
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzopyridine: a smart photo- and chemo-responsive substituent for polymers and supramolecular assemblies†
Hao
Ren
a,
Peng
Yang
 *a and
Françoise M.
Winnik
*bcd
*a and
Françoise M.
Winnik
*bcd
aKey Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, P. R. China. E-mail: yangpeng@snnu.edu.cn
bLaboratory of Polymer Chemistry, Department of Chemistry, PB 55, University of Helsinki, Helsinki, FI00140, Finland. E-mail: francoise.winnik@helsinki.fi
cInternational Center for Materials Nanoarchitectonics, National Institute for Material Science, 1-1 Namiki, Tsukuba 305-0044, Japan
dDepartment of Macromolecular Science, School of Graduate Studies, University of Osaka, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
First published on 7th September 2020
Abstract
Azo dyes, such as azobenzene, are able to convert absorbed light into motion or deformation on the macroscopic scale on the basis of their remarkable ability to undergo repeatedly and in 100% yield reversible trans-to-cis photoisomerization. Current needs for multiresponsive and fast photoswitches have led to the development of heteroaryl azo dyes, such as azopyridine. This remarkable azo compound combines the photoresponse of the azo chromophore with the chemistry of the pyridine ring, in particular its responsiveness to changes in pH and its ability to form hydrogen- and halogen-bonds. This mini-review summarizes key features of the photoisomerization of polymer-tethered azopyridine in aqueous media and describes a few recent research accomplishments in emerging areas that have benefited of the fast thermal cis-to-trans relaxation characteristics of azopyridinium or H-bonded azopyridine. It also discusses the effects of the photoisomerization of azopyridine on the thermoresponsive properties of azopyridine-tethered heat-sensitive polymers. Overall, azopyridine is a highly versatile actuator to consider when designing photo/multiresponsive polymeric materials.
1. Introduction
Azo compounds are commonly used actuators of photoresponsive polymer systems. They have a long history, dating from the turn of the 20th century when, shortly after the discovery of diazonium salts, azo dyes began to be produced industrially for the textile industry.1,2 Azo compounds exist in the trans (E) form and in the cis (Z) form. The two isomers can be interconverted by light. The isomerization involves a change in molecular geometry that is often accompanied by a change of the polarity or of other physical properties of the azo isomers. In the 1970s, polymer chemists introduced azobenzene compounds as light-driven triggers of specific changes in the physical properties of materials.3,4 Azobenzene remained the azo dye of choice for many years in view of its stability, tuneability, and reliability. Current trends towards miniaturization, which often require faster and more versatile photoswitches, led to the design and development of heteroaryl azo dyes, such as azopyridine (AzPy, Fig. 1a), among others. AzPy exhibits also all the chemical features associated with the pyridine nitrogen atom and offers chemical opportunities unattainable with azobenzene.5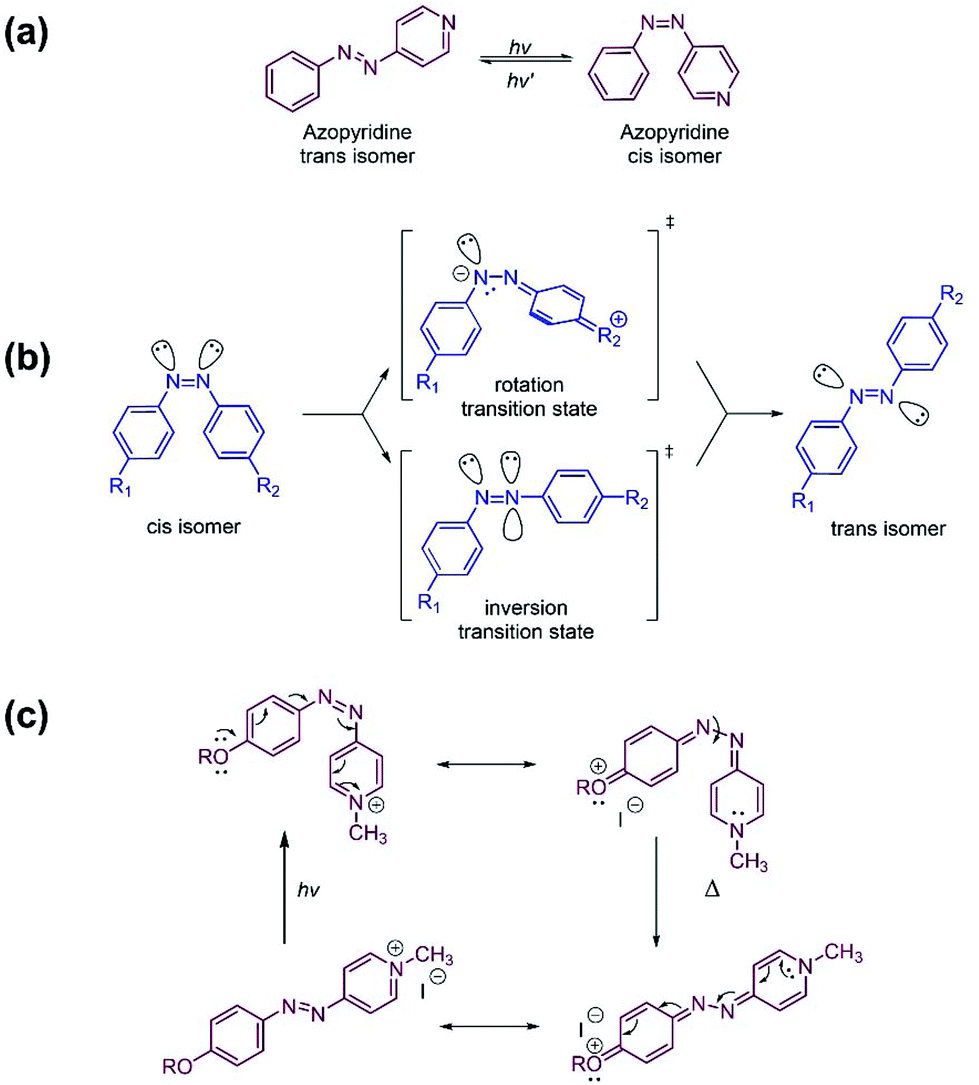 | ||
| Fig. 1 (a) Reversible photoisomerization of azopyridine; (b) mechanism of the cis-to-trans thermal relaxation of azobenzene, (c) mechanism of the cis-to-trans thermal isomerization of cis-azopyridinium facilitated by the push–pull effect. Adapted with modifications from ref. 11 (DOI: 10.3762/bjoc.8.113) under the terms of the Creative Commons Attribution License https://www.beilstein-journals.org/bjoc. | ||
