
Catalyst free removal of trithiocarbonate RAFT CTAs from poly(vinylpyridine)s using tris(trimethylsilyl)silane and light†
Brandon A.
Fultz
 ,
Drake
Beery
,
Brianna M.
Coia
,
Kenneth
Hanson
and
Justin G.
Kennemur
*
,
Drake
Beery
,
Brianna M.
Coia
,
Kenneth
Hanson
and
Justin G.
Kennemur
*
Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32303, USA. E-mail: jkennemur@fsu.edu
First published on 15th September 2020
Abstract
Tris(trimethylsilyl)silane (TTMSS) is introduced as a non-toxic and kinetically superior hydrogen source for the photo-driven reduction of trithiocarbonates from poly(methyl methacrylate), polystyrene, and poly(vinyl pyridine)s (PVP)s created through RAFT polymerization. No other reagents were necessary and quantitative reduction is achieved under UV- and blue-light irradiation. PVP reduction and sequestering of chain combination events is investigated for the first time.
Introduction
Reversible addition–fragmentation chain transfer (RAFT) has matured as a robust and versatile controlled radical polymerization (CRP) technique that is utilized for creating a large scope of polymers, copolymers, and macromolecular architectures.1 Although this technique presents many advantages, one notable disadvantage is the thiocarbonylthio functionalities installed on the polymer chain ends. These moieties cause coloration, reduced thermal stability, potential reactivity, unpleasant odor, and potential toxicity that ultimately limit the value and scalability of the resulting materials.2 Research continues to explore facile methods for the removal of these moieties. Of the many chemical transformations explored,3,4 replacing the thiocarbonylthio groups with a hydrogen has the advantage of “capping” the polymer chain with a strong and inert C–H bond. This strategy mitigates the disadvantages listed above and typically employs a degenerative chain transfer reaction on the thiocarbonylthio species in the presence of a highly reactive hydrogen source. Trialkylstannane reagents, such as tributylstannane (n-Bu3SnH), are an effective hydrogen source but suffer from tin byproducts that are toxic and difficult to remove.5,6 Chong et al. explored radical-induced reduction strategies initiated by azobisisobutyronitrile (AIBN) at higher temperatures (≥100 °C) using alternative and non-toxic hydrogen sources.6 Notable reducing agents of the highest activity included tris(trimethylsilyl)silane (TTMSS) and N-ethylpiperidine hypophosphite (EPHP) that were both capable of reducing trithiocarbonates (TTC) and dithioesters quantitatively within 2–4 h. While effective, AIBN was required and the use of high temperatures (>100 °C) is energy intensive for scalability of this process. Alagi et al. recently showed rapid removal of a variety of thiocarbonylthio groups at ambient temperatures using trialkylboranes in the presence of oxygen.7 While a majority of the resulting polymers were effectively capped with hydrogen, some peroxy and alkyl chain ends were also evident.Light-mediated removal of thiocarbonylthio end groups has recently received increased attention due to its facile approach and performance at ambient temperatures. Mattson et al. reported irradiation at 380 nm to remove TTC groups from poly(tert-butyl acrylate) over the course of 48 h while incorporating 10-phenylphenothiazine (PTH) as a photoredox catalyst in the presence of tributylamine and formic acid.8 Soon after, Discekici et al. utilized 465 nm irradiation and a photocatalyst, Eosin Y, to promote hydrogen replacement of TTC groups in the presence of hexylamine and tri-n-butylphosphine.9 Carmean et al. reported a photoinduced removal of TTC, dithiobenzoate (DTB), xanthate, and dithiocarbamate (DTC) agents using EPHP at ambient temperatures and without the need for additional initiators or photocatalysts.10 This method was effective for a variety of polymers (acrylamides, acrylates, methacrylates, styrenes, and pyrrolidones) with little degradation but also required long irradiation times (≥24 h) for complete removal of TTC, DTB, and DTC agents. Another notable conclusion from this study was that the concentration of EPHP had little effect on the reduction kinetics and the rate limiting step was concluded to be photolysis of the C–S bond connecting the thiocarbonylthio group to the polymer chain end.10 Uchiyama et al. recently investigated thermal and photo-driven xanthate, DTB, and TTC removal from poly(vinyl acetate) (PVAc), PS, and poly(methyl acrylate) (PMA).11 Using triphenylsilane (Ph3SiH) as a hydrogen source and n-dodecanethiol as a polarity reversal catalyst, quantitative removal of the CTAs under irradiation (λ = 365 nm, 0.70 mW cm−2) occurred after ≥75 h. Notably, they also showed rapid increase in reduction kinetics when irradiating with blue light (λ = 470 nm, 70 mW cm−2) in the presence of a photo-radical initiator, biphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO).
