroxyphenyl-substituted p-QM 1b without any base or acid promoter (Scheme 55a).85 This method provided facile and direct access to a wide range of 2,3-dihydrobenzofurans 248 and 249 with retention of the p-QM moiety. Notably, when C4-unsubstituted p-QM 1b was reacted with 247b in the presence of 1.0 equivalents of iodobenzene diacetate (PhI(OAc)2), all of the reactions proceeded readily to afford the corresponding spiro products with C4 iodination of the phenol ring. The phenyl iodonium ylides 247 played a dual role as both a reaction component and an oxidant in this transformation. The reaction pathway involves successive 1,6-nucleophilic addition, proton transfer, nucleophilic substitution, and oxidation (Scheme 55b).
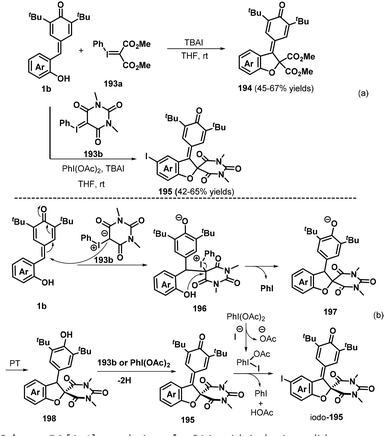 |
| | Scheme 55 [4 + 1] annulation of p-QMs with iodonium ylides. | |
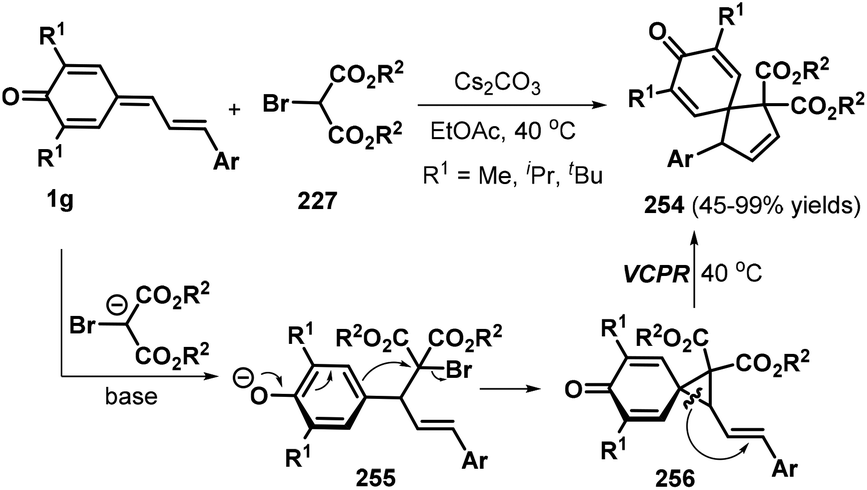
An interesting tandem [4 + 1] annulation between vinylogous p-QMs 1g and 2-bromomalonates 227 was disclosed by Yao, Lin, and co-workers. In the presence of Cs2CO3, the reaction proceeded via sequential 1,6-conjugate addition, cyclization, and vinylcyclopropane rearrangement (VCPR) to provide access to 26 examples of spiro[4.5]cyclohexadienones 254 in 45–99% yields.86 The authors also proposed a plausible mechanism for this transformation. Under basic conditions, 2-bromomalonate 227 is converted to a 2-bromomalonate carbanion, which engages in nucleophilic 1,6-conjugate addition to the vinylogous p-QM 1g to generate intermediate 255. This intermediate then undergoes an intramolecular ring closure reaction to form the key three-membered spiro intermediate 256, which smoothly undergoes VCPR to furnish the product 254 upon increasing the temperature to 40 °C (Scheme 56).
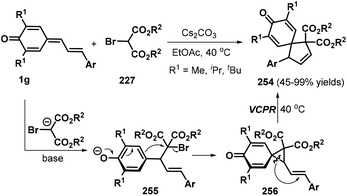 |
| | Scheme 56 [4 + 1] annulation of p-QMs with 2-bromomalonates. | |
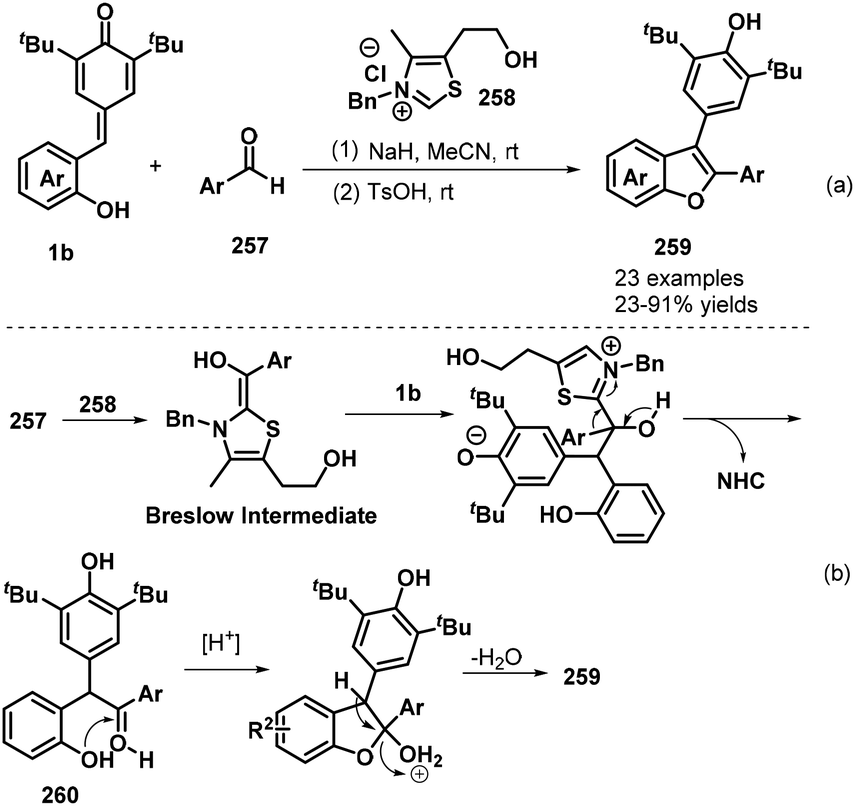
In subsequent work, the Anand group prepared 2,3-diarylbenzo[b]furans 259via an NHC-catalyzed 1,6-conjugate addition of aromatic aldehydes 257 to o-hydroxyphenyl-substituted p-QMs 1b followed by a TsOH-promoted dehydrative annulation reaction (Scheme 57a).87 This one-pot protocol proceeded well for various aldehydes bearing electron-poor, sterically hindered, and heteroaromatic substituents, although aliphatic aldehydes were very sluggish to react and resulted in only poor product yields. The one-pot [4 + 1] annulation involved two steps. The proposed mechanism involves generation of the Breslow intermediate from the aromatic aldehyde 257 and the NHC, 1,6-conjugate addition, regeneration of the NHC catalyst, oxo-cyclization, and dehydration (Scheme 57b).
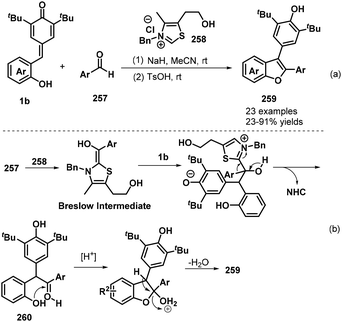 |
| | Scheme 57 NHC-Catalyzed synthesis of 2,3-diarylbenzo[b]furans. | |
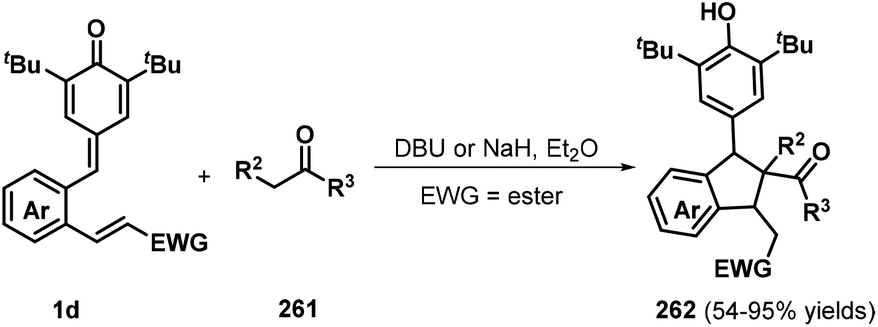
To further expand the variety and applicability of p-QMs, Yao, Huang, and a co-worker introduced a vinyl ester or vinyl sulfone into the phenyl ring of p-QMs to prepare o-vinyl ester or o-vinyl sulfone substituted p-QMs 1d and reacted these compounds with various carbon nucleophiles 261 in the presence of DBU or NaH, which furnished a wide range of functionalized indanes 262 with good yields (Scheme 58). Various carbon nucleophiles such as malonate esters, pentane-2,4-dione, indene-1,3-dione, 1,3-dimethylbarbituric acid, and acetophenone successfully participated in this reaction. In addition, isoindolines were obtained by reacting the p-QMs with p-toluenesulfonamide as a nitrogen nucleophile in the presence of a base, which proceeded via tandem 1,6-conjugate addition/1,4-Michael addition. The authors also attempted to use aniline and benzylamine as nitrogen nucleophiles to further extend the utility of this reaction, although these reactions were unsuccessful.88
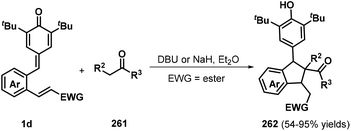 |
| | Scheme 58 Base-mediated synthesis of indanes. | |
2.2.4. 1,6-Addition/[3 + 2] annulation of p-QMs.
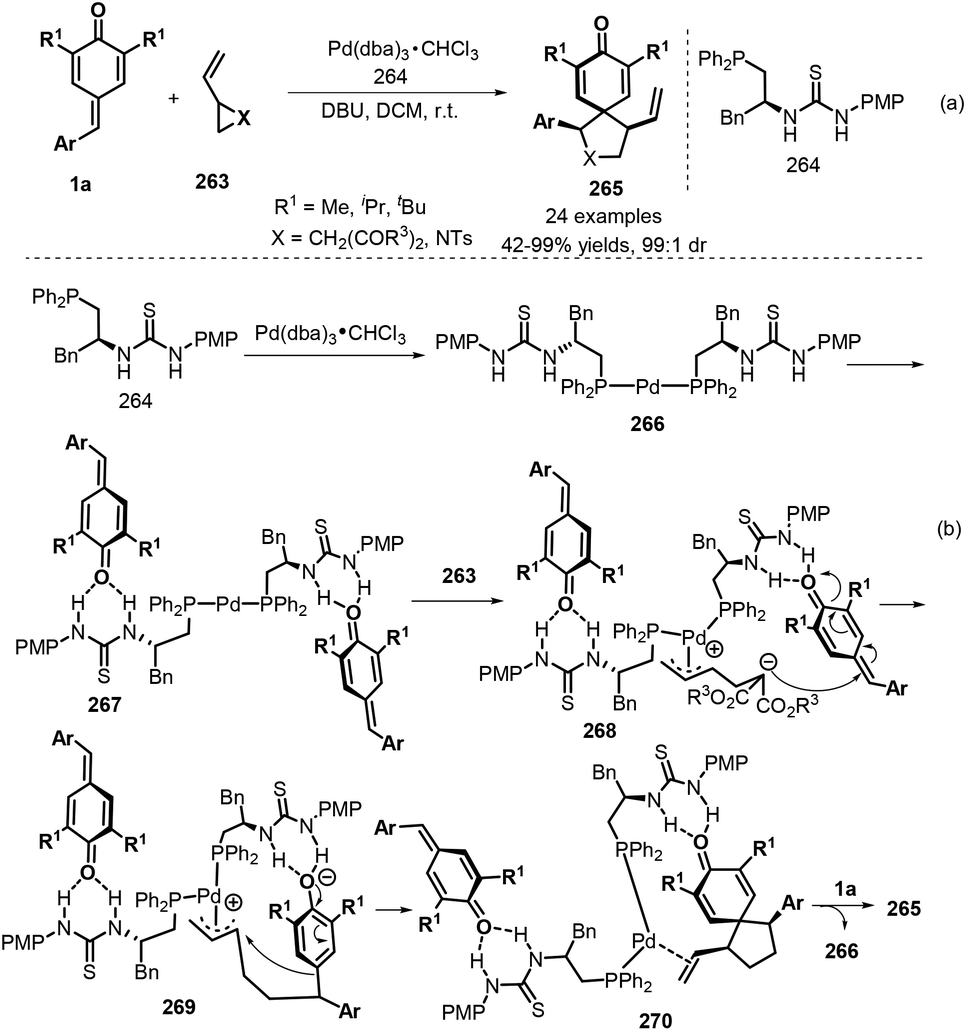
Spirocyclohexadienones are important structures that constitute the core unit of a myriad of natural products with a broad spectrum of biological activities, such as spirobacillene A, stepharine, and annosqualine.89 Furthermore, spirocyclohexadienones are versatile intermediates in the synthesis of important alkaloids and related products.90 In this context, [3 + 2] annulation reactions involving p-QMs have emerged as a useful strategy for the preparation of these compounds. Yao, Lin, and co-workers pioneered a new 1,6-addition/[3 + 2] annulation reaction using p-QMs 1a and vinylcyclopropanes 263, in which the reactivity and diastereoselectivity could be adequately controlled via a palladium/phosphine-thiourea co-catalytic system (Scheme 59a).91 A wide variety of vinylcyclopropanes and 1-tosyl-2-vinylaziridine underwent this reaction to afford spirocyclohexadienones 265 in high yields and diastereoselectivities. Mechanistically, phosphine–Pd(II) complex 266 is first formed from the palladium source and phosphine–thiourea, which then activates p-QM 1a through intermolecular hydrogen-bond interactions to generate complex 267 (Scheme 59b). Then, activated zwitterionic π-allylpalladium species 268 is produced via ring opening of vinylcyclopropane 263. Subsequent 1,6-conjugate addition of the carbanion moiety to p-QM 1a affords intermediate 269, followed by ring closure and de-aromatization to generate complex 270. Disassociation of this complex yields the product 265 and regenerates the catalyst 266 for the next catalytic cycle.