Initially designed for use in photoresponsive bulk materials, such as liquid crystalline films, a growing number of AzPy-based polymers are used in the form of multiresponsive nanoparticles, vesicles, etc., addressable independently by light, pH, and temperature, if the polymer is thermosensitive. In this mini-review, we describe the spectroscopy and photoisomerization of AzPy. In particular, we explain how the substitution pattern of the pyridine ring and the involvement of the pyridine nitrogen in non-covalent bonds affect the UV-Vis absorption spectra of trans- and cis-AzPy as well as the dynamics of the trans ↔ cis conversion with light or thermally. Pitfalls related to the fast dark relaxation of cis-AzPy will be discussed. In a second part, we present selected recent publications that take advantage of the distinct spectroscopic and chemical properties of AzPy derived from the presence of a pyridine ring. At the end of the mini-review (ESI†), readers will find a list of the AzPy-modified polymers prepared so far that includes their structure and molecular characteristics.
2. The photoisomerization of azopyridine and azopyridinium: notable features
Azopyridine (AzPy) (Fig. 1a) is interconverted reversibly between the trans and the cis isomer upon light absorption. Ultraviolet (UV) light irradiation converts the trans isomer to the less stable cis isomer. Irradiation with visible (Vis) light reverses the process, such that the trans isomer is recovered. The cis-to-trans conversion takes place also thermally in the absence of light. The exact mechanism of this dark back conversion is still under discussion. It is believed to lie between the two mechanisms originally proposed to account for the dark conversion of cis-azobenzene to trans-azobenzene: one entailing a rotation around the intermediate N–N bond6,7 and the other implicating an inversion and in-plane lateral shift through a linear transition state8,9 (Fig. 1b). The photoisomerization of AzPy occurs within a few nanoseconds through electronic and nuclear rearrangements of the azopyridine molecule. The dark relaxation time is much longer than the photoconversion time. It ranges from several hours to a few minutes.10Work carried out earlier with azobenzene (Azo) established that the cis-to-trans dark relaxation time decreases to a few seconds by introducing in the azobenzene molecule a substitution pattern that produces a “push–pull” electronic distribution, such as a dimethylamino group at the C4 position and a nitro group at the C4′ position of Azo.11,12 The cis-to-trans dark relaxation time reaches the desired microsecond time domain by replacing a phenyl ring of azobenzene with a pyridinium ring that is a more powerful electron-withdrawing group than –NO2.11 The strong charge transfer from the electron donor substituent on the benzene ring to the positively-charged nitrogen of the pyridinium ring,11,13 partially cleaves the N–N bond formed during the thermal cis-to-trans relaxation of AzPy+ (Fig. 1c). This facilitates and accelerates the recovery of the more stable trans state.13 Formation of non-bonding interactions between the nitrogen atom of AzPy and hydrogen- or halogen-bond donors, also strengthens the push–pull effect, such that the cis-to-trans dark relaxation time of H-bonded AzPy is ∼1 s, compared to several minutes or hours, in the case of neutral azopyridine in the absence of H-bond donor.10
Key features of the UV-Vis spectrum of AzPy and of the dynamics of the reversible trans-to-cis azo isomerization are illustrated here with data collected during a study of the AzPy-modified poly(N-isopropylacrylamide) (AzPy-PNIPAM) shown in Fig. 2, top.14 AzPy-PNIPAM is quaternized in acidic aqueous solutions. In neutral solutions (pH 7), the AzPy nitrogens are H-bonded to amide hydrogens of the PNIPAM repeat units. The H-bonds are broken under basic conditions (pH 10). Hence, this polymer gives us the opportunity to present data characteristic of AzPyH+, H-bonded AzPy, and free neutral AzPy. The polymer was prepared by reversible addition fragmentation chain transfer (RAFT) polymerization of NIPAM in the presence of a trithiocarbonate chain transfer agent.14 The trithiocarbonate group of the chain transfer agent is attached to one end of the polymer. Its characteristic absorption band centered at 310 nm (marked with a star) is visible in all UV absorption spectra presented below. It does not interfere with the photoisomerization and can be ignored.
