
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(vinyl acetate-co-ethylene) particles prepared by surfactant-free emulsion polymerization in the presence of a hydrophilic RAFT/MADIX macromolecular chain transfer agent†
James
Delorme
 a,
Olivier
Boyron
a,
Pierre-Yves
Dugas
a,
Pierre-Emmanuel
Dufils
b,
D. James
Wilson
b,
Vincent
Monteil
a,
Franck
D'Agosto
*a and
Muriel
Lansalot
*a
a,
Olivier
Boyron
a,
Pierre-Yves
Dugas
a,
Pierre-Emmanuel
Dufils
b,
D. James
Wilson
b,
Vincent
Monteil
a,
Franck
D'Agosto
*a and
Muriel
Lansalot
*a
aUniv Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS, UMR 5265, Chemistry, Catalysis, Polymers and Processes (C2P2), 43 Bd du 11 Novembre 1918, 69616 Villeurbanne, France. E-mail: franck.dagosto@univ-lyon1.fr; muriel.lansalot@univ-lyon1.fr
bSolvay Novecare, Research and Innovation Centre – Paris, 93306 Aubervilliers, France
First published on 16th October 2020
Abstract
Poly(acrylamide-co-acrylic acid) (P(AAm-co-AA)-X) was prepared by RAFT/MADIX and used as a hydrophilic macromolecular chain transfer agent (macroCTA) in the aqueous emulsion copolymerization of vinyl acetate (VAc) and ethylene. Stable latexes were obtained over a broad range of conditions with macroCTA contents ranging from 1 to 65 wt% (compared to the initial amount of VAc) and ethylene pressure from 10 to 100 bar. The different systems investigated generated latexes incorporating amorphous to semi-crystalline poly(vinyl acetate-co-ethylene) (P(VAc-co-E)) chains using macroCTA content as low as 1 wt% in the absence of additional surfactant. The particle nucleation mechanism was investigated with the help of kinetic studies using cryogenic transmission electronic microscopy (cryoTEM) and was consistent with the concepts underlying polymerization-induced self-assembly (PISA). High solids content latexes were finally targeted with a formulation more in line with industrial constraints (0.4 wt% macroCTA, 35 bar ethylene pressure, semi-batch conditions). A stable P(VAc-co-E) latex was produced exhibiting a solids content of 38 wt%. This work provides an easy access to a full range of alternative stabilization modes for P(VAc-co-E) latexes and potentially to new VAE and EVA products.
Introduction
Copolymers of ethylene and vinyl acetate (VAc) are part of a very important class of polymeric materials, known for a long time in industry, the first known copolymerization trials dating back to 1928.1 These copolymers can be produced in the presence of VAc either by reactive extrusion of polyethylene, or by copolymerization in solution, bulk or dispersed media under low to very high temperature and ethylene pressure (>150 °C and >1000 bar, respectively). In the case of copolymerization, this broad range of experimental conditions gives access to a broad range of copolymer compositions. Containing low (<30 wt%) to high content of ethylene (>70 wt%), the resulting poly(vinyl acetate-co-ethylene) (P(VAc-co-E)), also called VAE or EVA, respectively, can achieve completely amorphous to semi-crystalline properties. EVAs find applications in fuel additives, wire and cable insulation or foams, while VAEs are used for adhesive, paint and coating applications.2–5 In the case of VAEs, which are the main topic of this communication, ethylene actually acts as a plasticizing comonomer for PVAc, to counterbalance the rigid nature of the latter and bring more flexibility to the polymer backbone.1 Industrially, VAEs are mostly produced by aqueous emulsion polymerization at temperatures below 100 °C between 20 and 120 bar of ethylene pressure (mainly up to 30 bar).6 Whereas the patent literature on VAE synthesis is quite large, the number of papers available in the open literature is comparatively rather limited. This is likely due to the fact that special equipment is required to work with ethylene (high pressure reactor). In addition, as a gas, ethylene induces more complexity in the systems in terms of monomer partitioning and diffusion. The most extensive study was conducted by Scott et al.,3,7–10 who have screened in detail the impact of different parameters on semi-batch emulsion copolymerization of ethylene and vinyl acetate, such as the type and amount of the initiating system, of the buffer, and of the emulsifier. The addition of a co-solvent and the VAc feed rate were also considered, as well as the agitation and mixing phenomena and the reactor design. Experiments were mostly performed at low temperatures (20 and 40 °C) and pressures (15 to 35 bar) as the authors were aiming at increasing the ethylene content under mild conditions. This allowed the formation of copolymers containing up to 34 wt% of ethylene units. The large number of these tunable parameters and their interactions demonstrate the complexity of VAE syntheses. In this process, the surfactant is an important ingredient in the recipe since, after the composition of the final VAE itself, it defines to a large extent the final properties of the targeted product.11 Different types of surfactants have thus been investigated to produce VAE latexes. These include conventional anionic surfactants,3,7,12–15 mixtures of anionic and nonionic surfactants,7 protective colloids such as hydroxyethyl cellulose7 or poly(vinyl alcohol) (PVOH)9,11,16,17 or combinations of those.9,18,19 PEG-based macromolecules have also been investigated.11Among the different strategies employed, the use of hydrophilic PVOH as protective colloid is quite widespread. Indeed, resulting from partial hydrolysis of preformed PVAc, PVOH offers multiple stabilization possibilities depending on its VAc content and its microstructure, that can be blocky depending on the PVAc hydrolysis conditions,20–22 and consequently leads to various VAE products.
When PVOH is used for VAc emulsion polymerization, it somehow acts as a reactive stabilizer due to chain transfer reactions taking place in water along PVOH chains, followed by reinitiation of VAc polymerization.23–26 To the best of our knowledge, the occurrence of such reactions in the case of emulsion copolymerization of vinyl acetate and ethylene has been only briefly mentioned by Reynolds,1 but never evidenced. However, considering the similar reactivity of these two monomers (reactivity ratios, rVAc and rE, are both close to one, with a slight pressure dependence of rE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27–31), and the fact that VAc is significantly more water-soluble (26 g L−1 at 50 °C32) than ethylene (0.3 g L−1 at 4.5 bar to 4.6 g L−1 at 111 bar, at 75 °C33), one can safely assume that similar transfer reactions happen during VAE synthesis and that particle stabilization is most probably ensured via grafting reactions occurring along the PVOH backbone.1
27–31), and the fact that VAc is significantly more water-soluble (26 g L−1 at 50 °C32) than ethylene (0.3 g L−1 at 4.5 bar to 4.6 g L−1 at 111 bar, at 75 °C33), one can safely assume that similar transfer reactions happen during VAE synthesis and that particle stabilization is most probably ensured via grafting reactions occurring along the PVOH backbone.1
Reversible-deactivation radical polymerization (RDRP) techniques may advantageously be used in emulsion polymerization to form surfactant-free latexes.34,35 This approach is indeed part of the so-called polymerization-induced self-assembly (PISA) strategy,34–37 which takes advantage of the chain-end reactivity of solvophilic macromolecules obtained by RDRP for the polymerization of solvophobic monomers. Hydrophilic chains can thus be extended with hydrophobic monomers directly in water. The amphiphilic block copolymers formed in situ are not only excellent stabilizers for the particles produced simultaneously by emulsion polymerization, but also provide an interesting tool to tailor the particle surface through the composition of the hydrophilic segment.
