
Tailored covalent organic frameworks by post-synthetic modification
Huimin
Ding
,
Arindam
Mal
and
Cheng
Wang
 *
*
Sauvage Center for Molecular Sciences and Hubei Key Lab on Organic and Polymeric Optoelectronic Materials, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: chengwang@whu.edu.cn
First published on 2nd October 2019
Abstract
Covalent organic frameworks (COFs), constructed by strong covalent bonds based on reticular chemistry, hold great promise in diverse applications. Manipulating COFs with desired functional properties is of paramount significance. However, the standard solvothermal synthetic methodologies for COF synthesis do not always allow direct incorporation of functional groups into frameworks, due to the incompatibility with the building blocks or reaction conditions. In this regard, alternative approaches for post-synthetic modification (PSM) on a pre-established network provide an opportunity for tuning the functional properties of COFs while maintaining a fundamental framework structure. This review describes recent advances and scientific challenges in the area of post-synthetic methodologies to tailor the properties of COFs. Two different major PSM techniques, namely, covalent PSM and post-synthetic metal functionalization, will be discussed.
 Huimin Ding | Huimin Ding received her BS in Chemistry from Qufu Normal University in 2012. She pursued her PhD study under the supervision of Prof. Cheng Wang at Wuhan University and obtained her PhD degree in 2018, where she is currently carrying out postdoctoral research on 3D covalent organic frameworks. |
 Arindam Mal | Dr Arindam Mal completed his Master of Science at Indian Institute of Technology, Kharagpur (India) and PhD from CSIR-National Institute for Interdisciplinary Science and Technology (India). He is currently working as a postdoctoral research scholar of Wuhan University in Prof. Cheng Wang's research group under an international postdoctoral exchange fellowship program. His research focuses on 3D covalent organic frameworks. |
 Cheng Wang | Dr Cheng Wang obtained his BSc degree (2003) from Wuhan University and his PhD degree (2008) in organic chemistry from Institute of Chemistry, Chinese Academy of Sciences, under the supervision of Professor Deqing Zhang and Academician Professor Daoben Zhu. Following postdoctoral research with Professor Sir J. Fraser Stoddart in Northwestern University, he returned to China on March 2012 and started his new position as a full professor at College of Chemistry and Molecular Sciences, Wuhan University. His research focuses on functional microporous materials, including covalent organic frameworks and metal–organic frameworks. |
1. Introduction
Covalent organic frameworks (COFs) are a class of porous, crystalline materials made up of organic building units linked by strong covalent bonds to generate long-range ordered two-dimensional (2D)1–5 or three-dimensional (3D)6–10 network structures. The inherent characteristic structural features of COFs, such as pre-designable functionality and modular porous nature, have endowed this emerging material with interesting applications in gas storage and separation,11–13 catalysis,14–17 chemical sensing,18–22 optoelectronics,23–26 proton conductivity,27–29 energy storage30–33 and so on. Therefore, the incorporation of novel functional moieties into COFs, which can allow the resulting material to have the desired properties, has gained much attention over the past decade. However, sometimes, these functional groups could not be directly incorporated into the framework through the routine ‘bottom-up’ solvothermal synthetic strategy, due to the incompatibility with the building blocks or reaction conditions. Consequently, it is highly desirable to develop a simple and efficient method to overcome this issue.Post-synthetic modification (PSM),34 which can introduce new functional groups on a pre-established COF skeleton through chemical transformation or modification while retaining the underlying topology, provides a powerful method to construct functional COFs. Firstly, the characteristic strong covalent linkages in COFs offer structural robustness necessary for PSM techniques. Secondly, the judicious selection of building blocks allows the integration of linkers with pendant reactive groups or metal binding sites into COFs in predetermined positions for PSM. Thirdly, the high crystallinity of COFs endows the ability to implement a PSM process and results in materials which can be well characterized. Thanks to the efforts of many groups, significant progress has been achieved in constructing COFs with structural diversity and complexity via PSM strategies.
In recent times, a few interesting reviews on PSM of COFs have been reported,35,36 indicating the importance of this research area. However, a concise discussion covering the most important achievements of PSM strategies with more focused classifications was still unavailable. In this review, we summarize the research progress in PSM of COFs over the past decade, by classifying the functionalization strategy into two parts, covalent PSM and post-synthetic metal functionalization. According to different modification sites, the former can be further subdivided as the covalent PSM of linkers and post-synthetic conversion of linkages, where the latter can be understood via coordinating with the functional groups on the backbone. Lastly, a brief perspective on the challenges and possibilities of this field will be covered.
2. Covalent post-synthetic modification
Covalent PSM of COFs is considered to be the most significant strategy, which involves functional group inter-conversion through different covalent reactions. In this part, we will summarize the covalent PSM in two different sections, covalent PSM of linkers and linkages.2.1. Covalent post-synthetic modification of linkers
Starting from a COF with predesigned reactive groups, functional moieties can be covalently attached to the COF backbone by reacting with linkers while retaining the original framework. In this part, we present the main organic reactions used for COF PSM functionalization.Furthermore, to avoid the instability of boronate ester COFs, Jiang and co-workers synthesized an ultra-stable imine-based COF (TPB-DMTP) with alkyne groups.40 By performing a post-synthetic CuAAC click reaction, organocatalytic sites were anchored onto the pore channels to generate [(S)-Py]X%-TPB-DMTP-COFs, which can be used as an efficient heterogeneous catalyst in Michael addition reaction (Fig. 1). For example, the quantitative conversion of cyclohexanone and β-nitrostyrene was achieved in 12 hours with enantioselectivity (92% e.e.) and diastereoselectivity (90/10 d.r.). Very recently, Shustova and co-workers integrated corannulene π-bowls (πBs) into COFs by using this reaction while preserving porosity and crystallinity.41 Interestingly, the πBs-integrated COF exhibited 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold enhancement in conductivity compared to the initial framework.
000-fold enhancement in conductivity compared to the initial framework.
 | ||
Fig. 1 (a) Post-synthetic modification of [HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C]X-TPB-DMTP-COFs with chiral functional groups via a CuAAC reaction. (b) Channel-wall structure of [(S)-Py]0.17-TPB-DMTP-COF. The space between the catalytic sites is required for the face-on stacking of nitrostyrene over the catalytic sites and plays a role in controlling the activity of the catalysts. (c) [HCC]X-TPB-DMTP-COFs can be used as catalysts in asymmetric Michael reactions. Reproduced from ref. 40, with permission from Springer Nature, Copyright 2015. C]X-TPB-DMTP-COFs with chiral functional groups via a CuAAC reaction. (b) Channel-wall structure of [(S)-Py]0.17-TPB-DMTP-COF. The space between the catalytic sites is required for the face-on stacking of nitrostyrene over the catalytic sites and plays a role in controlling the activity of the catalysts. (c) [HCC]X-TPB-DMTP-COFs can be used as catalysts in asymmetric Michael reactions. Reproduced from ref. 40, with permission from Springer Nature, Copyright 2015. | ||
Another type of click reaction, the thiol–ene reaction, has also been extensively applied in PSM of COFs. Dichtel and co-workers have demonstrated that a functionalized 3D COF can be constructed by the combination of a tandem truncation-functionalization strategy with thiol–ene reaction.42 Firstly, via co-condensation of tetrahedral and truncated trigonal monomers, a boroxine-linked COF has been synthesized with a 22% allyl-loading throughout the lattice. Succeedingly, the as-prepared allyl functionalized COF has been subjected to quantitative thiol–ene reaction with propanethiol to produce an alkane-thiol modified COF, maintaining the crystallinity and permanent porosity. In another work, Ma and co-workers synthesized a vinyl-functionalized COF (COF-V) and subsequently modulated the structure and function via the same reaction (Fig. 2).43 The resulting sulfur-based COF-S-SH exhibited a strong binding affinity for Hg2+, with high uptake capacity of 1350 mg g−1. This performance was better than the previously reported thiol and thioether-functionalized adsorbents for mercury removal, which can be attributed to densely populated thiol groups (over 90%) within ordered mesopores. Very recently, they integrated perfluoroalkyl groups on COF-V for altering its water-repellent properties.44 Although only 4% of the vinyl groups were involved in the PSM process, the small number of perfluoroalkyl groups on the resulting COF significantly increased the hydrophobicity, with a static water contact angle of about 167°.
