
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chaperone-like chiral cages for catalyzing enantio-selective supramolecular polymerization†‡
Yu
Wang§
 a,
Yibin
Sun§
a,
Peichen
Shi
a,
Matthew M.
Sartin
a,
Xujing
Lin
a,
Pei
Zhang
a,
Hongxun
Fang
a,
Pixian
Peng
a,
Zhongqun
Tian
a and
Xiaoyu
Cao
*ab
a,
Yibin
Sun§
a,
Peichen
Shi
a,
Matthew M.
Sartin
a,
Xujing
Lin
a,
Pei
Zhang
a,
Hongxun
Fang
a,
Pixian
Peng
a,
Zhongqun
Tian
a and
Xiaoyu
Cao
*ab
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
bKey Laboratory of Chemical Biology of Fujian Province, Xiamen University, Xiamen 361005, China. E-mail: xcao@xmu.edu.cn; Fax: +86 592 2186628; Tel: +86 592 2185862
First published on 30th July 2019
Abstract
Cage catalysis has emerged as an important approach for mimicking enzymatic reactions by increasing the reaction rate and/or product selectivity of various types of covalent reactions. Here, we extend the catalytic application of cage compounds to the field of non-covalent molecular assembly. Acid-stable chiral imine cages are found to catalyze the supramolecular polymerization of porphyrins with an accelerated assembling rate and increased product enantioselectivity. Because the imine cages have a stronger interaction with porphyrin monomers and a weaker interaction with porphyrin assemblies, they can fully automatically detach from the assembled products without being consumed during the catalytic process. We reveal the kinetics of the auto-detachment of cages and the chirality growth of the assemblies using spectroscopic characterization studies. We find that the passivation groups attached to the cages are important for maintaining the structural stability of the cages during catalyzed assembly, and that the steric geometries of the cages can profoundly affect the efficiency of chiral regulation. This strategy demonstrates a new type of catalytic application of cage compounds in the field of molecular assembly, and paves the way to controlling supramolecular polymerization through a catalytic pathway.
1 Introduction
Cage compounds are discrete assemblies with sophisticated and well-defined local environments,1–3 making them an exceptional model for studying the fundamentals of molecular recognition4–10 and many related applications.11–16 For instance, cage catalysis has recently emerged as an important approach for mimicking enzymatic reactions by increasing the reaction rate and/or product selectivity of various types of reactions.17–21 As functional porous materials, cage compounds possess unique solution-processability for homogeneous catalysis,1 complementary with heterogeneous supramolecular catalysis through metal–organic frameworks22–24 or covalent organic frameworks.25–27 Extensive studies of cage catalysis have been conducted by using both metal–organic28–31 and pure organic cages.32–35 The catalytic strategies are versatile, and they include encapsulating the reactants or introducing functional groups either inside36–38 or outside39 the cage cavities. All these strategies are designed to realize the essential feature of cage catalysis: cages co-assemble with the reactants, and automatically release the resulting products after facilitating the reactions. To this end, the initial interaction between the cage and reactants needs to be strong enough to control the reaction pathway and hence to increase the reaction rate and/or selectivity. Once the product forms, its interaction with the cage weakens, due to increased steric hindrance, resulting in the automatic separation of the cage and product. Because such an intricate, dynamic series of interactions is required, rationally developing new systems or new models of cage catalysis remains a challenge.40–43Here, we report a catalyzed assembly strategy to increase the assembly rate and enantio-selectivity of the supramolecular polymerization of porphyrins by using acid-stable chiral organic cages. A comparison between this strategy and the reported cage catalysis for covalent reactions is depicted in Fig. 1A and B. Both strategies share two important features, namely the increased efficiency and/or selectivity of a reaction or an assembly process, and the automatic release of products after the reaction. However, in the present study, cages facilitate the formation of non-covalent assemblies instead of covalent molecules, similar to the way chaperones assist the assembly of biomolecules.44,45 In addition to showing the chaperone-like behavior of cage compounds, we reveal the kinetics of the auto-detachment of cages and the chirality growth of the assemblies using spectroscopic characterization studies. Through comparison experiments with several cages and small molecules, we demonstrate that the passivation groups attached to the cages are important for maintaining the structural integrity of the cages during catalyzed assembly, and that their steric geometry can profoundly impact the chiral transfer between cages and assemblies.
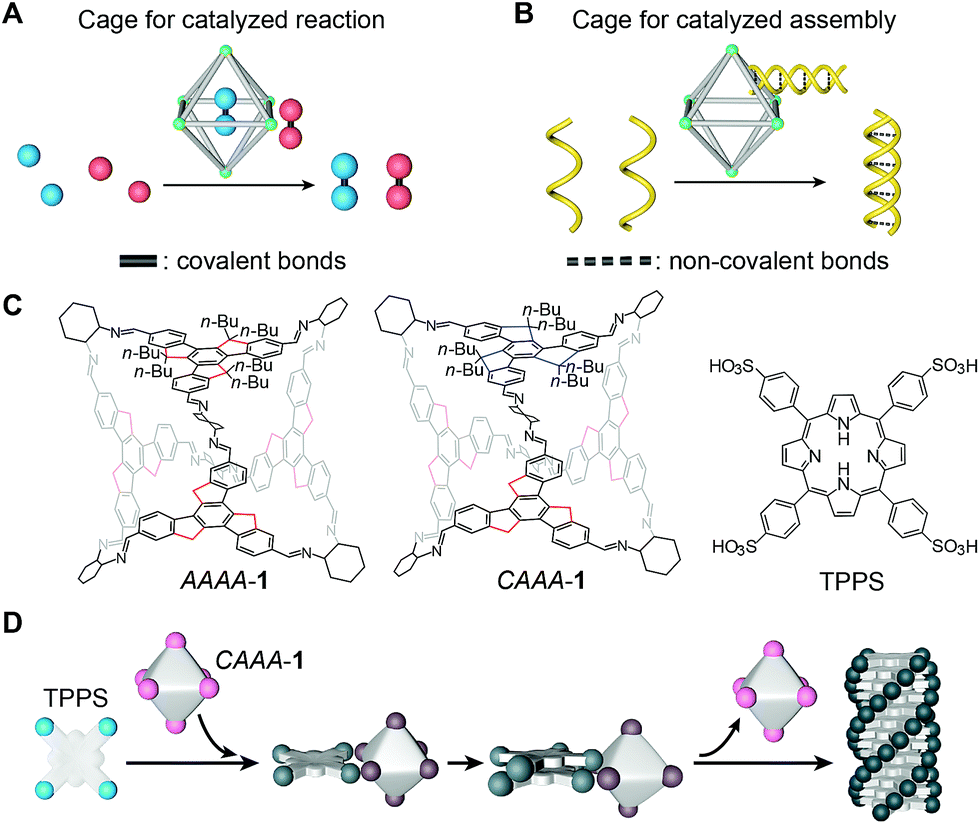 | ||