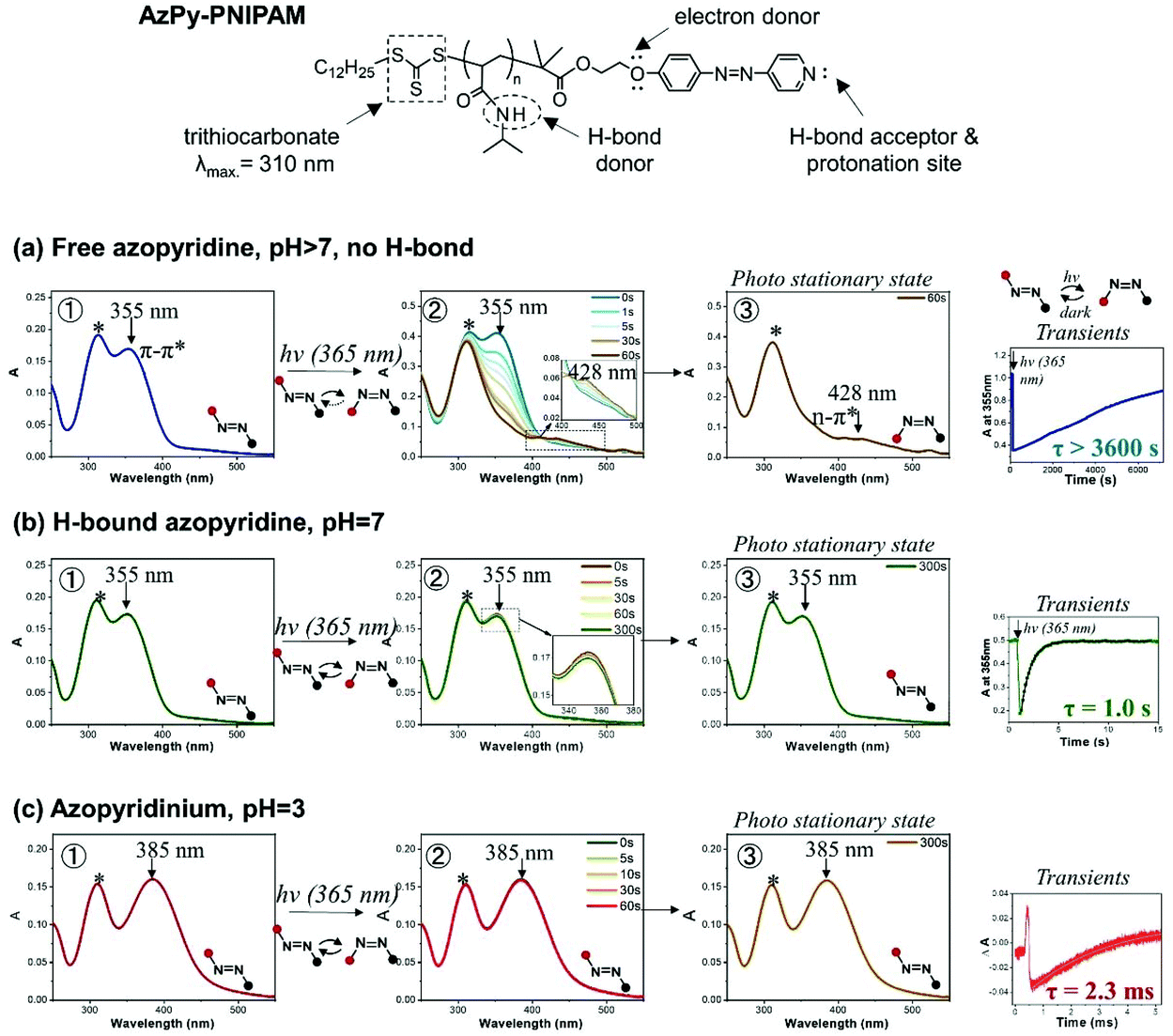 | ||
| Fig. 2 Chemical structure of AzPy-PNIPAM (top section); photophysical properties of azopyridine, (a) in solutions of pH 7 or above, in the absence of non-bonding interactions; (b) under conditions where the azopyridine is involved in a hydrogen or halogen bond; and (c) in acidic solutions where azopyridine is protonated. Adapted with permission from ref. 14 Copyright (2019) American Chemical Society. | ||
Fig. 2 is divided in three horizontal lines devoted to a: spectra of AzPy in a solution of pH > 7 in the absence of H-bonds; b: H-bonded AzPy; and c: protonated AzPy. Each line contains three UV-Vis spectra (1, 2, and 3) and a smaller frame displaying transients. Frames 1 on each line show the UV-Vis spectra of trans-AzPy prior to irradiation, frames 2 present changes of the UV-Vis spectra of trans-AzPy under continuous irradiation at 365 nm. Frames 3 are the UV-Vis spectra of AzPy under irradiation when the photostationary state is reached.