We successfully undertook several studies based on this concept employing hydrophilic polymer chains obtained by reversible addition–fragmentation chain transfer (RAFT) and macromolecular design by interchange of xanthates (MADIX) in order to produce industrially relevant surfactant-free formulation for the production of film forming latex such as poly(meth)acrylics,38–41 poly(vinylidene fluoride)42 and poly(vinylidene chloride).43–45
In the present paper, we apply this strategy for the first time to the emulsion copolymerization of ethylene and vinyl acetate for the production of VAE, building on our recent works on the polymerization of ethylene by free radical polymerization in water under mild conditions (i.e., temperature <90 °C and ethylene pressure <250 bar),46–48 and on PVAc49,50 and polyethylene (PE)51 particle synthesis mediated by macromolecular chain transfer agents (macroCTAs). The reactivity of these two monomers restricts the choice of potential macroCTAs to hydrophilic polymer chains carrying dithiocarbonate or dithiocarbamate end groups. Here, a poly(acrylamide-co-acrylic acid) (P(AAm-co-AA)-X) containing an equimolar ratio of AA and AAm was prepared by RAFT/MADIX and employed in emulsion polymerization as hydrophilic macroCTA (Scheme 1). The ethylene pressure and the macroCTA content were varied to first validate the strategy and then identify the potential of the system. Analysis of the obtained latexes was performed to characterize the particles by dynamic light scattering (DLS) and cryogenic transmission electronic microscopy (cryoTEM), but also the copolymer chains by differential scanning calorimetry (DSC), thermogravimetry (TGA), infrared (IR) and nuclear magnetic resonance (NMR) spectroscopy. Eventually, conditions were identified to isolate stable VAE latexes under industrially relevant experimental conditions and the process (batch or semi-batch) was adjusted to achieve high solids content. To the best of our knowledge, this is the first time that reactive macromolecules obtained by RDRP (RAFT/MADIX here) are used to form VAE latexes.
 | ||
| Scheme 1 Preparation of poly(vinyl acetate-co-ethylene) particles by emulsion polymerization using P(AAm-co-AA)-X as macroCTA. | ||
Experimental
Materials
Acrylic acid (AA, Aldrich, 99.8%), acrylamide (AAm, SNF, 50 wt% solution), ammonium persulfate (APS, Aldrich, >98%), ethylene (E, 99.95%, Air Liquide), Rhodixan A1 (Solvay, 95%), hydroxymethane sulfonic acid sodium salt dihydrate (NaFS, Aldrich, >98%), sodium persulfate (NaPS, 98+%, Aldrich) and ethanol (VWR, 96%) were used as received. Vinyl acetate (VAc, 99+%, Aldrich) was purified by removing the inhibitor by filtration with aluminum oxide. Water was deionized before use (Purelab ClassicUV, Elga LabWater).Synthesis of poly(acrylamide-co-acrylic acid) (P(AAm-co-AA)-X macroCTA
In a typical procedure, water (58 g), ethanol (70 g), Rhodixan A1 (25 g), AA (4.84 g) and AAm (14.35 g, 50 wt% solution) were introduced in a glass reactor, equipped with a mechanical stirrer and a condenser. After deoxygenation by nitrogen bubbling, the mixture was heated to 40 °C. Then, aqueous solutions of APS (13.74 g, 16 wt%), and NaFS (4.87 g, 1 wt%) were introduced shot-wise. At the same time a solution of AA (236.9 g)/AAm (703.35 g, 50 wt%) and NaFS (34.66 g, 1.25 wt%) were introduced in the reactor for 240 minutes and 695 minutes respectively. After 300 minutes, the mixture was heated to 60 °C for 300 minutes and then cooled down to room temperature. Conversion of both monomers was complete (as evidenced by HLPC – data not shown), and the final polymer (Scheme 2) was obtained as a 36 wt% aqueous solution. The dried polymer was characterized by aqueous size exclusion chromatography (Mn ≈ 5300 g mol−1 and Đ = 1.3).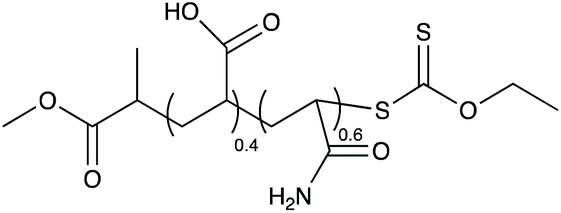 | ||
| Scheme 2 Structure of P(AAm-co-AA)-X macroCTA. | ||
Batch emulsion copolymerizations of vinyl acetate and ethylene
Caution: Polymerizations involve high pressures and explosive gas.Copolymerizations (Table 1) were performed in a 160 mL stainless steel autoclave (from Parr Instrument Co.) equipped with safety valves and a stirrer. An intermediate 1.5 L tank filled with ethylene was used to charge the reactor. The tank was cooled down to −20 °C to liquefy ethylene at 35 bar. Then the intermediate tank was isolated and heated to reach up to 300 bar of ethylene pressure. The initiator NaPS, macroCTA solution, VAc and water were introduced in a Schlenk tube. The volume of this mixture was always equal to 50 mL. Then the mixture was deoxygenated by argon bubbling for 30 min and introduced into the reactor through a cannula. The reactor was heated up to 77 °C beforehand and freed from oxygen by successive vacuum-argon cycles. Afterwards, ethylene was introduced until the desired pressure was reached, which marked the beginning of the polymerization. It is important to note that for all the polymerizations, the ethylene pressure value refers to the pressure set at the beginning of the polymerization. Pressure was not maintained constant during the polymerization, which induced depletion of ethylene from the reaction medium over time. The stirring speed was always set to 400 rpm. After the desired polymerization time, the reactor's jacket was cooled down by water. The reactor was then slowly degassed until reaching atmospheric pressure to collect the latex.
