
Tetracoordinate borates as catalysts for reductive formylation of amines with carbon dioxide†
Xiaolin
Jiang
ab,
Zijun
Huang
ac,
Mohamed
Makha
a,
Chen-Xia
Du
d,
Dongmei
Zhao
*b,
Fang
Wang
*a and
Yuehui
Li
 *a
*a
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute of LICP, Center for Excellence in Molecular Synthesis, Lanzhou Institute of Chemical Physics (LICP), Chinese Academy of Sciences, Lanzhou 730000, P.R. China. E-mail: fwang@licp.cas.cn; yhli@licp.cas.cn
bKey Laboratory of Structure-Based Drug Design & Discovery of Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, P.R. China. E-mail: dongmeiz-67@163.com
cCollege of Chemistry and Chemical Engineering, Hunan Institute of Engineering, Xiangtan 411104, P.R. China
dCollege of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, P.R. China
First published on 1st July 2020
Abstract
We report sodium trihydroxyaryl borates as the first robust tetracoordinate organoboron catalysts for reductive functionalization of CO2. These catalysts, easily synthesized from condensing boronic acids with metal hydroxides, activate main group element–hydrogen (E–H) bonds efficiently. In contrast to BX3 type boranes, boronic acids and metal-BAr4 salts, under transition metal-free conditions, sodium trihydroxyaryl borates exhibit high reactivity of reductive N-formylation toward a variety of amines (106 examples), including those with functional groups such as ester, olefin, hydroxyl, cyano, nitro, halogen, MeS–, ether groups, etc. The over-performance to catalyze formylation of challenging pyridyl amines affords a promising alternative method to the use of traditional formylation reagents. Mechanistic investigation supports electrostatic interactions as the key for Si/B–H activation, enabling alkali metal borates as versatile catalysts for hydroborylation, hydrosilylation, and reductive formylation/methylation of CO2.
Introduction
Boron-based catalysts such as BF3, HB(C6F5)2 (Piers borane), B(C6F5)3, Corey–Bakshi–Shibata oxazaborolidine reduction catalysts, boron nitrides, boronic acids, borinic acids, etc. are widely used in organic synthesis and/or industrial applications.1–4 In procedures using boron-based catalysts and related borylation reactions, tetradentate borates are known as key intermediates formed due to the Lewis acidity of boron atoms originating from the existence of empty 2p orbitals.5–7 Here, these borate intermediates are treated as active species assisting the chemical reactions (Fig. 1a).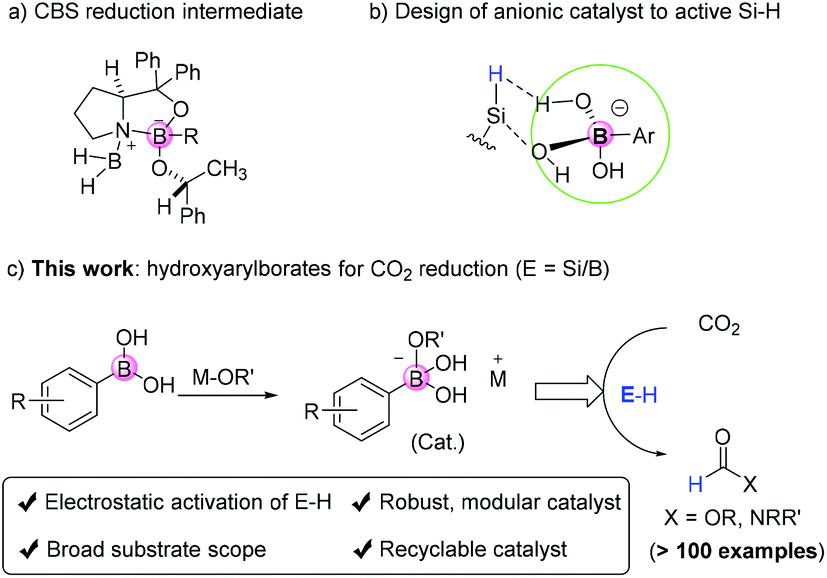 | ||
| Fig. 1 Metal hydroxyaryl borates for activation of E–H to selectively reduce CO2. | ||
CO2 reduction with functionalization for the preparation of formamides and methylated amines represents one of the most important ways to incorporate CO2 into organic molecules as C1 building blocks. Other than the use of transition metal catalysts,8–12 non-covalent weak interactions such as van der Waals interactions and hydrogen and halogen bondings are widely involved weak forces in organo-activation of chemical bonds.13–15 In this respect, although quite many types of transition metal-free catalysts were shown to be active in the title reaction,16 the principle governing transition metal-free catalysis mainly relies on acid–base interactions between the catalyst and the reductant. For instance, FLPs (Frustrated Lewis Pairs) are used as efficient and successful catalysts to activate small molecules including even dihydrogens.17,18 Therefore, the use of strong acids or bases tends to be the preferred choice to activate main group element–H (E–H) bonds. However, these known systems often suffer from limited substrate scope drawbacks, especially for acidic or basic groups containing substrates (e.g. phenolic, pyridyl amines).
Electrostatic catalysis refers to substrate activation by catalysts via electrostatic interactions.19–21 This strategy is a late emerging approach to activate the B–H bond with the recent report by Hirao and Kinjo et al. on elegant reduction of ketone/aldehyde/CO2 using diazadiborinine compounds as pre-catalysts.22 However, to the best of our knowledge, there is no previous report on the use of metal borates for Si–H activation and reductive functionalization of CO2, even though they contain multiple functional sites such as acidic protons, basic oxygen atoms, and aryl groups possibly useful for catalysis. Our design strategy stems from the use of aryl bronate base catalysts to activate Si–H through electrostatic interactions, which in turn reacts with acidic CO2 to form the silyl formate reagent required for N-formylation (Fig. 1b). Such electrostatic interactions induce hydride transfer to affect hydrosilylation of CO2 and form silyl formates. Herein, we demonstrate the use of transition metal-free arylborates, well-known Suzuki coupling reagents, for efficient catalytic reduction of CO2 and selective N-formylation/methylation of a broad scope of aromatic and aliphatic amines (Fig. 1c).