| Fig. 1 Schematic illustration and molecular structures of the cage-catalyzed molecular assembly. (A) Illustration of the cage-catalyzed strategy for covalent reactions. Coupling reactions can occur either inside or outside the cage cavity (still having interactions with the cage) as indicated by the red and blue balls, respectively. (B) Illustration of the cage-catalyzed strategy for non-covalent molecular assemblies. A cage assists the supramolecular recognition of two helical polymer chains, showing a novel catalytic mode for forming non-covalent bonds, in a way that resembles chaperone-assisted protein assembly. (C) Molecular structures of two isomers of cage 1, i.e., CAAA-1 and AAAA-1, and the porphyrin TPPS. In the cage structures, butyl groups are only shown on one of four faces, for clarity. In complete structures, each sp3 carbon of the truxene backbones is connected to two n-butyl groups, i.e., 24 butyl groups for a cage. Rotation patterns along the three sp3 carbons of the truxene backbones are either clockwise (C) or anticlockwise (A) when viewed from outside the octahedra, as indicated by the blue (C) and red (A) lines. (D) Schematic illustration of enantioselective supramolecular polymerization of TPPS catalyzed by CAAA-1. Note that the models show a reversible process on the cage vertices as indicated by the color change from pink to brown, and then back to pink in the final step. This illustrates how we can monitor the different stages of the catalyzed assembly process, using spectroscopy, and confirm the recovery of the cage after it catalyzes the formation of TPPS assemblies. | ||
2 Results and discussion
2.1 Molecular structures and schematic procedures of catalyzed assembly
Chiral imine cages were synthesized as previously reported,46via the condensation of four butylated truxene faces and six (R,R)-1,2-cyclohexanediamine (CHDA) chiral vertices (structures shown in Fig. 1C and the reaction schematic shown in Fig. S1A‡). Condensation at room temperature generates two isomers of face-rotating octahedra, which are named after the directionalities of the exterior faces as AAAA-1 and CAAA-1. These cages possess excellent stability, allowing us to isolate them by chiral high-performance liquid chromatography (HPLC) and their individual structures have been confirmed by single-crystal X-ray diffraction (CCDC# 1406534 and 1406540).46 The isolated T-symmetric AAAA-1 and C3-symmetric CAAA-1 have very similar structures, except for the small differences in the arrangements of the butyl chains. These similarities enable close comparison between the complexes when they are used as catalysts. The structures of imine cages used for comparison experiments, including isomers of cage 2 with (S,S)-CHDA vertices, cage 3 with phenyl faces, cage 4 with ethylated truxene faces, and cage 5 with ethylenediamine (EDA) vertices, are shown in Fig. S1B–E.‡Traditional supramolecular polymerization of tetrakis(4-sulfonatophenyl)-porphyrin (TPPS, structure shown in Fig. 1C) was selected as a representative non-covalent assembly system47–55 to be catalyzed by chiral cages. Supramolecular polymerization of TPPS has previously been regulated through a co-assembly method by adding oppositely charged chiral auxiliaries, such as amines56 and amino acids.57 Learning from the cage catalysts for covalent reactions, we consider that a stronger interaction with the reactant and weaker interaction with the product are the key to achieving the self-release of cages, and thus allows the recycling of cages after catalysis.
We envisioned that the chiral truxene imine cage 1 might be a potential candidate for catalyzing the enantioselective supramolecular polymerization of TPPS, because the chiral imine groups in cage 1 are less basic than the amine groups in (R,R)-CHDA, and hence they exhibit weaker interactions with the sulfonic groups in TPPS. Therefore, unlike the co-assembly of (R,R)-CHDA and TPPS,56 cage 1 and TPPS might not co-assemble in the final product, which is a necessary condition for achieving catalyzed assembly. In addition, the butyl chains on the outer surfaces of the cages provide stronger steric hindrance to the TPPS assemblies over the monomers,46,58 thus causing relatively stronger interactions with monomers and weaker interactions with assemblies (detailed in Fig. S2‡). This could lead to the automatic release of products from the cages after the enantioselective polymerization of TPPS (Fig. 1D).
2.2 Spectroscopic characterization of chiral supramolecular polymerization
To induce supramolecular polymerization, we add ethyl acetate, with or without imine cages, to the stock solution of TPPS in methanol to attain a solvent ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ethyl acetate:methanol, v/v). The self-assembly of TPPS occurs very slowly when the concentration of TPPS is 10 μM, showing no perceptible aggregation in two weeks. By contrast, when an equivalent amount of CAAA-1 was introduced into this system, obvious precipitation was observed in 48 hours. Circular dichroism (CD) and UV-vis spectroscopy were used to show the catalytic behavior of CAAA-1 for regulating the enantioselective supramolecular polymerization of TPPS.
:1 (ethyl acetate:methanol, v/v). The self-assembly of TPPS occurs very slowly when the concentration of TPPS is 10 μM, showing no perceptible aggregation in two weeks. By contrast, when an equivalent amount of CAAA-1 was introduced into this system, obvious precipitation was observed in 48 hours. Circular dichroism (CD) and UV-vis spectroscopy were used to show the catalytic behavior of CAAA-1 for regulating the enantioselective supramolecular polymerization of TPPS.