| Latex | macroCTAa (wt %) | P (bar) | [VAc] (mol L−1) | Time (min) | SCb (%) | PCb (%) | Z ave (nm) | PdIc | VAcd (wt%) | VAce (mol%) | X VAc (%) | T g (°C) | X c (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| For all experiments, T = 77 °C.a With respect to VAc amount.b Solid content (SC) and polymer content (PC), with PC = SC − amount of non-volatile species (in %).c Determined by DLS.d Weight content of VAc units in formed P(VAc-co-E) determined by averaging the values from the relevant analyses (see ESI†).e Molar content of VAc units in formed P(VAc-co-E) determined by averaging the values from the relevant analyses (see ESI†).f VAc conversion calculated from eqn (1).g Determined by DSC.h Degree of crystallinity (Xc) calculated from eqn (2).i The latex was not stable after 24 h.j Experiment run with P(AAm-co-AA), after removal of the xanthate chain end in P(AAm-co-AA)-X via peracetic acid treatment.k A large proportion of aggregates was present at the end of the polymerization (14.9 wt%). | |||||||||||||
| L1i | 1 | 0 | 1.09 | 240 | 8.0 | 7.7 | 150 | 0.03 | 100 | 100 | 79 | 42.8 | — |
| L2 | 1 | 10 | 1.09 | 240 | 9.4 | 9.1 | 145 | 0.02 | 88.7 | 77.1 | 89 | 30.8 | — |
| L3 | 1 | 35 | 1.09 | 240 | 9.7 | 9.3 | 158 | 0.02 | 80.6 | 57.5 | 80 | 7.7 | — |
| L4 | 1 | 50 | 1.09 | 240 | 11.4 | 11.2 | 148 | 0.02 | 70.0 | 43.2 | 83 | −12.0 | 1.2 |
| L5 | 1 | 100 | 1.09 | 240 | 13.9 | 13.7 | 165 | 0.03 | 41.7 | 19.0 | 61 | −35.7 | 9.1 |
| L6 | 0 | 35 | 1.09 | 60 | 5.8 | 5.7 | 505 | 0.10 | 85.0 | 64.9 | 50 | 10.3 | — |
| L3*j | 1 | 35 | 1.09 | 240 | 8.7 | 8.4 | 564 | 0.13 | 74.5 | 48.9 | 67 | 4.0 | — |
| L21 | 10 | 50 | 1.09 | 5 | 1.3 | 0.2 | — | — | — | — | — | — | — |
| L22 | 10 | 50 | 1.09 | 10 | 3.3 | 2.2 | 55 | 0.3 | — | — | — | 8.2 | — |
| L23 | 10 | 50 | 1.09 | 20 | 8.1 | 7.0 | 59 | 0.1 | 79.5 | 55.8 | 58 | 8.4 | — |
| L10 | 10 | 50 | 1.09 | 240 | 12.5 | 11.4 | 100 | 0.2 | 54.4 | 28.3 | 69 | −33.0 | 6.6 |
| L24 | 1 | 35 | 1.09 | 15 | 7.4 | 7.1 | 170 | 0.16 | 82.0 | 59.7 | 61 | 18.5 | — |
| L25 | 1 | 35 | 1.09 | 30 | 8.5 | 8.3 | 158 | 0.02 | 89.5 | 72.5 | 78 | 14.1 | — |
| L26 | 1 | 35 | 1.09 | 60 | 8.4 | 8.2 | 161 | 0.03 | 84.7 | 63.1 | 72 | 11.5 | — |
| L27 | 1 | 35 | 2.18 | 60 | 19.0 | 18.6 | 165 | 0.04 | 87.9 | 70.5 | 88 | 19.4 | — |
| L28 | 1 | 35 | 3.25 | 60 | 29.3 | 28.8 | 184 | 0.02 | 86.0 | 66.6 | 90 | 19.1 | — |
| L29k | 1 | 35 | 4.32 | 60 | 36.4 | 35.7 | 220 | 0.08 | 86.0 | 66.6 | 82 | 16.4 | — |
When kinetic studies were undertaken an experiment was carried out for each investigated time as no withdrawal of samples was technically possible during the polymerization.
Semi-batch emulsion copolymerizations of vinyl acetate and ethylene
For this process (Table 2), the reactor was set up the same way as for the batch process, but the preparation of the reaction mixture differed. In a first Schlenk tube were introduced a desired amount of initiator, macroCTA solution, water and VAc (mVAc). The volume in this Schlenk tube was always equal to 50 mL. The solution was then degassed under argon for 30 min. In a second Schlenk tube was introduced a desired amount of VAc (2mVAc for L30 and 3mVAc for L31). This medium was also degassed under argon for 30 min. The polymerization was launched using the same protocol as for the batch process. Then, every hour, mVAc was injected as a single shot into the reactor using a HPLC pump (flow rate = 10 mL min−1). Note that the ability of the pump to precisely deliver the corresponding quantity in the pressurized reactor has been checked. There were 2 and 3 injections for L30 and L31, respectively, during the polymerization. After the last addition, the polymerization was allowed to proceed for one more hour, setting the total reaction time to 3 and 4 hours, respectively. The polymerization was stopped as described for the batch process.| Latex | macroCTAa (wt%) | [VAc] (mol L−1) | SCb (%) | PCb (%) | Z ave (nm) | PdIc | VAcd (wt%) | VAce (mol%) | X VAc (%) | T g (°C) |
|---|---|---|---|---|---|---|---|---|---|---|
| For all experiments, T = 77 °C.a With respect the VAc amount.b Solid content (SC) and polymer content (PC), with PC = SC − amount of non-volatile species (in %).c Determined by DLS.d Weight content of VAc units in formed P(VAc-co-E) determined by averaging the values from the relevant analyses (see ESI†).e Molar content of VAc units in formed P(VAc-co-E) determined by averaging the values from the relevant analyses (see ESI†).f VAc conversion calculated from eqn (1).g Determined by DSC.h VAc was added in 2 shots.i VAc was added in 3 shots. | ||||||||||
| L30h | 1 | 3.25 | 29.6 | 29.2 | 143 | 0.04 | 82.3 | 59.8 | 90.6 | 9.9 |
| L31i | 0.4 | 4.32 | 38.3 | 38.0 | 266 | 0.09 | 89.3 | 72.5 | 90.5 | 14.8 |
Characterization
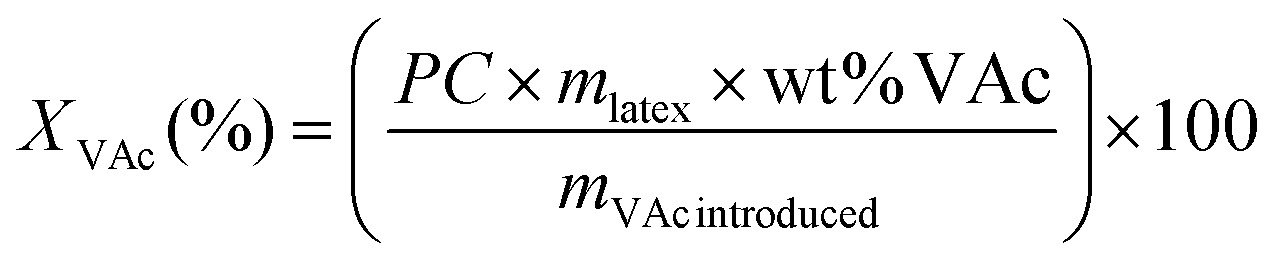 | (1) |
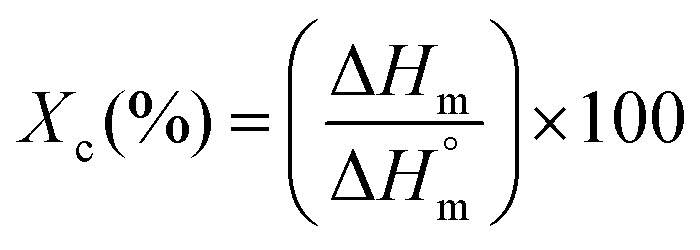 | (2) |
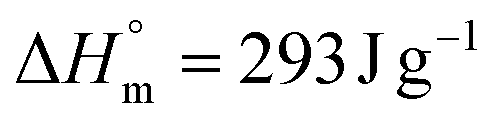 as standard enthalpy of polyethylene.
as standard enthalpy of polyethylene.
A partial least squares (PLS) method was previously constructed from a series of EVA in order to correlate the composition with the complete IR spectra. 26 EVA samples with well know VAc content were selected as standards to construct the chemometric model. TQ Analyst software from Thermo Fisher Scientific was used for the calibration and for the quantification of the composition of our EVA samples (see ESI†).