Results and discussion
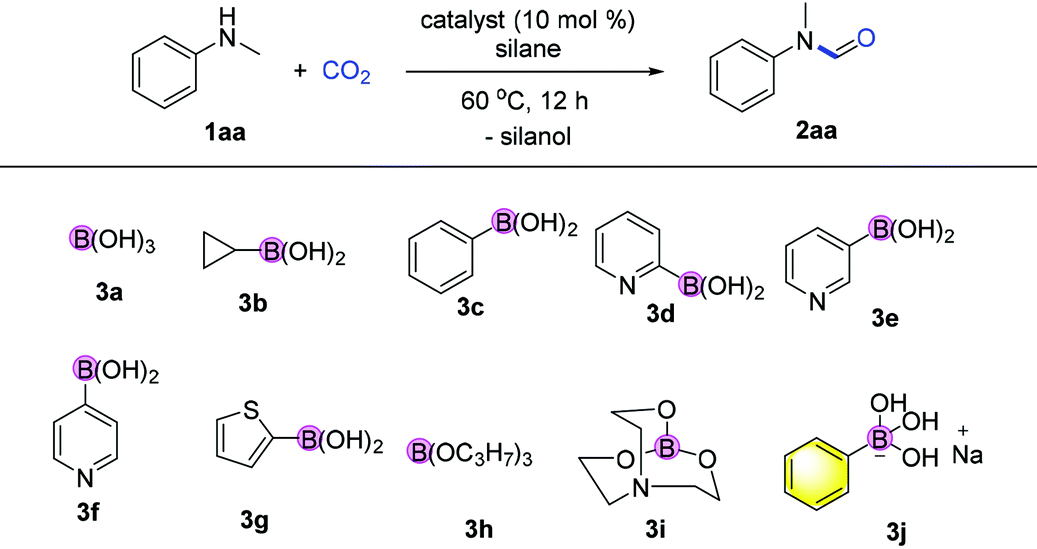
Formates and formamides are ever present functionalities in many drug intermediates, fine chemicals, polymer materials, etc.23 From the multitude of methods for the preparation of formamides, direct N-H formylation using CO2 in the presence of reductants represents the most viable approach.24–27 Generally, selective formylation of primary amines is always challenging because of possible formation of di-formylated or methylated by-products, and moreover occurrence of serious over-reduction or side-reactions of functional groups in complex amine substrates. In our continuing interest on CO2 utilization and of using hydrosilane as the reductant28–32 and boronic acid as the catalyst,33 we considered the use of boron-containing compounds as the catalyst for the N-formylation of amines.Initial investigation of N-formylation of aniline with CO2 in the presence of hydrosilanes was examined (Table 1). Surprisingly, we found that boronic acids 3a–c were ineffective for N-formylation even though aromatic boronic acids were used for the hydrosilylation reduction of amides.34 Boronic acids bearing pyridyl groups were beneficial, although only low yields were obtained (Table 1, entries 5–7). The exploration of other aromatic heterocyclic boronic acids and borate esters was unsuccessful (Table 1, entries 8–10). To our delight, when ionic sodium trihydroxyphenyl borate 3j was used, moderate reactivity could be achieved (Table 1, entry 11). The increase in temperature to 100 °C led to a higher yield of 88%, while lower yields were obtained when other types of silanes or solvents were used (Table 1, entries 11–16). We believe that the aggregation and shielding of hydridic silanes around the catalyst induce hydride transfer to CO2. Furthermore, the choice of hydrophobic diglyme as the solvent is effective for the adsorption of CO2 in the presence of amines.35 Notably, such a kind of catalyst working under heating conditions in polar solvents implies special activation mode different from that of the known σ-hole catalysis.36 Notably, when 1.0 gram of N-methylaniline was used as the substrate, the desired product 2aa was obtained in 86% yield (1.1 gram) under the optimized conditions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | Entry | Catalyst | Yieldb (%) |
| a Reaction conditions: 1aa (0.2 mmol, 1 equiv.), catalyst (0.02 mmol, 10 mol%), phenylsilane (0.2 mmol, 1 equiv.), CO2 (0.25 MPa) in diglyme (diethylene glycol dimethyl ether, 1 mL) at 60 °C for 12 h. b Yields determined based on GC analysis using n-hexadecane as an internal standard. c Reaction carried out at 100 °C. d Reaction carried out using 2 equiv. of phenylsilane at 100 °C. e Using 5 equiv. of PMHS (polymethylhydrosiloxane) at 100 °C for 12 h. f Reaction carried out at 100 °C with THF (tetrahydrofuran) as the solvent. g Toluene as the solvent. | |||||
| 1 | — | 0 | 9 | 3h | <3 |
| 2 | 3a | 0 | 10 | 3i | <3 |
| 3 | 3b | 0 | 11 | 3j | 48 |
| 4 | 3c | 0 | 12c | 3j | 68 |
| 5 | 3d | 9 | 13d | 3j | 88 |
| 6 | 3e | 5 | 14e | 3j | 25 |
| 7 | 3f | 0 | 15f | 3j | 20 |
| 8 | 3g | <3 | 16g | 3j | Trace |
As shown in the work of Hall et al., aromatic borates (such as sodium trihydroxyphenyl borates and phenoxy-dialkoxy borates) were elegantly applied into base-free Pd-catalysed Suzuki coupling reactions.37,38 Here, we highlight that the use of such compounds as catalysts for E–H bond activation has never been reported nor discussed before this work.39 Metal borates are known as core structures of natural products (e.g. boromycin),40 Lewis acid catalysts,41,42 and boron “ate” complex related reaction intermediates.43,44 However, borates are unstable/reactive intermediates in these reactions.
Hence, we embarked on a systematic study probing the catalytic activity of various borates to confirm the reliability of catalysis using borates, and ultimately further discover more efficient catalysts. As shown in Fig. 2, a variety of sodium aromatic borates were conveniently prepared by in situ mixing aromatic boronic acids with NaOH or sodium alkoxides.45,46 It is interesting that different reactivities were observed depending on the counterbase used. Specifically, when MeONa, EtONa, tBuONa or PhONa was used as the counter base (catalyst 3k–3n), the yields ranging from 34 to 68% were obtained, with catalyst 3l from EtONa giving better reactivity. Meanwhile, ortho-substitution by alkyl groups on the aryl ring influenced the reactivity significantly with methyl substitution to form 3o giving the highest yield of the desired product of 65%. meta or para-Substitution with the methyl group decreased the reactivity (47–58% yields).