UV-Vis absorption spectra of trans-AzPy and trans-AzPyH+: (frame 1, lines a, b, and c)
The absorption band centered at 355 nm in the UV-Vis spectra of AzPy in solutions of pH ≥ 7 and H-bonded AzPy is attributed to the azo π–π* transition. This band is shifted to 385 nm in the spectrum of AzPyH+ (line c). The red-shift results from the strong charge transfer from the alkoxy substituent on the benzene ring to the positively-charged nitrogen of the pyridinium ring.11,13Spectral changes prompted by the photoisomerization of AzPy and AzPyH+
It is normal practice for researchers in the field of photoresponsive materials to monitor the changes of the UV-Vis absorption spectrum of a chromophore continuously irradiated with a light of wavelength causing the desired photoresponse. This simple experiment confirms that the desired photoproduct is obtained. It also gives access to the photoreaction kinetics under various conditions. The test was performed in the case of trans-AzPy-PNIPAM in aqueous solutions of pH 3, 7, and 10 subjected to continuous irradiation at 365 nm.Continuous irradiation with a 365 nm light of free trans-AzPy (pH > 7, frame 2, line a) results in the decrease of the absorption band centered at 355 nm and the appearance of a weak band centered at 428 nm characteristic of the cis-isomer, confirming the successful formation of cis-AzPy. Complete trans-to-cis conversion takes ∼60 s under the experimental conditions selected (frame 3, line a). Irradiation of the solution of H-bonded AzPy (pH 7, frame 3, line b), even for as long as 5 min, does not affect the UV-Vis spectrum. The UV-Vis spectrum recorded at the photostationary state is identical to that of trans-AzPy. Similarly, irradiation of the AzPyH+ solution of pH 3 (frame 3, line c) for up to 5 min has no effect on the sample UV spectrum.
Given the fast dark relaxation of cis-azopyridinium and H-bonded AzPy, it is possible that the dark cis-to-trans relaxation of AzPy+ (pH 3) or H-bonded AzPy (pH 7) occurs so fast that the transient formation of the cis isomer cannot be detected by standard UV-Vis absorption spectroscopy. The recording time of a spectrum with the spectrophotometer we used is 5 s or more. Since the measurement is done after UV light irradiation, as soon as the cis-to-trans isomerization is complete, the trans-isomer is reconverted (within a few ns for pH 3 and ∼1 s for pH 7) to the cis, etc. In other words, the UV-Vis spectrometer is “blind” to the intermediate formation of the cis isomer, and the UV-Vis spectra recorded during the entire irradiation time are identical.
By measuring the transients generated upon flash irradiation at 365 nm of the trans-AzPy solutions it is possible to access the kinetics of the cis-to-trans dark relaxation from the millisecond to the second timescale. The dark half-lifes of AzPy in aqueous solutions of pH 3, 7, and 10 were, respectively, 2.3 ms, 1.0 s, and >1 h (Fig. 2), in agreement with previous reports.11
Several research teams have investigated the photoirradiation of AzPy covalently-linked to block copolymers consisting a polymethacrylate AzPy block and a water-soluble block, such as poly(ethylene oxide), poly(N-isopropylacrylamide), or poly(2-dimethylaminoethyl methacrylate) (ESI†) in aqueous solutions or in mixed water/THF solutions.15–21 They monitored the progress of the photoirradiation by UV-Vis absorption spectroscopy as discussed above and included representative spectra in the articles or their ESI.† In several cases, the spectra reported were similar to those shown in Fig. 2b and c, namely the absorption band assigned to the π–π* transition did not decrease upon irradiation of the trans AzPy isomer.
3. Chemical properties of azopyridines and their applications in responsive polymeric materials