To the best of our knowledge, there have been no studies on the reduction of thiocarbonylthio reagents from poly(2-vinylpyridine) (P2VP) systems and poly(4-vinylpyridine) (P4VP) that are receiving increased interest for association of variety of guests through pyridine coordination.12 Due to complications with copper association on PVPs during atom transfer radical polymerization (ATRP),13–15 RAFT has become the CRP of choice to produce PVPs and a study focused on the facile removal of their CTA end groups is needed. While early studies showed success using DTB CTA for the RAFT synthesis of P2VP and P4VP,16 a large number of recent manuscripts, including ours,17 have used TTC CTAs to produce both isomers with good control.18–29
The investigation of CTA removal from PVPs is warranted due to the aromaticity of the pyridine pendant groups. This is analogous to PS which has been highly studied and is consistently problematic in earlier reports. A major issue stems from the benzylic radical chain end produced upon scission of the C–S bond on PS and its competitive termination through combination versus hydrogen abstraction. This results in the appearance of high molar mass shouldering in size exclusion chromatography (SEC) analysis and concludes significant chain coupling (doubling in molar mass) during the reduction reactions. Additionally, the benzylic radicals are comparatively stable. Photoinduced end group removal is therefore sluggish and often requires days of irradiation with non-quantitative removal.10 PVPs are also prone to termination through combination.30,31 Investigating whether similar complications occur with reduction of PVPs was the initial motivation for this study. However, we were also intrigued by the recent yet promising results of Ph3SiH as a hydrogen source for photo-driven reductions (both UV and blue light).11 Alternatively, TTMSS is a non-toxic hydrosilane that exhibited much faster reduction kinetics in the thermally driven reduction methods.6 Herein we introduce TTMSS as a powerful and rapid reducing agent, requiring no additional reagents, for photo-driven reduction of PMMA, PS, P2VP and P4VP for the first time.
Results and discussion
All polymers in this study were synthesized through RAFT using 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDPA) as the CTA and their characterizations are summarized in Table 1 (ESI†). UV-visible (UV-Vis) spectroscopy was used to measure λmax of the π → π* and n → π* absorption of CDPA (Fig. S2†) and each polymer bearing the TTC end group (Fig. S46 and S47†) in addition to molar absorptivities (Table S1†). PMMA-TTC (Mn = 6.1 kg mol−1, Đ = 1.08) was chosen for initial photoinduced reduction due its high reactivity in previous studies (Table 1, entry 1).10 PMMA-TTC was dissolved with TTMSS (15 equiv. to TTC) in THF (5.5 mM TTC) and irradiated (λ = 365 nm, 1.0 mW cm−2) at a fixed distance of 2.5 cm from the emission source. UV-visible (UV-Vis) spectroscopy was used to measure the loss in the π → π* absorbance over time (Fig. S3†). For the studies throughout, the reduction reaction was considered complete (>99% TTC removal) once the measured absorbance decreased to <0.1 O.D. Complete reduction of PMMA-TTC occurred in 1 h and the time needed to reach half of the original PMMA-TTC absorbance (t1/2) was 12 min. To confirm successful TTC removal, PMMA-H was purified and analyzed by 1H NMR (Fig. S3†). The absence of dodecyl methylene signals associated with TTC (3.28–3.20 ppm) confirms quantitative reduction of the PMMA chain ends (Fig. S4 and S5†). SEC analysis of PMMA-H revealed only a slight shifting of the curve to higher elution time as a result of the loss of the TTC group and no observable broadening or shouldering of the peak (Đ = 1.08) (Fig. S3†). Unlike PS, PMMA has a high propensity for disproportionation over combination.321H NMR revealed only trace olefin proton signals resulting from disproportionation (Fig. S5,† inset) and is notable since disproportionation is not observed by SEC analysis.| IDa | Polymer | M n,SEC (kDa) | M n,NMR (kDa) | Đ | Hydrogen source |
t
1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d (h) d (h) |
% TTC removale | Durationf (h) | After TTC removal | |
|---|---|---|---|---|---|---|---|---|---|---|
| M n,SEC | Đ | |||||||||
|
a All reactions were performed with hydrogen source:TTC of 15:1, ([TTS]0 = 5.5 mM) in THF and irradiated at 365 nm otherwise noted.
b A 50:50 solvent mixture of DMAC:toluene was used, and molecular weight was characterized via1H NMR end group analysis.
c Irradiated with 452 nm.
d
Values were determined as the irradiation time in which initial absorbance [TTC]0 was reduced by half.
e TTS removal was evaluated by UV-Vis spectroscopy and determined to be >99% when <0.1 OD was achieved.
f Irradiation time needed to reach <0.1 ABS determined by UV-Vis.
|
||||||||||
| 1 | PMMA-TTC | 6.1 | 5.9 | 1.08 | TTMSS | 0.2 | >99 | 1 | 5.8 | 1.08 |
| 2 | PMMA-TTC | 6.1 | 5.9 | 1.08 | EPHP | 2 | >99 | 18 | 6.2 | 1.14 |
| 3 | P2VP-TTC | 8.5 | 8.3 | 1.08 | TTMSS | 0.4 | >99 | 1.5 | 8.3 | 1.12 |
| 4b | P4VP-TTC | — | 7.6 | — | TTMSS | 0.3 | >99 | 1.25 | — | — |
| 5 | PS-TTC | 7.8 | 7.8 | 1.1 | TTMSS | 2.4 | >99 | 6.25 | 7.8 | 1.16 |
| 6 | P2VP-TTC | 8.5 | 8.3 | 1.08 | EPHP | 5 | >99 | 26.5 | 6.5 | 1.19 |
| 7b | P4VP-TTC | — | 7.6 | — | EPHP | 4 | >99 | 24 | — | — |
| 8c | P2VP-TTC | 8.5 | 8.3 | 1.08 | TTMSS | 0.6 | >99 | 1.25 | 8.1 | 1.10 |
We were encouraged by the greatly enhanced reduction kinetics of TTMSS (1 h) versus those reported using identical molar equivalents of EPHP on acrylic-TTC end groups (24 h) previously.10 To ensure this disparity in kinetics was not an artifact of our own experimental setup, we recreated the reduction of PMMA-TTC under the exact same conditions with the exception that TTMSS was replaced with EPHP (Table 1, entry 2). After an initial rapid decrease in the UV-Vis absorbance of TTC (t1/2 = 2 h), the reaction rate decreased and total loss of TTC absorption required 18 h (Fig. S6 and S7†). The t1/2 value for TTMSS (0.2 h) was an order of magnitude faster than EPHP (2 h) (Table 1). Interestingly, SEC analysis after reduction to PMMA-H using EPHP revealed some shouldering at higher molar mass (Đ = 1.14, Fig. S8†). The rate limiting step for EPHP reduction was proposed to be photolytic degradation of the C–S bond,10 however, such a drastic difference in reduction kinetics by changing the hydrogen source may indicate other mechanistic aspects need consideration.