A CD spectrum recorded immediately after mixing TPPS and CAAA-1 shows relatively weak and broad peaks at 340 nm and 400 nm (Fig. 2A), whereas the corresponding UV-vis spectrum shows the typical absorption band of the protonated TPPS monomer at 417 nm (Fig. 2B).56,59 The CD and UV-vis spectra of the TPPS and CAAA-1 mixture at 0 hours are dramatically different from the sum of their individual spectra (Fig. S3‡), indicating that the interaction between TPPS and CAAA-1 occurs immediately after mixing. After 48 hours, the formation of chiral TPPS H-assemblies was indicated by the strong exciton-coupled CD peaks at 400 nm and 453 nm and the corresponding UV-vis absorption peak at 419 nm (Fig. 2A and B). The chirality of the TPPS H-assemblies can be empirically assigned as right-handed based on the positive cotton effect in the CD spectrum.47,56,59 Meanwhile, the appearance of the typical peaks of CAAA-1 in the CD spectrum (340 nm) and the absorption spectrum (328 nm) recorded at 48 hours, suggests that CAAA-1 separated from the TPPS assemblies.
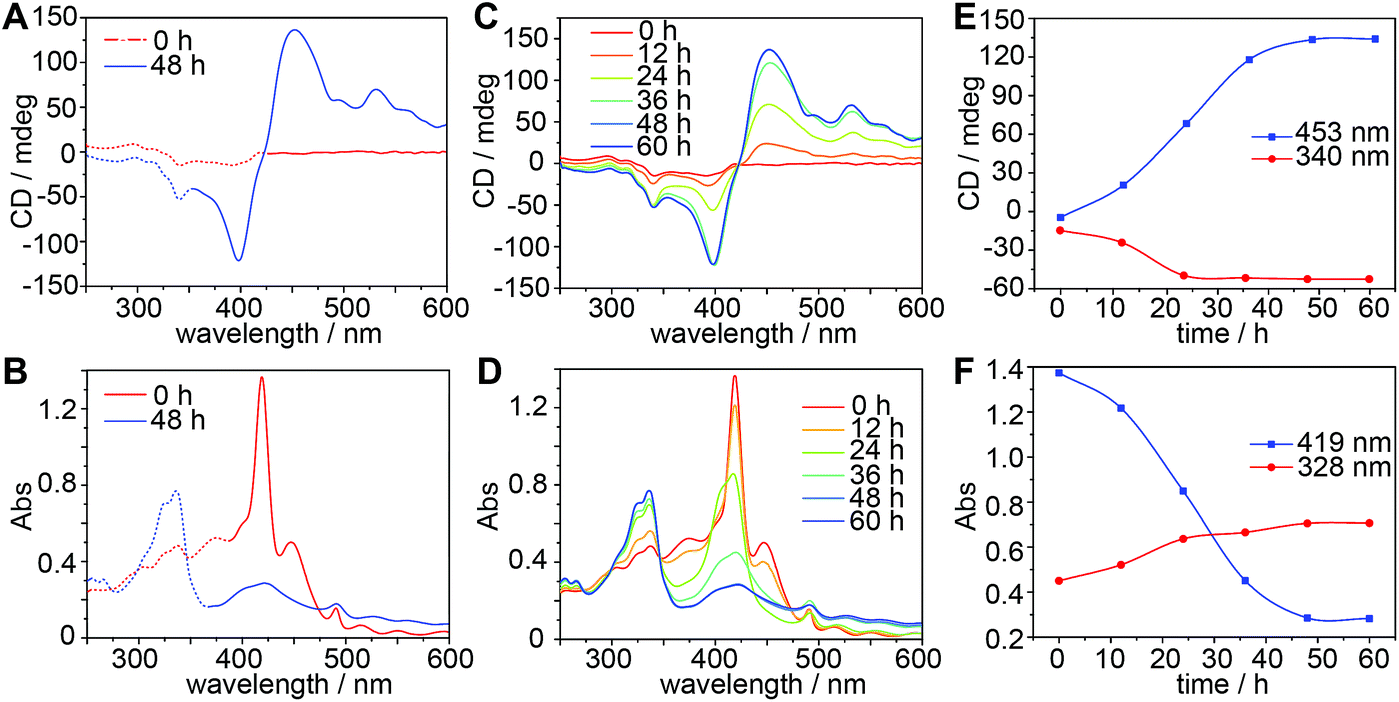 | ||
| Fig. 2 CD and UV-vis spectroscopic characterization of the catalyzed assembly of TPPS. (A and B) CD and UV-vis spectra of the equivalent mixture of TPPS and CAAA-1 (10 μM) at 0 and 48 hours. Solid and dashed lines indicate the regions generated by TPPS assemblies and CAAA-1, respectively. (C and D) Real-time CD and UV-vis monitoring of the kinetics of the catalyzed assembly. (E and F) Time-dependent intensities of the representative peaks of CD and UV-vis spectra extracted from (C) and (D). Intensity increase of the CD peak at 340 nm and the UV-vis peak at 328 nm indicates the growth of free CAAA-1, the intensity increase of the CD peak at 453 nm indicates the growth of chiral TPPS H-assemblies, and the intensity decrease of the UV-vis peak at 419 nm indicates the consumption of TPPS monomers. | ||
We further investigated the kinetics of the growth of TPPS assemblies and the release of the cage (Fig. 2C and D). As analyzed in Fig. 2E and F, the cage was almost completely released from TPPS after 24 hours. However, UV-vis spectra show continuously decreasing absorbance of the TPPS monomer at 417 nm, suggesting the further formation of TPPS assemblies from monomers and/or oligomers. During this process, the CD intensity of TPPS assemblies continues to grow until reaching its maximum at 48 hours. The chirality growth of the TPPS assemblies from 24 h to 48 h could be caused by the cage catalyst, and may also be facilitated by the previously formed chiral TPPS assemblies through self-propagation,60 as shown in Fig. S4.‡
To directly compare the catalyzed assembly with the self-assembly of TPPS, we increased the TPPS concentration from 10 μM to 50 μM. Under this condition, the self-assembly of TPPS took approximately 72 hours to show perceptible aggregation in solution (Fig. S5A‡). In contrast, after introducing 50 μM CAAA-1, obvious precipitation was observed in 30 min. UV-vis spectra show that TPPS molecules form J-assemblies in the self-assembly product and form H-assemblies when CAAA-1 is added (Fig. S5B‡), indicating that CAAA-1 can both accelerate the assembly of TPPS monomers and control the selectively of assembly products.