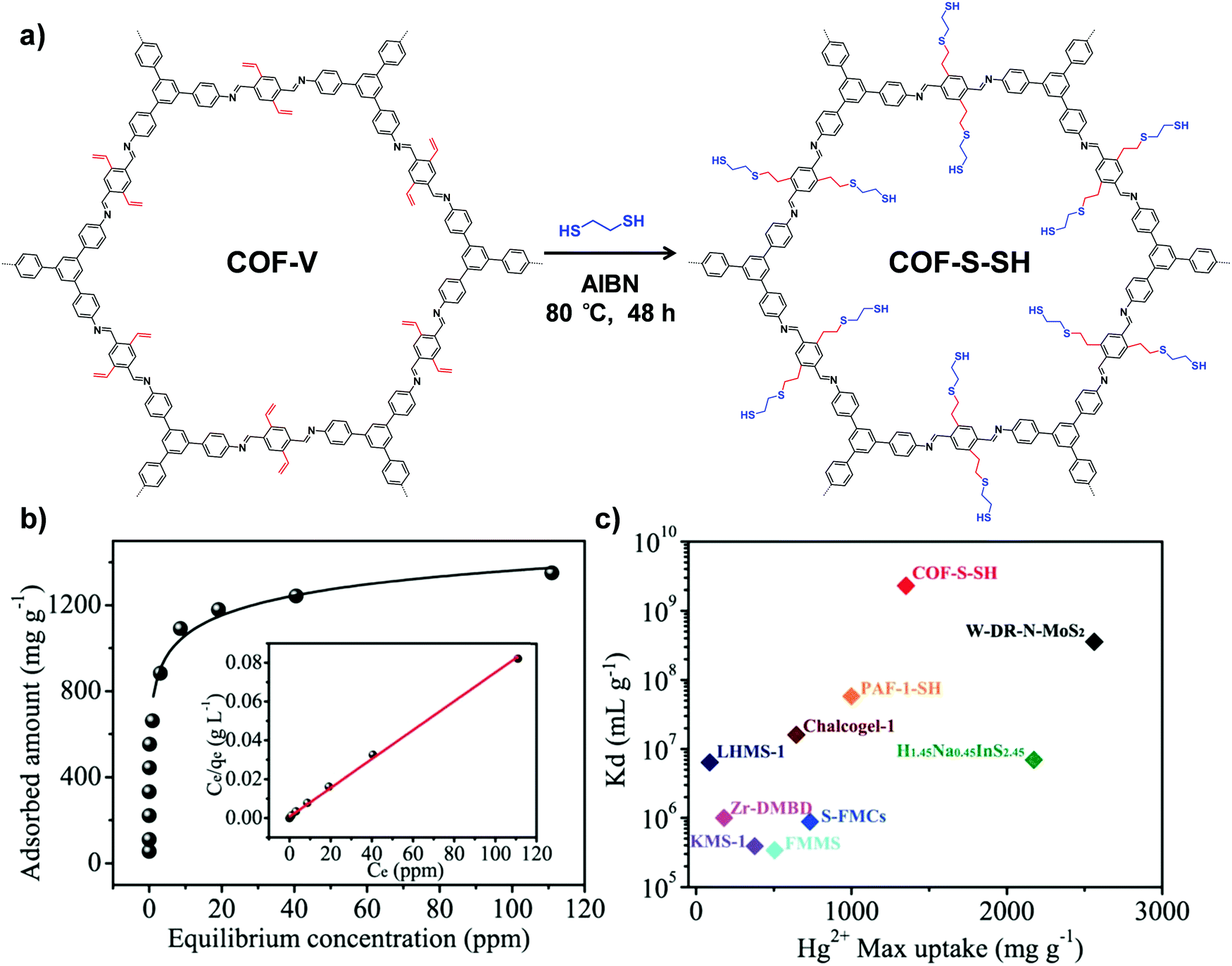 | ||
| Fig. 2 (a) Post-synthetic modification of COF-Vvia a thiol–ene click reaction. (b) Hg2+ adsorption isotherm for COF-S-SH. Inset shows the linear regression by fitting the equilibrium data with the Langmuir adsorption model. (c) Comparison of Hg2+ saturation uptake amount and Kd value for COF-S-SH with those of benchmark porous materials. Reproduced from ref. 43, with permission from American Chemical Society, Copyright 2017. | ||
Jiang and co-workers reported the design and synthesis of [HO]X%-H2P-COFs with hydroxyl groups which could react with succinic anhydride to generate a series of COFs ([HO2C]X%-H2P-COFs) bearing a tunable amount of carboxylic acid on the pore walls.45 The decreased pore size, together with carboxylic acid groups on [HO2C]X%-H2P-COFs enabled the superior performance of CO2 adsorption. Later on, Fang and co-workers reported a hydroxy-functionalized 3D COF which can be used to construct a carboxy-functionalized COF that exhibited selective and high metal-loading capability (0.71 mmol g−1/Nd3+, 0.72 mmol g−1/Sr2+ and 4.86 mmol g−1/Fe3+).46
Gao and co-workers have controllably synthesized a series of COFs with various contents of ester functionalities via a post-synthetic acylation reaction of the hydroxyl groups and carbonyl chloride.47 The attached CO2-philic units and N2-phobic units, i.e., 4-phenylazobenzoyl chloride and stilbenecarbonyl chloride could significantly affect the CO2 uptake and CO2/N2 selectivity of the COFs. Very recently, the same group synthesized a sulfonic acid-based COF via an etherification reaction with the phenolic hydroxyl moieties of the COF and 1,3-propane sultone.48 The modified COF having charged sulfonic acid groups was able to capture metal ions through the ion exchange process. In addition, the obtained ionic COF showed good catalytic activity in the epoxidation of olefins, with yields of 83%, 80%, and 62% for cyclohexene, cyclooctene, and 1-hexene, respectively.