Results and discussion
Preliminary comments
To validate the strategy consisting in using for the first time hydrophilic P(AAm-co-AA)-X to produce VAE particles, we decided to employ conditions that could be of industrial interest in terms of hydrophilic species content and cost, i.e. using low amount of macroCTA. We thus started by employing 1 wt% of macroCTA with respect to VAc content and varied the ethylene pressure from 0 to 100 bar. In a second step, and to get a better insight into this system the amount of macroCTA was varied between 1 to 65 wt%. These experiments were all carried out in batch. The switch to semi-batch conditions was the focus of the last part of our study.Whatever the conditions used in the study, the isolated polymers forming the latexes could not be dissolved in any solvent, which made their analysis by size exclusion chromatography impossible. This phenomenon can find its origins in different reasons. The strong difference in chemical nature between very hydrophilic P(AAm-co-AA)-X and the hydrophobic formed VAE chains is probably one of them, particularly when the content of macroCTA is high. In addition, the known ability of this copolymerization system to lead to branching1,3,18,52 and a possible grafting along the P(AAm-co-AA) backbone,1 similarly to what was reported with PVOH, may give rise to gel formation impeding complete solubilization. Nevertheless, the formation of translucent gels upon mixing with a mixture of tetrachloroethylene and deuterated benzene still allowed liquid NMR analysis. However, as the signals remained broad, additional characterization tools such as ATR-FTIR, TGA and DSC were used (and detailed in ESI†) in combination with NMR to determine relevant values for the composition of the formed chains.
Synthesis of poly(vinyl acetate-co-ethylene) latexes under different ethylene pressures using 1 wt% of macroCTA
Stable PVAc latex could be formed with 1 wt% of P(AAm-co-AA)-X after 4 h of polymerization as shown in Table 1 (entry L1). The Tg measured by DSC is 42.8 °C (Table 1 and Fig. 2), a bit higher that some literature values for PVAc (e.g., ∼30 °C,53 37 °C,9 41 °C54). Increasing the pressure of ethylene from 10 to 100 bar led to stable latexes too (entries L2 to L5, Table 1), the particle sizes measured by DLS remaining in the same range (145 to 165 nm) as the one of the PVAc latex (150 nm) showing the strong ability of P(AAm-co-AA)-X to stabilize the particles. PdIs remained low (<0.03) and additionally showed that P(AAm-co-AA)-X induced an efficient nucleation over a broad range of experimental conditions. DLS data were confirmed by cryo-TEM pictures (Fig. 1 for L3, L4 and L5, Table 1). The proof that VAE were formed for the first time using emulsion polymerization of ethylene and vinyl acetate mediated by P(AAm-co-AA)-X came from DSC measurements of the polymer formed (Fig. 2). Indeed, upon increase of the ethylene pressure, the Tg originally at 42.8 °C for PVAc, shifted towards lower values (down to −35.7 °C, L5 in Table 1) as a result of the copolymerization of VAc with ethylene, additionally evidenced by the appearance of a broad crystallization phenomenon when the ethylene pressure is equal or higher than 50 bar (Table 1, Xc). Indeed, Tg and crystallization phenomena both span a large range of temperatures (see Fig. S4 in ESI†) and this could depend on different phenomena. While VAc is introduced in the reactor at the beginning of the process, the ethylene pressure is a static pressure that decreases with the consumption of ethylene. Together with the high solubility of VAc in water compared to ethylene, this can induce a strong compositional drift during the copolymerization.14 Besides, the conversion of VAc decreases with the increase of the applied pressure and therefore with the incorporation of ethylene (XVAc = 89% at 10 bar and 61% at 100 bar, Table 1). This is a trend already observed in previous studies.7,55 According to Scott et al.7 limited VAc conversion would depend upon the composition of the copolymer produced: the higher the ethylene content in the copolymer, the lower the limiting VAc conversion. It is worth noting that the polymer obtained after drying of latex L1 (VAc homopolymerization) was rigid and brittle. As the fraction of ethylene increases in the copolymer, the material isolated after drying becomes more and more flexible, transparent and sticky.
 | ||
| Fig. 1 Cryo-TEM images of P(VAc-co-E) latexes synthesized with 1 wt% macroCTA, at different ethylene pressures: 35 bar (L3), 50 bar (L4) and 100 bar (L5). | ||
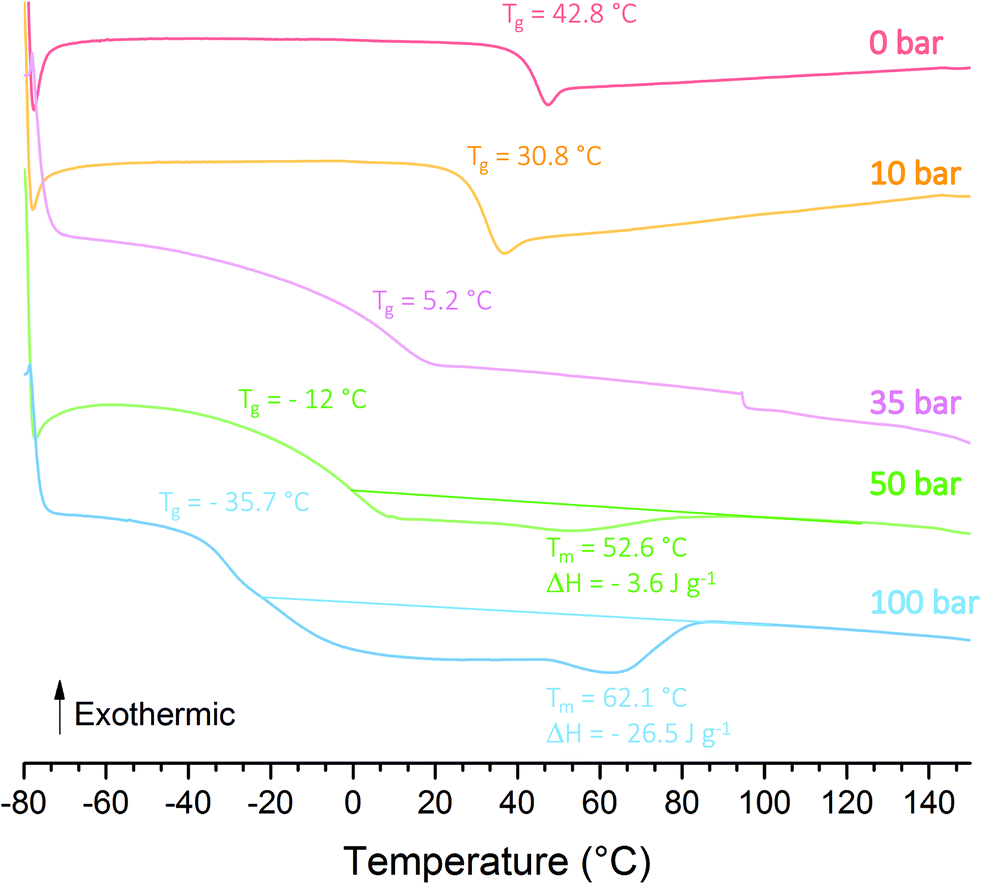 | ||
| Fig. 2 Thermograms of polymers from experiments L1 (0 bar), L2 (10 bar), L3 (35 bar), L4 (50 bar) and L5 (100 bar) (1 wt% macroCTA). | ||
These first successful and promising results show that the production of a broad range of VAE latexes with a very different composition (from 19.1 mol% (42 wt%) to 80.7 mol% (93 wt%) of VAc) and exhibiting various Tg (from 30.8 °C to −35.7 °C) was possible using the macroCTA strategy. This strategy is thus a simple way to achieve stable latexes with a minimum amount of macroCTA and to achieve not only VAE but also EVA latexes.