In addition, we have performed a concentration-dependent study on the cage catalysis as shown in Fig. S6.‡ In the systems with a consistent 10 μM concentration of TPPS, 10 μM CAAA-1 results in the highest CD intensity of the TPPS H-assemblies. Increasing the concentration of CAAA-1 decreases the CD intensity of TPPS assemblies, probably because a high concentration of the cage removes more protons from TPPS and thus causes stronger π–π interaction between TPPS molecules and relatively weakens the interaction between TPPS and CAAA-1, leading to a fast self-assembly process of TPPS with decreased chiral regulation from CAAA-1. For instance, aggregation in the 40 μM CAAA-1 system is obvious after 1 hour, whereas it takes over 12 hours for the 10 μM CAAA-1 system to show obvious aggregation. Decreasing the concentration of CAAA-1 also leads to a decreased CD intensity of TPPS assemblies as less CAAA-1 acts as the catalyst. We found the minimum concentration of the catalyst required to ensure the supramolecular polymerization of TPPS in our current experimental setup to be around 8 μM. When the concentration of CAAA-1 goes lower than this limitation, CAAA-1 starts to decompose, leading to poor reproducibility of the spectroscopic results. Note that this limitation is caused due to the hydrolytic decomposition of the cage, which is strongly related to the experimental conditions, especially the concentration of trace water in the system. We assume that the limitation could be further pushed towards lower concentrations if the trace amount of water can be reduced from the solvents and environments.
2.3 Recovery of the cage catalyst
To further confirm the self-detachment of the cage, we characterized the isolated precipitate and the supernatant of the assembly product (TPPS:CAAA-1 = 1:1, 10 μM) by CD, dynamic light scattering (DLS), and scanning electron microscopy (SEM). As shown in Fig. 3A, the CD spectra of the precipitate and the thoroughly stirred mixture overlap well in the region of TPPS assemblies, whereas the supernatant shows the typical spectrum of pure CAAA-1, without any signal from TPPS monomers or assemblies, suggesting that all TPPS assemblies have been precipitated from the solution. The CD spectrum of pure CAAA-1 at 10 μM is almost identical to that of the supernatant (Fig. 3B), which confirms the full recovery of the cage catalyst after 48 hours. DLS shows that the formed TPPS assemblies are over 100 nm (Fig. S7‡). The SEM image of the precipitate further confirms that TPPS molecules have been assembled into nanorods and nanosheets with a corresponding diameter or thickness, respectively, of about 100 nm (Fig. 3C). The SEM image of the supernatant shows the octahedral shape of the cage single crystals (Fig. 3D), proving the purity of the cage in the supernatant.
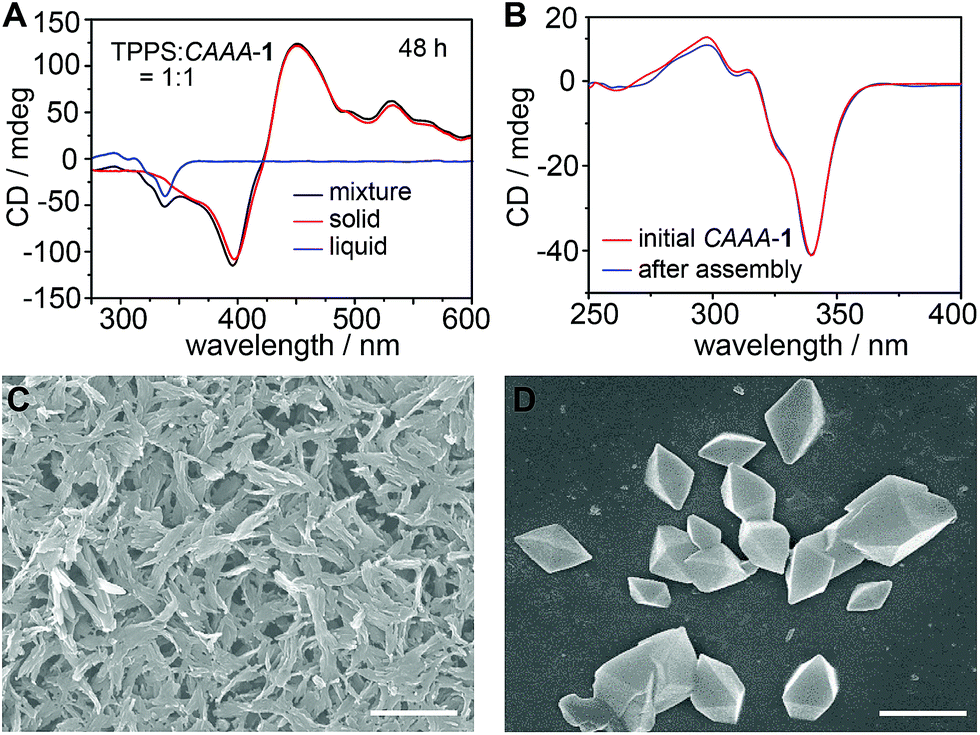 | ||
| Fig. 3 Auto-detachment of the cage catalyst revealed by CD and SEM characterization studies. (A) CD spectra of the precipitate (red), supernatant (blue), and thoroughly stirred mixture (black) of the assembly product of equivalent TPPS and CAAA-1 at 48 hours, showing that TPPS and CAAA-1 are completely separated into the precipitate and supernatant, respectively. (B) CD spectra of the initial CAAA-1 (10 μM) and the supernatant after catalyzing the assembly of TPPS, showing the complete auto-detachment of the cage catalyst. (C and D) SEM images of TPPS assemblies (C) in the precipitate and CAAA-1 crystals (D) obtained from the supernatant, showing completely different morphologies and their complete separation in the assembly product. Scale bars: 2 μm. | ||
Based on the above spectroscopic and SEM studies, we conclude that when the cage CAAA-1 is introduced into the solution of TPPS, it interacts with TPPS immediately and thus increases the efficiency of supramolecular polymerization in terms of the assembly rate and the enantioselectivity of the product. After the formation of chiral TPPS assemblies, all cage molecules are completely restored to their initial states and can catalyze another round of supramolecular polymerization of TPPS. Therefore, we consider CAAA-1 a chaperone-like self-detachable template for the enantioselective supramolecular polymerization of TPPS.