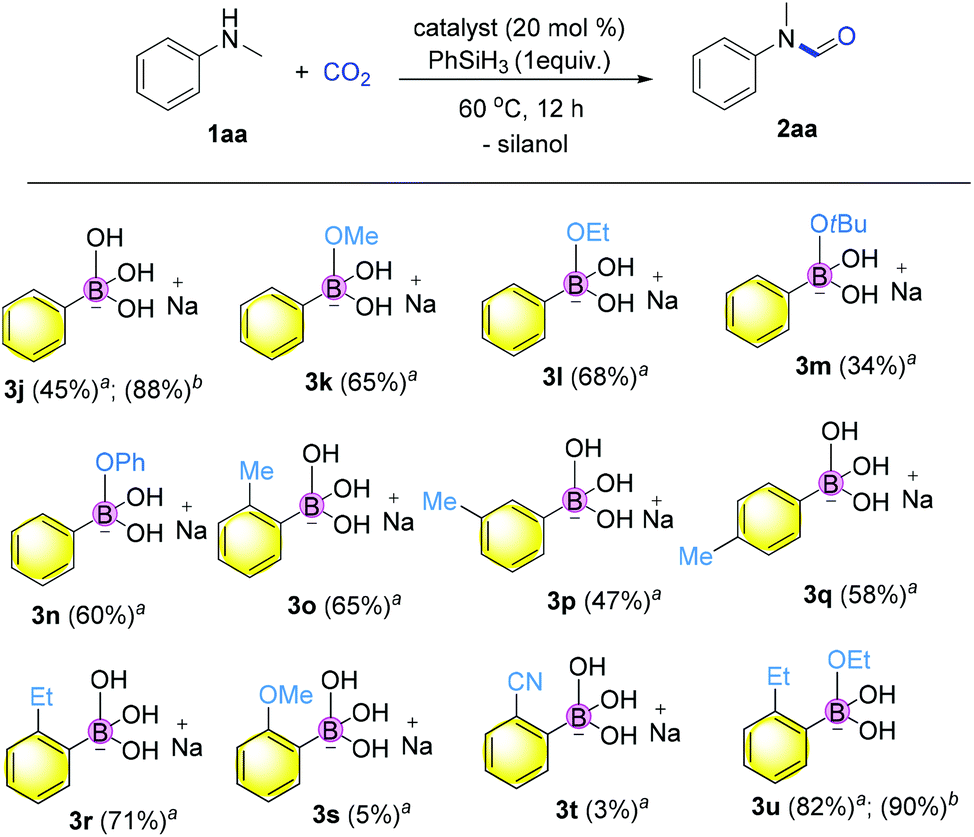 | ||
Fig. 2 Screening of the sodium arylborates. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1aa (0.2 mmol, 1 equiv.), catalyst (0.04 mmol, 20 mol%), phenylsilane (0.2 mmol, 1 equiv.), CO2 (0.25 MPa) in diglyme at 60 °C for 12 h. bPhenylsilane (0.4 mmol, 2 equiv.) at 100 °C for 12 h. Yield of 2aa shown in brackets was determined by GC analysis using n-hexadecane as an internal standard. The catalyst was prepared quantitatively from the in situ reaction of aryl boronic acids with the base. Reaction conditions: 1aa (0.2 mmol, 1 equiv.), catalyst (0.04 mmol, 20 mol%), phenylsilane (0.2 mmol, 1 equiv.), CO2 (0.25 MPa) in diglyme at 60 °C for 12 h. bPhenylsilane (0.4 mmol, 2 equiv.) at 100 °C for 12 h. Yield of 2aa shown in brackets was determined by GC analysis using n-hexadecane as an internal standard. The catalyst was prepared quantitatively from the in situ reaction of aryl boronic acids with the base. | ||
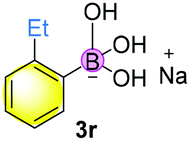
Finally, when catalyst 3u was used, the highest yield of 82% was achieved. When catalyst loading (10 mol%) was lowered at elevated temperature, higher yields were obtained for both 3j and 3u. Notably, in principle, an unlimited number of metal aromatic borate type compounds could be obtained by combining different aromatic boronic acids with metal hydroxides/alkoxides. To the best of our knowledge, metal borates have never been used for catalytic reduction reactions. We also compared the reactivity of other types of boron-containing compounds. As shown in Table 2, only the use of borate 3r led to high reactivity under similar conditions. Even the use of the strong base NaOH or strong Lewis acids such as BF3 or B(C6F5)3 resulted in poor reactivity for the desired reaction, with significant amounts of aniline starting materials being recovered.
|
|
||
|---|---|---|
| Entry | Catalyst | Yield (2aa) |
| Reaction conditions: 1aa (0.2 mmol, 1 equiv.), catalyst (0.04 mmol, 20 mol%), phenylsilane (0.2 mmol, 1 equiv.), CO2 (0.25 MPa) in diglyme (1 mL) at 60 °C for 12 h. Yields determined by GC analysis using n-hexadecane as an internal standard. | ||
| 1 |
|
71% |
| 2 |

|
NR |
| 3 |

|
Trace |
| 4 |
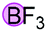
|
NR |
| 5 |
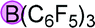
|
NR |
| 6 |
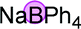
|
Trace |
| 7 |

|
16% |
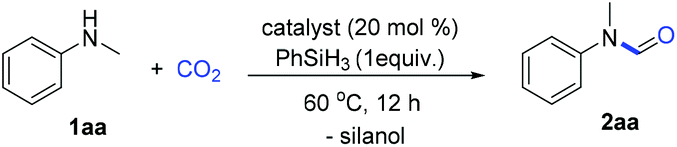
Substrate–catalyst activation via weak interactions could be tested by NMR techniques. Therefore, 1H NMR measurements of mixtures of CO2, PhSiH3 or N-Me aniline, individually with 3j were performed (see the ESI†). Initially, when 3j was added to PhSiH3, chemical shift changes of Si–H of PhSiH3 and C–H on the phenyl ring of 3j were not observed (Si–H at 4.03 ppm for PhSiH3). Control experiments suggested that borates could be stabilized by silane molecules. Specifically, without silane, CO2 reacts with the borate salt 3j to form free phenyl boronic acid and NaHCO3. In contrast, with PhSiH3, no decomposition of 3j occurred in the presence of CO2. This result suggests the occurrence of weak interactions (not able to be detected in the 1H NMR time scale) between silane and borate catalysts offering a protective shield to the borate catalyst from decomposition by CO2. In this respect, silane might be activated via the stabilization of the borate catalyst with the formation of the energized favored intermediate. Regarding the role of alkali metal cation (i.e. Na+), no influence on reactivity was observed in the reaction of N-Me aniline with 3j in the presence of other alkali metals such as CsCl, LiCl or KCl. Hence, we envision that the borate anion plays a key role in the activation of Si–H bonds.