A benchmark experiment further demonstrated the impact of the macroCTA on the particle stabilization. A polymerization was run at 35 bar in absence of macroCTA or any other surfactant (Table 1, L6). A latex was indeed obtained with a SC of 5.8 wt%. However, the particles were significantly larger (505 nm) and relatively polydisperse (PdI = 0.1). In absence of surfactant, the stability of this latex was only ensured by the charges coming from the initiator and the large particle sizes obtained are consistent with a rather poor efficiency of this stabilization mode. Similarly, large particle sizes were also obtained when the experiment was carried out with P(AAm-co-AA), after removal of the xanthate chain end in P(AAm-co-AA)-X, using the same conditions as for latex L3 (L3*, Table 1). The strong reduction in particle size and the obtainment of isometric stable particles for latex L3 (158 nm) definitively confirms the beneficial implication of the xanthate moieties in the stabilization of the obtained latexes.
Finally, 3 series of experiments were performed using 10, 20 and 65 wt% of P(AAm-co-AA)-X (latexes L7 to L20, Table S1,†Fig. 4 and Fig. S1†). In all cases, stable latexes were obtained, and systematic characterizations by DSC of the formed polymers allowed to draw for each P(AAm-co-AA)-X content used, the evolution of Tgversus the ethylene pressure (Fig. 3).
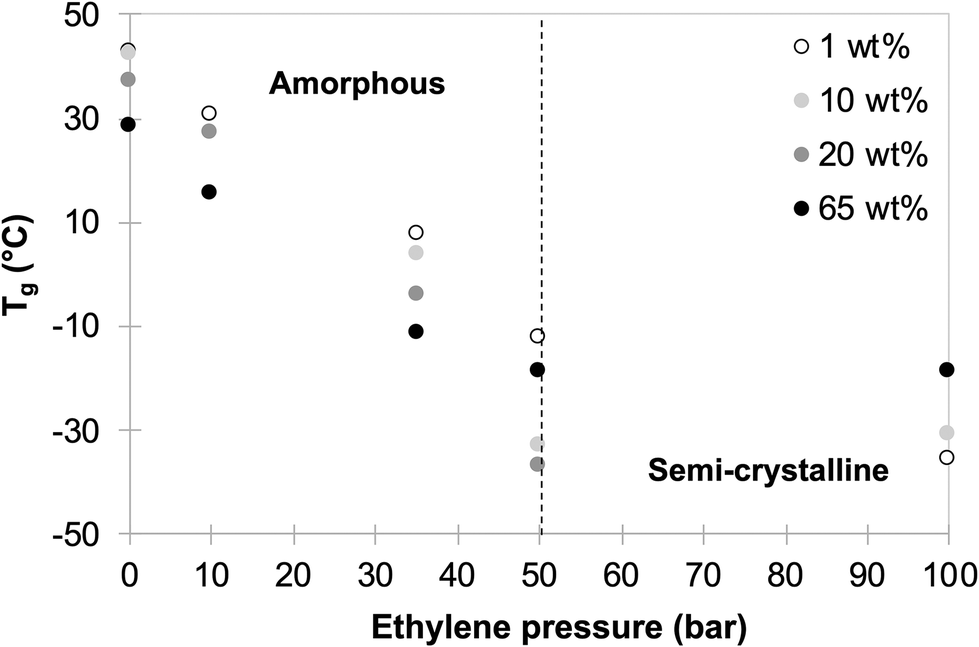 | ||
| Fig. 3 Variation of Tg as a function of ethylene pressure for different contents of macroCTA (1 to 65 wt% macroCTA/VAc). | ||
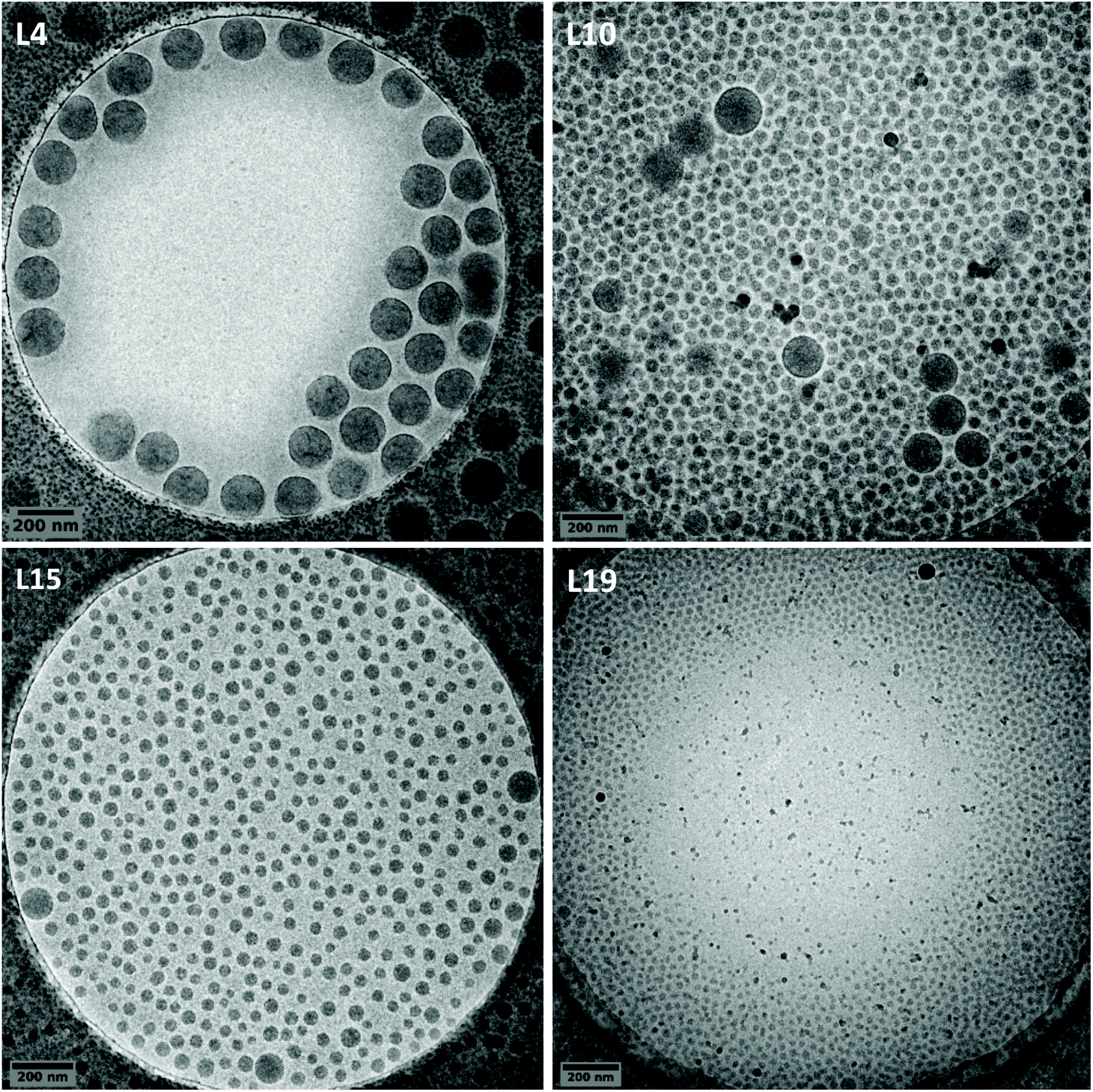 | ||
| Fig. 4 Cryo-TEM images of P(VAc-co-E) latexes synthesized at 50 bar of ethylene pressure, using varying amount of macroCTA: 1 wt% (L4), 10 wt% (L10), 20 wt% (L15) and 65 wt% (L19). Please note that the black dots are related to ice contamination. | ||
All these experiments demonstrate that a broad range of P(VAc-co-E) copolymers, from amorphous with tunable Tg to semi-crystalline, can be obtained in the form of stable latexes. As already observed in other studies,9,13 the increase of ethylene incorporation in P(VAc-co-E) copolymers clearly causes the decrease of Tg (see Fig. S2†). This confirms the robustness of this copolymerization system and that the range of potential accessible products and polymerization conditions can be expanded even more.