The hydroxyl groups present in COFs can also be applied in a nucleophilic substitution or addition reaction with halides and isothiocyanate, respectively. Gao and associates have demonstrated that phenol groups of pyrene-based COFs could take part in a post-synthetic Williamson ether reaction with (2-bromoethyl)triethylammonium bromide.49 The obtained ionic COF facilitated excellent heterogeneous catalytic performance for the N-formylation reactions of different amines. For example, N-methylanilines with electron-donating or electron-withdrawing groups were converted into the corresponding formamides with isolated yields over 88%.
Bein and co-workers grafted a fluorescent dye onto the framework of a hydroxyl-functionalized mesoporous COF by reacting with fluorescein-isothiocyanate (FITC).50 Even though the proportion of hydroxyl groups involved in a PSM process was low, the FITC-labelled COF exhibited strong green fluorescence which was absent in the case of the initial COF.
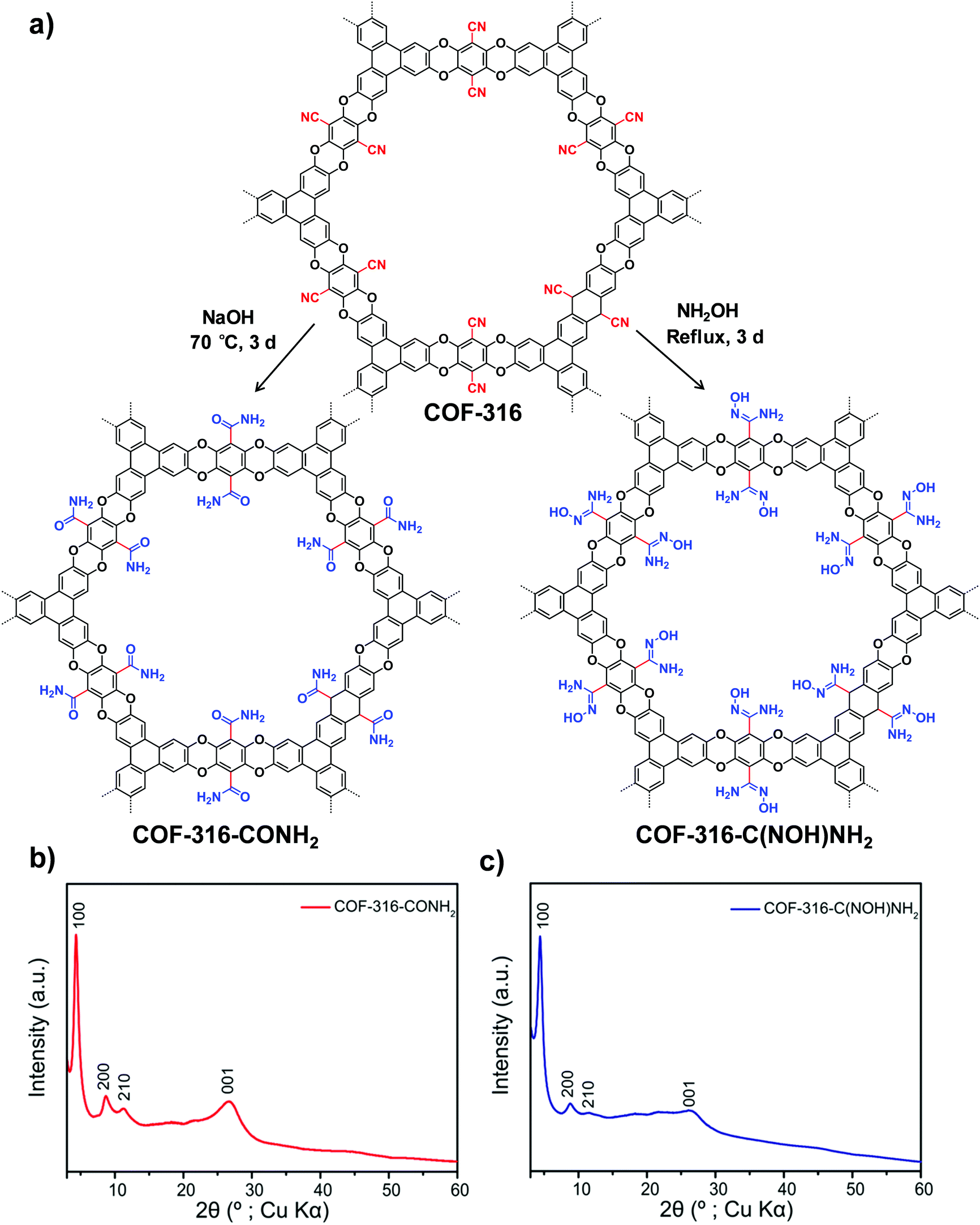 | ||
| Fig. 3 (a) Transformation of the nitrile-functionalized COF-316 to amide-functionalized COF (COF-316-CONH2) and amidoxime-functionalized COF (COF-316-C(NOH)NH2). PXRD patterns: (b) COF-316-CONH2, (c) COF-316-C(NOH)NH2. Reproduced from ref. 51, with permission from American Chemical Society, Copyright 2018. | ||
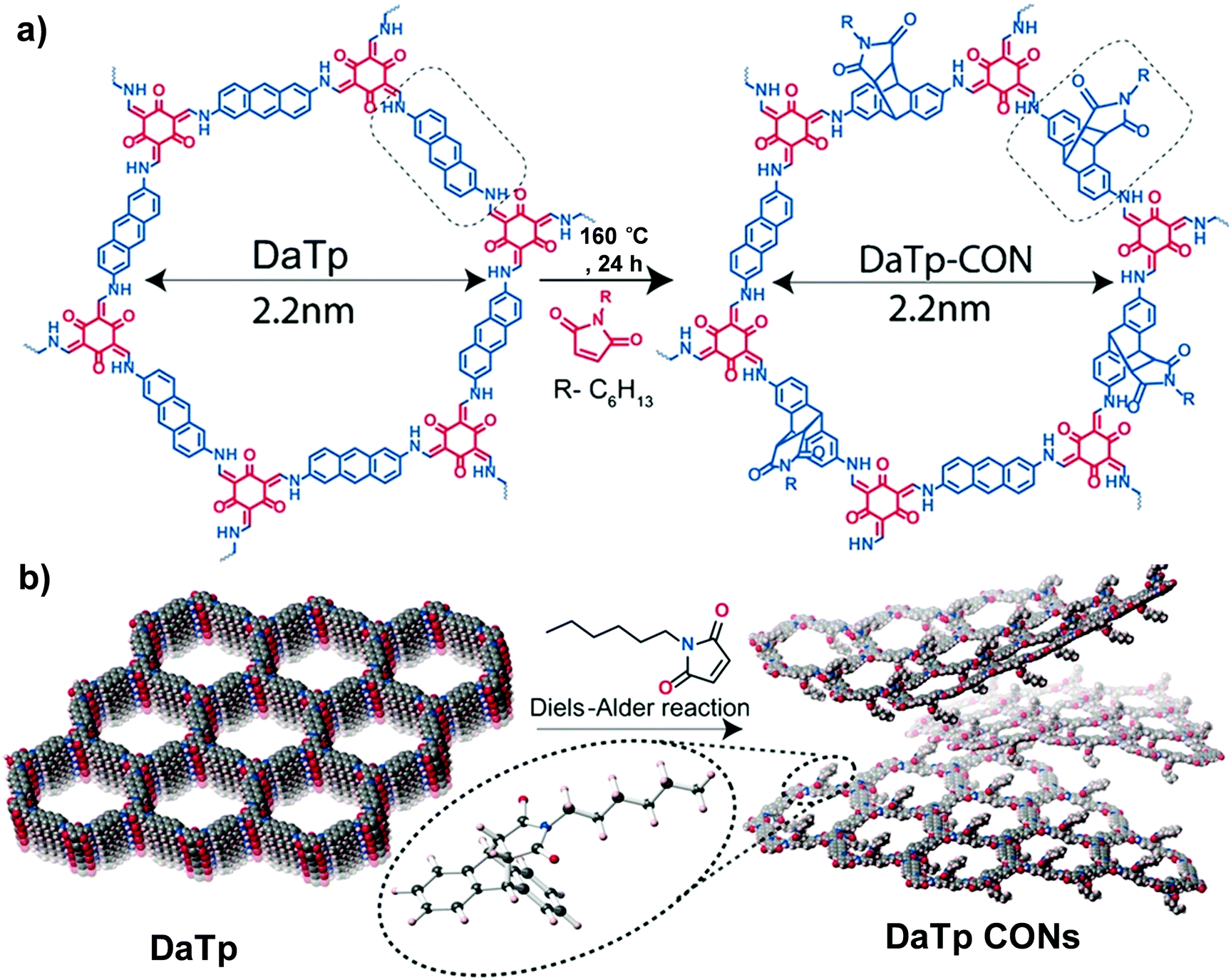 | ||
| Fig. 4 (a) Diels–Alder cycloaddition reaction of DaTp with N-hexylmaleimide to afford DaTp CONs. (b) Schematic representation of the exfoliation process. Reproduced from ref. 55, with permission from John Wiley and Sons, Copyright 2016. | ||
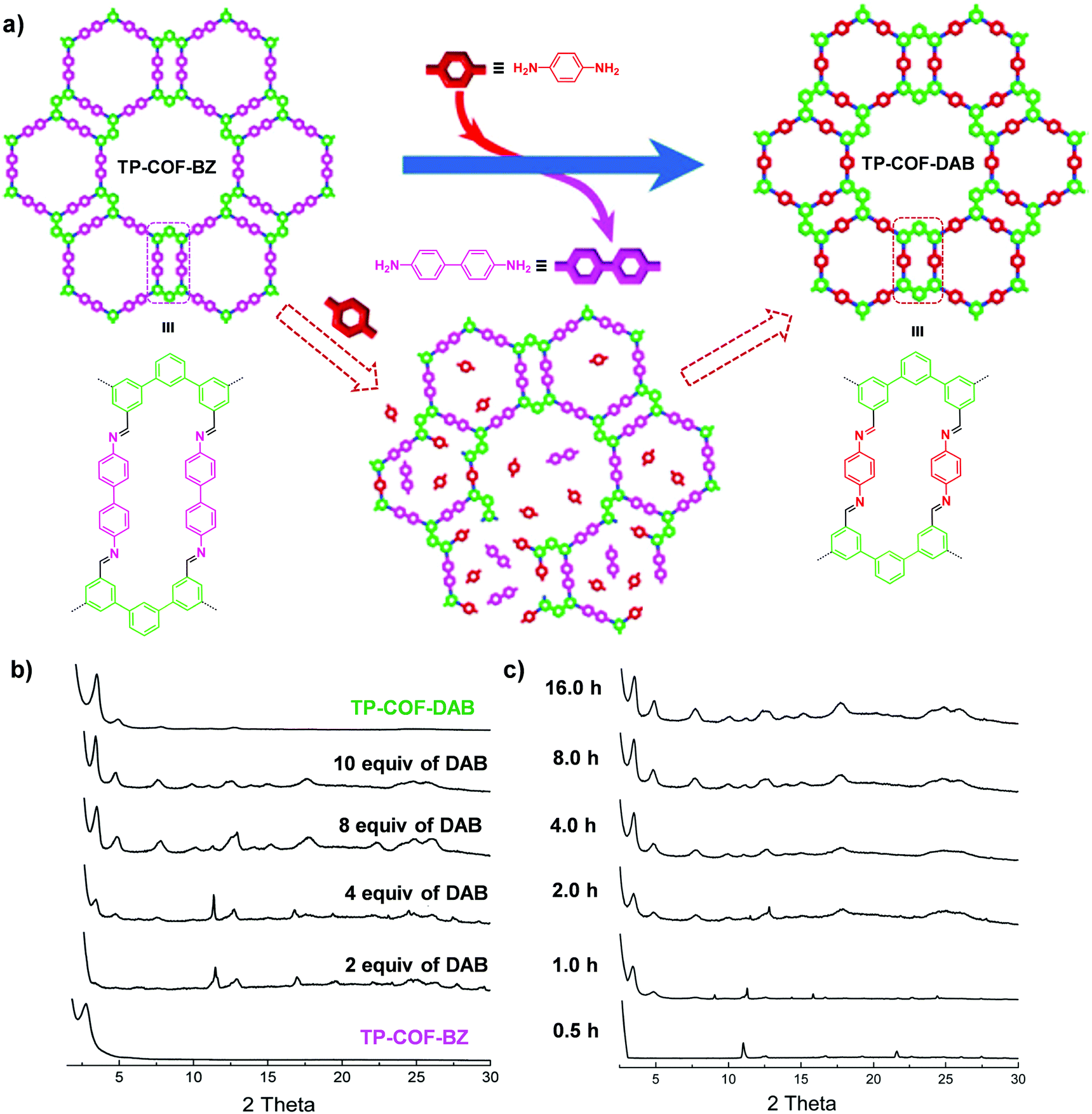 | ||
| Fig. 5 (a) Synthesis of TP-COF-DAB by post-synthetic linker exchange and the proposed process for the in situ transformation of TP-COF-BZ into TP-COF-DAB in the presence of 1,4-diaminobenzene. (b) Transformation from TP-COF-BZ to TP-COF-DAB in different equivalents of 1,4-diaminobenzene for 72 h at 120 °C indicated by PXRD patterns. (c) Time-dependent PXRD patterns of the samples prepared by heating a mixture of TP-COF-BZ and 1,4-diaminobenzene (10 equiv.). Reproduced from ref. 65, with permission from American Chemical Society, Copyright 2017. | ||