2.4 Acid stability of the truxene–CHDA imine cage
It is interesting that the absorption spectrum of CAAA-1 exhibits weak and broad peaks after being mixed with TPPS and that it reverts to its original, intense peak at 340 nm after the separation from TPPS (Fig. 2A and C). We conjectured that this reversible spectroscopic change is related to the interaction between the acid groups in TPPS and the imine groups in the cage, i.e., the protonation of the imine cage. To verify this conjecture, we studied the protonation of the cage under strong non-chromophore-containing trifluoromethanesulfonic acid (TFSA). As shown in Fig. 4A, upon adding 100 μM TFSA to 10 μM CAAA-1 in ethyl acetate, the CD spectrum red-shifts to 400 nm immediately due to the protonation of the imine groups. Moreover, an additional 100 μM KOH can bring the CD spectrum fully back to its original shape. Similar reversible spectroscopic changes are also found in the fluorescence measurement (Fig. S8‡). Titration of TFSA with a CAAA-1 solution gives a series of CD spectra showing the full range from the unprotonated to fully protonated states (Fig. 4B). The spectrum corresponding to a 1:4 ratio of cage:TFSA has a very similar shape to that of the spectrum of the 1:1 mixture of cage and TPPS (note that each TPPS monomer has four sulfonic groups), suggesting the partially protonated states of the cage during the initial stage of catalyzed assembly.
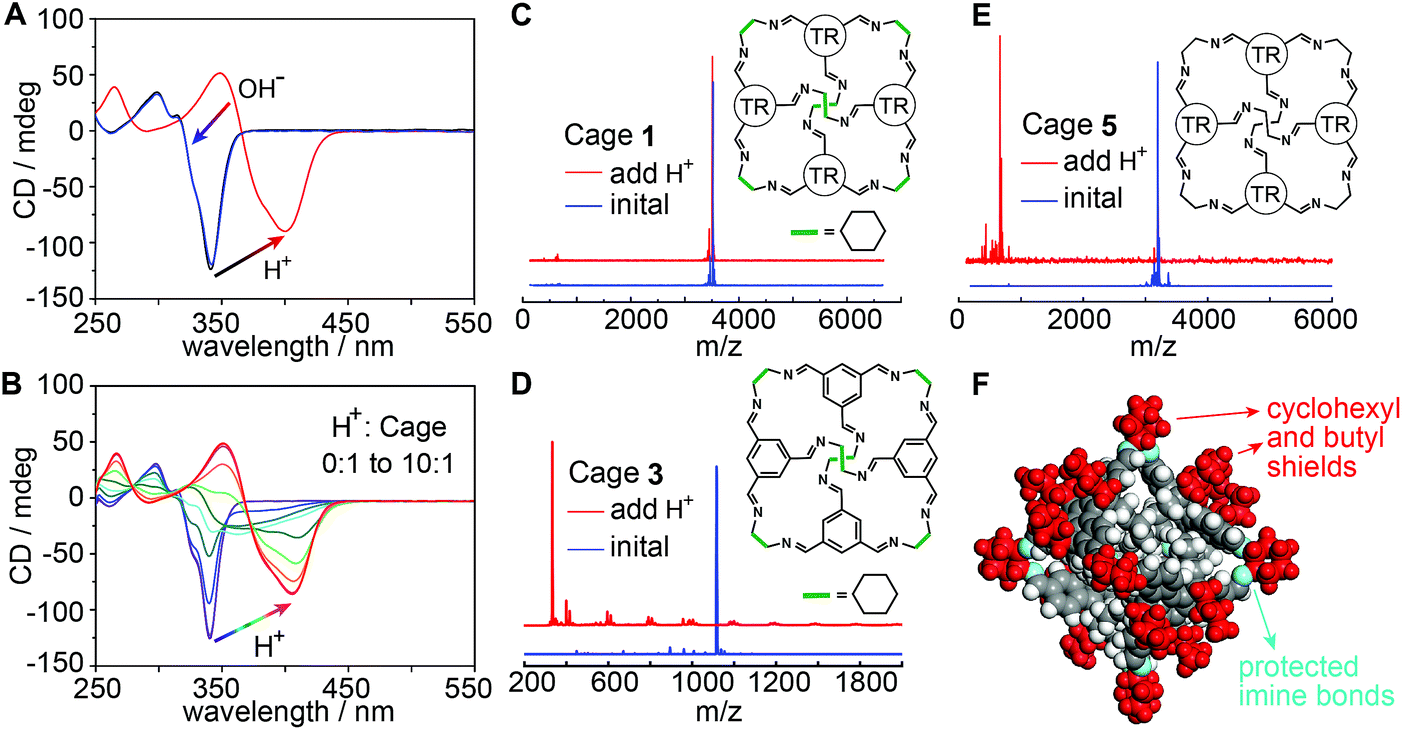 | ||
| Fig. 4 Extraordinary acid stability of the truxene imine cage. (A) CD spectra of CAAA-1 (black) upon the addition of TFSA acid (red) and further addition of KOH base (blue). The amount of added TFSA and KOH is 10 equivalents to the initial CAAA-1 (10 μM). (B) CD spectra of CAAA-1 upon titration with TFSA. Note that the spectrum corresponding to a TFSA:CAAA-1 ratio of 4:1 is very similar to the spectrum of the initial mixture of equivalent TPPS and CAAA-1 (red dashed line in Fig. 2A). (C–E) Mass spectra of CAAA-1 (10 μM) and two comparison imine cages 3 and 5 (10 μM) before (blue) and after (red) the addition of 10 equivalents of TFSA. TR groups in the inserted structures of 1 and 5 indicate the truxene faces with perpendicular butyl chains (detailed in Fig. 1C and S1‡). (F) Single-crystal structure of CAAA-1 showing that the imine bonds are shielded by the outer cyclohexyl and butyl groups. The comparison of more cages and imine compounds in Fig. S9 and S10‡ shows that both the cyclohexyl and butyl shields and the cage shape are necessary for the extraordinary acid stability of cage 1. | ||
Mass spectroscopy further confirms that CAAA-1 maintains the cage structure when the concentration ratio of TFSA to CAAA-1 is 10:1 in ethyl acetate (Fig. 4C), showing extraordinary acid stability over other imine cages and imine-containing molecules under the same conditions. In controlled experiments, we have synthesized imine cages with similar [4 + 6] structures by changing the butyl-substituted truxene faces in cage 1 into unsubstituted phenyl faces (cage 3 in Fig. 4D and S1C‡) or ethyl-substituted truxene faces (cage 4 in Fig. S1D‡). Both cages 3 and 4 are decomposed into fragments under the same TFSA treatment (Fig. 4D, S9A and B‡). A similar decomposition occurs for cage 5 (Fig. 4E), which has ethyl vertices instead of the cyclohexyl vertices in cage 1. Moreover, even though they consist of the same truxene and cyclohexyl components, the [1 + 1] condensation compound of butyl-substituted truxene and CHDA (structure shown in Fig. S10‡) still decomposes after the addition of TFSA, showing lower stability than the [4 + 6] cage counterpart. Imine bonds in all these structures have similar nucleophilicity, yet cage 1 exhibits the highest acid stability among them. This suggests that the cage shape and the passivating effects of cyclohexyl and butyl groups are both critical for protecting the imine bonds from decomposition (Fig. 4F), resembling the strategy of protecting soldiers with shields and a “testudo formation”.