To investigate RAFT agent removal from PVPs, we began with P2VP-TTC (Mn = 8.5 kg mol−1, Đ = 1.08) (Table 1, entry 3). Reduction of P2VP-TTC using TTMSS (15 equiv. to TTC) in THF (5.5 mM TTC) (Fig. 1a) resulted in complete reduction in 1.5 h (t1/2 = 24 min) (Fig. 1b and c) and was accompanied by a visible change in appearance from a yellow to colorless solution (Fig. S9†). 1H NMR analysis of P2VP-H (Fig. 1e and S10†) displayed a complete loss of TTC dodecyl methylene signals (∼3.2 ppm), indicating quantitative removal. SEC of P2VP-H (Fig. 1d) displayed a small increase in elution time and a slight broadening (Đ = 1.12) at the higher molar mass side of the peak, indicating some chain combination has occurred. However, the absence of pronounced shouldering suggests these events are not as prominent compared to PS (vide infra). Due to insolubility of P4VP-TTC (Mn = 7.6 kg mol−1) in THF, reduction kinetics were investigated in a 50:50 v/v mixture of N,N-dimethylacetamide (DMAC):toluene using TTMSS (15 equiv. to TTC) (Table 1, entry 4). UV-Vis analysis revealed slightly faster reduction than P2VP-TTC and complete after 1.25 h of irradiation (t1/2 = 20 min) (Fig. 2b and c). The loss of dodecyl TTC methylene signal by 1H NMR was also confirmed (Fig. 2d). Therefore, TTMSS is a fast and efficient hydrogen source to remove and replace the TTC end group on both P2VP and P4VP isomers.
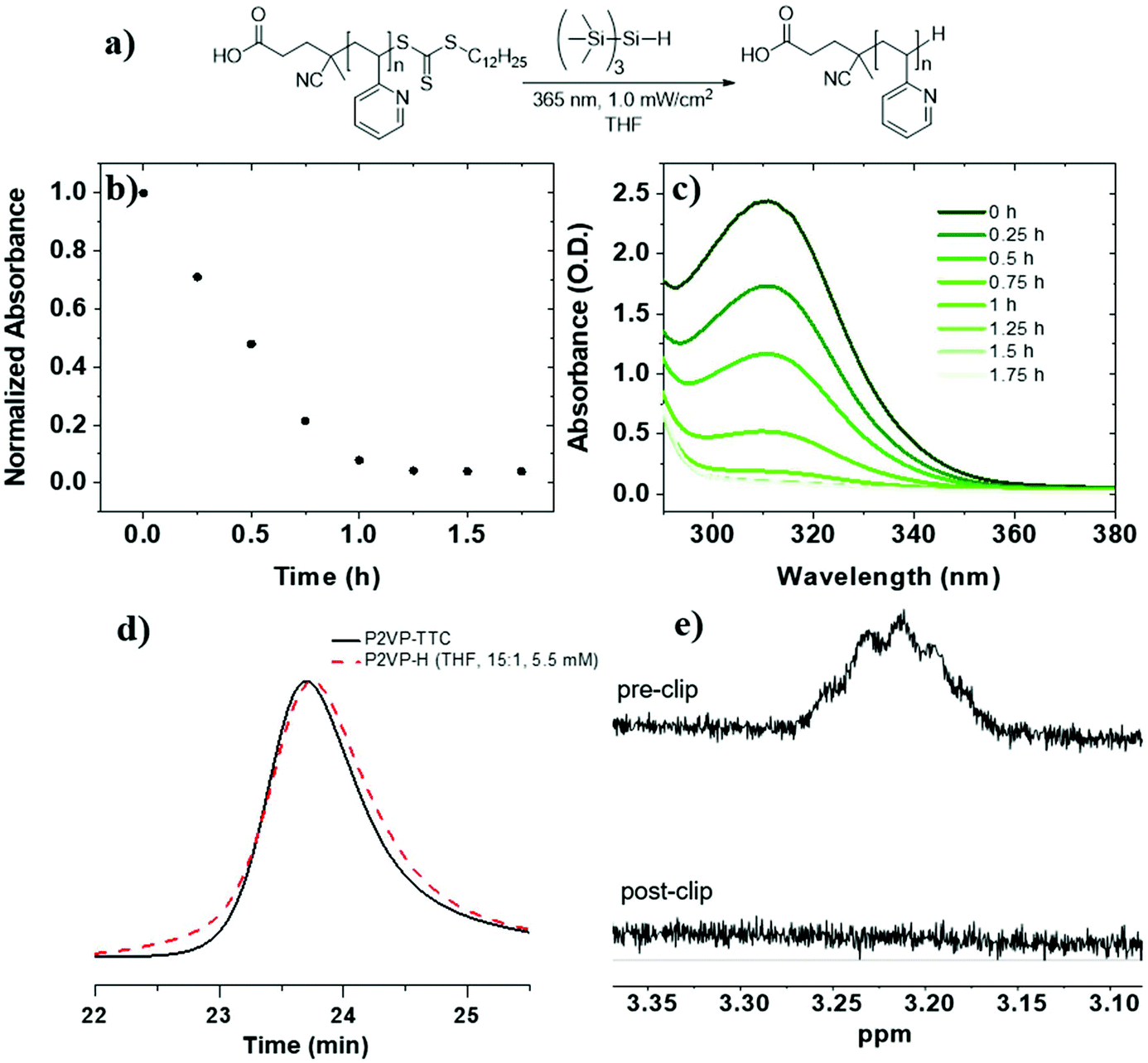 | ||