2.2. Post-synthetic conversion of linkages
Besides the linkers, the linkages of COFs can also take part in a PSM reaction. However, only the imine-linked COFs have been successfully transformed to the corresponding modified COFs by several reactions. In this part, we will summarize the recent progress of this PSM conversion in detail.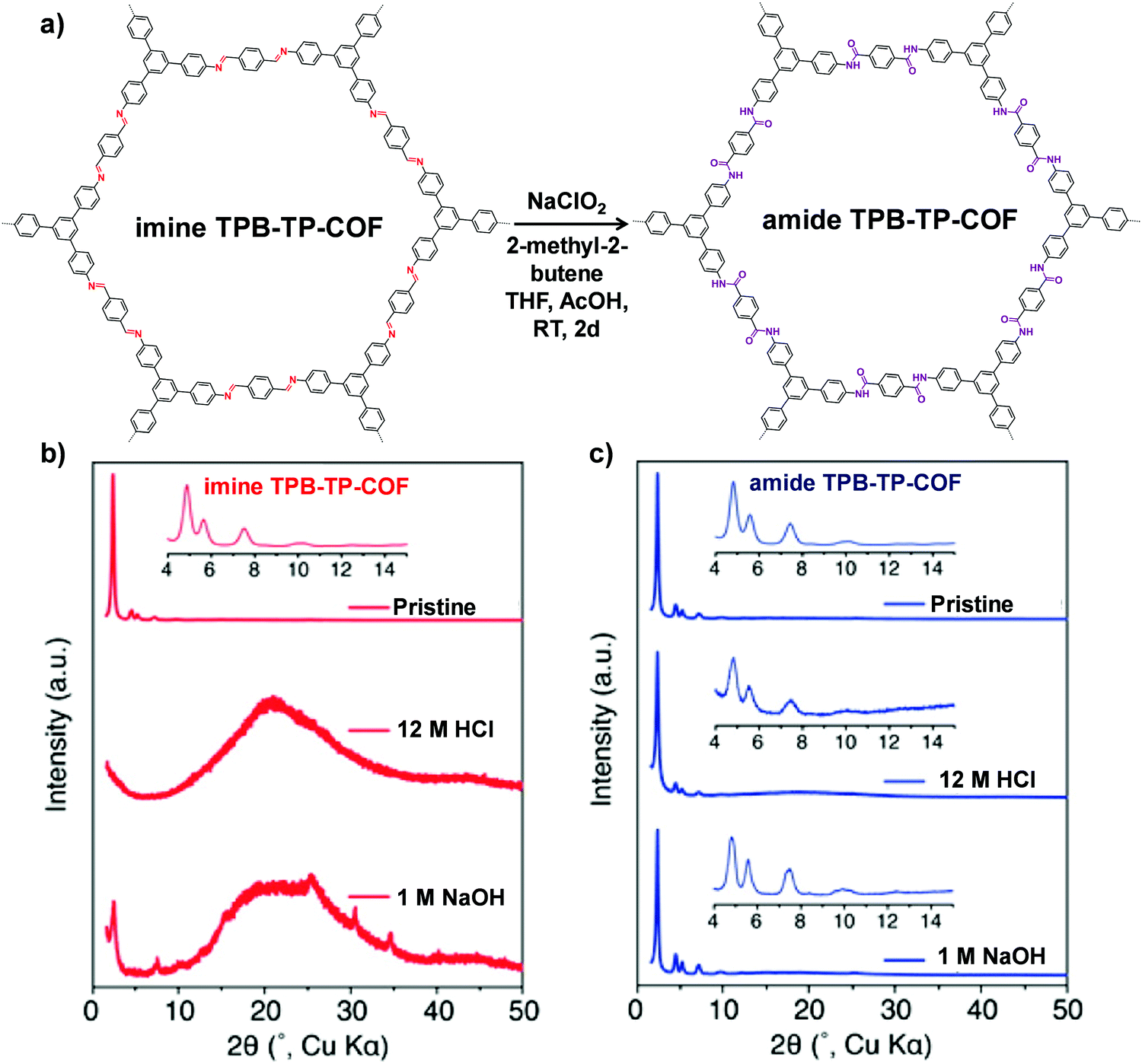 | ||
| Fig. 6 (a) Conversion of imine TPB-TP-COF to amide TPB-TP-COF under oxidative reaction conditions. Comparison of diffraction patterns of imine TPB-TP-COF (b) and amide TPB-TP-COF (c) after 24 h treatment in solutions. Reproduced from ref. 67, with permission from American Chemical Society, Copyright 2016. | ||
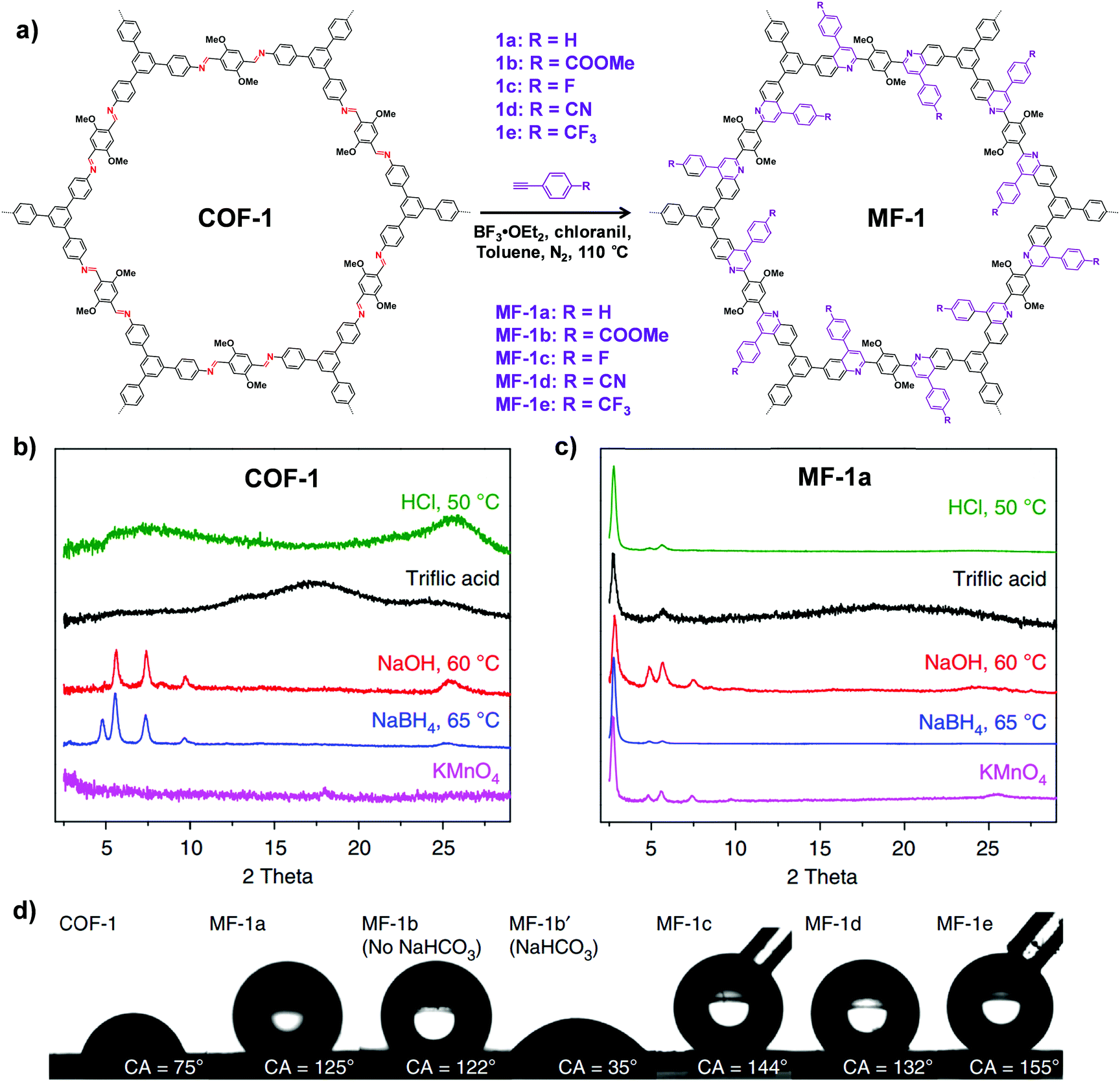 | ||
| Fig. 7 (a) Synthesis of the imine-linked COF-1 to quinoline-linked MF-1 by aza-Diels–Alder cycloaddition reaction. PXRD patterns of COF-1 (b) and MF-1a (c) after treatment with different conditions. (d) Contact angles of water droplets on the pressed pellet of COF-1 and MF-1a–e. Reproduced from ref. 69, with permission from Springer Nature, Copyright 2018. | ||
Analogously, Lotsch and co-workers also transformed an imine-linked COF into a thiazole-linked COF through cascade oxidation-cyclisation (Fig. 8).71 The elemental sulfur oxidized aromatic imines to thioamides, followed by oxidative cyclization to form a thiazole ring. The final COF displayed enhanced chemical and electron beam stability, enabling investigation of the exact framework structure in great detail by electron diffraction and transmission electron microscopy. It is worth mentioning that this work revealed the structural features of 2D COFs including grain boundaries and edge dislocations. Very recently, Baek and co-workers also converted an imine-linked COF into a benzoxazole-linked COF via post-cyclization–oxidation.72 Due to the irreversibility of the benzoxazole linkage, the converted COF still retained high crystallinity and porosity even under 12 M HCl(aq.) and 10 M NaOH(aq.) conditions.