2.5 Structural sensitivity to chiral transfer
We also perform the catalyzed assembly of TPPS by using the isomer AAAA-1, which has a very similar structure to CAAA-1, except for different rotational directions of one of the truxene faces and one of the related butyl groups. Therefore, we examine whether the steric geometry of truxene faces can influence the chiral transfer from the cage to TPPS assemblies. As shown in Fig. 5A and S11,‡ both AAAA-1 and CAAA-1 experience a similar protonated to deprotonated pathway during catalysis. However, AAAA-1 exhibits a much lower efficiency for controlling the chirality of TPPS assemblies than does CAAA-1. After 48 hours, AAAA-1 leads to chiral TPPS assemblies with a 10 mdeg peak in the CD spectrum, which is approximately 15 times lower than that generated by CAAA-1. In addition, we also synthesized imine cages with (S,S)-CHDA vertices, and isolated two cage products CCCC-2 and CCCA-2, which are enantiomers of AAAA-1 and CAAA-1, respectively. As expected, CCCC-2 and CCCA-2 generate mirror-like CD spectra compared with their enantiomers respectively (Fig. 5A).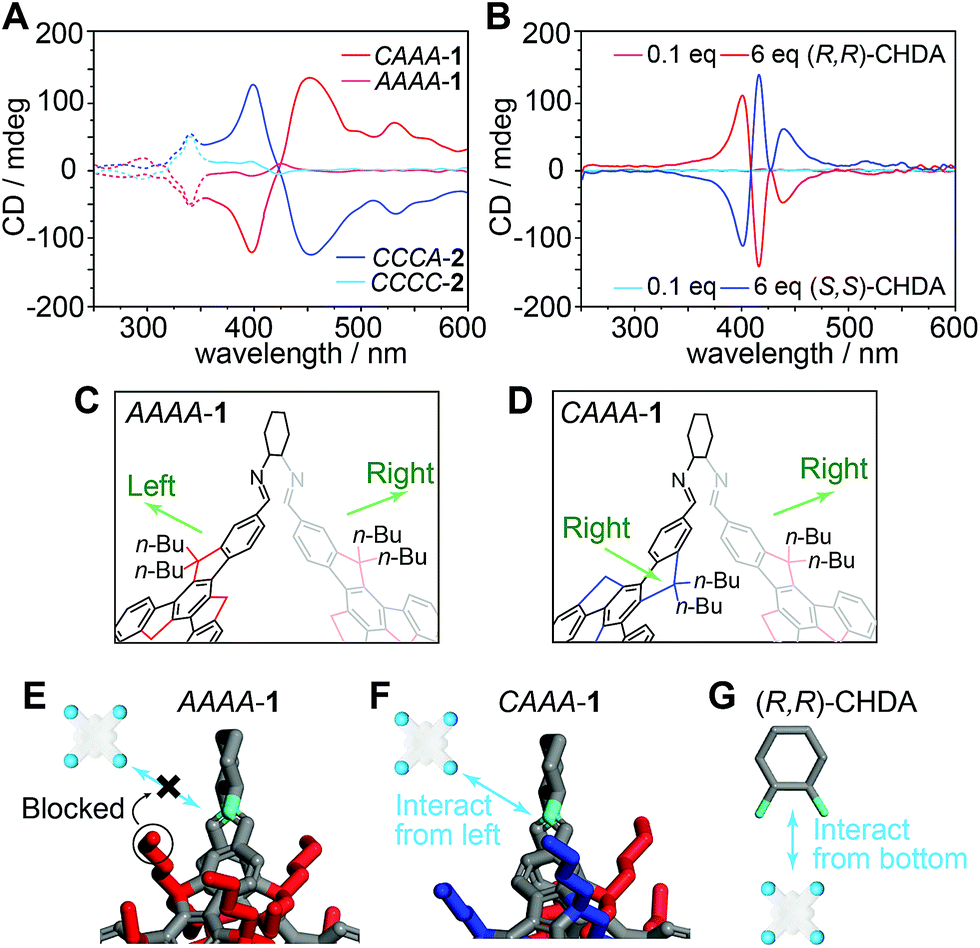 | ||
| Fig. 5 Structural sensitivity of chirality transfer from cages to TPPS assemblies. (A) CD spectra of TPPS assemblies catalyzed by truxene imine cages. Solid and dashed lines indicate the regions generated by TPPS assemblies and cages, respectively. CAAA-1 and AAAA-1 are formed from (R,R)-CHDA, whereas their mirror-image structures CCCA-2 and CCCC-2 are formed from (S,S)-CHDA. (B) CD spectra of TPPS co-assembled with (R,R)-CHDA and (S,S)-CHDA in different ratios. Note that the TPPS assemblies catalyzed by cage 1, which has (R,R)-CHDA on the cage vertices, exhibit opposite chiroptical activity to the co-assemblies of TPPS and (R,R)-CHDA. The same chiral inversion is found between cage 2 and (S,S)-CHDA. (C and D) Structural details of AAAA-1 and CAAA-1 obtained by single-crystal X-ray analysis, emphasizing different arrangements of butyl chains on the vertices. For a vertex connecting two anticlockwise truxene faces (C), the two closest butyl groups point in opposite directions; for a vertex connecting a clockwise truxene face and an anticlockwise one (D), the two closest butyl groups point in the same direction. (E–G) Diagrams showing the different models of the interaction between TPPS and the imine in cage 1 or the amine groups in (R,R)-CHDA. It is difficult for AAAA-1 to transfer its chirality to TPPS assemblies, due to strong steric hindrance in all directions. CAAA-1 transfers its chirality to TPPS assemblies through the interaction in the least hindered direction, i.e., the left. (R,R)-CHDA transfers its chirality to TPPS assemblies through the interaction in the least hindered direction, i.e., the bottom. | ||
Due to the dynamic nature of imine chemistry, it is possible that a tiny amount of cage 1 could hydrolyze and generate (R,R)-CHDA, and even we have not measured it through spectroscopic comparison (Fig. 3B). To make sure that the observed chiral catalysis is not an artifact phenomenon caused by partially hydrolyzed (R,R)-CHDA, we performed comparison experiments of the co-assembly of TPPS with (R,R)-CHDA in different concentrations. Since each CAAA-1 contains six (R,R)-CHDA vertices, 6, 1, and 0.1 equivalents of (R,R)-CHDA correspond to the fully hydrolyzed, partially hydrolyzed, and barely hydrolyzed conditions of CAAA-1. The results show that both six equivalents or one equivalent of (R,R)-CHDA leads to the opposite chirality compared to one equivalent of CAAA-1 (Fig. 5B and S12‡), whereas 0.1 equivalent of (R,R)-CHDA is insufficient to control the chirality of the TPPS assemblies. The chirality inversion between cages and their CHDA components rules out the assumption that the chiral regulation during catalyzed assembly is caused through chiral self-propagation by a trace amount of CHDA decomposed from cage vertices.