Moreover, in 1H NMR experiments, B–H formation was undetected unlike with the recent development of alkali metal hydridotriphenylborate catalysts for the hydroboration of CO2via B–H and B–OR interconversion.7 Therefore, we advance the involvement of weak interactions seemingly weaker than classical hydrogen bonding. Meanwhile, it is interesting to note that in the Pd-catalysed Suzuki coupling reaction of acyl chlorides or aryl iodides with sodium trihydroxyphenylborate, the presence of PhSiH3 strongly suppressed the C–C coupling reaction. This result implies that the interaction between silanes and borates influences the original coupling reaction (Fig. S1, see the ESI†).
To further understand the mechanism, density functional theory (DFT) calculations on the interaction of silanes with borates were examined. It is suggested that the weak attraction between borate catalysts and hydrosilanes is through Si–H⋯H–O, Si–H⋯H–C and Si⋯O interactions, I and II as illustrated in Fig. 3. The stabilized energy is in the range of 4–5 kcal mol−1 belonging to the energy strength of electrostatic interactions. Besides, the electrostatic potential map illustrated in Fig. 3 also suggests that the negatively charged hydride of phenylsilane interacts with the positively charged hydrogen atoms connected with oxygen atoms and carbon atoms in sodium aryl borates. This is also consistent with the results of NMR experiments that interactions weaker than hydrogen bonding could exist and were undetectable in the 1H NMR time scale. Besides, as mentioned above, the presence of the methyl group at the ortho-position of the boron (catalyst 3o) or the methoxy group on the boron atom (catalyst 3k) increased the reaction efficiency significantly. From the DFT study, it is clear that the CH3 moiety interacts with hydride of silane. The Si–H⋯H–O and Si–H⋯H–C distances are shortened in comparison with those in I and II. Moreover, the binding energies are −7.6, −5.7, and −6.2 kcal mol−1 for I-MeO, II-MeO, and I-Me, while they are −5.6 and −4.0 kcal mol−1 for I and II, respectively (Table S5†). These results demonstrate that the interactions between PhSiH3 and sodium aromatic borates are effectively strengthened by the introduction of the methoxy group on the boron atom or ortho-methyl substitution to boron, being beneficial for the activation of Si–H bonds for the key CO2 reduction step. The present results rationalize the experimental findings.
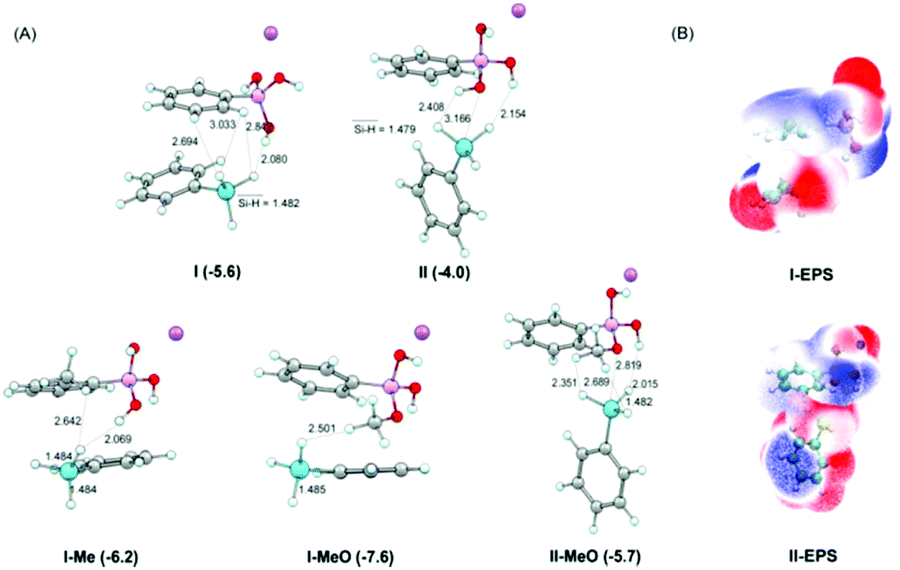 | ||
| Fig. 3 DFT-calculated interaction modes. (A) Optimized complex structures of sodium aryl borates (3j, 3k, and 3o) with phenylsilane at the M06-2X-D3/6-311G(d, p) level of theory in solvent. Values in parenthesis denote the binding energies (in kcal mol−1) calculated at the DSD-PBEP86/def2-TZVPP level in solvent. Bond lengths are in Å. (B) Electrostatic potential map for the catalyst–silane interaction. | ||
From the experimental results and theoretical studies, we propose that the ionic borate moiety of sodium phenyl trihydroxy borate could interact with hydrosilane selectively via electrostatic interactions (Fig. 4). This is a key step for CO2 reduction to give silyl formate intermediates, which further undergo an additive substitution reaction with amine substrates to give the desired formamide product (pathway A). Besides, the formation of carbamate intermediates followed by reduction to give the formamide product is also possible (pathway B). The use of borates to promote the activation of the Si–H or B–H bond is unique. In addition, it is suggested that the non-coordinating nature of tetrahedral borate makes them suitable for tasks that involve ion-pairing with sensitive organometallic complexes or reactive cationic intermediates.