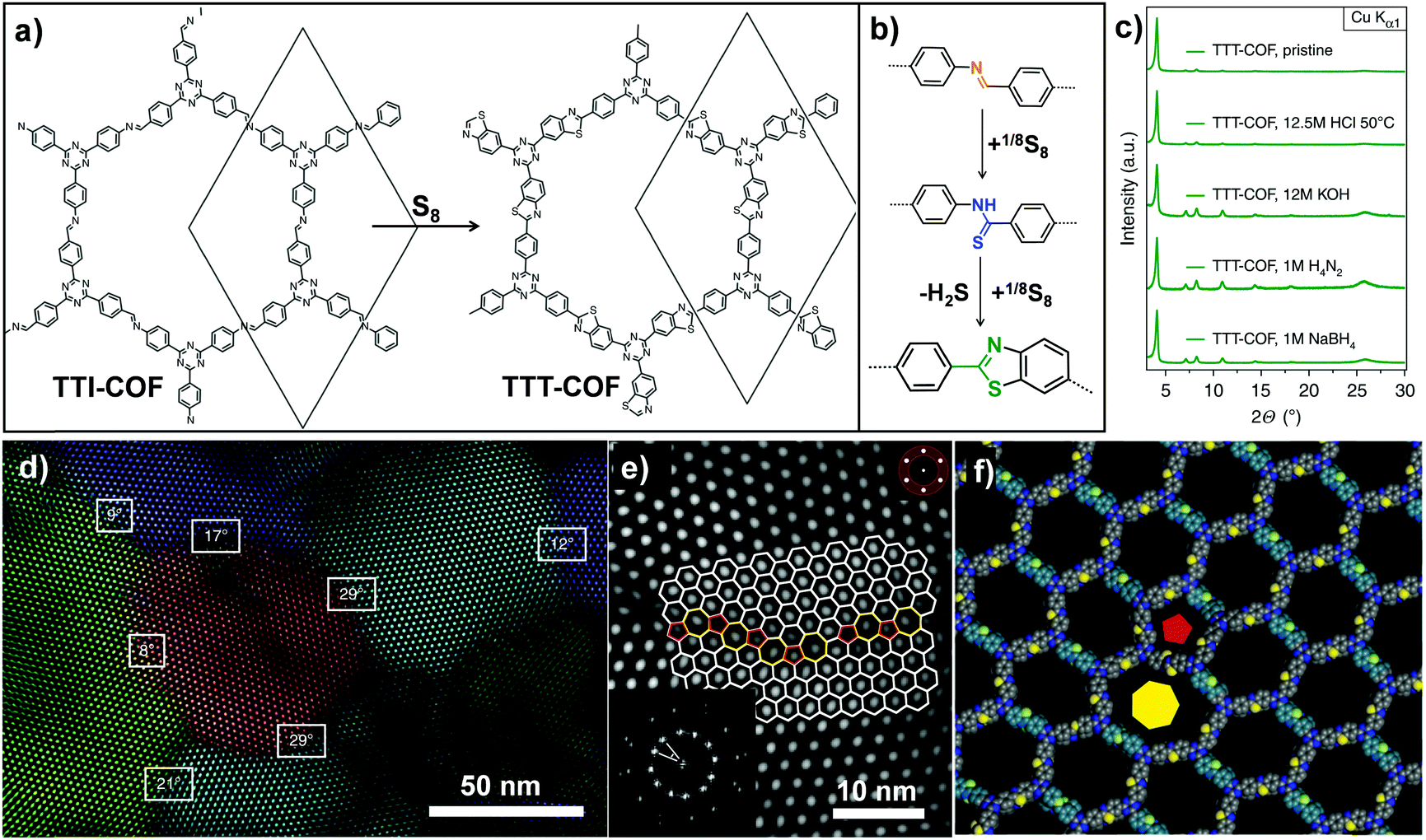 | ||
| Fig. 8 (a) Post-synthetic transformation of the imine-linked TTI-COF to thiazole-linked TTT-COF. (b) Linkage transformation from imine to thioamide and to thiazole. (c) The TTT-COF samples were exposed to various conditions for 16 h. (d) Grain boundaries and edge dislocations visible by TEM. (e) High angle grain boundary of crystallites showed the interface consisting of five, six and seven membered rings. (f) Modeling of dislocation along the pentagonal (red pentagon) and the heptagonal channel (yellow heptagon). Reproduced from ref. 71, with permission from Springer Nature, Copyright 2018. | ||
3. Post-synthetic metal-functionalization
COFs are the ideal candidates for metal-functionalization, because both the linkers and linkages consisting of electronegative elements such as N, O, S etc., can act as anchoring sites for metal binding. In a typical procedure, specific metallic units were incorporated into the as-prepared COFs by a coordination complexation reaction, affording metal-containing COFs with interesting properties. Based on the different probable interactions between metal ions and COF structures, the existing post-synthetic metal-functionalization methods can be classified into three classes: coordination with functional groups of linkers (a) or linkages (b) and post-synthetic demetallation or metal exchange (c).3.1. Post-synthetic metalation on organic linkers
COFs with multidentated ligands such as porphyrin, bipyridine, N,N′-bis(salicylidene)ethylenediamine (Salen) etc. can be utilized as matrices for metalation. In this section, we will focus on the post-synthetic metalation strategy of COFs possessing these linkers.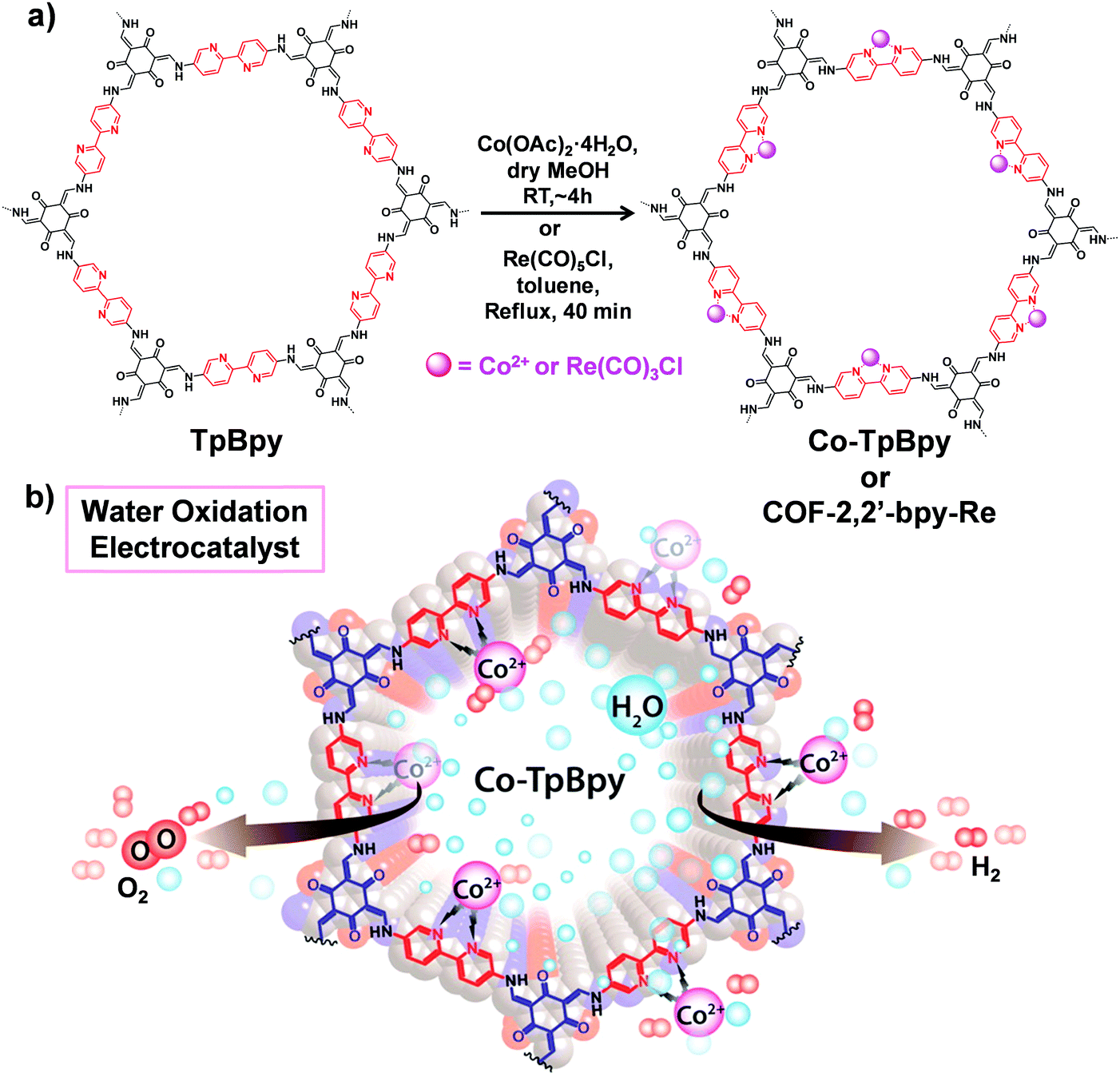 | ||
| Fig. 9 (a) Post-synthetic metalation of TpBpy with cobalt and rhenium moieties to afford Co-TpBpy and COF-2,2′-bpy-Re, respectively. (b) Utilizing Co-TpBpy as an oxygen-evolving catalyst. Reproduced from ref. 80, with permission from American Chemical Society, Copyright 2016. | ||
Via a similar post-metalation process, Marinescu and co-workers successfully incorporated the rhenium(I) tricarbonyl fragments into a β-ketoenamine linked COF with the maintenance of crystallinity and porosity.81 In another report, Huang and co-workers have designed a photoactive COF hybrid upon one-step post-synthetic incorporation of tricarbonylchloro(bipyridyl)Re complex into a 2D triazine COF.82 Recently, Cui and co-workers post-metalated 2,2′-bipyridine containing COFs to achieve metallo-bipyridyl COFs, which can be used as a heterogeneous catalyst in C–H borylation of arenes.83
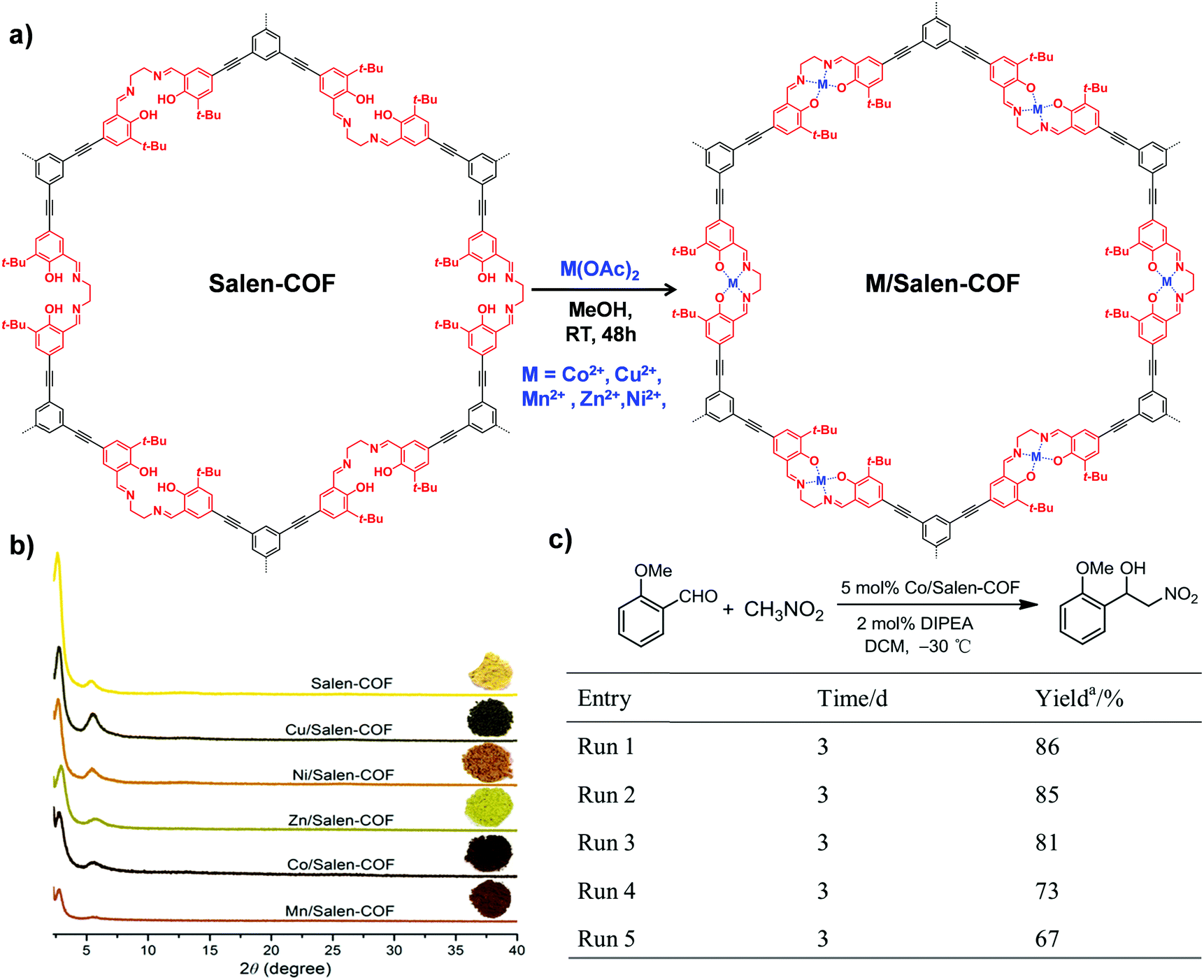 | ||
| Fig. 10 (a) The synthesis of M/Salen-COF by post-metalation of Salen-COF with various metal acetates. (b) Comparison of the PXRD patterns and colors of Salen-COF and M/Salen-COF. (c) Recycling tests of Co/Salen-COF in a Henry reaction. Reproduced from ref. 85, with permission from American Chemical Society, Copyright 2017. | ||