Kinetic studies followed by cryo-TEM
When varying the macroCTA amount in the aforementioned experiments, the characterizations by cryo-TEM of the different latexes obtained revealed that large and isometric particles (ca. 150 nm) were mainly obtained when the content of P(AAm-co-AA)-X was 1 wt% (Fig. 1), while a majority of very small particles (ca. 30 nm) were obtained when its content was increased to 65 wt% (L19 in Fig. 4 and L18 in Fig. S1†). This trend can be expected when increasing the amount of stabilizer. However, an intermediate regime was observed for 10 and 20 wt% of P(AAm-co-AA)-X, where bipopulated particles were obtained (Fig. 4), the trend being that the respective fraction of the large particles (ca. 150 nm) over the small ones (ca. 30 nm) varies according to the P(AAm-co-AA)-X content, rather independently of the ethylene pressure, the increase of which seems to only slightly increase the size of the large particles.These results can indeed be explained considering the nucleation mode of the particles. The small particles around 30 nm are consistent with the formation of micellar aggregates of amphiphilic block copolymers. Those particles would form and be stabilized by the expected mechanism depicted for PISA systems,36 in which the growth of the hydrophobic block in water by chain extension of P(AAm-co-AA)-X would lead to the self-assembly of the expected P(AAm-co-AA)-b-P(VAc-co-E)-X block copolymers. Unfortunately, the above-mentioned insolubility of the resulting copolymers did not allow their characterization by SEC analyses. Nevertheless, the very small size of the particles obtained is consistent with the formation and the self-assembly of block copolymers. This results in the shift of the polymerization locus from the aqueous phase to the core of the so-formed block copolymer particles. This nucleation mechanism appears to prevail when the amount of P(AAm-co-AA)-X used is high. On the other hand, the large particles may result from particle nucleation induced by much less P(AAm-co-AA)-b-P(VAc-co-E)-X block copolymers as a result of the use of lower amounts of P(AAm-co-AA)-X. P(AAm-co-AA)-b-P(VAc-co-E)-X formed would simply act as in situ formed stabilizers for the emulsion copolymerization of ethylene and VAc. In this particular case, the coalescence of the nucleated block copolymer particles to ensure efficient stabilization would lead to larger particles (ca. 150 nm). In addition, conventional emulsion copolymerization taking place in the presence of the originally formed small amount of P(AAm-co-AA)-b-P(VAc-co-E)-X cannot be overruled. It could take place at the same time and also lead to large particles. P(AAm-co-AA)-X content higher than 1 wt% and lower than 65 wt% would then reflect intermediate situations for which both nucleation mechanisms would co-exist.
In order to identify the underpinning nucleation mechanism, we undertook a cryo-TEM kinetic study for the emulsion copolymerization carried under 50 bar of ethylene pressure and using 10 wt% of P(AAm-co-AA)-X, for which bipopulated latex formed (L10, Fig. 4). As no samples could be withdrawn from the pressurized reactor, the kinetic study is thus the result of 4 similar experiments stopped after 5, 10, 20 and 240 min (latex L21, L22, L23 et L10, Table 1). The polymerization starts between 5 and 10 minutes and is fast before slowing down, as 60% of the final polymer content (11.4%) is reached in 20 minutes.
The cryo-TEM images corresponding to the experiments performed during 10, 20 and 240 min are presented in Fig. 5. After 10 min of polymerization (L22), small isometric particles of ca. 30 nm are observed exclusively. This is consistent with the formation of block copolymer aggregates depicted above. A few larger particles (around 100 nm) appear between 10 and 20 min (L23) that keep on growing in size (up to 150 nm) and amount, while the size of the small particles remain in the same range (<40 nm) between 20 and 240 min (L10). The most likely cause of the formation of these large particles is a phenomenon of coalescence between some micellar aggregates in order to reduce their interfacial area. The coalescence phenomenon will less and less take place when the initial amount of P(AAm-co-AA)-X is increased, ensuring the stability of all the aggregates.
 | ||
| Fig. 5 Cryo-TEM images photos of the latexes obtained during the kinetic study performed at 50 bar with 10 wt% of macroCTA: t = 10 min (L22), t = 20 min (L23) and t = 240 min (L10). | ||
This kinetic study confirmed the mechanistic picture we provided above for the nucleation of particles formed in our systems. It seems reasonable to assume that the particle nucleation exclusively occurs by self-assembly of block copolymers when the content of macroCTA is close to or slightly higher than 10 wt% (very low fraction of large particles).