 | ||
| Fig. 4 Proposed reaction mechanism. | ||
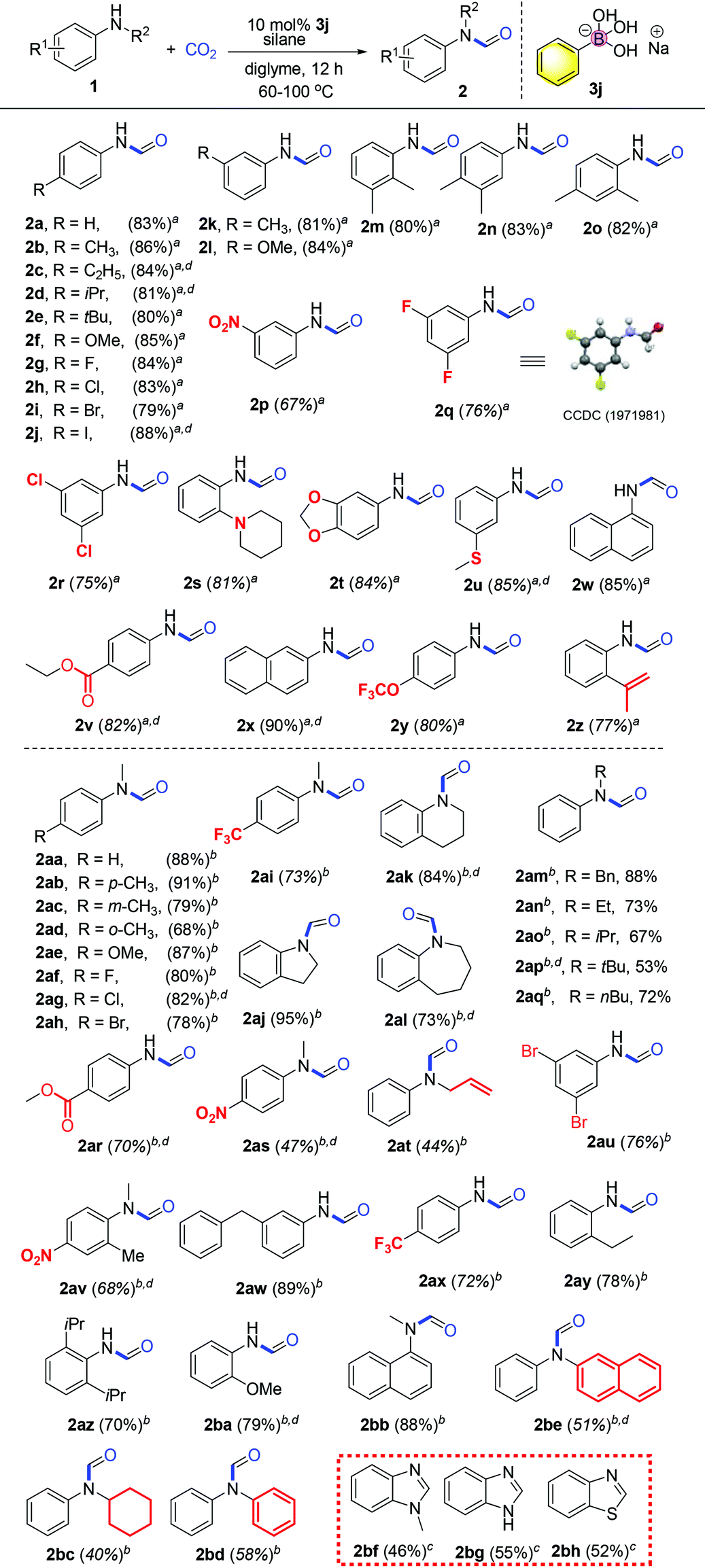
With the optimized reaction conditions in hand, investigation of the substrate scope was performed. Firstly, different aromatic aniline substrates were tested (Table 3). It was found that good to excellent reactivity and selectivity for the desired N-formylation reaction was achieved for most aromatic primary amines at 60 °C, with occasionally aromatic primary amines requiring 100 °C. Electron donating aromatic amines such as Me, Et, iPr, tBu, di-Me, MeS and OMe derivatives afforded the desired compounds (2b–f, 2k–o, and 2u) in 80–86% yields respectively. The reaction also underwent smooth conversion with electron-withdrawing aromatic amine substituents such as F, Cl, Br, I, CF3, 3,5-di-F, 3,5-di-Cl and NO2 to afford the desired compounds (2g–j and 2p–r) in 70–88% yields. To our surprise, the reaction gave the desired compounds 2z in 77% yields using allylamine without affecting the double bond which is often sensitive to reductants. Fortunately, this method was also successful in the N-formylation of amines which contain piperidyl (2s, 81% yield), oxygen heterocyclic (2t, 84%), ester (2v, 82%), trifluoromethoxy (2y, 80%) and naphthalene derivatives showing good to excellent reactivity (2w–x, 85–90% yields).
| a Reaction conditions: 1 (0.2 mmol, 1 equiv.), catalyst (0.02 mmol, 10 mol%), phenylsilane (0.4 mmol, 2 equiv.), CO2 (0.25 MPa) in diglyme (1 mL) at 60 °C for 12 h. b Phenylsilane (0.4 mmol, 2 equiv.) at 100 °C for 12 h. c Phenylsilane (0.4 mmol, 2 equiv.) at 100 °C for 24 h. d Yields determined based on GC analysis using n-hexadecane as an internal standard. Other yields refer to isolated yields. |
|---|
|
In light of the success with aromatic primary amine derivatives, we extended the scope to aromatic secondary amine substrates, as also presented in Table 3. To our delight, the reactions also underwent smooth conversion to afford the expected N-formylated product. Similar to aromatic primary amines, aromatic secondary amines also exhibit functional group tolerance and exhibit good to excellent reactivity. The reaction underwent smooth conversion regardless of electron-withdrawing and electron-donating substituents affording the desired compounds in good to excellent yields (2ab–ac, 2ae–ai, 73%–91%). Conversely, we find that the product conversion is slightly lowered with the ortho position substituted substrate, presumably due to steric hindrance 2ad (68% yield).
Heterocyclic secondary amines were also amenable for this transformation and converted to the corresponding carboxamide products in good to excellent yields (2aj–al, 73–95%). For alkylated secondary amines, we found that the length and branching of the carbon chain of the alkyl substituent on the N atom has influence on the yield. For instance, amine substrates with sterically hindered isopropyl or t-butyl substituents afford desired products with lower yields, while side reaction methylation products increase (2an–aq, 53–73%). Moderate yields were obtained when secondary amine substrates (substituted N-methylanilines) bearing electron withdrawing substituents (2ar–as, 47–70%). Notably, the presence of the nitro group often inhibits N-formylation using CO2 in previous reports. Substituted amines (anilines) with either electron donating or electron withdrawing groups afforded the desired products in moderate to good yields (2ba, 79%; 2au–2az, 68–89%). Secondary alkenyl amine allylphenylamine was also transformed into the corresponding formylated products in lower moderate yield (2at, 44%). Secondary amines bearing bulky substituents regarded incompatible for such transformations also afforded the desired products in moderate yields (2bc–be, 40–58%). Lastly, when the substrate is an ortho-diamine, the corresponding benzimidazole or benzothiazole compounds were formed in moderate yields (2bf, 46%; 2bg, 55%; 2bh, 52%). Here, it is important to note that at elevated temperature, the catalyst remained stable after the reaction showing the robustness of the borate catalyst in this catalytic system. With respect to the concept of green chemistry, the development of reusable heterogeneous catalysts is highly attractive.47 In our system, the catalyst recycling test shows that the catalytic activity remains high after reusing twice with the yields of the desired product obtained as 88%, 75%, and 70% respectively.