AzPy exhibits chemical features associated with the pyridine nitrogen atom summarized in Fig. 3. It is readily ionized, responsive to changes in pH, to the presence of CO2, and can be quaternized permanently with alkylating agents. The pyridine nitrogen is also a superb hydrogen- and halogen-bond acceptor. It has coordinative and chelating abilities towards inorganic species, both metal ions and metal nanoparticles (Fig. 3c). Chemical reactions and non-covalent modifications of the AzPy nitrogen atom perturb the electronic distribution of the AzPy chromophore and, consequently, affect the AzPy absorption spectrum and the kinetics of the dark cis-to-trans isomerization. Chemical reactions and non-covalent bonds of AzPy have been exploited extensively in the construction of light-responsive supermolecular assemblies of AzPy-polymers,22–24 oligomers, and small molecules.5,25 This aspect of AzPy has been exploited by materials chemists, who used AzPy as photoresponsive unit in supramolecular liquid crystalline materials,26–28 metal–organic frameworks,29–31 fibers,32–37 films,22,38,39 gels,40–43etc. In these materials, AzPy is incorporated in the main component as a compatible small molecule or linked to random or block copolymers, where one co-monomer carries an AzPy group.40–42,44 This area has been the object of several recent reviews.45,46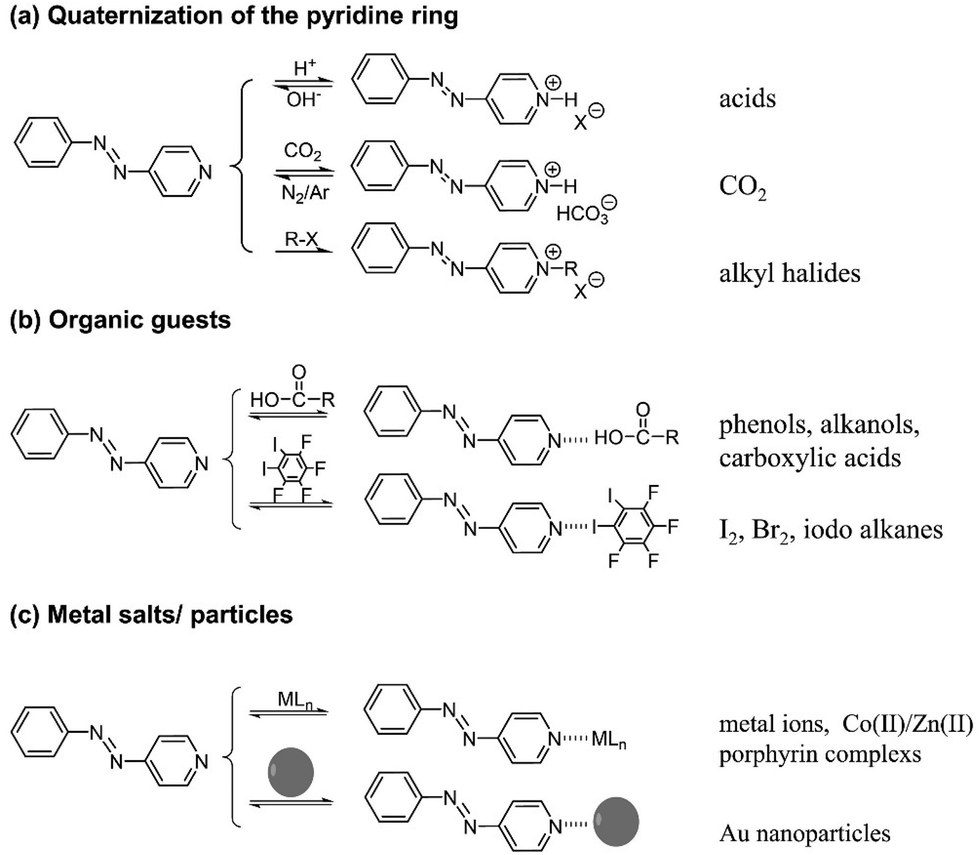 | ||
| Fig. 3 Chemical interactions/reactions of the pyridine nitrogen of AzPy. (a) Quaternization of the pyridine ring, (b) organic guests, (c) metal salts/particles. | ||
(a) Quaternization of the pyridine ring
Xue et al. demonstrated that CO2 is an excellent agent to acidify aqueous solutions of various copolymers of AzPy-PNIPAM rapidly and cleanly.15 In one example, the cloud point of the polymer, PNIPAM-co-PAzPy-co-PEGMA (see ESI†), in water increased from 49 °C to 62 °C after injection of CO2 gas, which was attributed to the increase of the polymer solubility in water upon acidification of the AzPy component caused by CO2 gas. Reversible and fast switch from a turbid to a clear sample was achieved for a solution of neutral solution of PNIPAM-co-PAzPy-co-PEGMA kept at 60 °C injected alternatively with CO2 and Ar.Aqueous solutions of AzPy-PNIPAM (structure shown in Fig. 2, top) respond to three orthogonal triggers:47 light, by virtue of the trans–cis isomerization of AzPy, pH via protonation of the pyridine nitrogen of AzPy, and temperature, due to the thermosensitivity of PNIPAM in water.48 Also, AzPy-PNIPAM self-assembles in water as it bears a dodecyl chain on one end and the AzPy group on the other. In acidic solution AzPyH+-PNIPAM forms micelles with a core of n-dodecyl chains surrounded by PNIPAM chains terminated by azopyridinium groups.14 In neutral and basic aqueous solutions, the polymer forms flower micelles with a core of n-dodecyl chains and AzPy, surrounded by a PNIPAM corona.14 The polymer undergoes a phase transition in water at a temperature (Tc, cloud point) that depends on the solution pH.47 The Tc value also changes upon light illumination, since the dipole moments of the trans-AzPy and cis-AzPy are different. An overview of the phenomena observed upon sequential application of the three triggers is given in Fig. 4. Application of heat to solutions of pH 7 and 10 causes the dehydration/collapse of the PNIPAM chain. The collapsed flower micelles aggregate into larger particles, observed macroscopically by the turbidity of the sample. A brief UV light irradiation converts trans-AzPy to cis-AzPy within a few seconds. The solution recovers its original transparency although the solution temperature is the same. The photoisomerization effectively brings the solution above its cloud point as a consequence of the change of the AzPy dipole. If the cis-AzPy solution of pH 7 is kept in the dark at the same temperature it recovers its turbidity as a consequence of the fast dark relaxation of cis-AzPy to trans-AzPy. Although the dark relaxation is fast (∼1 s), the turbidity recovery takes about 20 s. This time lag is a characteristic feature of the phase transition of aqueous PNIPAM solutions.48 The recovery of the solution of pH 10 is very slow (see the Absorbance vs. time plots in the pH 10 and 7 frames). A specific customized overall response can be designed through permutations of the order of the light, pH and heat triggers.