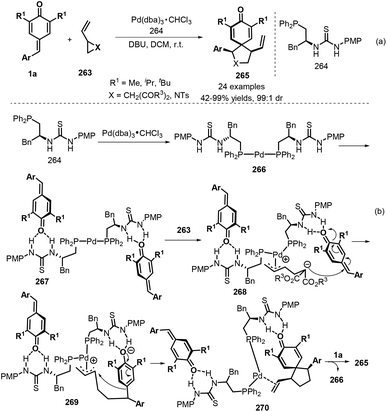 |
| | Scheme 59 [3 + 2] annulation of p-QMs with vinylcyclopropanes. | |
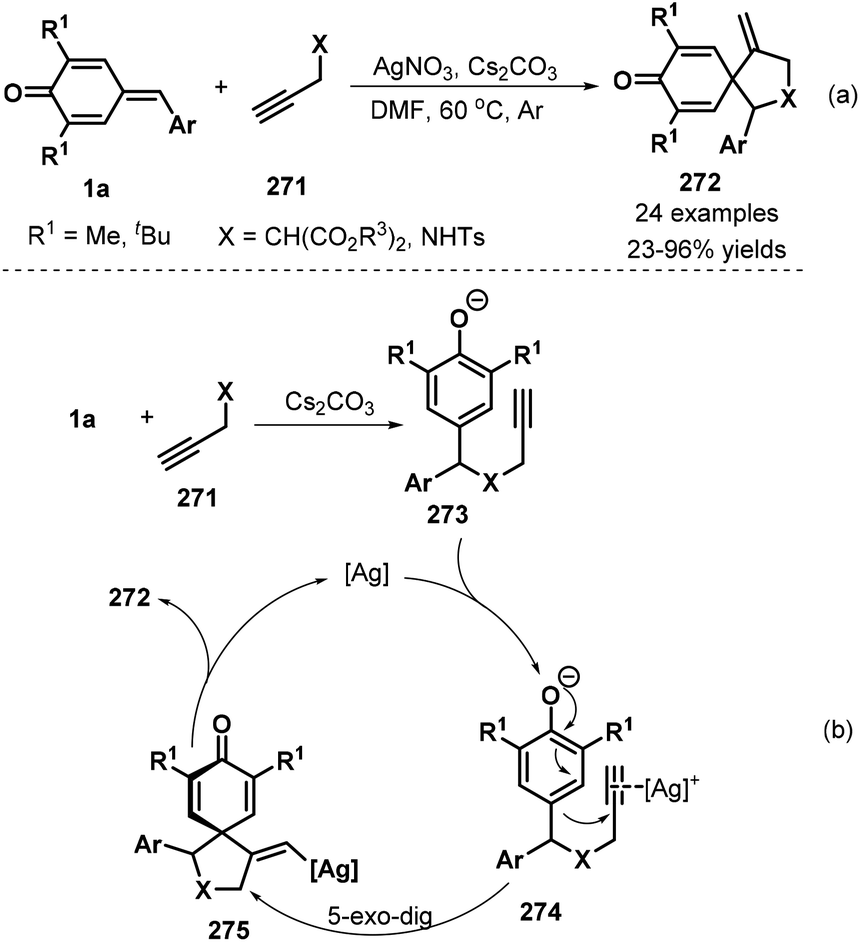
Following the above success, the same group also synthesized spirocyclohexadienones 272 using propargyl malonates 271 as the nucleophiles via silver-catalyzed cascade 1,6-addition/5-exo-dig cyclization (Scheme 60a).92 Various ester substituents, namely, dimethyl, diethyl, diisopropyl, di-tert-butyl, and dibenzyl, were compatible with this reaction. The procedure also tolerated propargyl p-toluenesulfonamide, albeit with low yield. However, internal alkynes did not undergo the reaction. The first step of this reaction is 1,6-addition of p-QM 1a to propargyl malonate 271 in the presence of Cs2CO3 to generate intermediate 273 (Scheme 60b). This intermediate coordinates with the silver catalyst to form intermediate 274, followed by 5-exo-dig cyclization to afford vinyl-silver intermediate 275, which is converted to spirocyclic product 272via protodemetalation with regeneration of the silver catalyst for the next catalytic cycle.
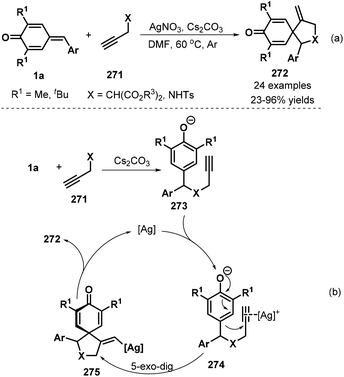 |
| | Scheme 60 [3 + 2] annulation toward spirocyclohexadienones. | |
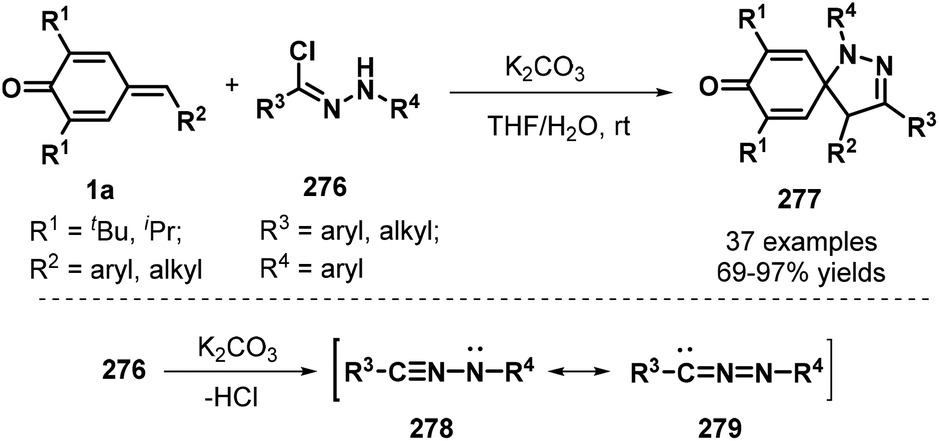
To further broaden the diversity of spirocyclohexadienones, Su and co-workers developed an impressive [3 + 2] cycloaddition between p-QMs 1a and hydrazonyl chlorides276 in the presence of K2CO3, which proceeded with complete regioselectivity to afford spiro-pyrazoline-cyclohexadienone products 277 in 69–97% yields (Scheme 61). The mechanistic pathway involves in situ generation of a nitrile imine with two resonance structures, namely, propargylic 278 and allenic 279, via the base-mediated dehydrochlorination of 276, followed by [3 + 2] cycloaddition.93
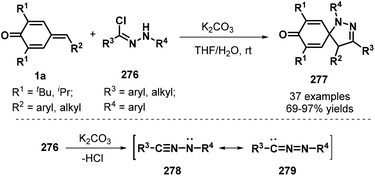 |
| | Scheme 61 [3 + 2] annulation of p-QMs with hydrazonyl chlorides. | |
2.2.5. 1,6-Addition/[4 + 2] annulation of p-QMs.
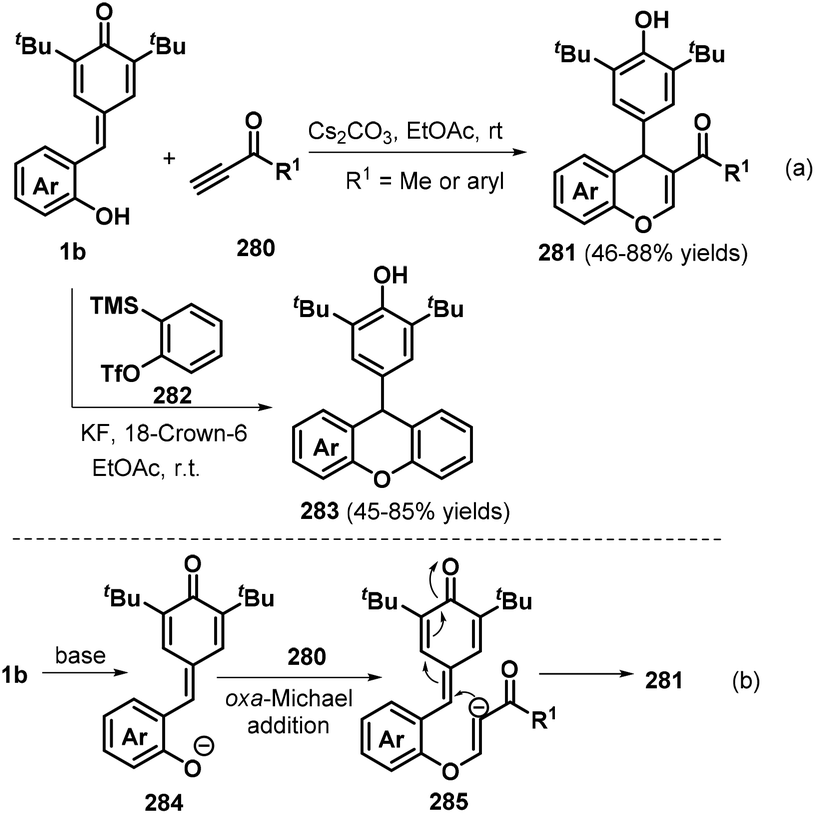
Chromanes and their derivatives constitute a privileged and fascinating class of oxygen-containing frameworks that are commonly found in numerous natural products, synthetic substances, and medicinally relevant compounds.94 To construct these versatile molecules, the Shi group pioneered a new Cs2CO3-promoted [4 + 2] cyclization between o-hydroxyphenyl-substituted p-QMs 1b and ynones 280, which efficiently delivered a range of functionalized chromenes 281 with generally good yields (Scheme 62a). The reaction also worked well upon replacing the ynone component with benzyne precursor 282 in the presence of KF and 18-crown-6 at room temperature, furnishing the corresponding functionalized xanthene 283 in good yields.95 Investigation of the reaction mechanism indicated that the [4 + 2] cyclization is initiated by the deprotonation of p-QM 1b under basic conditions to generate the strongly nucleophilic intermediate 284, which attacks ynone 280via nucleophilic addition to afford intermediate 285, followed by intramolecular 1,6-conjugate addition to yield the desired product 281 (Scheme 62b). Product 283 was formed via a similar pathway. Shortly after, He et al. reported a similar 1,6-addition/[4 + 2] annulation reaction between p-QMs 1b and benzyne precursor 282.96
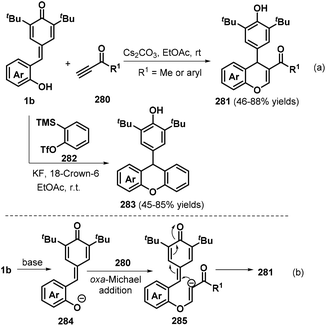 |
| | Scheme 62 [4 + 2] annulation of p-QMs with alkynes. | |
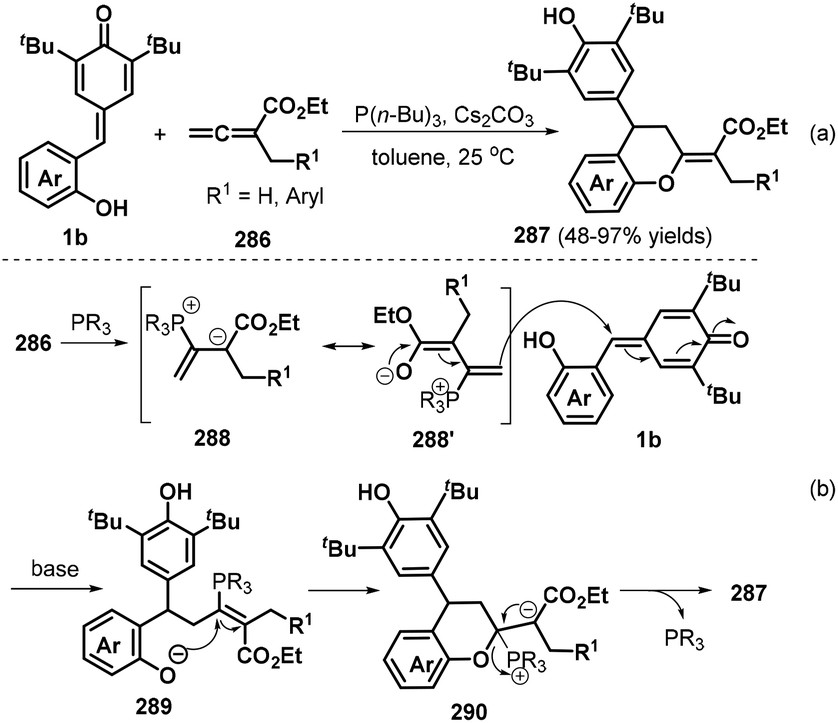
In a subsequent publication, the Shi group also documented an elegant phosphine-catalyzed [4 + 2] annulation between o-hydroxyphenyl-substituted p-QMs 1b and α-substituted allenoates 286 to afford a series of functionalized chroman derivatives 287 in 48–97% yields with complete (E) selectivity (Scheme 63a).97 When the R1 group of the allene component was ethyl or isopropyl, the reaction afforded a complex mixture of products. The proposed reaction mechanism involves activation of allene ester 286 by the phosphine catalyst to generate intermediate 288 and its isomer 288′, followed by 1,6-conjugate addition of the p-QM 1b to afford transient intermediate 289, which undergoes intramolecular nucleophilic addition and elimination of the phosphine catalyst to yield the final product 287 (Scheme 63b).
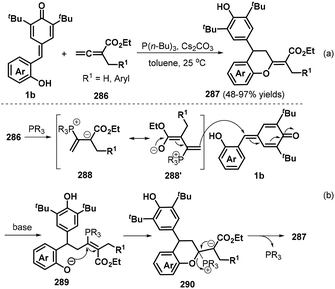 |
| | Scheme 63 Phosphine-catalyzed [4 + 2] annulation of p-QMs. | |
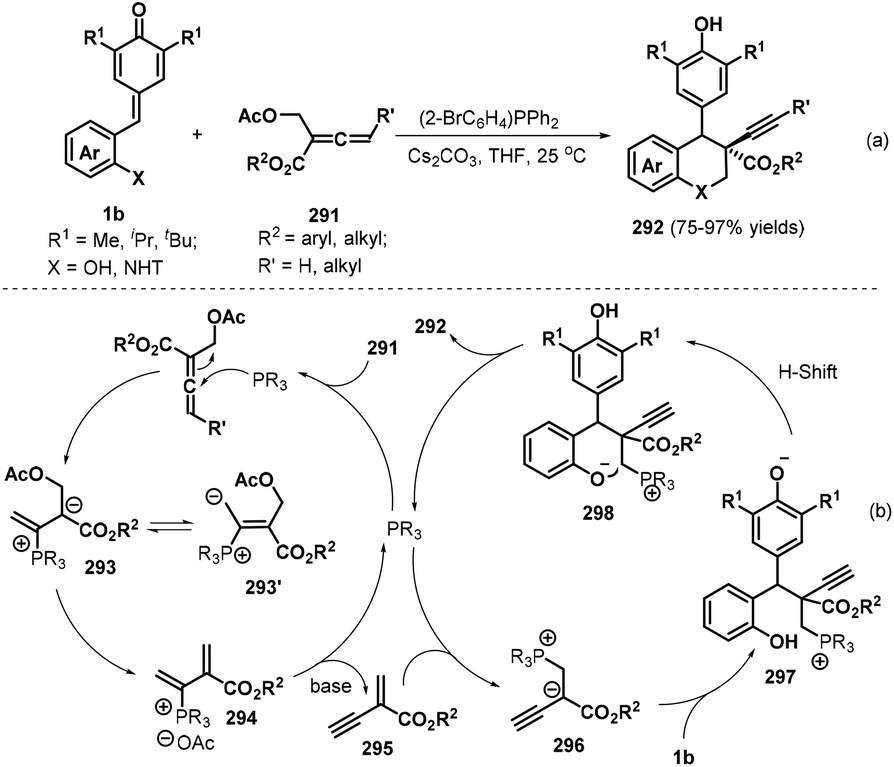
Almost simultaneously, Huang and co-workers reported another interesting phosphine-catalyzed [4 + 2] cycloaddition between p-QMs 1b and α-substituted allenoates 291, which provided 29 examples of chroman and tetrahydroquinoline derivatives 292 bearing an alkynyl-substituted quaternary carbon in up to 97% yield and 20:1 dr (Scheme 64a).98 Both internal and terminal alkynes could be constructed at the quaternary carbon center of the chroman products by using (2-BrC6H4)PPh2 (20 mol%) as the catalyst and Cs2CO3 (2.4 equivalents) as the base. Allenes containing an alkyl substituent and various aromatic rings in the ester moiety, including benzyl, o-fluorobenzyl, 3,5-bis-trifluoromethylbenzyl, and heteroaryl, were found to be well tolerated, with the desired products obtained in high yields with moderate dr values. Importantly, this domino process provided internal alkynes without loss of reactivity when γ-substituted allenes were used. According to the proposed mechanism, this reaction is initiated by nucleophilic attack of the phosphine catalyst on the α-substituted allenoate 291 to form intermediates 293 and 293′, followed by elimination of an acetate group and the phosphine catalyst under basic conditions to generate 1,3-enyne intermediate 295 (Scheme 64b). Subsequent nucleophilic addition of the phosphine catalyst to 295 produces intermediate 296, which undergoes 1,6-conjugate addition to p-QM 1b to afford intermediate 297. Finally, intermediate 297 is transformed into the target compound through proton transfer and nucleophilic cyclization with release of the phosphine catalyst.