3.2. Post-synthetic metalation with linkages
The COF linkages, such as imine and hydrazine bonds, can bind to specific metal ions. Early in 2011, Wang et al. constructed Pd/COF-LZU1via a simple post-synthetic metalation of imine-linked 2D COF (COF-LZU1) (Fig. 11).88 Owing to the eclipsed and ordered π-columnar structures, the palladium metals were sandwiched between the adjacent COF layers and coordinated with the imine bonds. Interestingly, the resulting Pd/COF-LZU1 can be used as a catalyst in the Suzuki–Miyaura coupling reaction (Fig. 11). In the presence of 0.5 mol% of Pd/COF-LZU1, the substituted aryl iodides afforded the cross-coupling products in high yields (96–98%). After this seminal work, many other reports appeared where metal integrated COFs have been utilized in heterogeneous catalysis.89–91 For example, Chen and co-workers have reported the immobilization of copper salts into the Schiff base centers of an imine-linked 2D COF with hydroxyl functionalities.92 Interestingly, the copper-modified COF displayed good reusability and stability toward selective oxidation of styrene to benzaldehyde.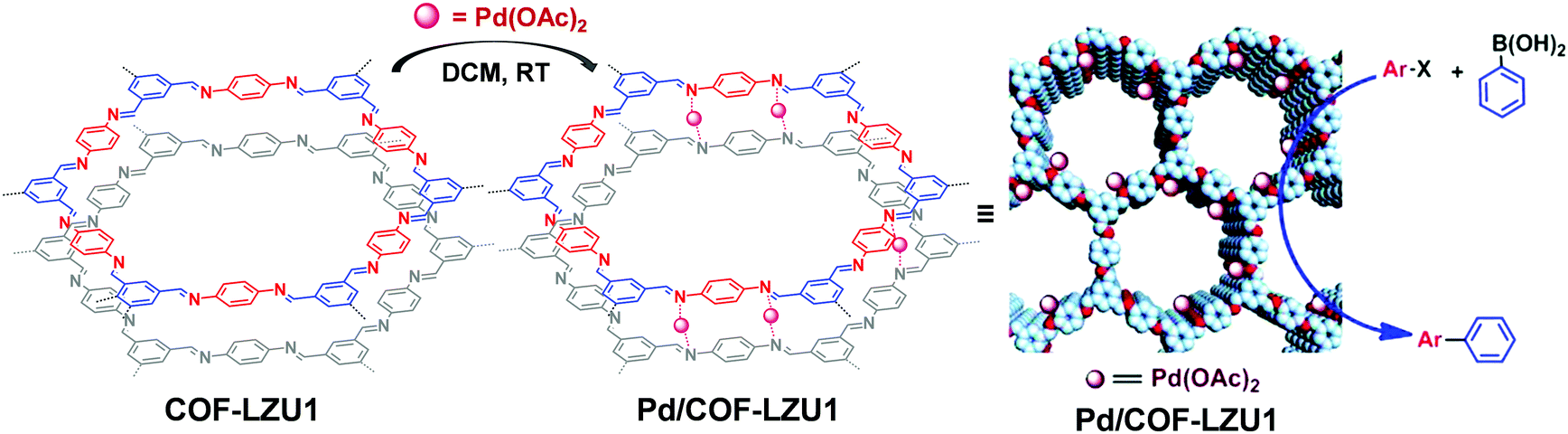 | ||
| Fig. 11 Post-synthetic metalation of COF-LZU1 with Pd(OAc)2, the resulting Pd/COF-LZU1 was used as a catalyst in a Suzuki–Miyaura coupling reaction. Reproduced from ref. 88, with permission from American Chemical Society, Copyright 2011. | ||
3.3. Post-synthetic demetallation or metal exchange
Similar to the post-synthetic deprotection strategy, post-synthetic demetallation involves the removal of metal ions from a metalated COF. Yaghi et al. have reported the construction of a 3D Cu(I) coordinated COF (COF-505), which can be demetalated by heating in a KCN solution (Fig. 12).93 Interestingly, upon removal of copper, the demetalated COF-505 showed more elastic properties with decreased effective Young's moduli of 1.3 GPa. It is worth mentioning that the metalation-demetalation and associated elasticity changes of COF-505 were found to be reversible upon reaction with Cu(I) ions.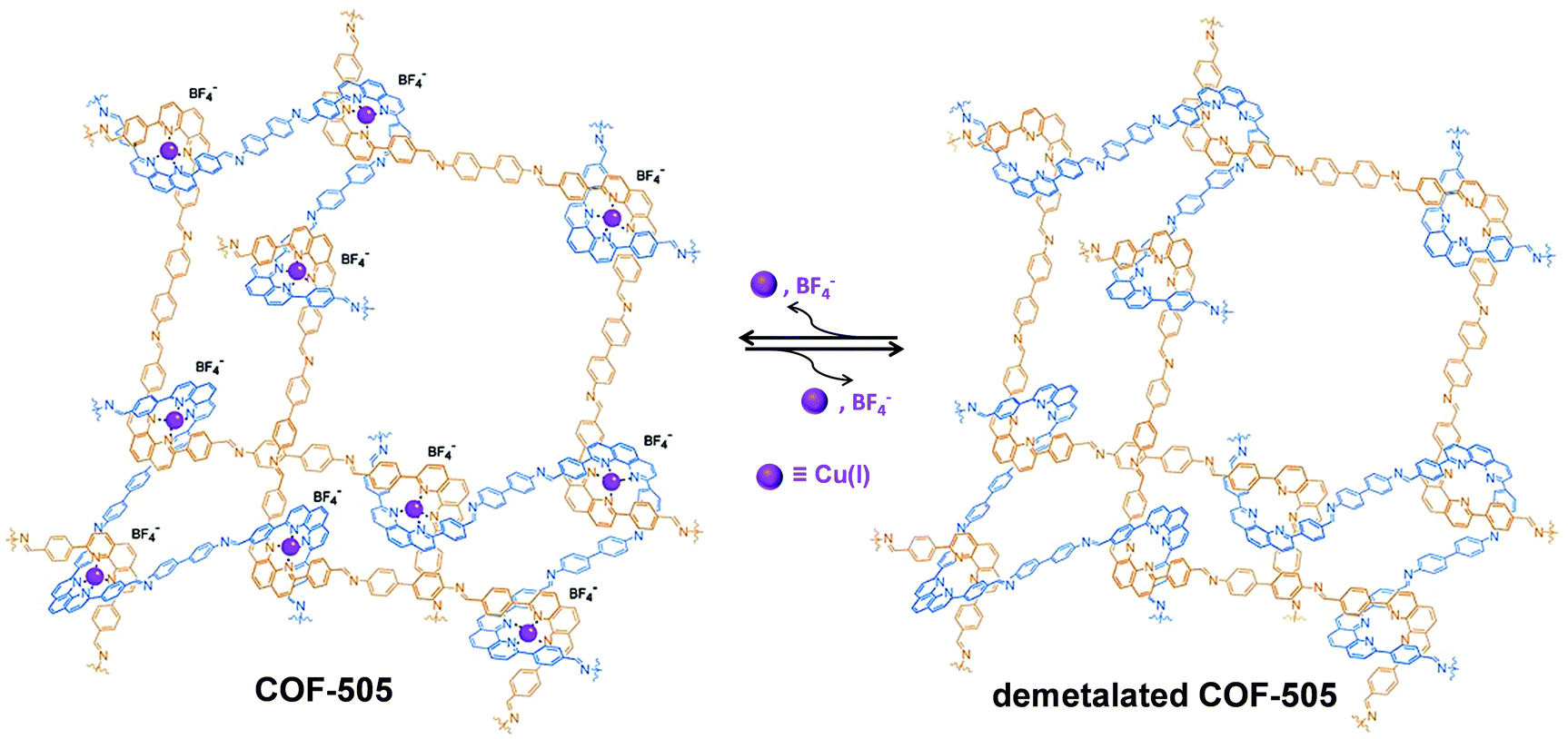 | ||
| Fig. 12 Post-synthetic demetalation and remetalation of COF-505. Reproduced from ref. 93, with permission from The American Association for the Advancement of Science, Copyright 2016. | ||
The post-synthetic metal exchange, associated with demetalation94 followed by remetalation with different metal ions, is another crucial PSM approach for COF functionalization. Recently, starting from Zn(salen) COF (CCOF-4), Cui et al. reported the construction of bimetallic COF (4-Cr-M′) by two-step post-synthetic metal exchange (Fig. 13), which showed advantageous catalytic properties in several asymmetric transformations.95 In particular, in the epoxidation reaction of alkene followed by ring opening of epoxide to amino alcohol, 4-Cr-Mn exhibited better catalytic activity compared to 4-Cr.