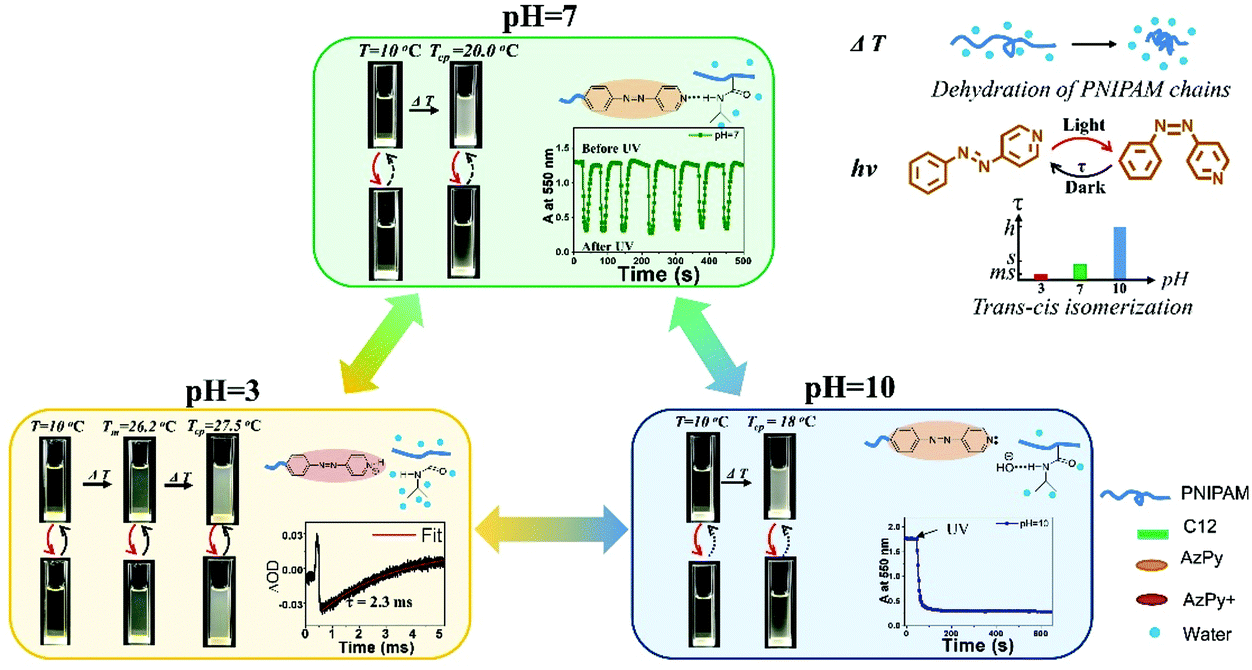 | ||
| Fig. 4 Heat-, pH- and UV-induced properties of AzPy-PNIPAM (Mn ∼ 7000 g mol−1), ΔOD: difference in the optical density before and after irradiation. Reprinted with permission from ref. 47 Copyright (2019) Royal Society of Chemistry. | ||
(b) Halogen- and hydrogen-bonds
Halogen bonds form between a polarized halogen atom, the halogen bond donor acting as electron acceptor and an electron donor or halogen acceptor, such as an amine or pyridine. Halogen bonds out-perform H-bonds and other non-covalent interactions in terms of strength, directionality, and tunability. The first halogen-bonded liquid crystals were reported in 2004 between non-mesomorphic 4-alkoxystilbazoles and iodopentafluorobenzene.49 Since then, various halogen-bond donating compounds were reported, including iodoacetylenes,50 and fluorinated aromatic derivatives.51 Chen et al. reported the first example of azopyridine containing halogen-bonded liquid crystals in 2014 between AzPy derivatives and either I2 or Br2. Photoinduced deformation-recovery of the liquid crystal phase upon alternate irradiation of UV and blue light was observed.52 The dark relaxation rate of halogen-bonded AzPy is usually on the order of days and rarely faster than 1 min. Nonetheless their strength and high directionality of halogen bonds are of interest to material scientists.In the early 2000's. T. Ikeda and coworkers reported that a liquid crystal network containing azobenzene can be repeatedly bent in a chosen direction upon irradiation with polarized light as a consequence of photo induced volume contraction.53 This discovery led to the development of microscale and nanoscale actuators and robots. In parallel, Song et al. reported that H-bonds between 4-(alkoxyphenylazo) pyridines (H-bond acceptors) and 4-octyloxylbenzoic acids (H-bond donors), form liquid crystalline phases, although by themselves they are non-mesomorphic.54 This observation proved to be a general phenomenon. Carboxylic acid/AzPy liquid crystal systems are superior to their azobenzene analogues as fast responsive dopants of liquid crystals.26,27,55,56 Unlike liquid crystals doped with azobenzene-derived mesogens, which bend on the macroscopic scale and keep their shape in the dark,57–59 heteroaryl azo chromophores such as AzPy can control the shape of films of a liquid crystal mesophase. This effect was demonstrated a few years ago in the case of a photoactive liquid crystalline polymer film doped with an azopyridine derivative.60 Photoirradiation of a trans-AzPy-doped liquid crystalline film yields the cis isomer and generates heat. The irradiated area heats up, easily reaching temperatures above the glass temperature of the film. Since AzPy is H-bonded to the carboxylic acid, the dark cis-to-trans relaxation is accelerated compared to neutral free AzPy (push–pull effect discussed above). The cis-AzPy probes that reside in film areas kept above Tg, are very mobile, which further facilitates the dark relaxation to the trans AzPy isomer. On the macroscopic scale, constant irradiation of such a film clamped at both ends causes the formation of a unidirectional wave that oscillates along the film and can transport small objects. This experiment provides convincing macroscopic evidence of the fast dark relaxation of cis H-bound AzPy.