| Fig. 1 (a) Reaction scheme for photoinduced (λ = 365 nm) removal of TTC from P2VP-TTC in THF ([TTC]0 = 5.5 mM) using TTMSS:TTC of 15:1. (b) Normalized absorbance at 309 nm as a function of time. (c) UV-Vis absorption spectra from aliquots taken at known time intervals throughout the reaction. (d) Normalized SEC-RI trace overlay (THF mobile phase, 23 °C) of P2VP-TTC before (black solid) and after (red dashed) the reaction. (e) Offset 1H NMR (CDCl3, 25 °C) spectra of the methylene proton signal (S-CH2-C11H23) before (top) and after (bottom) the reaction. | ||
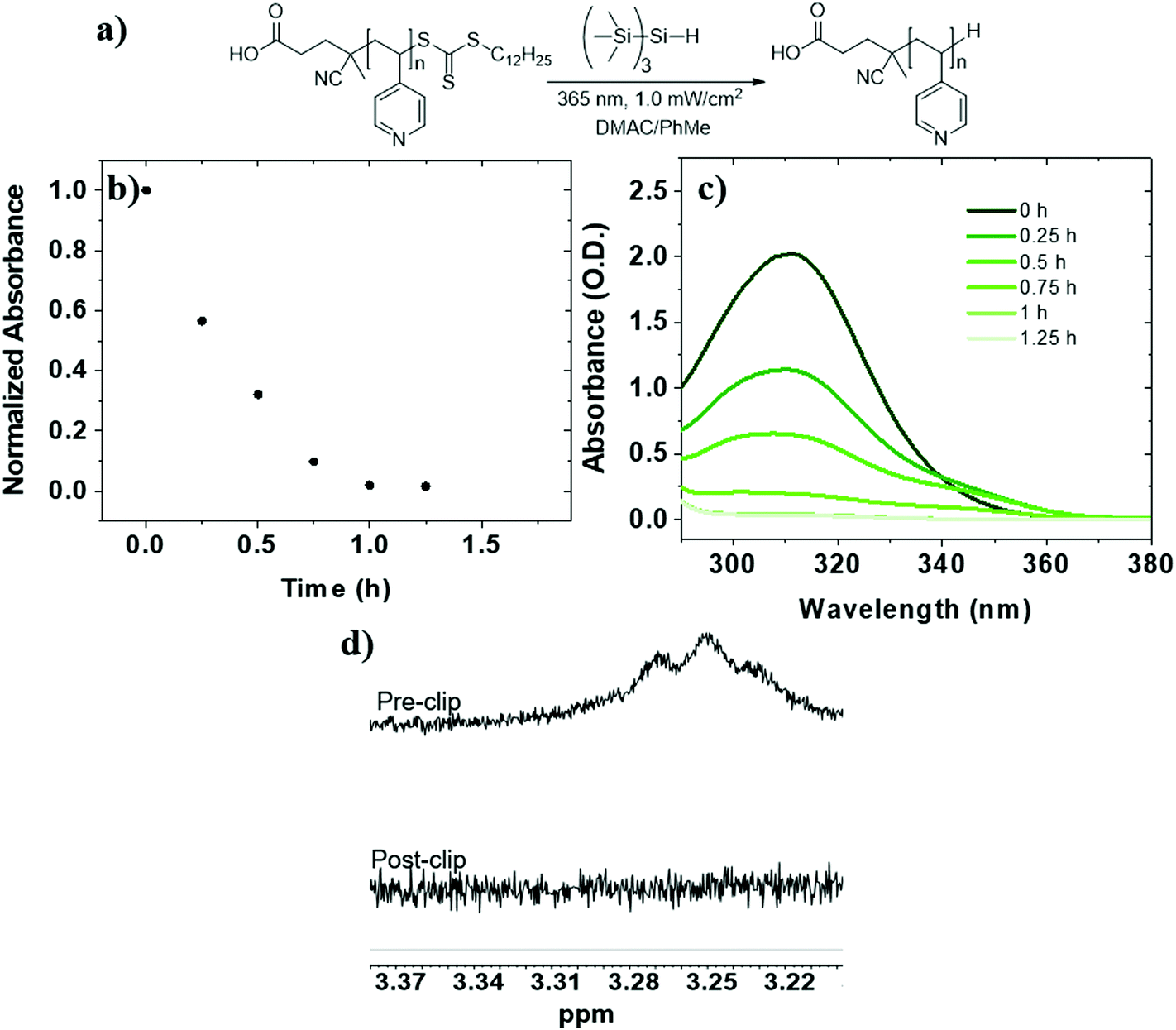 | ||
| Fig. 2 (a) Reaction scheme for photoinduced (λ = 365 nm) removal of TTC from P4VP-TTC in DMAC/toluene (50:50) ([TTC]0 = 5.5 mM) using TTMSS:TTC of 15:1. (b) Normalized absorbance at 309 nm as a function of time. (c) UV-Vis absorption spectra from aliquots taken at known time intervals throughout the reaction. (d) Offset 1H NMR (CDCl3, 25 °C) spectra of methylene proton signal (S-CH2-C11H23) before (top) and after (bottom) the reaction. | ||
Although the SEC trace for P2VP-H indicates minor chain combination events, we compared this outcome to a reduction on PS-TTC (Mn = 7.8 kg mol−1, Đ = 1.10) under identical conditions (Table 1, entry 5). UV-Vis analysis revealed complete reduction in 6.25 h (t1/2 = 2.4 h) (Fig. S11†) which is the fastest of any photo-driven reductions on PS to date. Successful removal of the TTC was again confirmed by 1H NMR (Fig. S11 and S12†). SEC analysis of PS-H revealed substantial shouldering at higher molar mass indicative of prominent chain coupling (Fig. S11†), consistent with previous reports. Therefore, we conclude P2VP to be much less prone to combination events in comparison to PS.