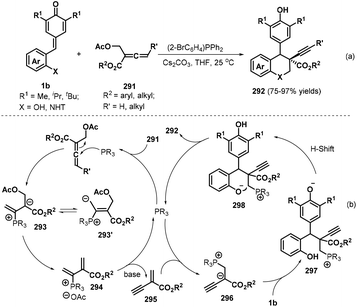 |
| | Scheme 64 [4 + 2] annulation of p-QMs with allenoates. | |
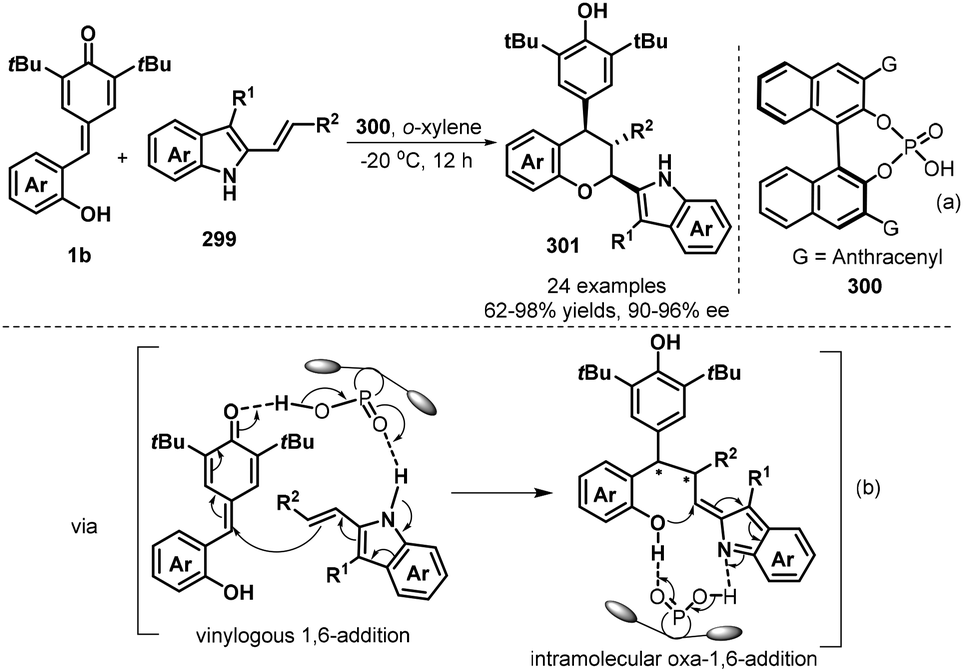
The emergence of organocatalysis opened a new opportunity in catalytic asymmetric [4 + 2] annulation of p-QMs for the synthesis of enantioenriched chromanes. In 2018, Shi and co-workers succeeded in realizing the first catalytic asymmetric [4 + 2] cyclization of p-QMs 1b with electron-rich alkenes 299 by using chiral phosphoric acid 300 (Scheme 65a).99 They discovered that a catalytic amount of chiral phosphoric acid facilitates the reaction of 1b with 3-alkyl-2-vinylindoles bearing various substituents efficiently, affording chiral chromanes products 301 in good yields (63–98%) with high diastereoselectivities (78:22→95:5 dr) and excellent enantioselectivities (90–96% ee). The mechanism is considered to undergo vinylogous 1,6-addition and oxa-1,6-addition. High diastereo- and enantioselectivities were controlled by the hydrogen-bonding network between chiral phosphoric acid 300 and substrates (Scheme 65b).
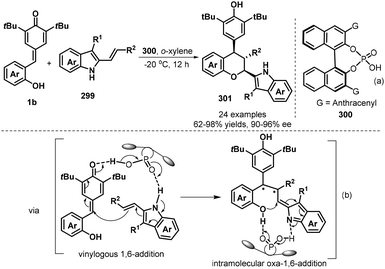 |
| | Scheme 65 Asymmetric [4 + 2] cyclization of p-QMs with 3-alkyl-2-vinylindoles. | |
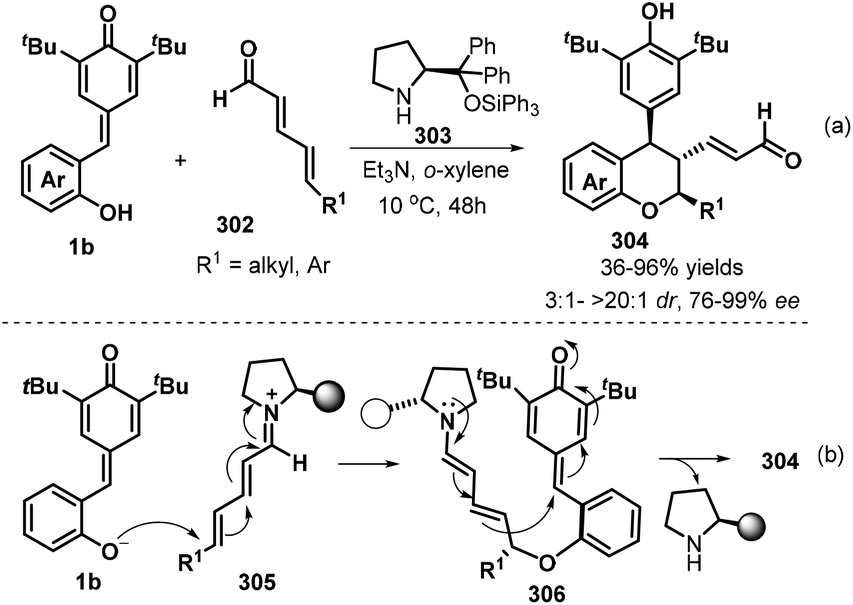
To enrich the library of chiral chromanes, Chatterjee and co-workers elegantly described a secondary amine catalyzed vinylogous double 1,6-addition strategy for the synthesis of enantioenriched poly-substituted chroman derivatives.100 Starting from p-QMs 1b and 2,4-dienal derivatives 302, use of chiral diphenylprolinol silyl ether 303 as a secondary amine catalyst facilitates their transformations in o-xylene in the presence of Et3N to generate chroman derivatives 304 with generally excellent yields (up to 96%) and excellent stereocontrol (up to >20:1 dr, >99% ee, Scheme 66a). A variety of δ-substituted unbiased 2,4-dienals, such as alkyl (ethyl, methyl, n-pentyl, phenylethyl, cyclohexyl) and aryl group, were successfully engaged in this protocol, in which δ-aryl substituted one seemed to be more reluctant to undergo this process as the low yield (36%) and enantioselectivity (76% ee) was obtained. The authors also conducted the late-stage application of some chromans with α,β-unsaturated enal functionality to introduce molecular complexity without loss of enantioselectivity. It is proposed that this reaction triggers by the intermolecular vinylogous iminium-ion based 1,6-addition (oxa-Michael) of the deprotonated oxygen anion of p-QMs 1b to the linear unbiased 2,4-dienal, producing intermediate 305, which is transformed into the targets via an intramolecular vinylogous dienamine-based another 1,6-addition (Scheme 66b).
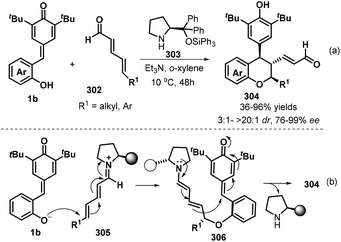 |
| | Scheme 66 Double 1,6-addition for the synthesis of chiral chromans. | |
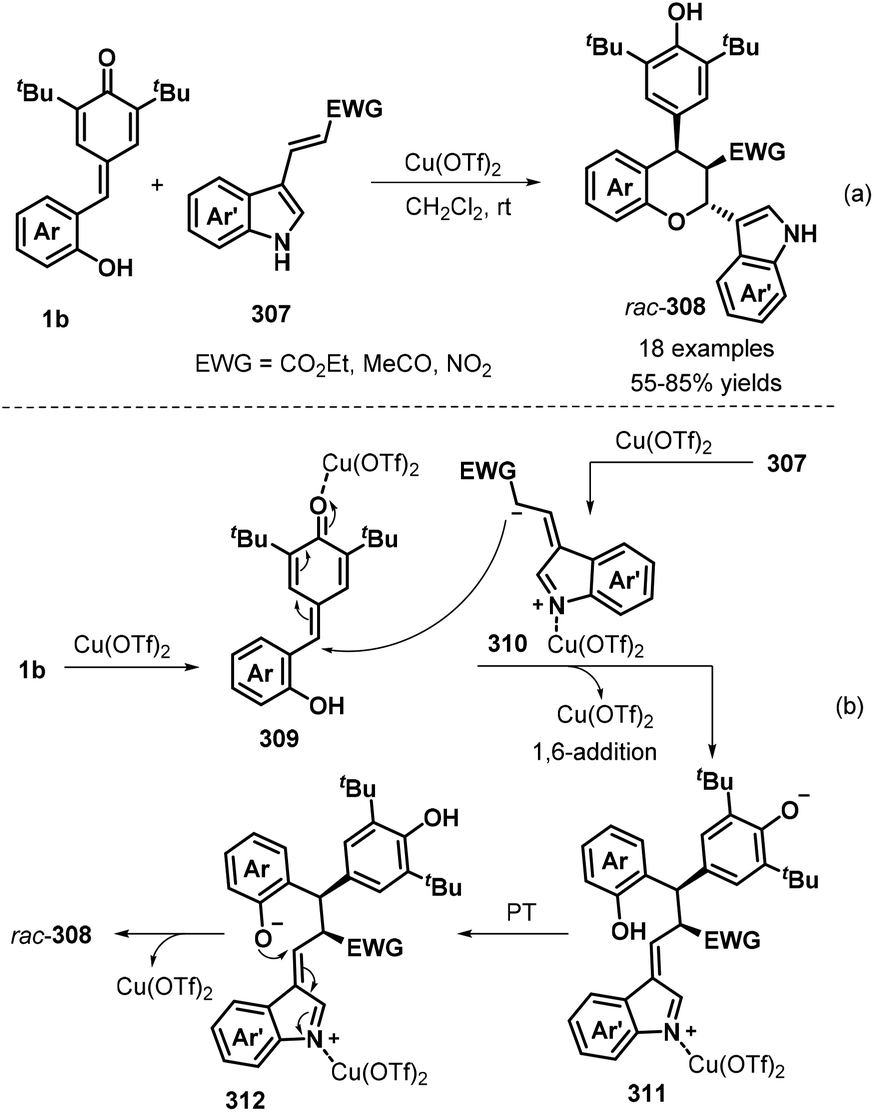
Furthermore, a novel [4 + 2] cycloaddition reaction between p-QMs 1b and electron-poor 3-vinylindoles 307 was investigated by Wang, Jiang, and co-workers (Scheme 67a).101 In the presence of 10 mol% Cu(OTf)2 in DCM at room temperature, the desired polysubstituted chromans 308 were obtained in moderate to good yields and poor diastereoselectivity. The reaction tolerated various substituents, including fluoro, chloro, bromo, cyano, methyl, and methoxy, in the C4 to C6 positions of the indole ring of the electron-poor 3-vinylindole component 307, furnishing the corresponding indole-chroman derivatives 308 in 55–85% yields. It is also worth noting that indoles containing α,β-unsaturated ketone and α,β-unsaturated nitroalkene moieties were well tolerated under these conditions, affording the corresponding products in 59% and 27% yields, respectively. Mechanistic studies revealed that Cu(OTf)2 activates 3-vinylindole 307 to form 1,5-dipolar intermediate 310, followed by sequential 1,6-addition of p-QM 1b in the presence of Cu(OTf)2, proton transfer, and intramolecular 1,4-addition to diastereoselectively afford the final chroman product (Scheme 67b).
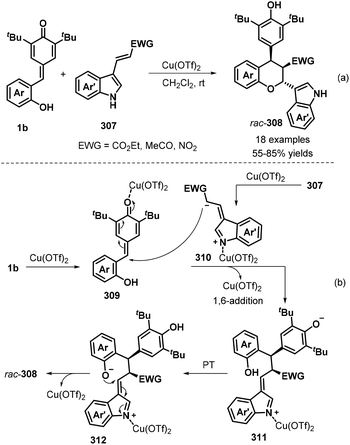 |
| | Scheme 67 [4 + 2] annulation toward chromans. | |
Given the importance of 4-arylchromanones in natural products and pharmaceuticals,102 in 2018, Mei and co-workers employed o-hydroxyphenyl-substituted p-QMs 1b and azlactones 313 as starting materials in a [4 + 2] annulation catalyzed by diphenyl phosphate as a Brønsted acid catalyst to construct highly functionalized 4-arylchromanones 314 in excellent yields and high diastereoselectivity (Scheme 68a).103 In this process, the Brønsted acid plays a dual role, both activating the two substrates and controlling the diastereoselectivity of the reaction via hydrogen-bonding interactions. In the same year, its asymmetric version was achieved by Li's group.104 An employment of chiral phosphoric acid catalyst 315 oriented the complete diastereoselectivity and high enantioselectivity to give enantioenriched 4-arylchromanones 314 in moderate to excellent yields under mild condition. A broad range of p-QMs 1b and azlactones 313 were well-tolerate. Two possible pathways were proposed by the author to explain the stereoselectivity, including (i) [4 + 2] cycloaddition between the enol tautomer of the azlactones and o-QM from 1b (path i) and (ii) 1,6-addition of the enol tautomer of the azlactones to p-QMs 1b. Also, the hydrogen-bonding interaction controls the diastereo- and enantio-selectivity of this asymmetric reaction (Scheme 68b).
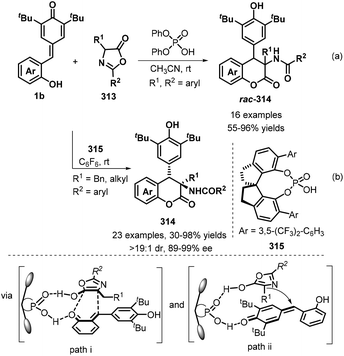 |
| | Scheme 68 [4 + 2] annulation toward 4-arylchromanones. | |
As an expansion of asymmetric synthesis of 4-arylchromanones, very recently, Zhao et al. investigated the asymmetric [4 + 2] annulation reaction between p-QMs 1b and α-isocyanoacetates 316 by using Ag(I)/dihydroquinidine-derived aminophosphine 317 catalyst (Scheme 69).105 The reaction tolerated a wide range of isocyanoacetates and p-QMs, giving rise to the desired 4-arylchromanones 318 in 67–78% yields with excellent diastereoselectivities (>20:1 dr) and good to excellent enantioselectivities (up to 94% ee). However, some less reactive α-substituted isocyanoacetates (R1 = 2-methylphenyl, benzyl and methyl) proved to be unsuited. The authors proposed a plausible transition-state model, which includes the coordination of 317/Ag catalytic system to isocyanides and hydrogen bonding of the protonated quinuclidine with the coordinated ketone oxygen atom.
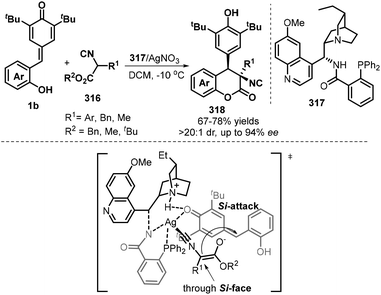 |
| | Scheme 69 Asymmetric synthesis of dihydrocoumarins. | |
Besides the above examples, an achiral [4 + 2] annulation between p-QMs 1b and homophthalic anhydrides 319 followed by esterification with TMSCHN2 in MeOH was found to deliver a wide range of 4-arylchromanones 320 in 44–99% yields and 77:23 to >95:5 dr (Scheme 70).106 The authors examined the substrate scope for the homophthalic anhydride component and demonstrated good tolerance to fluoro and methyl substituents at the C6 and C7 positions of the phenyl ring. The proposed reaction mechanism involves 1,6-addition, ring-opening of the cyclic anhydride, and esterification with TMSCHN2.