We further investigated a wide range of aliphatic amines (Table 4). Reactions with both aliphatic and benzylic primary and secondary amines afforded the corresponding formylated products in good to excellent yields, including substrates with bulky tBu group substitution. Notably, functional groups such as ether, nitrile, cyclopropyl, halogen, phenol and trifluoromethyl groups were well tolerated. Cyclohexamine, benzylamine, aminopropylbenzene and aminobutylbenzene were converted successfully to N-formylated derivatives in good to excellent yields (2bi, 81%; 2cj–cl, 61–88%). Cyclic heterocyclic amines aza-cyclohexane, oxazine and piperazines afforded the desired products in moderate to good yields (2bj–bo, 56–80%). Di-alkylated amine substrates, N-butyl amines, di-isopropylamines, N,N,N-trimethyl diaminoethanes and bis(2-cyanoethyl)amines were transformed in excellent yields (2bp–bs, 75–93%). N-Methyl benzylamine and derivatives bearing electron-withdrawing or electron-donating groups were also amenable for this transformation in good yields (2bt–cd, 72–92%). It is interesting to note that aminomethyl phenol 2bx undergoes chemoselective N-formylation smoothly. N-Substituted benzylamines with various alkyl groups (ethyl, isopropyl, butyl and benzyl) gave the corresponding N-formylated products in moderate yields with bulky t-butyl one at the lower end of the range (2cc, 2ce–cg, 41–87%). Reactions of chiral substrates (S)- and (R)-2-pyrrolidinemethyl-methyl ether (2cn, 2cm) proceeded smoothly with the chiral properties maintained in the product.
| a Reaction conditions: 1 (0.2 mmol, 1 equiv.), catalyst (0.02 mmol, 10 mol%), phenylsilane (0.3 mmol, 1.5 equiv.), CO2 (0.25 MPa) in diglyme (1 mL) at 60 °C for 12 h. b Phenylsilane (0.2 mmol, 1 equiv.). c Yields refer to isolated yield. Other yields determined based on GC analysis using n-hexadecane as an internal standard. |
|---|

|
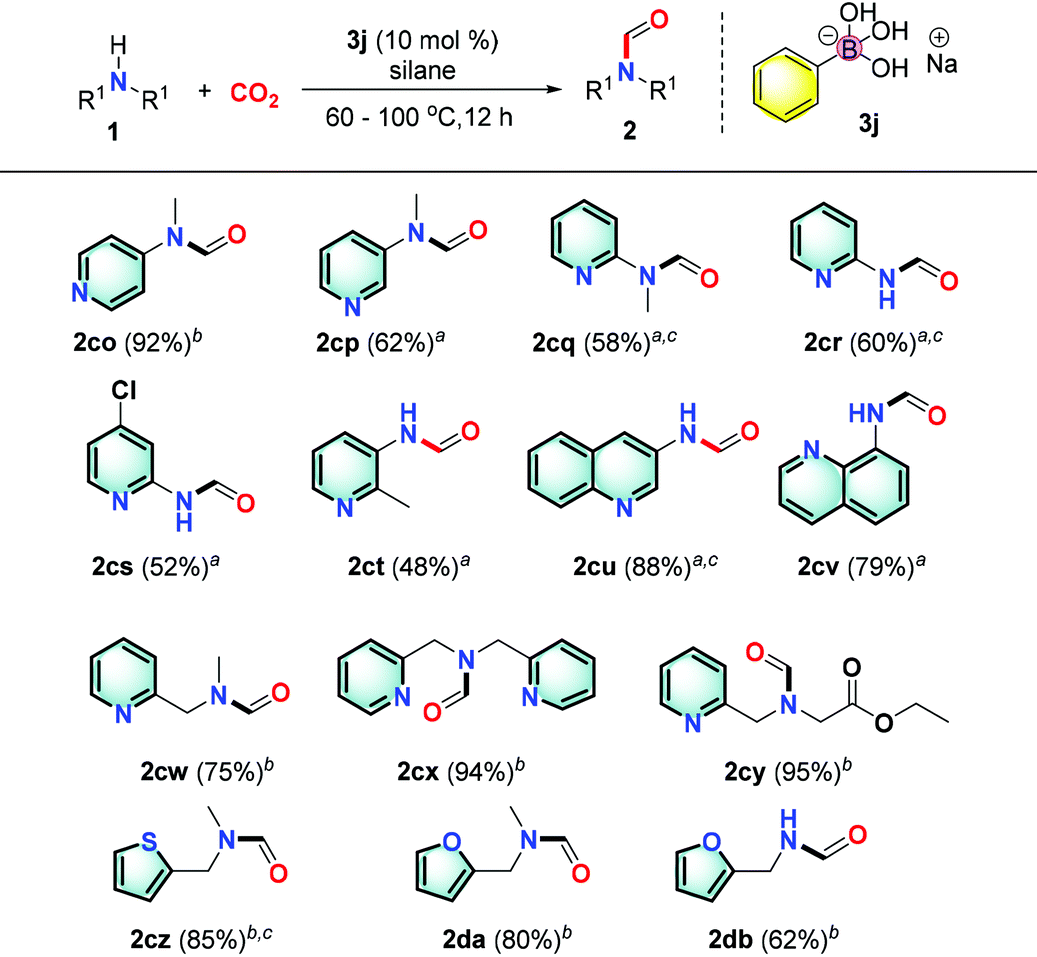
Pyridine based derivatives represent key motifs in drugs and fine chemicals including ligands for catalysis. It is noteworthy to mention that the presence of the pyridyl group is always problematic in catalytic transformations. Satisfactorily, we found that heterocyclic substrates such as substituted aminopyridine and derivatives could be transformed into the desired product in good to excellent yields (2co–ct, 48%–92%; Table 5). Notably, the Meyers formylating agent 2cq was produced in 58% yield.48a These results implied that a new mechanism pathway is involved to have the basic pyridine group well tolerated.
| a Reaction conditions: 1 (0.2 mmol, 1 equiv.), catalyst (0.02 mmol, 10 mol%), phenylsilane (0.3 mmol, 1.5 equiv.), CO2 (0.25 MPa) in diglyme (1 mL) at 60 °C for 12 h. b Phenylsilane (0.2 mmol, 1 equiv.). c Yields refer to isolated yield. Other yields determined based on GC analysis using n-hexadecane as an internal standard. |
|---|
|
Moreover, we successfully transformed more complex structures such as quinolinamine and pyridinemethanamine derivatives in excellent yields (2cu–cy, 75–95%). Also, the reaction of thiophenemethylamines or furarylmethylamines afforded the desired products in good yields (2cz–db, 62–85%).