For additional comparison, the use of EPHP as a hydrogen source was also explored for P2VP-TTC and P4VP-TTC under the same conditions used for TTMSS (Table 1, entries 6 and 7). EPHP reduction on P2VP and P4VP resulted in longer reaction times of 26.5 h (t1/2 = 5 h) and 24 h (t1/2 = 4 h), respectively, to reach complete removal of the TTC groups (Fig. S6†). The reduction of P4VP was, again, slightly faster than P2VP. 1H NMR analysis confirmed complete removal of the TTC groups (Fig. S13–S15†), while SEC analysis indicated a higher increase in dispersity (1.19) and shouldering (Fig. S16†). The final absorbance of P2VP-H and P4VP-H minimized at ∼0.12 O.D. and the reaction solution maintained a light yellow-orange color throughout. Interestingly, reductions performed on P2VP and P4VP with TTMSS resulted in a colorless solution (<0.1 O.D. in the visible region) and a white polymer upon workup. This suggests that EPHP may be interacting with the PVP systems to cause some discoloration.
Comparative reduction kinetics for each polymer were analyzed by plotting logarithmic concentration of reduced TTC groups (ln([CTA]0/[CTA]) as a function of time (Fig. 3). Kinetic studies were validated for each polymer with linear calibrations of [TTC]0 as a function of ABS (Fig. S38–S45†). Consistent with previous studies,10 the kinetics of reduction follow a quasi-first order trend with a linear progression at the onset followed by a monotonic increase at later reaction times. This deviation is more obvious for P2VP and P4VP versus PMMA and PS within the axis limits of Fig. 3, however, PS interestingly remains linear throughout (Fig. S17†). Nevertheless, the kinetic profiles clearly indicate that the rate of reduction follows the trend of (PMMA > P4VP > P2VP ≫ PS).
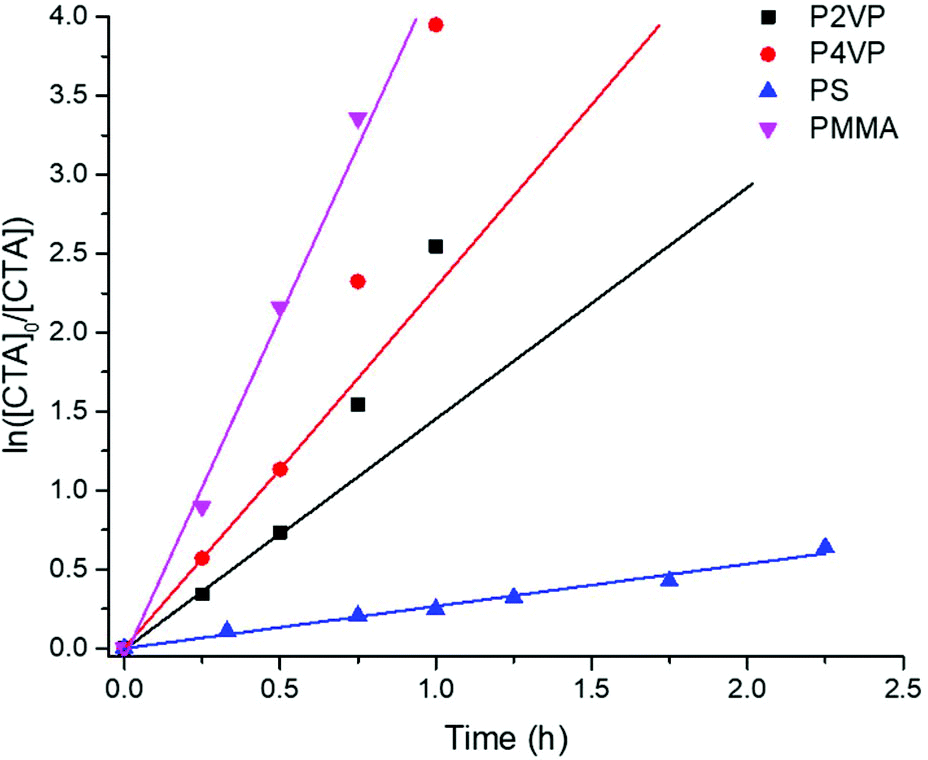 | ||
| Fig. 3 Logarithmic normalized concentrations of remaining TTS CTA (ln[CTA]0/[CTA]) as a function of time (h) using TTMSS (15 equiv. to CTA) in THF at 28 ± 3 °C ([CTA]0 = 5.5 mM) while irradiating with 365 nm light. While a linear first order kinetic profile exists for the initial stages of reduction, a monotonic increase is seen at increasing reaction times for P2VP and P4VP and suggests a pseudo-first order kinetic profile. | ||
Thermogravimetric analysis (TGA) was performed on PMMA-TTC and P2VP-TTC before and after reduction to determine if the TTC end group effects thermal stability. Congruent with earlier reports by Chong et al.,33 PMMA-TTC displays an early multistep decomposition (175 °C initial onset) when capped by a TTC end group (Fig. S18†). Removal of the TTC end group via TTMSS resulted in an observed 92 °C increase in thermal stability (271 °C onset). Alternatively, P2VP showed little difference in thermal stability regardless of end group (Fig. S19†).