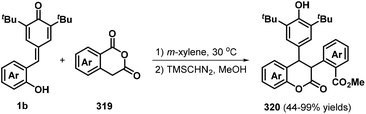 |
| | Scheme 70 [4 + 2] annulation of p-QMs with homophthalic anhydrides. | |
An efficient method for the synthesis of xanthenones and chromenes 322 using p-QMs 1b and β-functionalized ketones 321 as the starting materials was reported by the Kumar group in 2019 (Scheme 71).107 A series of xanthenones and chromenes 322 were obtained in good yields using 10 mol% Tf2NH as a Brønsted acid catalyst in 1,2-dichloroethane (DCE). A number of β-functionalized ketones 321 such as dimedone, cyclohexane-1,3-dione, 4-hydroxy-2H-chromen-2-one, and ethyl 3-oxobutanoate were suitable for this [4 + 2] annulation transformation.
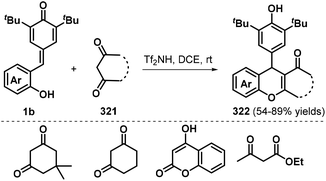 |
| | Scheme 71 [4 + 2] annulation of p-QMs with ketones. | |
Yuan, Yu, and co-workers elegantly orchestrated a stereoselective TsOH-catalyzed [4 + 2] annulation between p-QMs 1b and cyclic enamides 323 under mild conditions to obtain a variety of skeletally diverse chroman-containing heterocyclic compounds 324 in 75% to >99% yields and 2:1 to >20:1 dr (Scheme 72a).108 This protocol tolerated a wide range of cyclic enamides 323, such as 3,4-dihydronaphthalen-1-yl, chromen-4-yl, thiochromen-4-yl, inden-3-yl, 6,7-dihydro-5H-benzo[7]annulen-9-yl, cyclopentenyl, and cycloheptenyl derivatives. The proposed mechanistic pathway involves sequential 1,6-addition and oxa-Mannich reaction. Impressively, the chiral phosphoric acid-catalysed [4 + 2] annulation of these substrates enantioselectively provided a series of acetamido-substituted tetrahydroxanthenes 324 bearing three adjacent stereogenic centers as revealed by Li's group (Scheme 72b).109 Use of BINOL-based chiral phosphoric acid 325 as an efficient catalyst facilitates the generation of tetrahydroxanthenes in 60–99% yields with high diastereo- and enantioselectivities (up to >99:1 dr and 98% ee). Both electron-donating and electron-withdrawing substituents of p-QMs 1b on the phenyl ring and substituents on the aryl moiety of enamides were well tolerated in this asymmetric reaction.
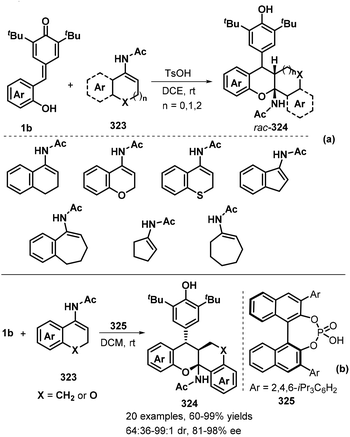 |
| | Scheme 72 [4 + 2] annulation of p-QMs with cyclic enamides. | |
The spirochroman skeleton is a core structural motif present in a range of biologically active molecules and natural products.110 The [4 + 2] annulation between o-hydroxyphenyl-substituted p-QMs and exocylic electron-deficient olefins is one of the most efficient and direct processes for synthesizing functionalized spirochromans. Zhou, Yu, and co-workers reported a mild Et3N-catalyzed [4 + 2] annulation between o-hydroxyphenyl-substituted p-QMs 1b and isoxazolone-derived olefins 326 (Scheme 73a). This reaction proceeded smoothly to generate new syn-spiroisoxazolonechromans 327 in 70–89% yield.111 Notably, aryl- and heteroaryl-substituted unsaturated isoxazolones reacted with complete diastereoselectivity to afford the target products (>99:1 dr), while a cyclohexyl-substituted isoxazolone exhibited a lower yield and diastereoselectivity (2.1:1 dr). Cyclic barbiturate-based olefins 328 also successfully participated in this [4 + 2] annulation reaction to provide a series of new chroman-spirobarbiturates 329 in 90–99% yields and >99:1 dr (Scheme 73b).112 This transformation tolerated a variety of functionalities to provide access to highly functionalized products. The mechanisms of both reactions are expected to involve oxa-Michael reaction and 1,6-addition.
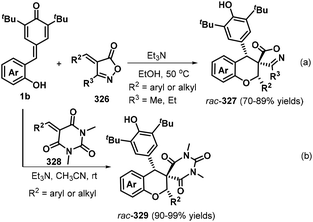 |
| | Scheme 73 Synthesis of spirochromans. | |
Interestingly, Wang's group succeeded in developing an enantioselective formal [4 + 2] annulation of 5-alkenyl thiazolones 330 with p-QMs 1b by employing a dipeptide-based phosphonium salt 331 as phase transfer catalyst, leading to 27 examples of chiral functionalized spiro-chroman-thiazolone compounds 332 bearing three contiguous stereocenters with good yields, excellent diastereo- and enantioselectivities (>20:1 dr and up to >99.9% ee, Scheme 74a).113 Of note, thienyl-substituted thiazolone could work smoothly to access the spirocyclic product in 81% yield but with moderate ee value. The authors proposed the plausible transition state models for the formation of major isomers, which includes oxa-Michael addition and subsequent 1,6-addition. The high enantioselectivity mainly relies on the hydrogen-bonding and ion–pair interactions between the catalyst 331 and generated phenolate anion (Scheme 74b).
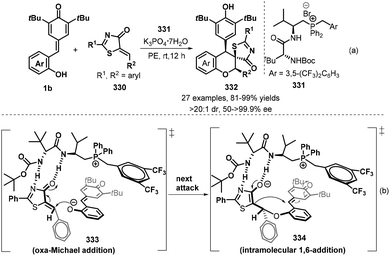 |
| | Scheme 74 Enantioselective construction of chroman-thiazolones. | |
As an important class of O,N-heterocycles, the 1,3-benzoxazine framework is found in a wide range of synthetic compounds exhibiting broad-spectrum biological activities. To obtain these targets, the Shi group synthesized a series of benzoxazin-2-ones 336 with varying degrees of substitution in 38–95% yields through the acid- and base-tunable [4 + 2] annulation between p-QMs 1b and isocyanates 335.114 The choice of Brønsted acid or Brønsted base catalysis was dependent on the isocyanate substituent. 1,1′-Binaphthyl-2,2′-diyl phosphate (BiNPO4H) was found to be an effective Brønsted acid catalyst for the [4 + 2] annulation using isocyanates bearing arylsulfonyl groups, whereas DIPEA was more suitable for isocyanates containing aryl groups, resulting in benzoxazin-2-ones 336 with good to excellent yields. This catalytic [4 + 2] annulation tolerated a variety of electronically distinct substituents at various positions of the phenyl rings of both p-QMs 1b and isocyanates 335 (Scheme 75).
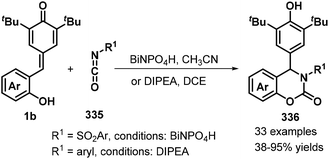 |
| | Scheme 75 [4 + 2] annulation of p-QMs with isocyanates. | |
Subsequent work by Zhao and co-workers revealed that readily available ethyl arylimidates 337 can serve as effective [4 + 2] annulation substrates for the direct synthesis of 2,4-diaryl-1,3-benzoxazines 338 in 37–98% yields (Scheme 76).115 Using 30 mol% FeCl3 as the catalyst, a diverse range of p-QMs and ethyl arylimidates possessing various substituents, including hydrogen, methoxy, methyl, fluoro, chloro, bromo, nitro, and chloromethyl moieties, cyclized smoothly to form the desired 1,3-benzoxazine products. Ethyl arylimidates with electron-donating groups on the phenyl ring generally afforded higher yields than those bearing electron-withdrawing groups.
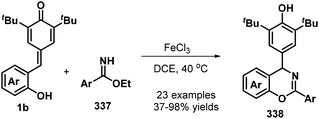 |
| | Scheme 76 Synthesis of 2,4-diaryl-1,3-benzoxazines. | |
2.2.6. 1,6-Addition/[4 + 3] annulation of p-QMs.
As a class of versatile nitrogen–carbon–carbon synthetic building blocks, vinyl aziridines are broadly applied in numerous annulation reactions to construct aza-heterocycles via zwitterionic π-allyl metal intermediates under metal catalysis.116 Very recently, the Shi group accomplished the first iridium-catalyzed [4 + 3] annulation between vinyl aziridines 339 and o-hydroxyphenyl-substituted p-QMs 1b, providing a diastereoselective protocol for the synthesis of seven-membered benzoxazepines 340 in 40–96% yields and 7:3 to >95:5 dr (Scheme 77).117 This transformation tolerated electron-rich, neutral, and electron-poor substituents on the benzenesulfonyl group of the vinyl aziridine component 339 to smoothly furnish products 340 with high diastereoselectivities and generally reasonable yields. Various substituents linked to the olefin moiety, such as cyclic, methyl, phenyl, and ester groups, favored this cyclization. The reaction process is believed to involve in situ generation of a zwitterionic π-allyl iridium species, oxa-allylic addition, and aza-1,6-nucleophilic addition.
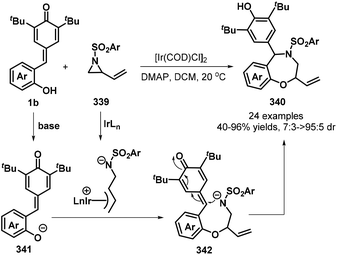 |
| | Scheme 77 [4 + 3] annulation of vinyl aziridines with p-QMs. | |
As an extension of the NHC-catalysis, Enders's group revealed that use of a chiral NHC precursor 344 in the presence of Et3N accelerates enantioselective [4 + 3] cycloaddition of p-QMs 1b with isatin-derived enals 343, leading to spirocyclic oxindole-ε-lactones 345 in high yields (73–93%) and high stereoselectivities (up to 97:3 er, Scheme 78a).118 This new protocol involves a 1,6-addition of the homoenolate equivalent intermediates to the hydroxy donor-1,6-Michael acceptors. Gratifyingly, the isatin-derived enals 343 with varying nitrogen protecting groups (N-methyl, N-ethyl, N-benzyl, N-allyl and N-aryl) all proceeded smoothly and gave products in good yields with high diastereoselectivities and enantiomeric ratios. At the beginning, the addition of the NHC catalyst to the isatin-derived enal gives the Breslow intermediate 346, which reacts with the p-QMs 1b by 1,6-Michael addition to give the adduct 347. Tautomerization of 347 leads to the acyl azolium intermediate 348. The final intramolecular lactonization of 348 furnishes the product 345 and regenerates the NHC catalyst (Scheme 78b). At the same time, Li's group have also developed a similar NHC-catalysis for the synthesis of the same products by adopting NaOAc as a base and DCE as a solvent at room temperature.119
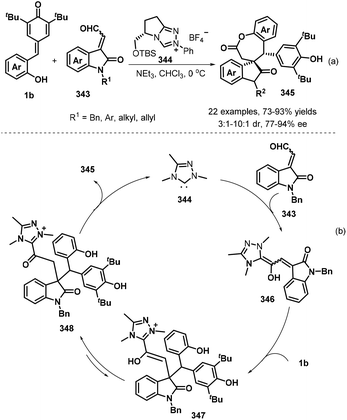 |
| | Scheme 78 Asymmetric synthesis of spiro-oxindole-ε-lactones. | |
2.2.7. 1,6-Addition/bicyclization of p-QMs.
Bicyclization reactions have emerged as an efficient and reliable synthetic tool for the construction of polycyclic ring systems of chemical and biomedical interest, enabling the synergistic utilization of several reactive sites in starting materials to directly establish two new rings in a single step.120 Such transformations possess good bond-forming/annulation efficiency and afford high levels of structural complexity. In recent years, the widespread utilization of bicyclization reactions has paved the way to assembling various fused carbocyclic and heterocyclic molecules that are not accessible via conventional methods.121 By exploiting a bicyclization strategy, Tu, Jiang, and co-workers recently established a new and selective silver/Brønsted acid dual synergistic catalytic system involving β-alkynyl ketones 349 and o-hydroxyphenyl-substituted p-QMs 1b, which permitted the preparation of 25 examples of spiro[chromane-2,1′-isochromene] derivatives 350 in generally good yields and excellent diastereoselectivity (Scheme 79a).122 Notably, the β-alkynyl ketone 349 bearing an n-butyl group on the alkynyl moiety was a suitable substrate, providing the corresponding product as a sole diastereoisomer in 50% yield. However, the β-alkynyl ketone bearing a 1-naphthyl group did not react. The authors found that the key step in the formation of these products is the in situ generation of methyleneisochromenes 351 with a nucleophilic site. A reasonable mechanism for this spiroketalization was proposed (Scheme 79b). Initially, silver-catalyzed 6-endo-dig oxo-cyclization of β-alkynyl ketone 349 generates an isobenzopyrylium intermediate, followed by proton transfer to afford isochromene 351. Next, BiNPO4H-catalyzed 1,6-addition between 351 and 1b yields intermediate 352, which undergoes intramolecular oxa-nucleophilic addition and subsequent proton transfer to afford the final product.
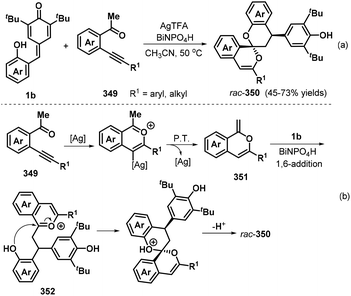 |
| | Scheme 79 Bicyclization of p-QMs with β-alkynyl ketones. | |
Interestingly, the above reaction was also conducted under silver/scandium bimetallic synergistic catalysis, which proceeded in a completely different direction to afford 17 examples of unexpected benzo[c]xanthenes 353 in 55–83% yields (Scheme 80a).123 A wide variety of β-alkynyl ketones possessing a diverse set of substituents at various positions of the arylalkynyl (R1) moiety were perfectly tolerated, providing good product yields in all cases. On the basis of control experiments, the authors postulated that the corresponding 3-arylnaphthalen-1-ol 355 behaves as a key intermediate in this transformation. Mechanistically, silver/scandium-co-catalyzed 6-endo-dig cyclization of the β-alkynyl ketone could generate intermediate 354, followed by proton transfer and tautomerization to afford 1-naphthol 355 (Scheme 80b). Next, scandium-catalyzed 1,6-addition of 355 to 1b yields the adduct intermediate 356, which undergoes proton transfer and intramolecular oxa-nucleophilic addition to furnish intermediate 357. Finally, intermediate 358 is transformed into product 353via deprotonation and dehydration. Subsequently, the Shi group developed a similar protocol for the synthesis of spiroacetal derivatives 360 in excellent yields and good diastereoselectivities from alkynyl benzyl alcohols 359 with Ph3PAuCl as the catalyst (Scheme 81).124 Au-Catalyzed in situ generation of electron-rich vinyl ether intermediate 361 is vital for this transformation, which then undergoes [4 + 2] annulation to yield the spiroacetal product.