Synthesis of poly(vinyl acetate-co-ethylene) latexes using an ethylene pressure of 35 bar and 1 wt% macroCTA targeting high solids content
In the last part of the present study, we focused on conditions that could be industrially extrapolated. In that respect, the ethylene pressure was set to 35 bar, pressure commonly used in industry to prepare VAE by emulsion copolymerization. In addition, a particular effort was put on targeting high solids content latexes (>30 wt%) using a low amount of P(AAm-co-AA)-X that was fixed here to 1 wt%, like in L3. However, this experiment led after 4 hours to solids content of 9.7% only. A kinetic study was thus performed in order to better comprehend the evolution of the solids content during the polymerization. Again, the kinetic study relied on 4 similar experiments, stopped after 15, 30, 60 and 240 min (latex L24, L25, L26 et L3, Table 1).Indeed, the consumption of VAc is very fast (61% consumed in 15 min, solids content of 7.4%) and seems to be rapidly levelling off at ca. 80% after 30 min. The decrease of the Tg value observed with time (from 18.5 °C after 15 min to 7.7 °C after 240 min, Table 1) shows that after 30 min, mainly ethylene is polymerized and the solids content does not vary much anymore. This trend, linked to the composition drift, was already observed in the kinetic study above, when using 10 wt% P(AAm-co-AA)-X under 50 bar ethylene pressure and in the literature.14
The compositional drift is unavoidable when using batch conditions and semi-batch is a way around it7,14 (see below). Nevertheless, based on these first observations, the amount of VAc was further varied from 1.09 mol L−1 to 4.32 mol L−1 to assess its impact on the final solids content. Since the solids content does not vary much after VAc has been consumed, the polymerization time was fixed to 1 h (L27, L28 and L29, Table 1). The three latexes show solids content of 8.2, 18.6 and 28.8 wt%, respectively, with Tg that are consistent with the formation of VAE (Table 1). L27 and L28 were stable at the end of polymerization and under storage. However, L29 exhibited 15% coagulum when recovered from the reactor and a strong exotherm (+5.5 °C) was detected in the early stages of the polymerization. As expected, for the stable systems (L27 and L28), VAc was largely consumed (87.5% and 90%, respectively). Eventually, the Tg increase observed (from 11.5 °C in L26 to 19.1 °C in L28, Table 1) is consistent with an increase of VAc content in the copolymer formed. As a result, the strategy that consists in increasing the amount of VAc in these batch emulsion copolymerizations can produce higher solids content VAE latex. This is however associated with a risk of exotherm potentially leading to the destabilization of the latex, which is redhibitory from an industrial point of view. In addition, as already mentioned, a compositional drift caused by a rapid consumption of VAc in the very beginning of the copolymerization cannot be controlled. For these reasons, a semi-batch process was investigated. In this process, the starting amount of VAc was the one used for L3 (Table 1). The polymerization was started, and the same amount of VAc was added as one shot every hour. The polymerization was stopped 1 hour after the last VAc addition (L30 and L31, with two and three additions of VAc, respectively, Table 2).
Comparison of L28 and L30, for which the same overall amount of VAc was employed, shows the influence of the process (batch vs. semi batch) on the size of the particles (143 nm and 184 nm for L30 and L28, respectively) and also on the composition of the copolymer (Tg of 9.9 and 19.1 °C for L30 and L28, respectively). For a similar final SC (ca. 29.5 wt%), more ethylene can be incorporated with the semi-batch process.
Encouraged by the previous improvements and in a last attempt to both decrease the amount of the macroCTA employed in the formulation and further increase the solids content, L31 was performed using 0.4 wt% of P(AAm-co-AA)-X and 3 additions of VAc, targeting a final VAc content equivalent to 4.32 mol L−1 (like in the batch experiment L29). A strong drop of the ethylene pressure was observed, corresponding to almost 17 bar. As already mentioned, the pressure is not maintained at 35 bar over the course of the polymerization. The concentration of ethylene in the reaction medium therefore varies greatly over time, probably here again leading to drifts in compositions. However, again a stable VAE latex was successfully obtained with a final solids content of 38.3% and a Tg of 14.8 °C suggesting that with controlled injections of both monomers, this copolymerization system would easily be turned into a powerful tool to produce poly(vinyl acetate-co-ethylene) copolymers with tunable compositions in emulsion.
Conclusions
Vinyl acetate/ethylene copolymerization was performed in aqueous emulsion using a P(AAm-co-AA) hydrophilic macromolecular chain transfer agent prepared by MADIX/RAFT in water.By varying the macroCTA contents (1, 10, 20, 65 wt% compared to the initial amount of VAC) and the ethylene pressure (10, 35, 50, 100 bar), a wide range of P(VAc-co-E) latexes (from VAE to EVA) were obtained. In particular, conditions requiring very low amount of hydrophilic P(AAm-co-AA)-X macroCTA, i.e. 1 wt%, to produce stable latexes for ethylene pressures spanning from 10 to 100 bar, were identified. Depending on ethylene pressure, copolymers from amorphous to semi-crystalline were obtained. Indeed, the increase in ethylene pressure led to a decrease of the Tg from 42.8 °C for pure PVAc to −35.7 °C at 100 bar. With the help of kinetic studies performed by cryo-TEM analyses of the same experiments stopped after different polymerization times, the particle nucleation mechanism was investigated at 50 bar using 10 wt% of macroCTA. Those conditions indeed led to a final bipopulated latex. Small particles (ca. 30 nm) first probably form according to the well-known PISA process leading to very small particles, which then partly coalesce to generate a small fraction of larger particles (ca. 150 nm). The fraction of the latter is much less when the macroCTA content employed is higher (20 wt%) since the system can then accommodate better the stabilization of the created surface area.
With a view to testing conditions more in line with industrial constraints, the final part of the study focused on reaching high solids content for an ethylene pressure of 35 bar, a pressure that is commonly used in the industry to produce VAE latexes. Switching from batch to semi-batch conditions, with successive shot additions of VAc, stable dispersions could be produced with only 0.4 wt% of macroCTA, and exhibiting a solids content of 38 wt%. To our knowledge, this work is the first study involving RAFT polymerization in the synthesis of poly(vinyl acetate-co-ethylene) in emulsion polymerization. Through the additional use of macroCTA different from P(AAm-co-AA), this work provides an easy access to a full range of alternative stabilization modes for P(VAc-co-E) latexes and potentially to new VAE and EVA products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Solvay for permission to publish this work.Notes and references
- G. E. J. Reynolds, Br. Polym. J., 1969, 1, 233–238 CrossRef CAS.
- A. M. Henderson, IEEE Electr. Insul. Mag., 1993, 9, 30–38 Search PubMed.
- P. J. Scott, A. Penlidis and G. L. Rempel, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 403–426 CrossRef CAS.
- J. K. Fink, in Handbook of Engineering and Speciality Thermoplastics: Polyolejns and Styrenics, ed. J. K. Fink, Scrivener Publishing LLC, 2010, pp. 187–209, DOI:10.1002/9780470881712.ch7.
- A. Zarrouki, E. Espinosa, C. Boisson and V. Monteil, Macromolecules, 2017, 50, 3516–3523 CrossRef CAS.
- E. Frauendorfer, M. Babar, T. Melchin and W.-D. Hergeth, in Polymer Reaction Engineering of Dispersed Systems: Volume II, ed. W. Pauer, Springer International Publishing, Cham, 2018, pp. 183–214, DOI:10.1007/12_2017_22.
- P. J. Scott, A. Penlidis and G. L. Rempel, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2205–2230 CrossRef CAS.
- P. J. Scott, A. Penlidis and G. L. Rempel, Chem. Eng. Sci., 1994, 49, 1573–1583 CrossRef CAS.
- P. J. Scott, A. Penlidis, G. L. Rempel and A. D. Lawrence, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 539–555 CrossRef CAS.
- P. J. Scott, A. Penlidis and G. L. Rempel, Polym. React. Eng., 1995, 3, 93–130 CrossRef CAS.
- B. A. Gruber, M. S. Vratsanos and C. D. Smith, Macromol. Symp., 2000, 155, 163–170 CrossRef CAS.
- N. Gospodinova, L. Terlemezyan, M. Mihailov, U. Men Han and K. Ben Du, Eur. Polym. J., 1992, 28, 961–967 CrossRef CAS.
- N. Gospodinova, T. Zlatkov and L. Terlemezyan, Polymer, 1998, 39, 2583–2588 CrossRef CAS.