(c) Metal complexation of AzPy-tethered to polymers
A copolymer consisting of an ethylhexyl methacrylate block and an AzPy-methacrylate block (PEHMA-co-PMAzPy, see ESI†) was shown to form a complex with the cobalt(II) Schiff base (CoS). The complex formed with a surprisingly large apparent binding constant. Moreover, upon complexation to CoS, the AzPy lost its ability to photoisomerize. The two effects were attributed to the formation of a micro-suspension consisting of small polymer domains that entrap complexed CoS dispersed in a large pool of solvent.294. Conclusion and outlook
This mini-review summarized the chemical and photophysical properties of azopyridine. It emphasized the distinct features of this chromophore, compared to the well-known azobenzene, that result from the association of the azo (PhNNPh) chromophore with the versatile chemistry of the pyridine group. From the chemical viewpoint, azopyridine is pH-sensitive, unlike azobenzene. It reacts reversibly with acids, both aqueous acids/bases and CO2 gas. The nitrogen atom of AzPy is a strong acceptor of hydrogen- and halogen-bonds with H-donors such as phenols, carboxylic acids, or alcohols, and halogen-bond donors such as I2, Br2, and activated iodobenzenes. AzPy can also form complexes with metals, such as Co(II). The chemical properties of AzPy were exploited by polymer chemists in many ways. Only a few examples are given in this review, since the topic has been reviewed extensively over the last few years. The truly unique feature of AzPy relates to its photophysical properties, more specifically to the rate of the cis-to-trans thermal (dark) relaxation. This rate ranges from hours when the molecule is neutral to milliseconds when it is quaternized. When the nitrogen atom of the AzPy chromophore is H- or halogen-bonded, the cis-to-trans dark relaxation takes a few seconds. The temporal space of AzPy has been studied by physicists and spectroscopists, but mostly overlooked, or misinterpreted, by polymer chemists used to design experiments with azobenzenes, for which the dark cis-to-trans isomerization rate is fixed for a given molecule and usually long, compared to the speed of common light sources and spectrophotometers. It is only recently that the wide temporal scale of the dark relaxation of cis-AzPy was used by materials and polymer chemists, driven by the need for fast tuneable photoswitches.Further challenges need to be overcome for AzPy to expand its scope beyond photoswitches to areas such as nanomedicine, biology, separation methods, responsive interfaces. Tunability of the AzPy light absorption spectrum from the UV to the near IR would give access to chromophores that absorb light within the spectral window desirable in medical imaging. A few studies in this direction are underway, such as the use of halogen-bond based supramolecules consisting of an azopyridine and an azobenzene in tandem,44 or heteroaryl azo dyes with two or more heteroatoms.5 In the chemical direction, it would be useful to design ways to incorporate AzPy into gel networks, especially hydrogels, without adverse effect on the dark relaxation rate, for use as healable gels for example.
We are convinced that in the near future, the controlled dark relaxation of cis-AzPy will bring about new or improved applications in various fields through interdisciplinary teams of theorists, spectroscopists, chemists, biologists, and material scientists. We hope that this minireview will be an inspiration to polymer chemists in academia and industry by bringing to their attention the unique properties of AzPy and that it will intensify research on multiresponsive materials and their applications in the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. R. acknowledges China Postdoctoral Science Foundation (Grant No. 2019M653859XB), National Natural Science Foundation of China (No. 21905166) and Natural Science Basic Research Program of Shaanxi (No. 2020JQ-406) for partly support this work. FMW acknowledges the Natural Sciences and Engineering Research Council of Canada for partial support of this work as well as the Osaka University (Japan) International Joint Research Promotion Program.References
- H. Zollinger, Color chemistry: syntheses, properties, and applications of organic dyes and pigments, John Wiley & Sons, New Jersey, 2003 Search PubMed.
- H. Zollinger, Azo and diazo chemistry: aliphatic and aromatic compounds, Interscience Publishers, New York, 1961 Search PubMed.
- S. Shinkai, T. Nakaji, Y. Nishida, T. Ogawa and O. Manabe, J. Am. Chem. Soc., 1980, 102, 5860–5865 CrossRef CAS.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4176 CrossRef CAS.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- J. Binenboym, A. Burcat, A. Lifshitz and J. Shamir, J. Am. Chem. Soc., 1966, 88, 5039–5041 CrossRef CAS.
- E. R. Talaty and J. C. Fargo, Chem. Commun., 1967, 2, 65–66 RSC.
- D. Schulte-Frohlinde, Justus Liebigs Ann. Chem., 1958, 612, 138–152 CrossRef.
- R. J. W. Le Fèvre and J. Northcott, J. Chem. Soc., 1953, 867–870 RSC.
- K. Bujak, H. Orlikowska, J. G. Małecki, E. Schab-Balcerzak, S. Bartkiewicz, J. Bogucki, A. Sobolewska and J. Konieczkowska, Dyes Pigm., 2019, 160, 654–662 CrossRef CAS.
- J. García-Amorós and D. Velasco, Beilstein J. Org. Chem., 2012, 8, 1003–1017 CrossRef.
- A. Goulet-Hanssens and C. J. Barrett, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3058–3070 CrossRef CAS.
- J. García-Amorós, W. A. Massad, S. Nonell and D. Velasco, Org. Lett., 2010, 12, 3514–3517 CrossRef.
- H. Ren, X. Qiu, Y. Shi, P. Yang and F. M. Winnik, Macromolecules, 2019, 52, 2939–2948 CrossRef CAS.
- X. Xue, F. Qing, W. Huang, H. Yang, Q. Jiang, L. Zhou and B. Jiang, Acta. Polym. Sin., 2018, 1175–1183 CAS.
- Y. Wang, G. Shen, J. Gao, G. Zou and Q. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 415–421 CrossRef CAS.
- W. Yuan, W. Guo, H. Zou and J. Ren, Polym. Chem., 2013, 4, 3934 RSC.
- J. Wei, Z. Yan, L. Lin, J. Gu, Z. Feng and Y. Yu, React. Funct. Polym., 2013, 73, 1009–1014 CrossRef CAS.
- T. Michinobu, R. Eto and K. Shigehara, Kobunshi Ronbunshu, 2011, 68, 195–197 CrossRef CAS.
- L. Lin, Z. Yan, J. Gu, Y. Zhang, Z. Feng and Y. Yu, Macromol. Rapid Commun., 2009, 30, 1089–1093 CrossRef CAS.