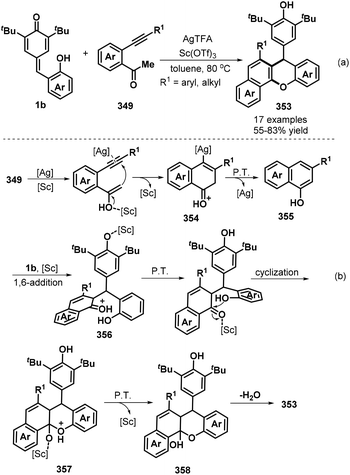 |
| | Scheme 80 Silver/scandium-co-catalyzed bicyclization of p-QMs. | |
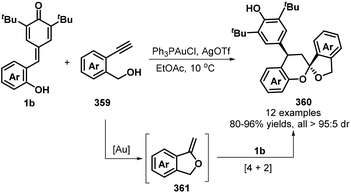 |
| | Scheme 81 Oxa-[4 + 2] cyclization toward spiroacetals. | |
In 2017, Zhao and co-workers established an impressive catalyst-controlled reaction between p-QM aryl esters 1h and activated isocyanides 362 to afford two new types of skeletally diverse heterocycles with high chemo- and stereoselectivity (Scheme 82).125 The combination of Ag2O (5 mol%) and PPh3 (10 mol%) as a catalytic system led to tricyclic ketals 363, whereas oxazole-containing triarylmethanes 364 were produced in good to excellent yields via an unexpected carbon–carbon bond cleavage when the catalytic system was switched to Cu(OAc)2 (10 mol%)/dppp (20 mol%). Variation of the ester moiety in both substrates was well tolerated, and both alkyl and aryl substituents resulted in excellent selectivities. The authors postulated that the key species is intermediate 365, which is highly reactive and spontaneously undergoes subsequent reaction, although this intermediate was not directly observed.
 |
| | Scheme 82 Catalyst-controlled reaction of p-QMs with isocyanides. | |
Anand and co-workers exploited 2-alkenylated p-QMs 1e with Me3SiN3366 as substrates to establish a copper-catalyzed 1,6-conjugate addition reaction followed by intramolecular click cycloaddition, which provided access to a wide range of functionalized 1,2,3-triazole-fused isoindolines 367 in moderate to good yields (Scheme 83).126 Azide 368 was confirmed to be a key intermediate by NMR and IR spectroscopy.
 |
| | Scheme 83 Copper-catalyzed bicyclization of p-QMs with TMSN3. | |
2.3. Other reaction types of p-QMs
Recently, Shi and co-workers established the first [4 + 2] cyclization/retro-Mannich reaction cascade between o-hydroxyphenyl-substituted p-QMs 1b and vinyl benzoxazinanones 288 to realize carbon–carbon double bond cleavage (Scheme 84a).127 A series of vinyl benzoxazinanones 369 bearing various substituents, such as fluoro, chloro, bromo, methyl, and methoxy groups, at different positions (C5, C6, C7, and C8) were all applicable in this reaction, providing the products 370 in good to excellent yields. Two plausible reaction pathways were proposed (Scheme 84b). First, palladium-containing 1,4-dipolar compound 371 could be generated via decarboxylation of vinyl benzoxazinanone 369. In pathway a, the [4 + 2] cyclization occurs via a domino 1,6-addition/allylation dearomatization sequence. In pathway b, owing to the small isomerization energy of 6.7 kcal mol−1, p-QM 1b is transformed into o-QM 1b′, followed by [4 + 2] cyclization via a domino 1,4-addition/allylation dearomatization sequence. Both pathways would afford the same [4 + 2] annulation product as transient intermediate 372, which rapidly undergoes a retro-Mannich reaction to yield final product 370.
 |
| | Scheme 84 Catalytic CC bond cleavage of p-QMs. | |
3. Conclusions
In summary, various categories of reactions involving p-QMs, such as 1,6-addition, annulation, radical-induced cyclization, and radical–radical cross-coupling, have been explored in this review. The numerous examples of successful transformations expounded herein clearly demonstrate that p-QMs can serve as highly reactive and versatile synthons for the direct formation of multiple carbon–carbon and carbon–heteroatom bonds by acting as Michael acceptors or radical acceptors. The reactions highlighted in this review are expected to find great application in the fields of organic chemistry and pharmaceutical chemistry to provide more efficient access to important building blocks in organic synthesis, drug discovery, and materials science. Despite the tremendous advances and achievements made so far, there remain many opportunities and challenges for further exploration in this field. For example, 1,6-nucleophilic addition and annulation reactions of p-QMs using reactive oxygen-containing nucleophiles or S-, P-, or N-centered compounds as radical donors, especially with enantioselective control, have yet to be comprehensively investigated. Furthermore, the introduction of highly reactive substituents to prepare new types of p-QMs and further exploration of their reactivity profiles via 1,6-nucleophilic addition and/or annulation reactions would be of considerable interest. The substrate dependence also remains an important issue for some of the presented reactions.
Conflicts of interest
There is no conflicts of interest.
Acknowledgements
We are grateful for financial support from the NSFC (No. 21871112 and 21971090).
Notes and references
-
(a) K.-I. Takao, T. Sasaki, T. Kozaki, Y. Yanagisawa, K.-I. Tadano, A. Kawashima and H. Shinonaga, Syntheses and Absolute Stereochemistries of UPA0043 and UPA0044, Cytotoxic Antibiotics Having a p-Quinone-methide Structure, Org. Lett., 2001, 3, 4291–4294 CrossRef CAS PubMed;
(b) R. Jansen, K. Gerth, H. Steinmetz, S. Reinecke, W. Kessler, A. Kirschning and R. Müller, Elansolid A3, a Unique p-Quinone Methide Antibiotic from Chitinophaga sancti, Chem. – Eur. J., 2011, 17, 7739–7744 CrossRef CAS PubMed;
(c) Y. Yuan, H. Men and C. Lee, Total Synthesis of Kendomycin: A Macro–C–Glycosidation Approach, J. Am. Chem. Soc., 2004, 126, 14720–14721 CrossRef CAS PubMed;
(d) A. B. Smith, E. F. Mesaros and E. A. Meyer, J. Am. Chem. Soc., 2006, 128, 5292–5299 CrossRef CAS PubMed;
(e) S. M. Kupchan, A. Karim and C. Marcks, Evolution of a Total Synthesis of (−)-Kendomycin Exploiting a Petasis–Ferrier Rearrangement/Ring-Closing Olefin Metathesis Strategy, J. Am. Chem. Soc., 1968, 90, 5923–5924 CrossRef CAS PubMed.
- G. J. Leary, Quinone methides and the structure of lignin, Wood Sci. Technol., 1980, 14, 21–34 CrossRef CAS.
- A. A. Larsen, Catecholamine Chemical Species at the Adrenergic Receptors, Nature, 1969, 224, 25–27 CrossRef CAS PubMed.
-
(a) D. Hamels, P. M. Dansette, E. A. Hillard, S. Top, A. Vessières, P. Herson, G. Jaouen and D. Mansuy, Ferrocenyl Quinone Methides as Strong Antiproliferative Agents: Formation by Metabolic and Chemical Oxidation of Ferrocenyl Phenols, Angew. Chem., Int. Ed., 2009, 48, 9124–9126 CrossRef CAS PubMed;
(b) G. B. Messiano, T. da Silva, I. R. Nascimento and L. M. Lopes, Biosynthesis of antimalarial lignans from Holostylis reniformis, Phytochemistry, 2009, 70, 590–596 CrossRef CAS PubMed.
- Q. Zhou, Y. Qu, J. B. Mangrum and X. Wang, DNA Alkylation with N-Methylquinolinium Quinone Methide to N2-dG Adducts Resulting in Extensive Stops in Primer Extension with DNA Polymerases and Subsequent Suppression of GFP Expression in A549 Cells, Chem. Res. Toxicol., 2011, 24, 402–411 Search PubMed.
- P. Wang, Y. Song, L. Zhang, H. He and X. Zhou, Quinone Methide Derivatives: Important Intermediates to DNA Alkylating and DNA Cross-linking Actions, Curr. Med. Chem., 2005, 12, 2893–2913 CrossRef CAS PubMed.
-
(a) W.-D. Chu, L.-F. Zhang, X. Bao, X.-H. Zhao, C. Zeng, J.-Y. Du, G. Zhang, F.-X. Wang, X.-Y. Ma and C.-A. Fan, Asymmetric Catalytic 1,6-Conjugate Addition/Aromatization of para-Quinone Methides: Enantioselective Introduction of Functionalized Diarylmethine Stereogenic Centers, Angew. Chem., 2013, 125, 9399 CrossRef;
(b) L. Caruana, F. Kniep, T. K. Johansen, P. H. Poulsen and K. A. Jørgensen, A New Organocatalytic Concept for Asymmetric α-Alkylation of Aldehydes, J. Am. Chem. Soc., 2014, 136, 15929 CrossRef CAS PubMed.
-
(a) L.-L. Zhang, X. Zhou, P.-F. Li, Z.-T. Liu, Y. Liu, Y. Sun and W.-J. Li, Asymmetric synthesis of chromene skeletons via organocatalytic domino reactions of in situ generated ortho-quinone methide with malononitrile and β-functionalized ketone, RSC Adv., 2017, 7, 39216–39220 RSC;
(b) Z.-P. Zhang, K.-X. Xie, C. Yang, M. Li and X. Li, Asymmetric Synthesis of Dihydrocoumarins through Chiral Phosphoric Acid-Catalyzed Cycloannulation of para-Quinone Methides and Azlactones, J. Org. Chem., 2018, 83, 364–373 CrossRef CAS PubMed;
(c) C. Ma, Y. Huang and Y. Zhao, Stereoselective 1,6-Conjugate Addition/Annulation of para-Quinone Methides with Vinyl Epoxides/Cyclopropanes, ACS Catal., 2016, 6, 6408–6412 CrossRef CAS;
(d) S. Kim, Y. Kitano, M. Tada and K. Chiba, Alkylindan synthesis via an intermolecular [3 + 2] cycloaddition between unactivated alkenes and in situ generated p-quinomethanes, Tetrahedron Lett., 2000, 41, 7079–7083 CrossRef CAS.
-
(a) W. Li, X. Xu, P. Zhang and P. Li, Recent Advances in the Catalytic Enantioselective Reactions of para-Quinone Methides, Chem. – Asian J., 2018, 13, 2350–2359 CrossRef CAS PubMed;
(b) L. Caruana, M. Fochi and L. Bernardi, The Emergence of Quinone Methides in Asymmetric Organocatalysis, Molecules, 2015, 20, 11733–11764 CrossRef CAS PubMed;
(c) P. Chauhan, U. Kaya and D. Enders, Advances in Organocatalytic 1,6-Addition Reactions: Enantioselective Construction of Remote Stereogenic Centers, Adv. Synth. Catal., 2017, 359, 888–912 CrossRef CAS;
(d) A. Parra and M. Tortosa,
para-Quinone Methide: a New Player in Asymmetric Catalysis, ChemCatChem, 2015, 7, 1524–1526 CrossRef CAS.
-
(a) S. H. Cho, J. Yoon and S. Chang, Intramolecular Oxidative C–N Bond Formation for the Synthesis of Carbazoles: Comparison of Reactivity between the Copper-Catalyzed and Metal-Free Conditions, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed;
(b) K. Xu, Y. Hu, S. Zhang, Z. Zha and Z. Wang, Direct Amidation of Alcohols with N-Substituted Formamides under Transition-Metal-Free Conditions, Chem. – Eur. J., 2012, 18, 9793–9797 CrossRef CAS PubMed;
(c) S. Cai, Y. Xu, D. Chen, L. Li, Q. Chen, M. Huang and W. Weng, Visible-Light-Enabled Decarboxylative Sulfonylation of Cinnamic Acids with Sulfonylhydrazides under Transition-Metal-Free Conditions, Org. Lett., 2016, 18, 2990–2993 CrossRef CAS PubMed;
(d) G. C. Senadi, B.-C. Guo, W.-P. Hu and J.-J. Wang, Iodine-promoted cyclization of N-propynyl amides and N-allyl amides via sulfonylation and sulfenylation, Chem. Commun., 2016, 52, 11410–11413 RSC;
(e) J. Wen, W. Wei, S. Xue, D. Yang, Y. Lou, C. Gao and H. J. Wang, Metal-Free Oxidative Spirocyclization of Alkynes with Sulfonylhydrazides Leading to 3-Sulfonated Azaspiro[4,5]trienones, J. Org. Chem., 2015, 80, 4966–4972 CrossRef CAS PubMed.
-
(a) C. B. Rao, B. Chinnababu and Y. Venkateswarlu, An Efficient Protocol for Alcohol Protection Under Solvent-and Catalyst-Free Conditions, J. Org. Chem., 2009, 74, 8856–8858 CrossRef CAS PubMed;
(b) P. K. Khatri, S. L. Jain and B. Sain, Ultrasound-Promoted Oxidation of Sulfides with Hydrogen Peroxide under Catalyst-Free Conditions, Ind. Eng. Chem. Res., 2011, 50, 701–704 CrossRef CAS;
(c) A. Kumar, M. Kumar and K. Gupta, Catalyst-free hydroarylation of in situ generated ortho-quinone methide (o-QM) with electron rich arenes in water, Green Chem., 2012, 14, 2677–2681 RSC;
(d) Y. Yang, L. Tang, S. Zhang, X. Guo, Z. Zha and Z. Wang, Catalyst-free sulfonylation of activated alkenes for highly efficient synthesis of mono-substituted ethyl sulfones in water, Green Chem., 2014, 16, 4106–4109 RSC;
(e) J. Jiang, Y. Wu, R. Wang, Y. He, C. Xu, W. Deng, Z. Zeng and J. Xiang, A direct catalyst-free and stereoselective sulfonylation of propargyl esters for efficient synthesis of (Z)-β-sulfonyl enoates in water, Tetrahedron, 2016, 72, 7895–7900 CrossRef CAS;
(f) B. Li, Y. Li, L. Yu, X. Wu and W. Wei, Metal-free sulfonylation of quinones with sulfonyl hydrazides in water: Facile access to mono-sulfonylated hydroquinones, Tetrahedron, 2017, 73, 2760–2765 CrossRef CAS.
- A. S. Jadhav and R. V. Anand, 1,6-Conjugate addition of zinc alkyls to para-quinone methides in a continuous-flow microreactor, Org. Biomol. Chem., 2017, 15, 56–60 RSC.
- N. Molleti and J. Y. Kang, Synthesis of Diaryl Diazaphosphonates via 1,6-Hydrophosphonylation of p-Quinone Methides with N-Heterocyclic Phosphine–Thioureas, Org. Lett., 2017, 19, 958–961 CrossRef CAS PubMed.
- T. Liu, J. Liu, S. Xia, J. Meng, X. Shen, X. Zhu, W. Chen, C. Sun and F. Cheng, Catalyst-Free 1,6-Conjugate Addition/Aromatization/Sulfonylation of para-Quinone Methides: Facile Access to Diarylmethyl Sulfones, ACS Omega, 2018, 3, 1409–1415 CrossRef CAS PubMed.
- X.-Y. Guan, L.-D. Zhang, P.-S. You, S.-S. Liu and Z.-Q. Liu, 1,6-Conjugate sulfonylation of para-quinone methides: An expedient approach to unsymmetrical gem-diarylmethyl sulfones, Tetrahedron Lett., 2019, 60, 244–247 CrossRef CAS.
- K. Chen, W.-J. Hao, S.-J. Tu and B. Jiang, Metal-free synthesis of triarylated (Z)-nitrones via H2O-mediated 1,3-dipolar transfer under aerobic conditions, Green Chem., 2019, 21, 675–683 RSC.
-
(a) T. P. Pathak and M. S. Sigman, Applications of ortho-Quinone Methide Intermediates in Catalysis and Asymmetric Synthesis, J. Org. Chem., 2011, 76, 9210 CrossRef CAS PubMed;
(b) N. J. Willis and C. D. Bray,
ortho-Quinone Methides in Natural Product Synthesis, Chem. – Eur. J., 2012, 18, 9160 CrossRef CAS PubMed;
(c) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, The Domestication of ortho-Quinone Methides, Acc. Chem. Res., 2014, 47, 3655 CrossRef CAS PubMed;
(d) M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury,
ortho-Quinone methide (o-QM): a highly reactive, ephemeral and versatile intermediate in organic synthesis, RSC Adv., 2014, 4, 55924 RSC;
(e) A. A. Jaworski and K. A. Scheidt, Emerging Roles of in Situ Generated Quinone Methides in Metal-Free Catalysis, J. Org. Chem., 2016, 81, 10145 CrossRef CAS PubMed;
(f) Z. Wang and J. Sun, Recent Advances in Catalytic Asymmetric Reactions of o-Quinone Methides, Synthesis, 2015, 47, 3629 CrossRef;
(g) L. Caruana, M. Fochi and L. Bernardi, The Emergence of Quinone Methides in Asymmetric Organocatalysis, Molecules, 2015, 20, 11733 CrossRef CAS PubMed;
(h) A. Parra and M. Tortosa,
para-Quinone Methide: a New Player in Asymmetric Catalysis, ChemCatChem, 2015, 7, 1524 CrossRef CAS;
(i) P. Chauhan, U. Kaya and D. Enders, Advances in Organocatalytic 1,6-Addition Reactions: Enantioselective Construction of Remote Stereogenic Centers, Adv. Synth. Catal., 2017, 359, 888 CrossRef CAS.