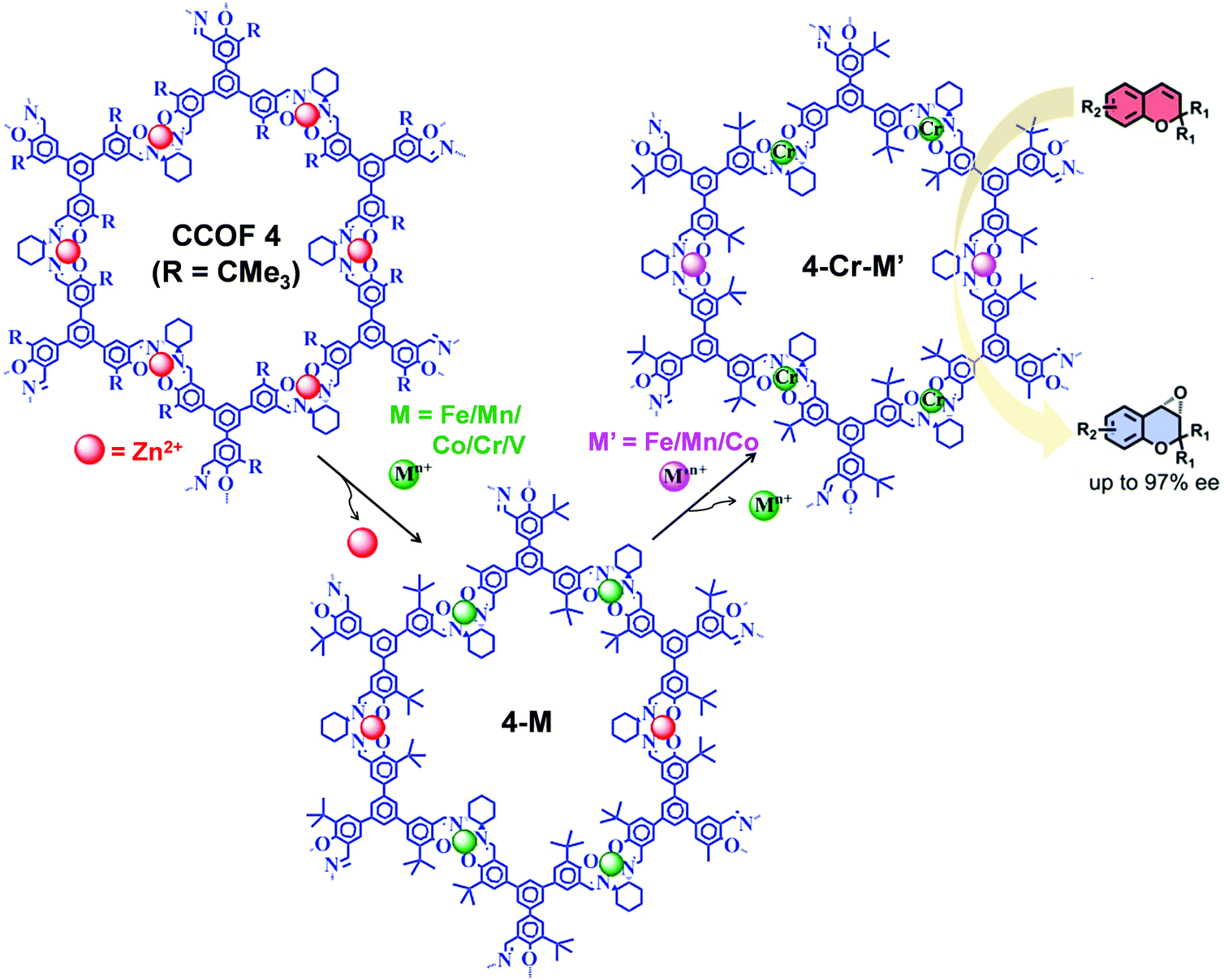 | ||
| Fig. 13 Synthesis of 4-Cr-M′ from CCOF-4via consecutive post-synthetic metal exchange reactions. 4-Cr-M′ as heterogeneous catalysts for the synthesis of amino-alcohol in sequential reactions starting from epoxidation reaction of alkene followed by ring-opening of epoxide. Reproduced from ref. 95, with permission from American Chemical Society, Copyright 2017. | ||
4. Conclusions and outlook
Over the last decade, rapid progress has been witnessed in tailor-made functionalities and properties of COFs due to the emergence of various post-synthetic modification protocols. This review presents a comprehensive collection of the state-of-the-art PSM strategies for COF functionalization. We believe that in future, the vast array of both covalent chemical transformations and metal-functionalization protocols will offer an excellent opportunity for accessing COF materials with increased complexity and advanced functions.Nevertheless, a large number of challenges need to be addressed in the field of functional exploration of COFs by PSM approaches. For example, vast post-synthetic options, including new methodologies and new reactions, remain to be further attempted. Apart from this, one vital point to be noted is the degree of post-synthetic conversion, which typically stays far from quantitative. Considering that the high concentration of functionalities in the COF framework endows them with superior performance, the next great challenge lies ahead to increase the contents of functional modules on the COF skeleton. Another issue to be looked at is the spatial location of the modified functional groups in COFs, as the functionalities present on the COF surface or the inside pores may result in different performances, especially in gas selectivity and catalytic activity. Thus, the exploration of functional group distributions in COFs is of great significance to establish the mechanistic pathways of adsorption or catalytic properties. Besides, PSM-programming for more complex multi-component COF architectures remains as the ultimate goal in order to realize the enormous potential of COFs in various fields. We still have a long way to go and an expanding research area in PSM of COFs for desired functionalities can be imagined.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21772149, 21572170 and 21905212), the Funds for Creative Research Groups of Hubei Province (2017CFA002) and the Fundamental Research Funds for the Central Universities.References
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS.
- Y. Jin, Y. Hu and W. Zhang, Nat. Rev. Chem., 2017, 1, 0056 CrossRef.
- S.-L. Cai, K. Zhang, J.-B. Tan, S. Wang, S.-R. Zheng, J. Fan, Y. Yu, W.-G. Zhang and Y. Liu, ACS Macro Lett., 2016, 5, 1348–1352 CrossRef CAS.
- R.-R. Liang and X. Zhao, Org. Chem. Front., 2018, 5, 3341–3356 RSC.
- F. Haase, K. Gottschling, L. Stegbauer, L. S. Germann, R. Gutzler, V. Duppel, V. S. Vyas, K. Kern, R. E. Dinnebier and B. V. Lotsch, Mater. Chem. Front., 2017, 1, 1354–1361 RSC.
- C. Gao, J. Li, S. Yin, G. Lin, T. Ma, Y. Meng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 9770–9775 CrossRef CAS.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS.
- X. Ma and T. F. Scott, Chem. Commun., 2018, 1, 98 CrossRef.
- Y.-B. Zhang, J. Su, H. Furukawa, Y. Yun, F. Gandara, A. Duong, X. Zou and O. M. Yaghi, J. Am. Chem. Soc., 2013, 135, 16336–16339 CrossRef CAS.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS.
- H. Fan, A. Mundstock, A. Feldhoff, A. Knebel, J. Gu, H. Meng and J. Caro, J. Am. Chem. Soc., 2018, 140, 10094–10098 CrossRef CAS.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- S. Yan, X. Guan, H. Li, D. Li, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 2920–2924 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS.
- Q. Sun, B. Aguila and S. Ma, Mater. Chem. Front., 2017, 1, 1310–1316 RSC.
- Q. Gao, X. Li, G.-H. Ning, K. Leng, B. Tian, C. Liu, W. Tang, H.-S. Xu and K. P. Loh, Chem. Commun., 2018, 54, 2349–2352 RSC.
- G. Lin, H. Ding, D. Yuan, B. Wang and C. Wang, J. Am. Chem. Soc., 2016, 138, 3302–3305 CrossRef CAS.
- R. Xue, H. Guo, T. Wang, L. Gong, Y. Wang, J. Ai, D. Huang, H. Chen and W. Yang, Anal. Methods, 2017, 9, 3737–3750 RSC.
- L. Ma, X. Feng, S. Wang and B. Wang, Mater. Chem. Front., 2017, 1, 2474–2486 RSC.
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS.
- M. Dogru and T. Bein, Chem. Commun., 2014, 50, 5531–5546 RSC.
- D. D. Medina, T. Sick and T. Bein, Adv. Energy Mater., 2017, 7, 1700387 CrossRef.
- H. Ding, J. Li, G. Xie, G. Lin, R. Chen, Z. Peng, C. Yang, B. Wang, J. Sun and C. Wang, Nat. Commun., 2018, 9, 5234 CrossRef.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2009, 48, 5439–5442 CrossRef CAS.