- K. Han, W. Su, M. Zhong, Q. Yan, Y. Luo, Q. Zhang and Y. Li, Macromol. Rapid Commun., 2008, 29, 1866–1870 CrossRef CAS.
- X. Li, S. Ma, J. Hu, Y. Ni, Z. Lin and H. Yu, J. Mater. Chem. C, 2019, 7, 622–629 RSC.
- H. Liu, T. Kobayashi and H. Yu, Macromol. Rapid Commun., 2011, 32, 378–383 CrossRef CAS.
- H. Yu, H. Liu and T. Kobayashi, ACS Appl. Mater. Interfaces, 2011, 3, 1333–1340 CrossRef CAS.
- H. M. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- M. Hagar, M. Hagar, H. A. Ahmed, H. A. Ahmed and O. A. Alhaddad, Liq. Cryst., 2019, 46, 1440–1451 CrossRef CAS.
- M. Alaasar and C. Tschierske, Liq. Cryst., 2019, 46, 124–130 CrossRef CAS.
- Y. Chen, H. Yu, M. Quan, L. Zhang, H. Yang and Y. Lu, RSC Adv., 2015, 5, 4675–4680 RSC.
- T. Suzuki, T. Moriya, R. Endo and N. Iwasaki, Polym. Chem., 2017, 8, 761–768 RSC.
- S. Dahmane, A. Lasia and Y. Zhao, Macromol. Chem. Phys., 2006, 207, 1485–1491 CrossRef CAS.
- L. Cui, S. Dahmane, X. Tong, L. Zhu and Y. Zhao, Macromolecules, 2005, 38, 2076–2084 CrossRef CAS.
- K. Aoki, M. Nakagawa and K. Ichimura, J. Am. Chem. Soc., 2000, 122, 10997–11004 CrossRef CAS.
- K. Aoki, M. Nakagawa and K. Ichimura, Chem. Lett., 1999, 11, 1205–1206 CrossRef.
- Y. Chen, M. Quan, H. Yu, L. Zhang, H. Yang and Y. Lu, RSC Adv., 2015, 5, 31219–31225 RSC.
- W. Zhou and H. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 2154–2159 CrossRef CAS.
- W. Zhou, T. Kobayashi, H. Zhu and H. Yu, Chem. Commun., 2011, 47, 12768–12770 RSC.
- W. Zhou and H. Yu, RSC Adv., 2013, 3, 22155–22159 RSC.
- Y. Chen, S. Huang, T. Wang and H. Yu, Macromolecules, 2020, 53, 1486–1493 CrossRef CAS.
- H. Zhang, R. Hao, J. K. Jackson, M. Chiao and H. Yu, Chem. Commun., 2014, 50, 14843–14846 RSC.
- Y. Ni, X. Li, J. Hu, S. Huang and H. Yu, Chem. Mater., 2019, 31, 3388–3394 CrossRef CAS.
- G. Liu, J. Sheng, H. Wu, C. Yang, G. Yang, Y. Li, R. Ganguly, L. Zhu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 6467–6473 CrossRef CAS.
- G. Liu, J. Sheng, W. L. Teo, G. Yang, H. Wu, Y. Li and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 16275–16283 CrossRef CAS.
- Y. Chen, X. Feng, Z. Sun, D. Wang, T. Yang, Z. Zhao, L. Li, X. Wang and H. Yu, Chem. – Asian J., 2018, 13, 2781–2785 CrossRef CAS.
- X. Tong, Y. Qiu, X. Zhao, B. Xiong, R. Liao, H. Peng, Y. Liao and X. Xie, Soft Matter, 2019, 15, 6411–6417 RSC.
- H. Yu, J. Tang, Y. Feng and W. Feng, Chin. J. Polym. Sci., 2019, 37, 1183–1199 CrossRef CAS.
- J. M. Abendroth, O. S. Bushuyev, P. S. Weiss and C. J. Barrett, ACS Nano, 2015, 9, 7746–7768 CrossRef CAS.
- H. Ren, X. Qiu, Y. Shi, P. Yang and F. M. Winnik, Polym. Chem., 2019, 10, 5080–5086 RSC.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS.
- H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 16–17 CrossRef CAS.
- L. González, N. Gimeno, R. M. Tejedor, V. Polo, M. B. Ros, S. Uriel and J. L. Serrano, Chem. Mater., 2013, 25, 4503–4510 CrossRef.
- M. Saccone, M. Spengler, M. Pfletscher, K. Kuntze, M. Virkki, C. Wölper, R. Gehrke, G. Jansen, P. Metrangolo, A. Priimagi and M. Giese, Chem. Mater., 2018, 31, 462–470 CrossRef.
- Y. Chen, H. Yu, L. Zhang, H. Yang and Y. Lu, Chem. Commun., 2014, 50, 9647–9649 RSC.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS.
- X. Z. Song, J. X. Li and S. W. Zhang, Liq. Cryst., 2003, 30, 331–335 CrossRef CAS.
- V. A. Mallia, P. S. Antharjanam and S. Das, Liq. Cryst., 2003, 30, 135–141 CrossRef CAS.
- D. C. Rogness, P. J. Riedel, J. R. Sommer, D. F. Reed and K. N. Wiegel, Liq. Cryst., 2006, 33, 567–572 CrossRef CAS.
- Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145–145 CrossRef CAS.
- H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149–2180 CrossRef CAS.
- H. Yu, Prog. Polym. Sci., 2014, 39, 781–815 CrossRef CAS.
- A. H. Gelebart, D. J. Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632–636 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0py01093f |
| This journal is © The Royal Society of Chemistry 2020 |