- B.-Q. Wang, Q. Luo, Q. Xiao, J. Lin and S.-J. Yan, Synthesis of Quinone Methide Substituted Neonicotinoid Derivatives via 1,6-Conjugate Addition of N-Benzyl Nitro Ketene Aminals with para-Quinone Methides Accompanying Oxidation, ACS Sustainable Chem. Eng., 2017, 5, 8382–8389 CrossRef CAS.
- A. K. Gupta, S. Ahamad, N. K. Vaishanv, R. Kantc and K. Mohanan, Base-mediated 1,6-conjugate addition of the Seyferth–Gilbert reagent to para-quinone methides, Org. Biomol. Chem., 2018, 16, 4623 RSC.
-
(a) N. K. Vaishanv, A. K. Gupta, R. Kant and K. Mohanan, Fluoride-Mediated α-Selective 1,6-Conjugate Addition of Allenic Esters to p-Quinone Methides, J. Org. Chem., 2018, 83, 8759–8767 CrossRef CAS PubMed;
(b) V. R. Reddy, N. Maripally, R. Mutyala, J. B. Nanubolu and R. Chandra, DMAP catalysed vinylogous Rauhut–Currier reaction of allenoates with para-quinone methides, Tetrahedron Lett., 2018, 59, 2631–2635 CrossRef CAS.
- D. Roy and G. Panda, Base-Mediated 1,6-Aza-Michael Addition of Heterocyclic Amines and Amides to para-Quinone Methides Leading to Meclizine-, Hydroxyzine- and Cetirizine-like Architectures, Synthesis, 2019, 51, 4434–4442 CrossRef CAS.
- S. Gao, X. Xu, Z. Yuan, H. Zhou, H. Yao and A. Lin, 1,6-Addition Arylation of para-Quinone Methides: An Approach to Unsymmetrical Triarylmethanes, Eur. J. Org. Chem., 2016, 3006–3012 CrossRef CAS.
- S. Mahesh, G. Kant and R. V. Anand, B(C6F5)3 catalysed 1,6-conjugate
allylation of para-quinone methides: expedient access to allyl diarylmethanes, RSC Adv., 2016, 6, 80718–80722 RSC.
- Z.-P. Zhang, N. Dong and X. Li, Bismuth-catalyzed allylation of para-quinone methides, Chem. Commun., 2017, 53, 1301–1304 RSC.
- S. Mahesh and R. V. Anand, B(C6F5)3 catalysed reduction of para-quinone methides and fuchsones to access unsymmetrical diaryl- and triarylmethanes: elaboration to beclobrate, Org. Biomol. Chem., 2017, 15, 8393 RSC.
- B. Huang, Y. Shen, Z. Mao, Y. Liu and S. Cui, Metathesis Reaction of Diazo Compounds and para-Quinone Methides for C–C Double Bond Formation: Synthesis of Tetrasubstituted Alkenes and Quinolinones, Org. Lett., 2016, 18, 4888–4891 CrossRef CAS PubMed.
- K.-X. Xie, Z.-P. Zhang and X. Li, Bismuth Triflate-Catalyzed Vinylogous Nucleophilic 1,6-Conjugate Addition of para-Quinone Methides with 3-Propenyl-2-silyloxyindoles, Org. Lett., 2017, 19, 6708–6711 CrossRef CAS.
- A. S. Jadhav, Y. A. Pankhade, R. Hazra and R. V. Anand, 1,6-Hydroolefination and Cascade Cyclization of p-Quinone Methides with Styrenes: Total Synthesis of (±)-Isopaucifloral F, J. Org. Chem., 2018, 83, 10107–10119 CrossRef CAS PubMed.
- B. M. Sharma, D. R. Shinde, R. Jain, E. Begari, S. Satbhaiya, R. G. Gonnade and P. Kumar, Unravelling the Nucleophilicity of Butenolides for 1,6-Conjugate Addition to p-Quinone Methides: A Direct Access to Diversely Substituted Butenolide-Derived Diarylmethanes, Org. Lett., 2018, 20, 2787–2791 CrossRef CAS PubMed.
- J. Rathoda, B. M. Sharmaa, P. S. Malic and P. Kumar, Tf2NH-Catalyzed 1,6-Conjugate Addition of Vinyl Azides with p-Quinone Methides: A Mild and Efficient Method for the Synthesis of β-Bis-Arylamides, Synthesis, 2017, 49, 5224–5230 CrossRef.
- S. R. Shirsath, G. H. Shinde, A. C. Shaikh and M. Muthukrishnan, Accessing α-Arylated Nitriles via BF3·OEt2 Catalyzed Cyanation of para-Quinone Methides Using tert-Butyl Isocyanide as a Cyanide Source, J. Org. Chem., 2018, 83, 12305–12314 CrossRef CAS PubMed.
- R. Li, Y. Chen, K. Jiang, F. Wang, C. Lu, J. Nie, Z. Chen, G. Yang, Y.-C. Chen, Y. Zhao and C. Ma, B(C6F5)3-Catalyzed redox-neutral β-alkylation of tertiary amines using p-quinone methides via borrowing hydrogen, Chem. Commun., 2019, 55, 1217–1220 RSC.
-
(a)
E. W. Colvin, Silicon Reagents in Organic Synthesis, Academic Press, London, 1988 Search PubMed;
(b)
Chemistry of Organosilicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley-VCH, New York, 2001, vol. 3 Search PubMed;
(c) W. Bains and R. Tacke, Silicon chemistry as a novel source of chemical diversity in drug design, Curr. Opin. Drug Discovery Dev., 2003, 6, 526 CAS;
(d) G. A. Showwell and J. S. Mills, Chemistry challenges in lead optimization: silicon isosteres in drug discovery, Drug Discovery Today, 2003, 8, 551 CrossRef;
(e) M. Mortensen, R. Husmann, E. Veri and C. Bolm, Synthesis and applications of silicon-containing α-amino acids, Chem. Soc. Rev., 2009, 38, 1002 RSC.
- A. López, A. Parra, C. Jarava-Barrera and M. Tortosa, Copper-catalyzed silylation of p-quinone methides: new entry to dibenzylic silanes, Chem. Commun., 2015, 51, 17684–17687 RSC.
- C. Yang, S. Gao, H. Yao and A. Lin, Rhodium-Catalyzed Hydroacylation of para-Quinone Methides with Salicylaldehydes: An Approach to α,α-Diaryl-2-Hydroxy Acetophenones, J. Org. Chem., 2016, 81, 11956–11964 CrossRef CAS PubMed.
- Z. Liu, H. Xu, T. Yao, J. Zhang and L. Liu, Catalyst-Enabled Chemodivergent Construction of Alkynyl- and Vinyl-Substituted Diarylmethanes from p-Quinone Methides and Alkynes, Org. Lett., 2019, 21, 7539–7543 CrossRef CAS PubMed.
- R. Zhuge, L. Wu, M. Quan, N. Butt, G. Yang and W. Zhang, Palladium-Catalyzed Addition of Arylboronic Acids to para-Quinone Methides for Preparation of Diarylacetates, Adv. Synth. Catal., 2017, 359, 1028–1036 CrossRef CAS.
- B. Yang, W. Yao, X.-F. Xia and D. Wang, Mn-Catalyzed 1,6-conjugate addition/aromatization of para-quinone methides, Org. Biomol. Chem., 2018, 16, 4547 RSC.
- B. Xiong, G. Wang, C. Zhou, Y. Liu, W. Xu, W.-Y. Xu, C.-A. Yang and K.-W. Tang, Base-Catalyzed Stereoselective 1,6-Conjugated Addition/Aromatization of P(O)–H Compounds with para-Quinone Methides, Eur. J. Org. Chem., 2019, 3273–3282 CrossRef CAS.
- B. Zhang, L. Liu, S. Mao, M.-D. Zhou, H. Wang and L. Li, Base-Catalyzed 1,6-Hydrophosphonylation of p-Quinone Methides with Diphenylphosphane Oxide/Phosphites, Eur. J. Org. Chem., 2019, 3898–3907 CrossRef CAS.
- Y. N. Aher and A. B. Pawar, Catalyst- and reagent-free 1,6-hydrophosphonylation of p-quinone methides: a practical approach for the synthesis of diarylmethyl phosphine oxides, Org. Biomol. Chem., 2019, 17, 7536–7546 RSC.
- C. Jiang, Y. Chen, G. Huang, C. Ni, X. Liu and H. Lu, Scandium(III)-Catalysed Decarboxylative Addition of β-Ketoacids to para-Quinone Methides: Evidence for 1,6-Addition and Base-Assisted Decarboxylation Tandem Process, Asian J. Org. Chem., 2019, 8, 257–260 CrossRef CAS.
- For some selected reviews on nucleophilic organocatalysis with NHCs, see:
(a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed;
(b) J. Mahatthananchai and J. W. Bode, On the Mechanism of N-Heterocyclic Carbene-Catalyzed Reactions Involving Acyl Azoliums, Acc. Chem. Res., 2014, 47, 696 CrossRef CAS PubMed;
(c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485 CrossRef CAS PubMed;
(d) S. J. Ryan, L. Candish and D. W. Lupton, Acyl anion free N-heterocyclic carbene organocatalysis, Chem. Soc. Rev., 2013, 42, 4906 RSC;
(e) J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, A Continuum of Progress: Applications of N-Hetereocyclic Carbene Catalysis in Total Synthesis; Anwendungen der Katalyse mit N-heterocyclischen Carbenen in Totalsynthesen, Angew. Chem., Int. Ed., 2012, 51, 11686 (
Angew. Chem.
, 2012
, 124
, 11854
) CrossRef CAS PubMed;
(f) X. Bugaut and F. Glorius, Organocatalytic umpolung: N-heterocyclic carbenes and beyond, Chem. Soc. Rev., 2012, 41, 3511 RSC.
- P. Arde and R. V. Anand, Expedient access to unsymmetrical triarylmethanes through N-heterocyclic carbene catalysed 1,6-conjugate addition of 2-naphthols to para-quinone methides, RSC Adv., 2016, 6, 77111–77115 RSC.
- S. Santra, A. Porey and J. Guin, 1,6-Conjugate Addition of 1,3-Dicarbonyl Compounds to para-Quinone Methides Enabled by Noncovalent N-Heterocyclic Carbene Catalysis, Asian J. Org. Chem., 2018, 7, 477–486 CrossRef CAS.
- P. Goswami, G. Singh and R. V. Anand,
N-Heterocyclic Carbene Catalyzed 1,6-Conjugate Addition of Me3Si-CN to para-Quinone Methides and Fuchsones: Access to α-Arylated Nitriles, Org. Lett., 2017, 19, 1982–1985 CrossRef CAS PubMed.
- P. Goswami, S. Sharma, G. Singh and R. V. Anand, Bis(amino)cyclopropenylidene Catalyzed Rauhut–Currier Reaction between α,β-Unsaturated Carbonyl Compounds and para-Quinone Methides, J. Org. Chem., 2018, 83, 4213–4220 CrossRef CAS PubMed.
- Y. Shen, J. Qi, Z. Mao and S. Cui, Fe-Catalyzed Hydroalkylation of Olefins with para-Quinone Methides, Org. Lett., 2016, 18, 2722–2725 CrossRef CAS PubMed.
- J. Qi, Z. Deng, B. Huanga and S. Cui, Metal-free α-alkylation of alcohols with para-quinone methides, Org. Biomol. Chem., 2018, 16, 2762 RSC.
- S. G. More and G. M. Suryavanshi, Metal-free, radical 1,6-conjugated addition of cyclic ethers with para-quinone
methides (p-QMs), Org. Biomol. Chem., 2019, 17, 3239–3248 RSC.
- J. Yu, H. Sheng, S. Wang, Z. Xu, S. Tang and S. Chen, Copper-catalyzed radical cascades of para-quinone methides with AIBN and H2O via α-cyanoalkylation by C–C bond cleavage: new access to benzofuran-2(3H)-ones, Chem. Commun., 2019, 55, 4578–4581 RSC.
- M. Ke and Q. Song, Copper-Catalyzed 1,6-Hydrodifluoroacetylation of para-Quinone Methides at Ambient Temperature with Bis(pinacolato)diboron as Reductant, Adv. Synth. Catal., 2017, 359, 384–389 CrossRef CAS.
- Y.-N. Zhao, Y.-C. Luo, Z.-Y. Wang and P.-F. Xu, A new approach to access difluoroalkylated diarylmethanes via visible-light photocatalytic cross-coupling reactions, Chem. Commun., 2018, 54, 3993 RSC.
- W. Zhang, C. Yang, Z.-P. Zhang, X. Li and J.-P. Cheng, Visible-Light-Triggered Cyanoalkylation of para-Quinone Methides and Its Application to the Synthesis of GPR40 Agonists, Org. Lett., 2019, 21, 4137–4142 CrossRef CAS PubMed.
- Q.-Y. Wu, G.-Z. Ao and F. Liu, Redox-neutral tri-/difluoromethylation of para-quinone methides with sodium sulfonates, Org. Chem. Front., 2018, 5, 2061–2064 RSC.
- K. G. Ghosh, P. Chandu, S. Mondal and D. Sureshkumar, Visible-light mediated trifluoromethylation of p-quinone methides by 1,6-conjugate addition using pyrylium salt as organic photocatalyst, Tetrahedron, 2019, 75, 4471–4478 CrossRef CAS.
- J. Guo, G. Huang, Q. Wu, Y. Xie, J. Weng and G. Lu, An efficient approach to access 1,1,2-triarylethanes enabled by the organo-photoredox-catalyzed decarboxylative addition reaction, Org. Chem. Front., 2019, 6, 1955–1960 RSC.
- Q.-Y. Wu, Q.-Q. Min, G.-Z. Ao and F. Liu, Radical alkylation of para-quinone methides with 4-substituted Hantzsch esters/nitriles via organic photoredox catalysis, Org. Biomol. Chem., 2018, 16, 6391–6394 RSC.
- S. Santra, A. Porey, B. Jana and J. Guin,
N-Heterocyclic carbenes as chiral Brønsted basecatalysts: a highly diastereo- and enantioselective 1,6-addition reaction, Chem. Sci., 2018, 9, 6446–6450 RSC.
- Y. Wang, K. Wang, W. Cao, X. Liu and X. Feng, Diastereo- and Enantioselective 1,6-Conjugate Addition of 2-Azaarylacetamides to para-Quinone Methides, Org. Lett., 2019, 21, 6063–6067 CrossRef CAS PubMed.
- D. Wang, Z.-F. Song, W.-J. Wang and T. Xu, Highly Regio- and Enantioselective Dienylation of p-Quinone Methides Enabled by an Organocatalyzed Isomerization/Addition Cascade of Allenoates, Org. Lett., 2019, 21, 3963–3967 CrossRef CAS.
- A. Eitzinger, M. Winter, J. Schorgenhumer and M. Waser, Quaternary β2,2-amino acid derivatives by asymmetric addition of isoxazolidin-5-ones to para-quinone methides, Chem. Commun., 2020, 56, 579–582 RSC.
- M. Chen and J. Sun, Catalytic Asymmetric N-Alkylation of Indoles and Carbazoles via 1,6-Conjugate Addition of Aza-para-Quinone Methides, Angew. Chem., Int. Ed., 2017, 56, 4583–4587 CrossRef CAS PubMed.