- X. Meng, H.-N. Wang, S.-Y. Song and H.-J. Zhang, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
- Z. Meng, A. Aykanat and K. A. Mirica, Chem. Mater., 2019, 31, 819–825 CrossRef CAS.
- C. Montoro, D. Rodriguez-San-Miguel, E. Polo, R. Escudero-Cid, M. Luisa Ruiz-Gonzalez, J. A. R. Navarro, P. Ocon and F. Zamora, J. Am. Chem. Soc., 2017, 139, 10079–10086 CrossRef CAS.
- C.-Y. Lin, D. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2018, 30, 1703646 CrossRef.
- X. Zhan, Z. Chen and Q. Zhang, J. Mater. Chem. A, 2017, 5, 14463–14479 RSC.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- H. Liao, H. Wang, H. Ding, X. Meng, H. Xu, B. Wang, X. Ai and C. Wang, J. Mater. Chem. A, 2016, 4, 7416–7421 RSC.
- V. Valtchev, G. Majano, S. Mintova and J. Perez-Ramirez, Chem. Soc. Rev., 2013, 42, 263–290 RSC.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- Y. Yusran, Q. Fang and S. Qiu, Isr. J. Chem., 2018, 58, 971–984 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Nat. Commun., 2011, 2, 536 CrossRef.
- L. Chen, K. Furukawa, J. Gao, A. Nagai, T. Nakamura, Y. Dong and D. Jiang, J. Am. Chem. Soc., 2014, 136, 9806–9809 CrossRef CAS.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS.
- A. M. Rice, E. A. Dolgopolova, B. J. Yarbrough, G. A. Leith, C. R. Martin, K. S. Stephenson, R. A. Heugh, A. J. Brandt, D. A. Chen, S. G. Karakalos, M. D. Smith, K. B. Hatzell, P. J. Pellechia, S. Garashchuk and N. B. Shustova, Angew. Chem., Int. Ed., 2018, 57, 11310–11315 CrossRef CAS.
- D. N. Bunck and W. R. Dichtel, Chem. Commun., 2013, 49, 2457–2459 RSC.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS.
- Q. Sun, B. Aguila, J. A. Perman, T. Butts, F.-S. Xiao and S. Ma, Chem, 2018, 4, 1726–1739 CAS.
- N. Huang, X. Chen, R. Krishna and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 2986–2990 CrossRef CAS.
- Q. Lu, Y. Ma, H. Li, X. Guan, Y. Yusran, M. Xue, Q. Fang, Y. Yan, S. Qiu and V. Valtchev, Angew. Chem., Int. Ed., 2018, 57, 6042–6048 CrossRef CAS.
- S. Zhao, B. Dong, R. Ge, C. Wang, X. Song, W. Ma, Y. Wang, C. Hao, X. Guo and Y. Gao, RSC Adv., 2016, 6, 38774–38781 RSC.
- H. Hu, Q. Yan, M. Wang, L. Yu, W. Pan, B. Wang and Y. Gao, Chin. J. Catal., 2018, 39, 1437–1444 CrossRef CAS.
- B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- S. Rager, M. Dogru, V. Werner, A. Gavryushin, M. Goetz, H. Engelke, D. D. Medina, P. Knochel and T. Bein, CrystEngComm, 2017, 19, 4886–4891 RSC.
- B. Zhang, M. Wei, H. Mao, X. Pei, S. A. Alshmimri, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 12715–12719 CrossRef CAS.
- Q. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Ma, Adv. Mater., 2018, 30, 1705479 CrossRef.
- D. Becker, B. P. Biswal, P. Kalenczuk, N. Chandrasekhar, L. Giebeler, M. Addicoat, S. Paasch, E. Brunner, K. Leo, A. Dianat, G. Cuniberti, R. Berger and X. Feng, Chem. – Eur. J., 2019, 25, 6562–6568 CrossRef CAS.
- M. S. Lohse, T. Stassin, G. Naudin, S. Wuttke, R. Ameloot, D. De Vos, D. D. Medina and T. Bein, Chem. Mater., 2016, 28, 626–631 CrossRef CAS.
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Angew. Chem., Int. Ed., 2016, 55, 15604–15608 CrossRef CAS.
- Y. Wu, Z. Zhang, S. Bandow and K. Awaga, Bull. Chem. Soc. Jpn., 2017, 90, 1382–1387 CrossRef CAS.
- S. Royuela, J. Almarza, M. J. Mancheno, J. C. Perez-Flores, E. G. Michel, M. M. Ramos, F. Zamora, P. Occon and J. L. Segura, Chem. – Eur. J., 2019, 25, 12394–12404 CrossRef CAS.
- Q. Jiang, Y. Li, X. Zhao, P. Xiong, X. Yu, Y. Xu and L. Chen, J. Mater. Chem. A, 2018, 6, 17977–17981 RSC.
- H.-S. Xu, S.-Y. Ding, W.-K. An, H. Wu and W. Wang, J. Am. Chem. Soc., 2016, 138, 11489–11492 CrossRef CAS.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8277–8285 CrossRef CAS.
- H.-L. Qian, Y. Li and X.-P. Yan, J. Mater. Chem. A, 2018, 6, 17307–17311 RSC.
- G. Zhang, M. Tsujimoto, D. Packwood, D. Nghia Tuan, Y. Nishiyama, K. Kadota, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2018, 140, 2602–2609 CrossRef CAS PubMed.
- M. C. Daugherty, E. Vitaku, R. L. Li, A. M. Evans, A. D. Chavez and W. R. Dichtel, Chem. Commun., 2019, 55, 2680–2683 RSC.
- Z. Miao, G. Liu, Y. Cui, Z. Liu, J. Li, F. Han, Y. Liu, X. Sun, X. Gong, Y. Zhai, Y. Zhao and Y. Zeng, Angew. Chem., Int. Ed., 2019, 58, 4906–4910 CrossRef CAS.
- C. Qian, Q.-Y. Qi, G.-F. Jiang, F.-Z. Cui, Y. Tian and X. Zhao, J. Am. Chem. Soc., 2017, 139, 6736–6743 CrossRef CAS.
- Z. Li, X. Ding, Y. Feng, W. Feng and B.-H. Han, Macromolecules, 2019, 52, 1257–1265 CrossRef.
- P. J. Waller, S. J. Lyle, T. M. O. Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15519–15522 CrossRef CAS.
- X. Hang, J. Huang, C. Yuan, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 892–895 CrossRef.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Nat. Commun., 2018, 9, 2998 CrossRef.
- P. J. Waller, Y. S. AlFaraj, C. S. Diercks, N. N. Jarenwattananon and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 9099–9103 CrossRef CAS.
- F. Haase, E. Troschke, G. Savasci, T. Banerjee, V. Duppel, S. Doerfler, M. M. J. Grundei, A. M. Burow, C. Ochsenfeld, S. Kaskel and B. V. Lotsch, Nat. Commun., 2018, 9, 2600 CrossRef.
- J.-M. Seo, H.-J. Noh, H. Y. Jeong and J.-B. Baek, J. Am. Chem. Soc., 2019, 141, 11786–11790 CrossRef CAS.
- S. J. Lyle, T. M. O. Popp, P. J. Waller, X. Pei, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11253–11258 CrossRef CAS.
- K. S. Suslick, P. Bhyrappa, J. H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283–291 CrossRef CAS.
- G. M. Eder, D. A. Pyles, E. R. Wolfson and P. L. McGrier, Chem. Commun., 2019, 55, 7195–7198 RSC.
- N. Keller, M. Calik, D. Sharapa, H. R. Soni, P. M. Zehetmaier, S. Rager, F. Auras, A. C. Jakowetz, A. Goerling, T. Clark and T. Bein, J. Am. Chem. Soc., 2018, 140, 16544–16552 CrossRef CAS.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705–8709 CrossRef CAS.
- K. Xu, Y. Dai, B. Ye and H. Wang, Dalton Trans., 2017, 46, 10780–10785 RSC.
- E. M. Johnson, R. Haiges and S. C. Marinescu, ACS Appl. Mater. Interfaces, 2018, 10, 37919–37927 CrossRef CAS.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- D. A. Popov, J. M. Luna, N. M. Orchanian, R. Haiges, C. A. Downes and S. C. Marinescu, Dalton Trans., 2018, 47, 17450–17460 RSC.
- S. Yang, W. Hu, X. Zhang, P. He, B. Pattengale, C. Liu, M. Cendejas, I. Hermans, X. Zhang, J. Zhang and J. Huang, J. Am. Chem. Soc., 2018, 140, 14614–14618 CrossRef CAS.
- X. Wu, X. Han, Y. Liu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2018, 140, 16124–16133 CrossRef CAS PubMed.
- C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 CrossRef CAS.
- L.-H. Li, X.-L. Feng, X.-H. Cui, Y.-X. Ma, S.-Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS.
- J. W. Crowe, L. A. Baldwin and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 10120–10123 CrossRef CAS.
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 15134–15137 CrossRef CAS PubMed.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS.
- T. Banerjee, F. Haase, G. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228–16234 CrossRef CAS.
- L. Chen, L. Zhang, Z. Chen, H. Liu, R. Luque and Y. Li, Chem. Sci., 2016, 7, 6015–6020 RSC.
- J. Romero, D. Rodriguez-San-Miguel, A. Ribera, R. Mas-Ballesté, T. F. Otero, I. Manet, F. Licio, G. Abellán, F. Zamora and E. Coronado, J. Mater. Chem. A, 2017, 5, 4343–4351 RSC.
- M. Mu, Y. Wang, Y. Qin, X. Yan, Y. Li and L. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 22856–22863 CrossRef CAS.
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gandara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS.
- Y. Liu, C. S. Diercks, Y. Ma, H. Lyu, C. Zhu, S. A. Alshmimri, S. Alshihri and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 677–683 CrossRef CAS.
- X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS.
| This journal is © the Partner Organisations 2020 |