Chong et al. showed that toluene can act as a weak, additional hydrogen source when used as a solvent during reduction.6 To investigate reduction of combination events on PS-TTC, we replicated entry 5 in Table 1 and replaced THF with toluene as the solvent. Indeed, suppression but not elimination of combination was observed by SEC (Fig. S20†). Additional attempts to reduce combination of PS-TTC by dilution ([TTC]0 = 2.25 mM) and increasing the stoichiometry of TTMSS:TTC (30:1) also showed little improvement (Fig. S20†). P2VP-TTC is only partially soluble in toluene. Instead, a mixed solvent system (75:25 v/v) of toluene:THF, respectively, was utilized to investigate suppression of combination events. The mixed solvent system under identical conditions as entry 3 in Table 1 resulted in slight improvement to the broadening of the SEC trace (Fig. 4, green trace) when compared to THF (blue trace). However, by increasing TTMSS:TTC (30:1), sequestering of combination events was achieved (Fig. 4, red trace). With this success, it was hypothesized that toluene was not necessary. By recreating a 30:1 TTMSS to TTC in pure THF, similar sequestering of combination events occurred, concluding that concentration of TTMSS is a critical parameter (Fig. S21†).
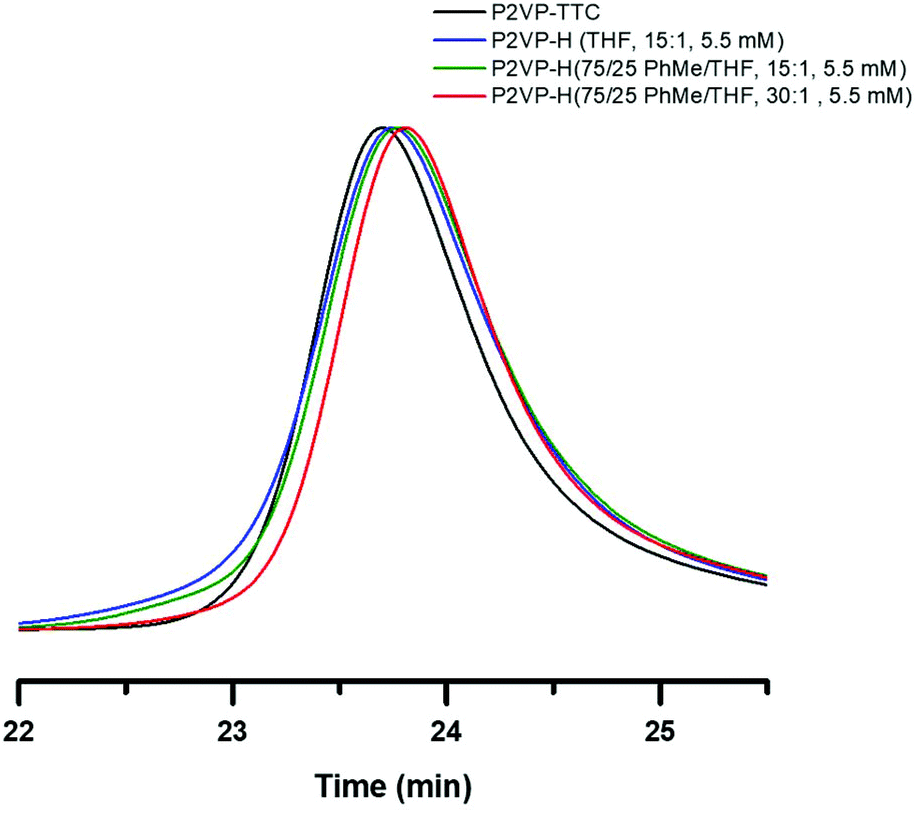 | ||
| Fig. 4 Normalized SEC-RI trace overlay (THF mobile phase, 23 °C) of P2VP-TTC (black) before photoinduced RAFT removal, P2VP-H (blue) post-removal RAFT removal using 15:1 ratio TTMSS:RAFT end respectively in THF at 5.5 mM, P2VP-H (green) post-removal RAFT removal using 15:1 ratio TTMSS:RAFT end respectively in a 75:25 mixed PhMe:THF solvent respectively at 5.5 mM, P2VP-H (red) post-removal RAFT removal using 30:1 ratio TTMSS:RAFT end respectively in a 75:25 mixed PhMe:THF solvent respectively at 5.5 mM. Concentrations are relative to the RAFT CTA chain end and performed under 365 nm light. | ||
Although using TTMSS with 365 nm irradiation is promising, this wavelength lies on the low energy edge of the π → π* absorption band of TTC (Fig. S47†). Alternatively, the lower energy n → π* transition, while much weaker in molar absorptivity, has a peak maximum at ∼450 nm (Fig. S47,† inset) and its excitation with blue light (452 nm) may promote reduction without photocatalysts. Using identical reaction conditions ([TTC]0 = 5.5 mM, TTMSS:TTC = 15:1), P2VP-TTC was subjected to 452 nm (<5 mW cm−2) irradiation (Fig. S22†) and aliquots were monitored by UV-Vis analysis. Under these conditions, complete reduction occurred in 1.25 h (Table 1, entry 8) and the spectral monitoring (Fig. S23†) shows a slow initial reduction rate (t1/2 = 0.6 h) that accelerates over time. We believe this acceleration is due to the higher heat produced from the LED light strip. When measuring the temperature of the reaction solution using UV light over time, it never exceeded 30 °C. However, the reaction solution under LED irradiation gradually warmed to 53 °C over the course of 1 h. Complete removal of the TTC groups and formation of P2VP-H was confirmed by 1H NMR (Fig. S23†). When comparing P2VP-TTC (Đ = 1.08) and P2VP-H (Đ = 1.10) by SEC, negligible peak broadening had occurred during the reduction (Fig. S24†). This was a promising result that indicates reduction through the n → π* excitation of TTC sequesters combination events for P2VP without the need for additional TTMSS or toluene. Future work aims to fully investigate this phenomenon through blue light-driven photoreduction of thiocarbonylthio groups using TTMSS.