- J.-R. Wang, X.-L. Jiang, Q.-Q. Hang, S. Zhang, G.-J. Mei and F. Shi, Catalytic Asymmetric Conjugate Addition of Indoles to para- Quinone Methide Derivatives, J. Org. Chem., 2019, 84, 7829–7839 CrossRef CAS PubMed.
- Y. Cheng, Z. Fang, Y. Jia, Z. Lu, W. Li and P. Li, Organocatalytic enantioselective conjugate addition of 2-naphthols to ortho-hydroxyphenyl substituted para-quinone methides: access to unsymmetrical triarylmethanes, RSC Adv., 2019, 9, 24212–24217 RSC.
- R. Torán, C. Vila, A. Sanz-Marco, M. C. Muñoz, J. R. Pedro and G. Blay, Organocatalytic Enantioselective 1,6-aza-Michael Addition of Isoxazolin-5-ones to p-Quinone Methides, Eur. J. Org. Chem., 2020, 627–630 CrossRef.
- T. Pan, P. Shi, B. Chen, D.-G. Zhou, Y.-L. Zeng, W.-D. Chu, L. He, Q.-Z. Liu and C.-A. Fan, CuH-Catalyzed Asymmetric 1,6-Conjugate Reduction of p-Quinone Methides: Enantioselective Synthesis of Triarylmethanes and 1,1,2-Triarylethanes, Org. Lett., 2019, 21, 6397–6402 CrossRef.
- R. Pan, L. Hu, C. Han, A. Lin and H. Yao, Cascade Radical 1,6-Addition/Cyclization of para-Quinone Methides: Leading to Spiro[4.5]deca-6,9-dien-8-ones, Org. Lett., 2018, 20, 1974–1977 CrossRef CAS PubMed.
- H.-D. Zuo, W.-J. Hao, C.-F. Zhu, C. Guo, S.-J. Tu and B. Jiang, Electrochemical Annulation–Iodosulfonylation of 1,5-Enyne-containing para-Quinone Methides (p-QMs) to Access (E)-Spiroindenes, Org. Lett., 2020 DOI:10.1021/acs.orglett.0c01470.
-
(a) X. Lv, F. Hu, K. Duan, S.-S. Li, Q. Liu and J. Xiao, Aromatization-Driven Cascade [1,5]-Hydride Transfer/Spirocyclization Promoted by Fluorinated Alcohols, J. Org. Chem., 2019, 84, 1833–1844 CrossRef CAS PubMed;
(b) L. Wang and J. Xiao, Hydrogen-Atom Transfer Reactions, Top. Curr. Chem., 2016, 374, 17 CrossRef;
(c) L. Wang and J. Xiao, C(sp3)–H bond functionalization by sequential hydride transfer/cyclization: electronic effect and steric effect controlled regioselectivity, Org. Chem. Front., 2016, 3, 635–638 RSC;
(d) L. Wang and J. Xiao, Advancement in Cascade [1,n]-Hydrogen Transfer/Cyclization: A Method for Direct Functionalization of Inactive C(sp3)-H Bonds, Adv. Synth. Catal., 2014, 356, 1137–1171 CrossRef CAS;
(e) M. C. Haibach and D. Seidel, C-H bond functionalization through intramolecular hydride transfer, Angew. Chem., Int. Ed., 2014, 53, 5010–5036 CrossRef CAS.
-
(a) G. Maas, Ruthenium-catalysed carbenoid cyclopropanation reactions with diazo compounds, Chem. Soc. Rev., 2004, 33, 183 RSC;
(b) R. Sambasivanand and Z. T. Ball, Screening Rhodium Metallopeptide Libraries “On Bead”: Asymmetric Cyclopropanation and a Solution to the Enantiomer Problem, Angew. Chem., Int. Ed., 2012, 51, 8568 CrossRef;
(c) F. G. Adly, M. G. Gardiner and A. Ghanem, Design and Synthesis of Novel Chiral Dirhodium(II) Carboxylate Complexes for Asymmetric Cyclopropanation Reactions, Chem. – Eur. J., 2016, 22, 3447 CrossRef CAS;
(d) C. Qin, V. Boyarskikh, J. H. Hansen, K. I. Hardcastle, D. G. Musaev and H. M. L. Davies, D2-Symmetric Dirhodium Catalyst Derived from a 1,2,2-Triarylcyclopropanecarboxylate Ligand: Design, Synthesis and Application, J. Am. Chem. Soc., 2011, 133, 19198 CrossRef CAS PubMed;
(e) H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S. F. Zhu and Q.-L. Zhou, Highly Enantioselective Copper- and Iron-Catalyzed Intramolecular Cyclopropanation of Indoles, J. Am. Chem. Soc., 2017, 139, 7697 CrossRef CAS PubMed.
- K. Gai, X. Fang, X. Li, J. Xua, X. Wu, A. Lin and H. Yao, Synthesis of spiro[2.5]octa-4,7-dien-6-one with consecutive quaternary centers via 1,6-conjugate addition induced dearomatization of para-quinone methides, Chem. Commun., 2015, 51, 15831–15834 RSC.
- S. Zhao, Y. Zhu, M. Zhang, X. Song and J. Chang, Base-Mediated Tandem 1,6-Addition/Cyclization/Isomerization Reactions between para-Quinone Methides and Benzyl Chlorides: Approaches to Diverse Frameworks at Each Cascade Stage, Synthesis, 2019, 51, 2136–2148 CrossRef CAS.
- Z. Yuan, X. Fang, X. Li, J. Wu, H. Yao and A. Lin, 1,6-Conjugated Addition-Mediated [2 + 1] Annulation: Approach to Spiro[2.5]octa-4,7-dien-6-one, J. Org. Chem., 2015, 80, 11123–11130 CrossRef CAS PubMed.
- X.-Z. Zhang, J.-Y. Du, Y.-H. Deng, W.-D. Chu, X. Yan, K.-Y. Yu and C.-A. Fan, Spirocyclopropanation Reaction of para-Quinone Methides with Sulfonium Salts: The Synthesis of Spirocyclopropanyl para-Dienones, J. Org. Chem., 2016, 81, 2598–2606 CrossRef CAS PubMed.
- S. B. Kale, P. K. Jori, T. Thatikonda, R. G. Gonnade and U. Das, 1,6-Conjugate-Addition-Induced [2 + 1] Annulation of para-Quinone Methides and Pyrazolones: Synthesis of Bis-Spiro Compounds with Contiguous Quaternary Spiro-Centers, Org. Lett., 2019, 21, 7736–7740 CrossRef CAS PubMed.
- For selected examples, see:
(a) P. H. Nguyen, T. N. A. Nguyen, T. T. Dao, H. W. Kang, D. T. Ndinteh, J. T. Mbafor and W. K. Oh, AMP-Activated Protein Kinase (AMPK) Activation by Benzofurans and Coumestans Isolated from Erythrina abyssinica, J. Nat. Prod., 2010, 73, 598–602 CrossRef CAS PubMed;
(b) Y. Lu, M. Kuang, G. P. Hu, R. B. Wu, J. Wang, L. Liu and Y.-C. Lin, Loddigesiinols G–J: α-Glucosidase Inhibitors from Dendrobium loddigesii, Molecules, 2014, 19, 8544–8555 CrossRef PubMed;
(c) R. R. Majinda, M. Motswaledi, R. D. Waigh and P. G. Waterman, Phenolic and Antibacterial Constituents of Vahlia capensis, Planta Med., 1997, 63, 268–270 CrossRef CAS;
(d) K. Nakashima, N. Abe, M. Oyama and M. Inoue, Yuccalides A–C, three new phenolic compounds with spiro-structures from the roots of Yucca gloriosa, Fitoterapia, 2016, 111, 154–159 CrossRef CAS.
- L. Liu, Z. Yuan, R. Pan, Y. Zeng, A. Lin, H. Yao and Y. Huang, 1,6-Conjugated addition-mediated [4 + 1] annulation: an approach to 2,3-dihydrobenzofurans, Org. Chem. Front., 2018, 5, 623–628 RSC.
- X.-M. Chen, K.-X. Xie, D.-F. Yue, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Catalyst-free synthesis of 2,3-dihydrobenzofurans through [4 + 1] cycloaddition of ortho-hydroxyphenylsubstituted para-quinone methides and sulfur ylides, Tetrahedron, 2018, 74, 600–605 CrossRef CAS.
- Y. Zhi, K. Zhao, C. von Essen, K. Rissanen and D. Enders, Synthesis of trans-disubstituted-2,3-dihydrobenzofurans by a formal [4 + 1] annulation between para-quinone methides and sulfonium salts, Org. Chem. Front., 2018, 5, 1348–1351 RSC.
- G. Singh, S. Kumar, A. Chowdhury and R. V. Anand, Base-Mediated One-pot Synthesis of Oxygen-Based Heterocycles from 2-Hydroxyphenyl-Substituted para-Quinone Methides, J. Org. Chem., 2019, 84, 15978–15989 CrossRef CAS PubMed.
- J. Zhou, G. Liang, X. Hu, L. Zhou and H. Zhou, Facile synthesis of 3-aryl 2,3-dihydrobenzofurans via novel domino 1,6-addition/O-alkylation reactions of para-quinone methides, Tetrahedron, 2018, 74, 1492–1496 CrossRef CAS.
- J.-P. Tan, P. Yu, J.-H. Wu, Y. Chen, J. Pan, C. Jiang, X. Ren, H.-S. Zhang and T. Wang, Bifunctional Phosphonium Salt Directed Enantioselective Formal [4 + 1] Annulation of Hydroxyl-Substituted para-Quinone Methides with α-Halogenated Ketones, Org. Lett., 2019, 21, 7298–7302 CrossRef CAS PubMed.
- K. Zielke, O. Kováč, M. Winter, J. Pospíšil and M. Waser, Enantioselective Catalytic [4 + 1]-Cyclization of ortho-Hydroxy para- Quinone Methides with Allenoates, Chem. – Eur. J., 2019, 25, 8163–8168 CrossRef CAS.
- Y.-J. Xiong, S.-Q. Shi, W.-J. Hao, S.-J. Tu and B. Jiang, A new dehydrogenative [4 + 1] annulation between para-quinone methides (p-QMs) and iodonium ylides for the synthesis of 2,3-dihydrobenzofurans, Org. Chem. Front., 2018, 5, 3483–3487 RSC.
- Z. Yuan, K. Gai, Y. Wu, J. Wu, A. Lin and H. Yao, Tandem 1,6-addition/cyclization/vinylcyclopropane rearrangement at low temperature under metal-free conditions: an approach to spiro[4.5]cyclohexadienones, Chem. Commun., 2017, 53, 3485–3488 RSC.
- G. Singh, P. Goswami, S. Sharma and R. V. Anand, A One-Pot Approach to 2,3-Diarylbenzo[b]furans through N-Heterocyclic Carbene-Catalyzed 1,6-Conjugate Addition Followed by Acid Mediated Dehydrative Annulation, J. Org. Chem., 2018, 83, 10546–10554 CrossRef CAS PubMed.
- Z. Feng, Z. Yuan, X. Zhao, Y. Huang and H. Yao, A [4 + 1] annulation of ortho-electrophile-substituted para-quinone methides for the synthesis of indanes and isoindolines, Org. Chem. Front., 2019, 6, 3535–3539 RSC.
-
(a) E. Gravel and E. Poupon, Biosynthesis and biomimetic synthesis of alkaloids isolated from plants of the Nitraria and Myrioneuron genera: an unusual lysine-based metabolism, Nat. Prod. Rep., 2010, 27, 32 RSC;
(b) Y.-S. Cai, Y.-W. Guo and K. Krohn, Structure, bioactivities, biosynthetic relationships and chemical synthesis of the spirodioxynaphthalenes, Nat. Prod. Rep., 2010, 27, 1840 RSC;
(c) Z. Jin, Amaryllidaceae and Sceletium alkaloids, Nat. Prod. Rep., 2005, 22, 111 RSC;
(d) E. M. Antunes, B. R. Copp, M. T. Davies-Coleman and T. Samaai, Pyrroloiminoquinone and related metabolites from marine sponges, Nat. Prod. Rep., 2005, 22, 6 RSC;
(e) H. Venkatesan, M. C. Davis, Y. Altas, J. P. Snyder and D. C. Liotta, Total Synthesis of SR 121463 A, a Highly Potent and Selective Vasopressin V2 Receptor Antagonist, J. Org. Chem., 2001, 66, 3653 CrossRef CAS PubMed.
-
(a) C.-X. Zhuo, W. Zhang and S.-L. You, Catalytic Asymmetric Dearomatization Reactions, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed;
(b) S. T. Roche and J. A. Porco Jr., Dearomatization Strategies in the Synthesis of Complex Natural Products, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed;
(c) R. Leon, A. Jawalekar, T. Redert and M. J. Gaunt, Catalytic enantioselective assembly of complex molecules containing embedded quaternary stereogenic centres from simple anisidine derivatives, Chem. Sci., 2011, 2, 1487 RSC;
(d) M.-Q. Jia and S.-L. You, Desymmetrization of cyclohexadienones viad-camphor-derived triazolium salt catalyzed intramolecular Stetter reaction, Chem. Commun., 2012, 48, 6363 RSC;
(e) J. Pereira, M. Barlier and C. Guillou, Formal Total Syntheses of Aspidosperma Alkaloids via a Novel and General Synthetic Pathway Based on an Intramolecular Heck Cyclization, Org. Lett., 2007, 9, 3101 CrossRef CAS PubMed.
- Z. Yuan, W. Wei, A. Lin and H. Yao, Bifunctional Organo/Metal Cooperatively Catalyzed [3 + 2] Annulation of para-Quinone Methides with Vinylcyclopropanes: Approach to Spiro[4.5]deca-6,9-diene-8-ones, Org. Lett., 2016, 18, 3370–3373 CrossRef CAS PubMed.
- Z. Yuan, L. Liu, R. Pan, H. Yao and A. Lin, Silver-Catalyzed Cascade 1,6-Addition/Cyclization of para-Quinone Methides with Propargyl Malonates: An Approach to Spiro[4.5]deca-6,9-dien-8-ones, J. Org. Chem., 2017, 82, 8743–8751 CrossRef CAS PubMed.
- Y. Su, Y. Zhao, B. Chang, X. Zhao, R. Zhang, X. Liu, D. Huang, K.-H. Wang, C. Huo and Y. Hu, [3 + 2] Cycloaddition of para-Quinone Methides with Nitrile Imines: Approach to Spiro-pyrazoline-cyclohexadienones, J. Org. Chem., 2019, 84, 6719–6728 CrossRef CAS PubMed.
-
(a) L.-G. Lin, H. Xie, H.-L. Li, L.-J. Tong, C.-P. Tang, C.-Q. Ke, Q.-F. Liu, L.-P. Lin, M.-Y. Geng, H. Jiang, W.-M. Zhao, J. Ding and Y. Ye, Naturally Occurring Homoisoflavonoids Function as Potent Protein Tyrosine Kinase Inhibitors by c-Src-Based High-Throughput Screening, J. Med. Chem., 2008, 51, 4419 CrossRef CAS PubMed;
(b) W. K. Brewster, D. E. Nichols, R. M. Riggs, D. M. Mottola, T. W. Lovenberg, M. H. Lewis and R. B. Mailmad, Trans-10,11-dihydroxy-5,6,6a,7,8,12b-hexahydrobenzo[a]phenanthridine: a highly potent selective dopamine D1 full agonist, J. Med. Chem., 1990, 33, 1756 CrossRef CAS PubMed;
(c) H. Bridi, G. C. Meirelles and G. L. von Poser, Structural diversity and biological activities of phloroglucinol derivatives from Hypericum species, Phytochemistry, 2018, 155, 203–232 CrossRef CAS PubMed;
(d) S. Emami and Z. Ghanbarimasir, Recent advances of chroman-4-one derivatives: Synthetic approaches and bioactivities, Eur. J. Med. Chem., 2015, 93, 539–563 CrossRef CAS PubMed;
(e) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, The Domestication of ortho-Quinone Methides, Acc. Chem. Res., 2014, 47, 3655–3664 CrossRef CAS PubMed.