- J. Wu, J. P. Tomba, J. K. Oh, M. A. Winnik, R. Farwaha and J. Rademacher, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5581–5596 CrossRef CAS.
- J. Guo, K. Y. Choi and F. J. Schork, Macromol. React. Eng., 2009, 3, 412–418 CrossRef CAS.
- F. P. Petrocelli and C. F. Cordeiro, Macromol. Symp., 2000, 155, 39–52 CrossRef CAS.
- I. Poljanšek, E. Fabjan, K. Burja and D. Kukanja, Prog. Org. Coat., 2013, 76, 1798–1804 CrossRef.
- N. Gupta, R. K. Srivastava, V. Choudhary, I. K. Varma and S. Patnaik, J. Therm. Anal. Calorim., 1999, 58, 509–515 CrossRef CAS.
- H. S. Mobarakeh and M. R. R. Daronkola, Iran. Polym. J., 2005, 14, 579–587 CAS.
- B. M. Budhlall, K. Landfester, D. Nagy, E. D. Sudol, V. L. Dimonie, D. Sagl, A. Klein and M. S. El-Aasser, Macromol. Symp., 2000, 155, 63–84 CrossRef CAS.
- B. M. Budhlall, K. Landfester, E. D. Sudol, V. L. Dimonie, A. Klein and M. S. El-Aasser, Macromolecules, 2003, 36, 9477–9484 CrossRef CAS.
- S. Aruldass, V. Mathivanan, A. R. Mohamed and C. T. Tye, J. Environ. Chem. Eng., 2019, 7, 103238 CrossRef CAS.
- C. M. Gilmore, G. W. Poehlein and F. J. Schork, J. Appl. Polym. Sci., 1993, 48, 1449–1460 CrossRef CAS.
- G. S. M. González, V. L. Dimonie, E. D. Sudol, H. J. Yue, A. Klein and M. S. El-Aasser, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 849–862 CrossRef.
- B. M. Budhlall, E. D. Sudol, V. L. Dimonie, A. Klein and M. S. El-Aasser, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3633–3654 CrossRef CAS.
- S. Carrà, A. Sliepcevich, A. Canevarolo and S. Carrà, Polymer, 2005, 46, 1379–1384 CrossRef.
- A. L. German and D. Heikens, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 2225–2232 CrossRef CAS.
- A. L. German and D. Heikens, Anal. Chem., 1971, 43, 1940–1944 CrossRef CAS.
- M. Rätzsch, W. Schneider and D. Musche, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 785–790 CrossRef.
- R. Van Der Meer, M. W. A. M. Aarts and A. L. German, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 1347–1357 CrossRef CAS.
- J. Filley, J. T. McKinnon, D. T. Wu and G. H. Ko, Macromolecules, 2002, 35, 3731–3738 CrossRef CAS.
- H. De Bruyn, R. G. Gilbert and B. S. Hawkett, Polymer, 2000, 41, 8633–8639 CrossRef.
- E. J. Bradbury, D. McNulty, R. I. Savage and E. E. McSweeney, Ind. Eng. Chem., 1952, 44, 211–212 CrossRef CAS.
- P. B. Zetterlund, S. C. Thickett, S. Perrier, E. Bourgeat-Lami and M. Lansalot, Chem. Rev., 2015, 115, 9745–9800 CrossRef CAS.
- J. Zhou, H. Yao and J. Ma, Polym. Chem., 2018, 9, 2532–2561 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS.
- J. L. de la Haye, I. Martin-Fabiani, M. Schulz, J. L. Keddie, F. D'Agosto and M. Lansalot, Macromolecules, 2017, 50, 9315–9328 CrossRef.
- I. Martín-Fabiani, J. L. de la Haye, M. Schulz, Y. Liu, M. Lee, B. Duffy, F. D'Agosto, M. Lansalot and J. L. Keddie, ACS Appl. Mater. Interfaces, 2018, 10, 11221–11232 CrossRef.
- I. Martín-Fabiani, D. K. Makepeace, P. G. Richardson, J. L. de la Haye, D. A. Venero, S. E. Rogers, F. D'Agosto, M. Lansalot and J. L. Keddie, Langmuir, 2019, 35, 3822–3831 CrossRef.
- V. Dehan, E. Bourgeat-Lami, F. D'Agosto, B. Duffy, A. Fortini, S. Hilton, K. Krassa, J. L. Keddie, M. L. Koh, M. Lansalot, M. Lee, J. L. de la Haye, I. Martin-Fabiani, C. Mantzaridis, D. P. Mazeffa, R. P. Sear, M. Schulz, M. Sibbald, B. Skerry and B. Thomas, Steel Constr., 2017, 10, 254–259 CrossRef.
- A. France, WO2019063445, 2019.
- Solvay SA, Centre National de la Recherche Scientifique, Université Pierre et Marie Curie (Paris 6), Université Claude Bernard Lyon 1, WO2013092587A1, 2013.
- E. Velasquez, J. Rieger, F. Stoffelbach, B. Charleux, F. D'Agosto, M. Lansalot, P.-E. Dufils and J. Vinas, Polymer, 2013, 54, 6547–6554 CrossRef CAS.
- E. Velasquez, J. Rieger, F. Stoffelbach, F. D'Agosto, M. Lansalot, P.-E. Dufils and J. Vinas, Polymer, 2016, 106, 275–284 CrossRef CAS.
- E. Grau, P.-Y. Dugas, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Angew. Chem., Int. Ed., 2010, 49, 6810–6812 CrossRef CAS.
- F. Brunel, G. Billuart, P.-Y. Dugas, M. Lansalot, E. Bourgeat-Lami and V. Monteil, Macromolecules, 2017, 50, 9742–9749 CrossRef CAS.
- G. Billuart, E. Bourgeat-Lami, M. Lansalot and V. Monteil, Macromolecules, 2014, 47, 6591–6600 CrossRef CAS.
- S. Binauld, L. Delafresnaye, B. Charleux, F. D'Agosto and M. Lansalot, Macromolecules, 2014, 47, 3461–3472 CrossRef CAS.
- P. Galanopoulo, P.-Y. Dugas, M. Lansalot and F. D'Agosto, Polym. Chem., 2020, 11, 3922–3930 RSC.
- C. Bergerbit, F. Baffie, A. Wolpers, P.-Y. Dugas, O. Boyron, M. Taam, M. Lansalot, V. Monteil and F. D'Agosto, Angew. Chem., 2020, 59, 10385–10390 CrossRef CAS.
- M. J. Wisotsky, A. E. Kober and I. A. Zlochower, J. Appl. Polym. Sci., 1971, 15, 1737–1742 CrossRef CAS.
- C. F. Cordeiro and F. P. Petrocelli, in Kirk–Othmer Encyclopedia of Chemical Technology, 2005, DOI:10.1002/0471238961.2209142503151804.a01.pub2.
- A. C. Mesquita, M. N. Mori and L. G. Andrade e Silva, Radiat. Phys. Chem., 2004, 71, 253–256 CrossRef CAS.
- G. Billuart, Free radical emulsion polymerization of ethylene, Université Claude Bernard Lyon 1, 2015 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01266a |
| This journal is © The Royal Society of Chemistry 2020 |