To further demonstrate the usefulness of this new method, the N-formylation of valuable pyridyl amines48b3co and primary amine 2t was carried out using the known non-catalytic methods49 to demonstrate the applicability of this work (Fig. 5). Notably, the presence of the pyridyl group is often problematic to the transition metal catalysed reactions that proceed via the formation of metal-H species. Most of the tested non-catalytic methods were unsuccessful and only trace amounts of the product were observed using ethyl formate for the formylation of 3co (Fig. 5). Also for the reaction of 2t, poorer yields were obtained compared to the approach of this work based on borate 3j and PhSiH3. The generality of this approach is unprecedented as shown above for substrate scope tolerance. Interestingly, in spite of the presence of the basic pyridyl group or the acidic phenolic group, good reactivity was maintained. This is in striking contrast to the known base–acid interaction strategy.
 | ||
| Fig. 5 Comparison results of borate-catalysis approach with non-catalytic methods. | ||
The control experiment for the reduction of CO2 in the absence of amines gave the silyl formate product in high yield (99%; Fig. 6A). At slightly higher temperatures, the reductive N-methylation using CO2 was also achieved with good reactivity (76–87 yields; Fig. 6B). To further prove the activation of main group element–hydrogen bonds using ionic borates, the catalytic formylation of amines using hydroborane was performed. Using HBpin as the reductant, the formylation of N-Me aniline proceeded smoothly giving the desired product in 89% yield. Similarly, when phenyl boronic acid or NaOH was used as the catalyst, much lower reactivity was observed (Fig. 6C).
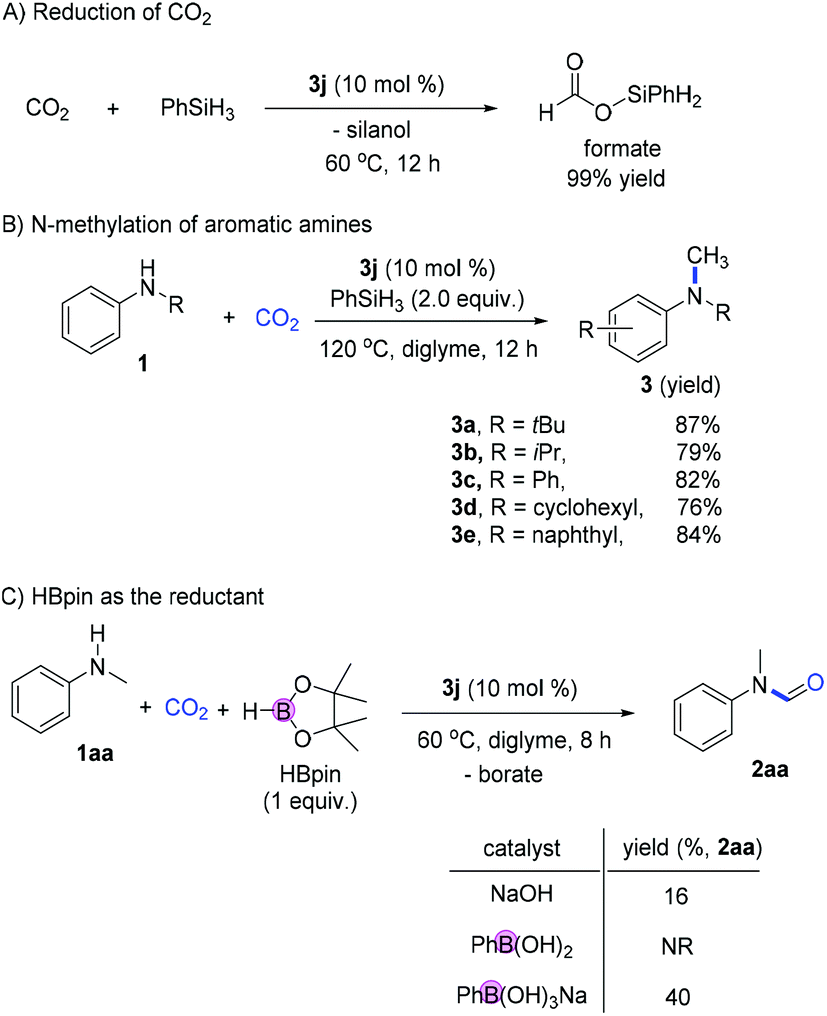 | ||
| Fig. 6 Further applications of reduction, N-methylation and activation of hydroborane. | ||
Conclusions
This work developed a general methodology of transition metal-free pathway based on alkali metal arylborate electrostatic catalysts for hydroborylation, hydrosilylation, and reductive formylation/methylation of CO2. Sodium trihydroxyphenylborate was found to be a superior catalyst in the activation of main group element–hydrogen bonds. This method of N-formylation is broad in scope applicable to both aromatic and aliphatic amine substrates (total 106 examples), especially the unexplored pyridyl amine substrates. Notably, this is the first example of using hydroxyborate for catalytic reductions of carbon dioxide for N-formylation/methylation reactions. Experimental data and DFT calculations show that the action of catalysts is presumably through the dual activation of Si and H atoms in hydrosilane as proposed in the mechanism.Experimental
General procedure for reductive formylation of amines with CO2
To a 4 mL sealing tube in a nitrogen-filled glovebox, the substrate (0.2 mmol), 3j and phenylsilane were added followed by addition of solvent diglyme (1 mL). Then, the tube was sealed, taken out of the glovebox and placed into the autoclave. The autoclave was sealed and purged three times with CO2 gas, then pressurized to 2.5 atm. Lastly, the autoclave was heated at 60–100 °C for 12 h with stirring. After the reaction finished, the autoclave was cooled to room temperature and the pressure was carefully released. The yield was determined by GC analysis or the product was purified by silica gel giving the isolated yield.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of this work by the NSFC (21633013 and 91745104) and the NSF of the Jiangsu Province (BK20180248).Notes and references
- R. Deloux and M. Srebnik, Chem. Rev., 1993, 93, 763–784 CrossRef.
- P. Eisenberger and C. M. Crudden, Dalton Trans., 2017, 46, 4874–4887 RSC.
- J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CAS.
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC.
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986–2012 CrossRef CAS PubMed.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS PubMed.
- D. Mukherjee, H. Osseili, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2016, 138, 10790–10793 CrossRef CAS PubMed and the literatures cited in.
- C. D. N. Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef PubMed.
- L. Zhang, Z. Han, X. Zhao, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2015, 54, 6186–6189 CrossRef CAS PubMed.
- T. V. Nguyen, W. J. Yoo and S. Kobayashi, Angew. Chem., Int. Ed., 2015, 54, 9209–9212 CrossRef CAS PubMed.