- G.-J. Mei, S.-L. Xu, W.-Q. Zheng, C.-Y. Bian and F. Shi, [4 + 2] Cyclization of para-Quinone Methide Derivatives with Alkynes, J. Org. Chem., 2018, 83, 1414–1421 CrossRef CAS PubMed.
- Z. Li, W. Wang, H. Jian, W. Li, B. Dai and L. He, Synthesis of 9-phenol-substituted xanthenes by cascade O-insertion/1,6-conjugate addition of benzyne with ortho-hydroxyphenyl substituted para-quinone methides, Chin. Chem. Lett., 2019, 30, 386–388 CrossRef CAS.
- F.-R. Yuan, F. Jiang, K.-W. Chen, G.-J. Mei, Q. Wu and F. Shi, Phosphine-catalyzed [4 + 2] cyclization of para-quinone methide derivatives with allenes, Org. Biomol. Chem., 2019, 17, 2361–2369 RSC.
- Y. Zhu, D. Wang and Y. Huang, Phosphine Sequentially Catalyzed Domino 1,6-Addition/Annulation: Access to Functionalized Chromans and Tetrahydroquinolines with an Ethynyl-Substituted All-Carbon Quaternary Center, Org. Lett., 2019, 21, 908–912 CrossRef CAS PubMed.
- X.-L. Jiang, S.-F. Wu, J.-R. Wang, G.-J. Mei and F. Shi, Catalytic Asymmetric [4 + 2] Cyclization of para-Quinone Methide Derivatives with 3-Alkyl-2-vinylindoles, Adv. Synth. Catal., 2018, 360, 4225–4235 CrossRef CAS.
- S. Roy, S. Pradhan, K. Kumar and I. Chatterjee, Asymmetric Organocatalytic Double 1,6-Addition: Rapid Access to Chiral Chromans with Molecular Complexity, Org. Chem. Front., 2020 10.1039/D0QO00354A.
- H.-M. Huang, X.-Y. Wu, B.-R. Leng, Y.-L. Zhu, X.-C. Meng, Y. Hong, B. Jiang and D.-C. Wang, Cu(II)-Catalyzed formal [4 + 2] cycloaddition between quinone methides (QMs) and electron-poor 3-vinylindoles, Org. Chem. Front., 2020, 7, 414–419 RSC.
-
(a) M. A. Brenzan, C. V. Nakamura, B. P. D. Filho, T. Ueda-Nakamura, M. C. M. Young, A. G. Correa, J. A. Junior, A. O. dos Santos and D. A. G. Cortez, Structure–activity relationship of (−)mammea A/BB derivatives against Leishmania amazonensis, Biomed. Pharmacother., 2008, 62, 651–658 CrossRef CAS PubMed;
(b) J. Sun, W. X. Ding, X. P. Hong, K. Y. Zhang and Y. Zou, Synthesis and antimicrobial activities of 4-aryl-3,4-dihydrocoumarins and 4-arylcoumarins, Chem. Nat. Compd., 2012, 48, 16–22 CrossRef CAS;
(c) X. F. Zhang, L. Xie, Y. Liu, J. F. Xiang, L. Li and Y. L. Tang, Molecular interaction and energy transfer between human serum albumin and bioactive component Aloe dihydrocoumarin, J. Mol. Struct., 2008, 888, 145–151 CrossRef CAS.
- Z. Cao, G.-X. Zhou, C. Ma, K. Jiang and G.-J. Mei, Brønsted Acid Catalyzed Domino 1,6-Addition/Intramolecular Cyclization-Reactions: Diastereoselective Synthesis of Dihydro-coumarin Frameworks, Synthesis, 2018, 50, 1307–1314 CrossRef CAS.
- Z.-P. Zhang, K.-X. Xie, C. Yang, M. Li and X. Li, Asymmetric Synthesis of Dihydrocoumarins through Chiral Phosphoric Acid-Catalyzed Cycloannulation of para-Quinone Methides and Azlactones, J. Org. Chem., 2018, 83, 364–373 CrossRef CAS PubMed.
- M.-X. Zhao, J. Xiang, Z.-Q. Zhao, X.-L. Zhao and M. Shi, Asymmetric synthesis of dihydrocoumarins via catalytic sequential 1,6-addition/transesterification of α-isocyanoacetates with para-quinone methides, Org. Biomol. Chem., 2020, 18, 1637–1646 RSC.
- J.-Y. Zhou, C. Ma, Y.-Z. Zhang, Q. Wu and F. Shi, Catalyst-free [4 + 2] cyclization of para-quinone methide derivatives with homophthalic anhydrides, Org. Biomol. Chem., 2018, 16, 9382–9389 RSC.
- S. Satbhaiya, N. S. Khonde, J. Rathod, R. Gonnade and P. Kumar, Tf2NH catalyzed 1,6-conjugate addition of 2-hydroxy-p-quinone methides with β-Functionalized Ketones: Access to 2,3,4,9-Tetrahydro-1H-xanthenones and 4H-Chromene Derivatives, Eur. J. Org. Chem., 2019, 3127–3133 CrossRef CAS.
- Z.-H. Wang, X.-Y. Zhang, Y. You, J.-Q. Zhao, M.-Q. Zhou, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Efficient construction of polycyclic chromans through 4-methylbenzenesulfonic acid mediated domino 1,6-addition/oxa-Mannich reaction of ortho-hydroxyphenyl substituted para-quinone methides and cyclic enamides, Tetrahedron, 2019, 75, 3456–3462 CrossRef CAS.
- G.-H. Yang, Q. Zhao, Z.-P. Zhang, H.-L. Zheng, L. Chen and X. Li, Asymmetric Cycloaddition of ortho-Hydroxyphenyl-Substituted para-Quinone Methides and Enamides Catalyzed by Chiral Phosphoric Acid, J. Org. Chem., 2019, 84, 7883–7893 CrossRef CAS PubMed.
-
(a) M. Matsukura, Y. Daiku, K. Ueda, S. Tanaka, T. Igarashi and N. Minami, Synthesis and antiarrhythmic activity of 2,2-dialkyl-1′-(N-substituted aminoalkyl)-spiro-(chroman-4,4′-imidazolidine)-2′,5′-diones, Chem. Pharm. Bull., 1992, 40, 1823–1827 CrossRef CAS PubMed;
(b) V. V. Kouznetsov, D. R. M. Arenas, F. Arvelo, J. S. B. Forero, F. Sojo and A. Munoz, 4-Hydroxy-3-methoxyphenyl Substituted 3-methyl-tetrahydroquinoline Derivatives Obtained Through Imino Diels-Alder Reactions as Potential Antitumoral Agents, Lett. Drug Des. Discovery, 2010, 7, 632–639 CrossRef CAS.
- Z. Ye, L. Bai, Y. Bai, Z. Gan, H. Zhou, T. Pan, Y. Yu and J. Zhou, High diastereoselective synthesis of spiro-isoxazolonechromans via domino oxa-Michael/1,6-addition reactions of ortho-hydroxyphenylsubstituted para-quinone methides with unsaturated isoxazolones, Tetrahedron, 2019, 75, 682–687 CrossRef CAS.
- M. Huo, J. Zhou, L. Bai, Q. Xu, Z. Zhou, H. Zhou and G. Liang, High diastereoselective synthesis of spiro-barbituratechromans via domino oxa-Michael/1,6-addition reactions of ortho-hydroxyphenylsubstituted para-quinone methides with barbiturate-based olefins, Tetrahedron, 2019, 75, 130752–130758 CrossRef.
- J.-P. Tan, H. Zhang, Z. Jiang, Y. Chen, X. Ren, C. Jiang and T. Wang, Enantioselective Construction of Spiro[chroman-thiazolones]: Bifunctional Phosphonium Salt-Catalyzed [2 + 4] Annulation between 5-Alkenyl Thiazolones and ortho-Hydroxyphenyl-Substituted para-Quinone Methides, Adv. Synth. Catal., 2020, 362, 1058–1063 CrossRef CAS.
- Y.-C. Cheng, C.-S. Wang, T.-Z. Li, F. Gao, Y. Jiao and F. Shi, Organocatalytic [4 + 2] cyclizations of para-quinone methide derivatives with isocyanates, Org. Biomol. Chem., 2019, 17, 6662–6670 RSC.
- J.-R. Zhang, H.-S. Jin, R.-B. Wang and L.-M. Zhao, Synthesis of 2,4-Diaryl-1,3-benzoxazines via FeCl3-Catalyzed Annulation of ortho-Hydroxyphenyl-Substituted para-Quinone Methides with Imidates, Adv. Synth. Catal., 2019, 361, 4811–4816 CrossRef CAS.
- For reviews on the chemistry of vinyl aziridines:
(a)
V. V. Fokin and P. Wu, Aziridines and Epoxides in Organic Synthesis (Yudin: Aziridines and Epoxides in Organic Synthesis O-BK) || Asymmetric Syntheses with Aziridinecarboxylate and Aziridinephosphonate Building Blocks, in Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, Germany, 2006, pp. 443–477 Search PubMed;
(b) D. J. Mack and J. T. Njardarson, Recent Advances in the Metal-Catalyzed Ring Expansions of Three- and Four-Membered Rings, ACS Catal., 2013, 3, 272–286 CrossRef CAS;
(c) H. Ohno, Synthesis and Applications of Vinylaziridines and Ethynylaziridines, Chem. Rev., 2014, 114, 7784–7814 CrossRef CAS PubMed;
(d) J.-J. Feng and J. Zhang, Synthesis of Unsaturated N-Heterocycles by Cycloadditions of Aziridines and Alkynes, ACS Catal., 2016, 6, 6651–6661 CrossRef CAS.
- F. Jiang, F.-R. Yuan, L.-W. Jin, G.-J. Mei and F. Shi, Metal-Catalyzed (4 + 3) Cyclization of Vinyl Aziridines with para-Quinone Methide Derivatives, ACS Catal., 2018, 8, 10234–10240 CrossRef CAS.
- Q. Liu, S. Li, X.-Y. Chen, K. Rissanen and D. Ender, Asymmetric Synthesis of Spiro-oxindole-ε-lactones through N-Heterocyclic Carbene Catalysis, Org. Lett., 2018, 20, 3622–3626 CrossRef CAS PubMed.
- W. Li, H. Yuan, Z. Liu, Z. Zhang, Y. Cheng and P. Li, NHC-Catalyzed Enantioselective [4 + 3] Cycloaddition of Ortho-Hydroxyphenyl Substituted para-Quinone Methides with Isatin-Derived Enals, Adv. Synth. Catal., 2018, 360, 2460–2464 CrossRef CAS.
- For selected reviews, see:
(a) A. Domling, W. Wang and K. Wang, Chemistry and Biology of Multicomponent Reactions, Chem. Rev., 2012, 112, 3083–3135 CrossRef CAS PubMed;
(b) B. Jiang, T. Rajale, W. Wever, S.-J. Tu and G. Li, Multicomponent Reactions for the Synthesis of Heterocycles, Chem. – Asian J., 2010, 5, 2318–2335 CrossRef CAS;
(c) R. A. Seibert and F. W. BERGSTROM, Phenylation of Some Hydrocarbons with a Phenyl Halide Activated by An Alkali Amide, J. Org. Chem., 1945, 10, 544–550 CrossRef CAS PubMed;
(d) T. Vlaar, B.-U. Maes, E. Ruijter and R.-V. Orru, Palladium-Catalyzed Migratory Insertion of Isocyanides: An Emerging Platform in Cross-Coupling Chemistry, Angew. Chem., Int. Ed., 2013, 52, 7084–7097 CrossRef CAS;
(e) B.-H. Rotstein, S. Zaretsky, V. Rai and A.-K. Yudin, Small Heterocycles in Multicomponent Reactions, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS.
- For selected examples, see:
(a) Z. Tang, Z. Liu, Y. An, R. Jiang, X. Zhang, C. Li, X. Jia and J. J. Li, Isocyanide-Based Multicomponent Bicyclization with Substituted Allenoate and Isatin: Synthesis of Unusual Spirooxindole Containing [5.5]-Fused Heterocycle, J. Org. Chem., 2016, 81, 9158–9166 CrossRef CAS PubMed;
(b) D. Wang, S. Liu, X.-C. Lan, A. Paniagua, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Tunable dimerization and trimerization of β-alkynyl ketones via silver catalysis for accessing spiro and dispiro compounds containing 1H-isochromene, Adv. Synth. Catal., 2017, 359, 3186–3196 CrossRef CAS;
(c) N.-N. Man, J.-Q. Wang, L.-M. Zhang, L.-R. Wen and M. Li, Chemo-, Regio-, and Stereoselective Construction of Core Skeleton of Furo[2,3-b]pyrrole via Multicomponent Bicyclization Reaction, J. Org. Chem., 2017, 82, 5566–5573 CrossRef CAS;
(d) B. Jiang, J. Li, Y.-Y. Pan, W.-J. Hao, G. Li and S.-J. Tu, Radical-enabled bicyclization cascades of oxygen-tethered 1,7-enynes leading to skeletally diverse polycyclic chromen-2-ones, Chin. J. Chem., 2017, 35, 323–334 CrossRef CAS;
(e) J. Li, W.-W. Zhang, X.-J. Wei, F. Liu, W.-J. Hao, S.-L. Wang, G. Li, S.-J. Tu and B. Jiang, Radical deaminative ipso-cyclization of 4-methoxyanilines with 1,7-enynes for accessing spirocyclohexadienone-containing cyclopenta[c]quinolin-4-ones, J. Org. Chem., 2017, 82, 6621–6628 CrossRef CAS PubMed.
- S. Liu, X.-C. Lan, K. Chen, W.-J. Hao, G. Li, S.-J. Tu and B. Jiang, Ag/Brønsted acid co-catalyzed spiroketalization of β-alkynyl ketones towards spiro[chromane-2,1′-isochromene] derivatives, Org. Lett., 2017, 19, 3831–3834 CrossRef CAS.
- K. Chen, S. Liu, D. Wang, W.-J. Hao, P. Zhou, S.-J. Tu and B. Jiang, Silver/Scandium cocatalyzed bicyclization of β-alkynyl ketones leading to benzo[c]xanthenes and naphtho[1,2-b]benzofurans, J. Org. Chem., 2017, 82, 11524–11530 CrossRef CAS PubMed.
- C.-S. Wang, Y.-C. Cheng, J. Zhou, G.-J. Mei, S.-L. Wang and F. Shi, Metal-Catalyzed Oxa,-[4 + 2] Cyclizations of Quinone Methides with Alkynyl Benzyl Alcohols, J. Org. Chem., 2018, 83, 13861–13873 CrossRef CAS PubMed.
- J.-Y. Liao, Q. Ni and Y. Zhao, Catalyst-Enabled Scaffold Diversity: Highly Chemo-and Stereoselective Synthesis of Tricyclic Ketals and Triarylmethanes, Org. Lett., 2017, 19, 4074–4077 CrossRef CAS PubMed.
- A. S. Jadhav, Y. A. Pankhade and R. V. Anand, Tandem One-Pot Approach To Access 1,2,3-Triazole-fused Isoindolines through Cu-Catalyzed 1,6-Conjugate Addition of Me3SiN3 to p-Quinone Methides followed by Intramolecular Click Cycloaddition, J. Org. Chem., 2018, 83, 8596–8606 CrossRef CAS PubMed.
- X.-L. Jiang, S.-F. Wu, J.-R. Wang, H. Lu, G.-J. Mei and F. Shi, The [4 + 2] cyclization/retro-Mannich reaction cascade of para-quinone methide derivatives with Pd-containing 1,4-dipoles, Org. Biomol. Chem., 2018, 16, 8395–8402 RSC.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Bo
Jiang
* and
Bo
Jiang