We scrutinize the structures of AAAA-1, CAAA-1, and their vertex component (R,R)-CHDA and propose a mechanism for their different catalytic efficiencies (Fig. 5C–F). Having a T symmetry, every CHDA vertex in AAAA-1 connects to two anticlockwise truxene faces with their butyl chains pointing in both left and right directions (Fig. 5C). Therefore, there is a strong steric hindrance in both directions when TPPS or TPPS assemblies try to interact with the chiral vertices (Fig. 5E). By contrast, in CAAA-1, there are three CHDA vertices connected to an anticlockwise and a clockwise truxene face, with their butyl chains pointing in the same direction (Fig. 5D). Thus, CAAA-1 can have a stronger interaction with TPPS and TPPS assemblies from the direction that is not blocked by the butyl chains (Fig. 5F). Compared with imine cage 1, (R,R)-CHDA has more basic amine groups and much less steric hindrance. Therefore, it forms co-assemblies with TPPS instead of showing catalytic behavior.56 In addition, when interacting with TPPS, (R,R)-CHDA has a large steric hindrance in the direction of the cyclohexyl group (Fig. 5G), which is different from that in a cage. This may result in different chiral selectivities between imine cages and their CHDA counterparts.
3 Conclusion
To summarize, we have found acid-stable chiral imine cages for catalyzing the enantioselective supramolecular polymerization of TPPS. During the catalysis, chiral imine cages regulate the chirality of TPPS assemblies and serve as spectroscopic probes to allow monitoring of the assembly and self-detachment kinetics. This study provides a strategy to construct chiral supramolecular polymers from achiral building blocks through cage catalysis, which also entails efforts towards extending the applications of cage catalysts from covalent interactions to non-covalent molecular assembly. The concept to design a stronger interaction with monomers and a weaker interaction with assemblies is general for the development of this new type of catalysis in the field of molecular assembly. In addition, the “testudo formation” strategy in constructing acid-stable imine cages may inspire other designs for applications that require stable cages.61,62Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank E. W. (Bert) Meijer, Li Zhang, Wei Zhang, and Hui Zhang for discussions. We also thank Liang Chen, Zhenghao Zhai, Shiyan Chen, and Xinchang Wang for assistance in experiments. This work is supported by the 973 Program (No. 2015CB856505), the NSFC (No. 91427304, 21573181, 91227111 and 21722304), the the Top-Notch Young Talents Program of China, and the Fundamental Research Funds for the Central Universities of China (No. 20720160050).Notes and references
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS PubMed.
- J. T. A. Jones, T. Hasell, X. Wu, J. Bacsa, K. E. Jelfs, M. Schmidtmann, S. Y. Chong, D. J. Adams, A. Trewin, F. Schiffman, F. Cora, B. Slater, A. Steiner, G. M. Day and A. I. Cooper, Nature, 2011, 474, 367–371 CrossRef CAS PubMed.
- R. E. Morris and X. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef CAS PubMed.
- T. Mitra, K. E. Jelfs, M. Schmidtmann, A. Ahmed, S. Y. Chong, D. J. Adams and A. I. Cooper, Nat. Chem., 2013, 5, 276–281 CrossRef CAS PubMed.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- T. Hasell, M. A. Little, S. Y. Chong, M. Schmidtmann, M. E. Briggs, V. Santolini, K. E. Jelfs and A. I. Cooper, Nanoscale, 2017, 9, 6783–6790 RSC.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2017, 56, 1244–1248 CrossRef CAS PubMed.
- G. Cecot, M. Marmier, S. Geremia, R. De Zorzi, A. V. Vologzhanina, P. Pattison, E. Solari, F. F. Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2017, 139, 8371–8381 CrossRef CAS PubMed.
- L. Chen, P. S. Reiss, S. Y. Chong, D. Holden, K. E. Jelfs, T. Hasell, M. A. Little, A. Kewley, M. E. Briggs, A. Stephenson, K. M. Thomas, J. A. Armstrong, J. Bell, J. Busto, R. Noel, J. Liu, D. M. Strachan, P. K. Thallapally and A. I. Cooper, Nat. Mater., 2014, 13, 954–960 CrossRef CAS PubMed.