- X. Frogneux, O. Jacquet and T. Cantat, Catal. Sci. Technol., 2014, 4, 1529–1533 RSC.
- H. Liu, Q. Mei, Q. Xu, J. Song, H. Liu and B. Han, Green Chem., 2017, 19, 196–201 RSC.
- (a) J. P. Guthrie, Chem. Biol., 1996, 3, 163–170 CrossRef CAS PubMed; (b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed; (c) H. Konishi, T. Y. Lam, J. P. Malerich and S. P. Nolan, Org. Lett., 2010, 12, 2028–2031 CrossRef CAS PubMed.
- (a) F. Niu, L. Zhang, S.-Z. Luo and W.-G. Song, Chem. Commun., 2010, 46, 1109–1111 RSC; (b) P. Nagorny and Z. Sun, Beilstein J. Org. Chem., 2016, 12, 2834–2848 CrossRef CAS PubMed.
- (a) S. Li, G. Li, W. Meng and H. Du, J. Am. Chem. Soc., 2016, 138, 12956–12962 CrossRef CAS PubMed; (b) W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS PubMed.
- X.-F. Liu, X.-Y. Li, C. Qiao, H.-C. Fu and L.-N. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS PubMed and the literatures cited in.
- X. Ren and H. Du, J. Am. Chem. Soc., 2016, 138, 810–813 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- S. Scheiner, Noncovalent Forces, Springer, 2015 Search PubMed.
- A. Barrozo, F. Duarte, P. Bauer, A. T. P. Carvalho and S. C. L. Kamerlin, J. Am. Chem. Soc., 2015, 137, 9061–9076 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- D. Wu, R. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Chem, 2017, 3, 134–151 CAS.
- T. M. E. Dine, D. Evans, J. Rouden and J. Blanchet, Chem. – Eur. J., 2016, 22, 5894–5898 CrossRef PubMed.
- O. Jacquet, C. D. N. Gomes, M. Ephritikhine and T. Cantat, J. Am. Chem. Soc., 2012, 134, 2934–2937 CrossRef CAS PubMed.
- L. Hao, Y. Zhao, B. Yu, Z. Yang, H. Zhang, B. Han, X. Gao and Z. Liu, ACS Catal., 2015, 5, 4989–4993 CrossRef CAS.
- H. Lv, Q. Xing, C. Yue, Z. Lei and F. Li, Chem. Commun., 2016, 52, 6545–6548 RSC.
- (a) W.-D. Li, D.-Y. Zhu, G. Li, J. Chen and J.-B. Xia, Adv. Synth. Catal., 2019, 361, 5098–5104 CrossRef CAS; (b) B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wang and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- Z. Huang, X. Jiang, S. Zhou, P. Yang, C.-X. Du and Y. Li, ChemSusChem, 2019, 12, 3054–3059 CrossRef CAS PubMed.
- H. Wang, Y. Dong, C. Zheng, C. A. Sandoval, M. Makha and Y. Li, Chem, 2018, 4, 2883–2893 CAS.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 9568–9571 CrossRef CAS PubMed.
- Y. Li, L.-Q. Lu, S. Das, S. Pisiewicz, K. Junge and M. Beller, J. Am. Chem. Soc., 2012, 134, 18325–18329 CrossRef CAS PubMed.
- Y. Li, S. Das, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2012, 134, 9727–9732 CrossRef CAS PubMed.
- Y. Li, J. A. Molina de La Torre, K. Grabow, U. Bentrup, K. Junge, S. Zhou, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 11577–11580 CrossRef CAS PubMed.
- K. Revunova and G. I. Nikonov, Dalton Trans., 2015, 44, 840–866 RSC.
- A. V. Rayer, P. D. Mobley, M. Soukri, T. R. Gohndrone, J. Tanthana, J. Zhou and M. Lail, Chem. Eng. J., 2018, 348, 514–525 CrossRef CAS.
- W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS PubMed.
- M. Paladino, M. Boghi and D. G. Hall, Eur. J. Org. Chem., 2019, 6566–6570 CrossRef CAS.
- (a) A. Modak, J. Mondal, M. Sasidharan and A. Bhaumik, Green Chem., 2011, 13, 1317–1331 RSC; (b) M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521–9532 CrossRef CAS PubMed.
- Sodium arylborates could also be conveniently prepared in situ by the reaction of boroxines with NaOH in aqueous solution, see the related important progress: K. Aelvoet, A. S. Batsanov, A. J. Blatch, C. Grosjean, L. G. F. Patrick, C. A. Smethurst and A. Whiting, Angew. Chem., Int. Ed., 2008, 47, 768–770 CrossRef CAS PubMed.
- J. D. White, M. A. Avery, S. C. Choudhry, O. P. Dhingra, B. D. Gray, M. C. Kang, S. C. Kuo and A. J. Whittle, J. Am. Chem. Soc., 1989, 111, 790–792 CrossRef CAS.
- C.-C. Chang, B.-S. Liao and S.-T. Liu, Synlett, 2007, 283–287 CAS.
- M. Uyanik, D. Nakashima and K. Ishihara, Angew. Chem., Int. Ed., 2012, 51, 9093–9096 CrossRef CAS PubMed.
- K. Yang, F. Zhang, T. Fang, G. Zhang and Q. Song, Angew. Chem., Int. Ed., 2019, 58, 13421–13426 CrossRef CAS PubMed.
- Z. He, Y. Hu, C. Xia and C. Liu, Org. Biomol. Chem., 2019, 17, 6099–6113 RSC.
- C. L. Fields, Thermochim. Acta, 1974, 8, 239–248 CrossRef CAS.
- A. N. Cammidge, V. H. M. Goddard, H. Gopee, N. L. Harrison, D. L. Hughes, C. J. Schubert, B. M. Sutton, G. L. Watts and A. J. Whitehead, Org. Lett., 2006, 8, 4071–4074 CrossRef CAS PubMed.
- H. Lv, W. Wang and F. Li, Chem. – Eur. J., 2018, 24, 16588–16594 CrossRef CAS PubMed.
- (a) D. Comins and A. I. Meyers, Synthesis, 1978, 403–404 CrossRef CAS; (b) C. W. Holyoke Jr., PCT Appl. No2018208595, 2018 Search PubMed.
- C. J. Gerack and L. McElwee-White, Molecules, 2014, 19, 7689–7713 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds and copies of 1H and 13C NMR for the new compounds. CCDC 1971981. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc01741h |
| This journal is © The Royal Society of Chemistry 2020 |