Conclusions
In summary, we have demonstrated the photoinitiated removal of TTC from PVP materials for the first time and learned the following: (1) reductions are kinetically faster than PS but slower than PMMA and P4VP is consistently faster that P2VP, (2) P2VP exhibits some chain combination events but they are much less prominent than PS, and (3) these combination events may be sequestered by either increasing the stoichiometry of the hydrogen source under 365 nm or using lower energy (452 nm) photoreduction. We have also introduced TTMSS as a kinetically superior hydrogen source that requires no additional reagents to quantitatively reduce TTC end groups from PMMA, PS, and PVP systems in the presence of light. The speed, versatility, and non-toxic nature of this reagent shows promise for efficient removal of TTC RAFT end groups. Expansion of these reductions to alternative RAFT CTAs, polymers, stoichiometries, and irradiation sources is currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgement is made to the Donors of the American Chemical Society Petroleum Research Fund (55378-DNI7) for partial support of this research. B. A. F. thanks Dr Edgar Gonzalez-Rodriquez for helpful discussions on TTMSS. We thank Dr Banghao Chen for training and use of the NMR Facility within the Department of Chemistry and Biochemistry at FSU.References
- S. Perrier, 50th Anniversary Perspective: RAFT Polymerization—A User Guide, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. Destarac, Industrial development of reversible-deactivation radical polymerization: is the induction period over?, Polym. Chem., 2018, 9, 4947–4967 RSC.
- H. Willcock and R. K. O'Reilly, End group removal and modification of RAFT polymers, Polym. Chem., 2010, 1, 149–157 RSC.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Bioapplications of RAFT Polymerization, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS.
- A. Postma, T. P. Davis, R. A. Evans, G. Li, G. Moad and M. S. O'Shea, Synthesis of Well-Defined Polystyrene with Primary Amine End Groups through the Use of Phthalimido-Functional RAFT Agents, Macromolecules, 2006, 39, 5293–5306 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Thiocarbonylthio end group removal from RAFT-synthesized polymers by radical-induced reduction, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- P. Alagi, N. Hadjichristidis, Y. Gnanou and X. Feng, Fast and Complete Neutralization of Thiocarbonylthio Compounds Using Trialkylborane and Oxygen: Application to Their Removal from RAFT-Synthesized Polymers, ACS Macro Lett., 2019, 8, 664–669 CrossRef.
- K. M. Mattson, C. W. Pester, W. R. Gutekunst, A. T. Hsueh, E. H. Discekici, Y. Luo, B. V. K. J. Schmidt, A. J. McGrath, P. G. Clark and C. J. Hawker, Metal-Free Removal of Polymer Chain Ends Using Light, Macromolecules, 2016, 49, 8162–8166 CrossRef CAS.
- E. H. Discekici, S. L. Shankel, A. Anastasaki, B. Oschmann, I.-H. Lee, J. Niu, A. J. McGrath, P. G. Clark, D. S. Laitar, J. R. de Alaniz, C. J. Hawker and D. J. Lunn, Dual-pathway chain-end modification of RAFT polymers using visible light and metal-free conditions, Chem. Commun., 2017, 53, 1888–1891 RSC.
- R. N. Carmean, C. A. Figg, G. M. Scheutz, T. Kubo and B. S. Sumerlin, Catalyst-Free Photoinduced End-Group Removal of Thiocarbonylthio Functionality, ACS Macro Lett., 2017, 6, 185–189 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Cooperative reduction of various RAFT polymer terminals using hydrosilane and thiol via polarity reversal catalysis, Chem. Commun., 2019, 55, 5327–5330 RSC.
- J. G. Kennemur, Poly(vinylpyridine) Segments in Block Copolymers: Synthesis, Self-Assembly, and Versatility, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker, S. J. Brooks and K. Matyjaszewski, Rational Selection of Initiating/Catalytic Systems for the Copper-Mediated Atom Transfer Radical Polymerization of Basic Monomers in Protic Media: ATRP of 4-Vinylpyridine, Macromolecules, 2006, 39, 6817–6824 CrossRef CAS.
- J. Xia, X. Zhang and K. Matyjaszewski, Atom Transfer Radical Polymerization of 4-Vinylpyridine, Macromolecules, 1999, 32, 3531–3533 CrossRef CAS.
- H. Wu, Y. Wan, W. Wang, Y. Wang, N. Zhou, W. Zhang, X. Li, Z. Zhang and X. Zhu, Hydrogen bonding promoting the controlled radical polymerization of 2-vinyl pyridine: supramonomer for better control, Polym. Chem., 2015, 6, 2620–2625 RSC.
- A. J. Convertine, B. S. Sumerlin, D. B. Thomas, A. B. Lowe and C. L. McCormick, Synthesis of Block Copolymers of 2- and 4-Vinylpyridine by RAFT Polymerization, Macromolecules, 2003, 36, 4679–4681 CrossRef CAS.
- B. A. Fultz, T. Terlier, B. Dunoyer de Segonzac, R. Verduzco and J. G. Kennemur, Nanostructured Films of Oppositely Charged Domains from Self-Assembled Block Copolymers, Macromolecules, 2020, 53, 5638–5648 CrossRef CAS.
- S. Qu, R. Liu, W. Duan and W. Zhang, RAFT Dispersion Polymerization in the Presence of Block Copolymer Nanoparticles and Synthesis of Multicomponent Block Copolymer Nanoassemblies, Macromolecules, 2019, 52, 5168–5176 CrossRef CAS.
- Y. Qi, I. I. Perepichka, Z. Song and S. K. Varshney, Synthesis and thermal properties of poly(vinylcyclohexane)-b-poly(4-vinylpyridine) diblock copolymers prepared via RAFT polymerization, e-Polymers, 2018, 18, 197–203 CAS.
- L. Juxiang, L. Mingchun, X. Meihua, S. Weifu and X. Wangchuan, Visible Light Induced RAFT Polymerization of 2−Vinylpyridine without Exogenous Initiators or Photocatalysts, Macromol. Chem. Phys., 2016, 217, 1777–1784 CrossRef.
- M. Faber, A. H. Hofman, K. Loos and G. ten Brinke, Highly Ordered Structure Formation in RAFT–Synthesized PtBOS–b–P4VP Diblock Copolymers, Macromol. Rapid Commun., 2016, 37, 911–919 CrossRef CAS.
- Z. Liu, G. Zhang, W. Lu, Y. Huang, J. Zhang and T. Chen, UV light-initiated RAFT polymerization induced self-assembly, Polym. Chem., 2015, 6, 6129–6132 RSC.
- L. Juxiang, L. Mingchun, X. Meihua and S. Weifu, Benzoyl Peroxide/2−Vinylpyridine Synergy in RAFT Polymerization: Synthesis of Poly(2−vinylpyridine) with Low Dispersity at Ambient Temperature, Macromol. Chem. Phys., 2015, 216, 1646–1652 CrossRef.
- X. He, Y. Qu, C. Gao and W. Zhang, Synthesis of multicompartment nanoparticles of a triblock terpolymer by seeded RAFT polymerization, Polym. Chem., 2015, 6, 6386–6393 RSC.
- Z. Özdemir, M. Topuzoğulları, İ.A İşoğlu and S. Dinçer, RAFT-mediated synthesis of poly(N-(2-hydroxypropyl)methacrylamide-b-4-vinylpyridine) by conventional and microwave heating, Polym. Bull., 2013, 70, 2857–2872 CrossRef.
- M. Dan, F. Huo, X. Zhang, X. Wang and W. Zhang, Dispersion RAFT polymerization of 4−vinylpyridine in toluene mediated with the macro–RAFT agent of polystyrene dithiobenzoate: Effect of the macro–RAFT agent chain length and growth of the block copolymer nano–objects, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1573–1584 CrossRef CAS.
- Y. Zhao, X. Shi, H. Gao, L. Zhang, F. Zhu and Q. Wu, Thermo- and pH-sensitive polyethylene-based diblock and triblock copolymers: synthesis and self-assembly in aqueous solution, J. Mater. Chem., 2012, 22, 5737–5745 RSC.
- L. Zhang, Q. Wang, P. Lei, X. Wang, C. Wang and L. Cai, Multiblock poly(4−vinylpyridine) and its copolymer prepared with cyclic trithiocarbonate as a reversible addition–fragmentation transfer agent, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2617–2623 CrossRef CAS.
- J. Božović-Vukić, H. T. Mañon, J. Meuldijk, C. Koning and B. Klumperman, SAN-b-P4VP Block Copolymer Synthesis by Chain Extension from RAFT-Functional Poly(4-vinylpyridine) in Solution and in Emulsion, Macromolecules, 2007, 40, 7132–7139 CrossRef.
- P. F. Onyon, The polymerization of 4-vinyl pyridine, Trans. Faraday Soc., 1955, 51, 400–412 RSC.
- W. I. Bengough and W. Henderson, The kinetics of the polymerization of 2-vinyl pyridine, Trans. Faraday Soc., 1965, 61, 141–149 RSC.
- G. G. Odian, Principles of polymerization, Wiley-Interscience, Hoboken, N.J., 4th edn, 2004 Search PubMed.
- B. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Thermolysis of RAFT-Synthesized Poly(Methyl Methacrylate), Aust. J. Chem., 2006, 59, 755–762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01104e |
| This journal is © The Royal Society of Chemistry 2020 |