- M. Zhang, M. L. Saha, M. Wang, Z. Zhou, B. Song, C. Lu, X. Yan, X. Li, F. Huang, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 5067–5074 CrossRef CAS PubMed.
- L. Zhang, L. Xiang, C. Hang, W. Liu, W. Huang and Y. Pan, Angew. Chem., Int. Ed., 2017, 56, 7787–7791 CrossRef CAS PubMed.
- C. Zhao, Q. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS PubMed.
- T. Hasell, M. Miklitz, A. Stephenson, M. A. Little, S. Y. Chong, R. Clowes, L. Chen, D. Holden, G. A. Tribello, K. E. Jelfs and A. I. Cooper, J. Am. Chem. Soc., 2016, 138, 1653–1659 CrossRef CAS PubMed.
- S. Jiang, L. Chen, M. E. Briggs, T. Hasell and A. I. Cooper, Chem. Commun., 2016, 52, 6895–6898 RSC.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- J. L. Bolliger, A. M. Belenguer and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 7958–7962 CrossRef CAS PubMed.
- M. Otte, ACS Catal., 2016, 6, 6491–6510 CrossRef CAS.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- L. Cai, S. Li, D. Yan, L. Zhou, F. Guo and Q. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS PubMed.
- S. M. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. L. Xamena, V. V. Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- C. Sun, G. Skorupskii, J.-H. Dou, A. M. Wright and M. Dincă, J. Am. Chem. Soc., 2018, 140, 17394–17398 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- X. Wang, X. Han, J. Zhang, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2016, 138, 12332–12335 CrossRef CAS PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- S. Chen, K. Li, F. Zhao, L. Zhang, M. Pan, Y. Fan, J. Guo, J. Shi and C. Su, Nat. Commun., 2016, 7, 13169 CrossRef CAS PubMed.
- W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, J. Am. Chem. Soc., 2018, 140, 2821–2828 CrossRef CAS PubMed.
- J. Jiao, C. Tan, Z. Li, Y. Liu, X. Han and Y. Cui, J. Am. Chem. Soc., 2018, 140, 2251–2259 CrossRef CAS PubMed.
- G. Bianchini, G. La Sorella, N. Canever, A. Scarso and G. Strukul, Chem. Commun., 2013, 49, 5322–5324 RSC.
- Q. Zhang and K. Tiefenbacher, J. Am. Chem. Soc., 2013, 135, 16213–16219 CrossRef CAS PubMed.
- Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197 CrossRef CAS PubMed.
- L. Catti, Q. Zhang and K. Tiefenbacher, Synthesis, 2016, 48, 313–328 CAS.
- S. H. A. M. Leenders, R. Gramage-Doria, B. De Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- B. Mondal, K. Acharyya, P. Howlader and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1709–1716 CrossRef CAS PubMed.
- P. T. Smith, B. P. Benke, Z. Cao, Y. Kim, E. M. Nichols, K. Kim and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 9684–9688 CrossRef CAS PubMed.
- L. R. Holloway, P. M. Bogie, Y. Lyon, C. Ngai, T. F. Miller, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2018, 140, 8078–8081 CrossRef CAS PubMed.
- M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073–2082 CrossRef CAS PubMed.
- G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233–2243 CrossRef CAS PubMed.
- L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed.
- M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS PubMed.
- Y. Wang, H. Lin, L. Chen, S. Ding, Z. Lei, D. Liu, X. Cao, H. Liang, Y. Jiang and Z. Tian, Chem. Soc. Rev., 2014, 43, 399–411 RSC.
- Y. Wang, H. Lin, S. Ding, D. Liu, L. Chen, Z. Lei, F. Fan and Z. Tian, Sci. Sin.: Chim., 2012, 42, 525–547 CAS.
- X. Wang, Y. Wang, H. Yang, H. Fang, R. Chen, Y. Sun, N. Zheng, K. Tan, X. Lu, Z. Tian and X. Cao, Nat. Commun., 2016, 7, 12469–12476 CrossRef PubMed.
- J. M. Ribó, J. Crusats, F. Sagués, J. Claret and R. Rubires, Science, 2001, 292, 2063–2066 CrossRef.
- F. J. M. Hoeben, M. Wolffs, J. Zhang, S. De Feyter, P. Leclère, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 9819–9828 CrossRef CAS PubMed.
- M. Wolffs, F. J. M. Hoeben, E. H. A. Beckers, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 13484–13485 CrossRef CAS PubMed.
- R. Randazzo, A. Mammana, A. D'Urso, R. Lauceri and R. Purrello, Angew. Chem., Int. Ed., 2008, 47, 9879–9882 CrossRef CAS PubMed.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. de Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- A. Romeo, M. A. Castriciano, I. Occhiuto, R. Zagami, R. F. Pasternack and L. M. Scolaro, J. Am. Chem. Soc., 2014, 136, 40–43 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- Y. Rong, P. Chen and M. Liu, Chem. Commun., 2013, 49, 10498–10500 RSC.
- L. Zhang, Y. Tian and M. Liu, Phys. Chem. Chem. Phys., 2011, 13, 17205–17209 RSC.
- Y. Wang, H. Fang, I. Tranca, H. Qu, X. Wang, A. J. Markvoort, Z. Tian and X. Cao, Nat. Commun., 2018, 9, 488 CrossRef PubMed.
- S. M. Vlaming, R. Augulis, M. C. A. Stuart, J. Knoester and P. H. M. van Loosdrecht, J. Phys. Chem. B, 2009, 113, 2273–2283 CrossRef CAS PubMed.
- R. Lauceri, G. F. Fasciglione, A. D'Urso, S. Marini, R. Purrello and M. Coletta, J. Am. Chem. Soc., 2008, 130, 10476–10477 CrossRef CAS PubMed.
- T. Jiao, L. Chen, D. Yang, X. Li, G. Wu, P. Zeng, A. Zhou, Q. Yin, Y. Pan, B. Wu, X. Hong, X. Kong, V. M. Lynch, J. L. Sessler and H. Li, Angew. Chem., Int. Ed., 2017, 56, 14545–14550 CrossRef CAS.
- F. J. Rizzuto, W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 11502–11509 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Jean-Marie Lehn on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc02412c |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |