
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium catalyzed template-assisted distal para-C–H olefination†
Uttam
Dutta
abc,
Sudip
Maiti
a,
Sandeep
Pimparkar
abc,
Siddhartha
Maiti
a,
Lawrence R.
Gahan
d,
Elizabeth H.
Krenske
 *d,
David W.
Lupton
*bc and
Debabrata
Maiti
*ab
*d,
David W.
Lupton
*bc and
Debabrata
Maiti
*ab
aDepartment of Chemistry, IIT Bombay, Powai, Mumbai 400076, India. E-mail: dmaiti@chem.iitb.ac.in
bIITB-Monash Research Academy, IIT Bombay, Powai, Mumbai 400 076, India
cSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: david.lupton@monash.edu
dSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: e.krenske@uq.edu.au
First published on 21st June 2019
Abstract
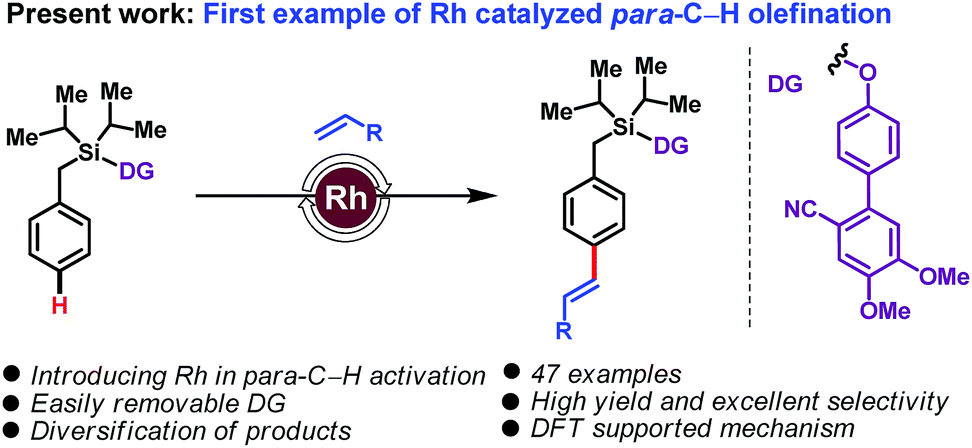
Rhodium catalysis has been extensively used for ortho-C–H functionalization reactions, and successfully extended to meta-C–H functionalization. Its application to para-C–H activation remains an unmet challenge. Herein we disclose the first example of such a reaction, with the Rh-catalyzed para-C–H olefination of arenes. The use of a Si-linked cyanobiphenyl unit as a traceless directing group leads to highly para-selective arene–olefin couplings.
The transformation of carbon–hydrogen (C–H) bonds into diverse classes of carbon–carbon (C–C) and carbon–heteroatom (C–X) bonds is a cornerstone of organic synthesis. There is intense interest in the discovery of new strategies for regioselective C–H functionalization.1 A daunting challenge is imposed by the innate inertness of C–H bonds combined with the subtle reactivity differences among the C–H bonds of a given substrate. Directing group (DG)-assisted transition metal-catalyzed C–H activation has proven a successful strategy for regioselective C–H functionalizations in a general and predictable manner.2 Most commonly coordination of a directing group to a transition metal to form a kinetically and thermodynamically stable 5- or 6-membered metallacycle is used to achieve ortho-C–H functionalization. In sharp contrast, distal C–H activation of meta3 and para4 sites is more challenging. In particular, para-C–H activation, which entails the formation of large macrocyclophane type metallacyclic intermediates, has remained elusive.5 In a recent breakthrough, palladium-catalyzed systems employing a carefully designed ‘D-shaped’ directing group/linker template, based on a cyanobiphenyl motif, led to the first examples of distal para-C–H olefinations and acetoxylations.5,6 Subsequent modifications of the 1st generation DGs through steric and electronic tuning led to 2nd generation DGs capable of effecting para-selective silylations7 and acylations.8
To the best of our knowledge, for template assisted para-selective functionalization palladium catalysis has been employed so far; albeit, other transition metals are also known to deliver para-selective functionalization relying on steric and electronic governance.5–9 As part of our ongoing interest in C–H functionalization, we have now translated this reaction into the realm of rhodium catalysis and we report here the first example of a Rh-catalyzed para-C–H olefination. Existing Rh-catalyzed approaches to C–H activation,10 using Rh(I)/Rh(III) redox cycles, are complementary to the Pd(0)/Pd(II) or Pd(II)/Pd(IV) cycles prevalent in palladium catalysis. The use of Rh offers benefits over Pd: (a) in contrast to Pd catalysis, which usually requires super-stoichiometric quantities of silver salts, Rh catalysis can be performed with alternative, often cheaper, oxidants; (b) compared with Pd catalysis, which employ monoprotected amino acids (MPAA) as ligands, the different coordination environment of Rh is expected to provide advantageous opportunities for stereoselective synthesis; and (c) importantly, Rh-catalysis does not require use of hexafluoroisopropanol (HFIP), often unavoidable in Pd-catalysed distal C–H activation. With these thoughts in mind, we set about examining a Rh-catalyzed, DG-assisted distal para-C–H olefination, as shown in Scheme 1.
 | ||
| Scheme 1 Rh-catalyzed para-C–H olefination. | ||
We commenced with the olefination of toluene scaffold DG1 by ethyl acrylate (Scheme 2). Our first attempt, using [Rh(COD)Cl]2 (5 mol%) as catalyst, N-Ac-Gly-OH (10 mol%) as ligand, and AgOAc (3 equiv.) as oxidant, was unsuccessful. However, use of copper trifluoroacetate [Cu(TFA)2] as oxidant with V2O5 as a co-oxidant provided the desired para-olefinated product in 30% yield. Encouraged by this initial result, we examined how the outcome could be improved by modifying the DG (Scheme 2). Analysis of cyano-based DGs (DG1–DG5) showed that the presence of an electron-withdrawing fluorine substituent (DG2) diminished the yield to 15% whereas an electron donating methoxy group (DG3) elevated the yield to 38%. By further enhancing the electron richness of the DG, the piperonal derivative DG4 afforded a 42% yield of the olefinated product. The dimethoxy-substituted DG5 gave a further improvement in yield, to 62%, with 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 para selectivity. The strong σ-donating DGs DG6–DG8 failed to provide any of the desired olefinated products. A range of different tethers, containing carbonyl (T1), sulfonyl (T2), and silyl (T3) linkers, were tested, as was a nitrile-free biphenyl template (T4); only the silyl based template T3 successfully delivered the desired olefinated product under the Rh-catalyzed conditions. These results indicate that the combination of sterically bulky silyl linker, nitrile group, and alkoxy groups present in DG5 is crucial for obtaining good yields of the para-olefinated product.
:1 para selectivity. The strong σ-donating DGs DG6–DG8 failed to provide any of the desired olefinated products. A range of different tethers, containing carbonyl (T1), sulfonyl (T2), and silyl (T3) linkers, were tested, as was a nitrile-free biphenyl template (T4); only the silyl based template T3 successfully delivered the desired olefinated product under the Rh-catalyzed conditions. These results indicate that the combination of sterically bulky silyl linker, nitrile group, and alkoxy groups present in DG5 is crucial for obtaining good yields of the para-olefinated product.
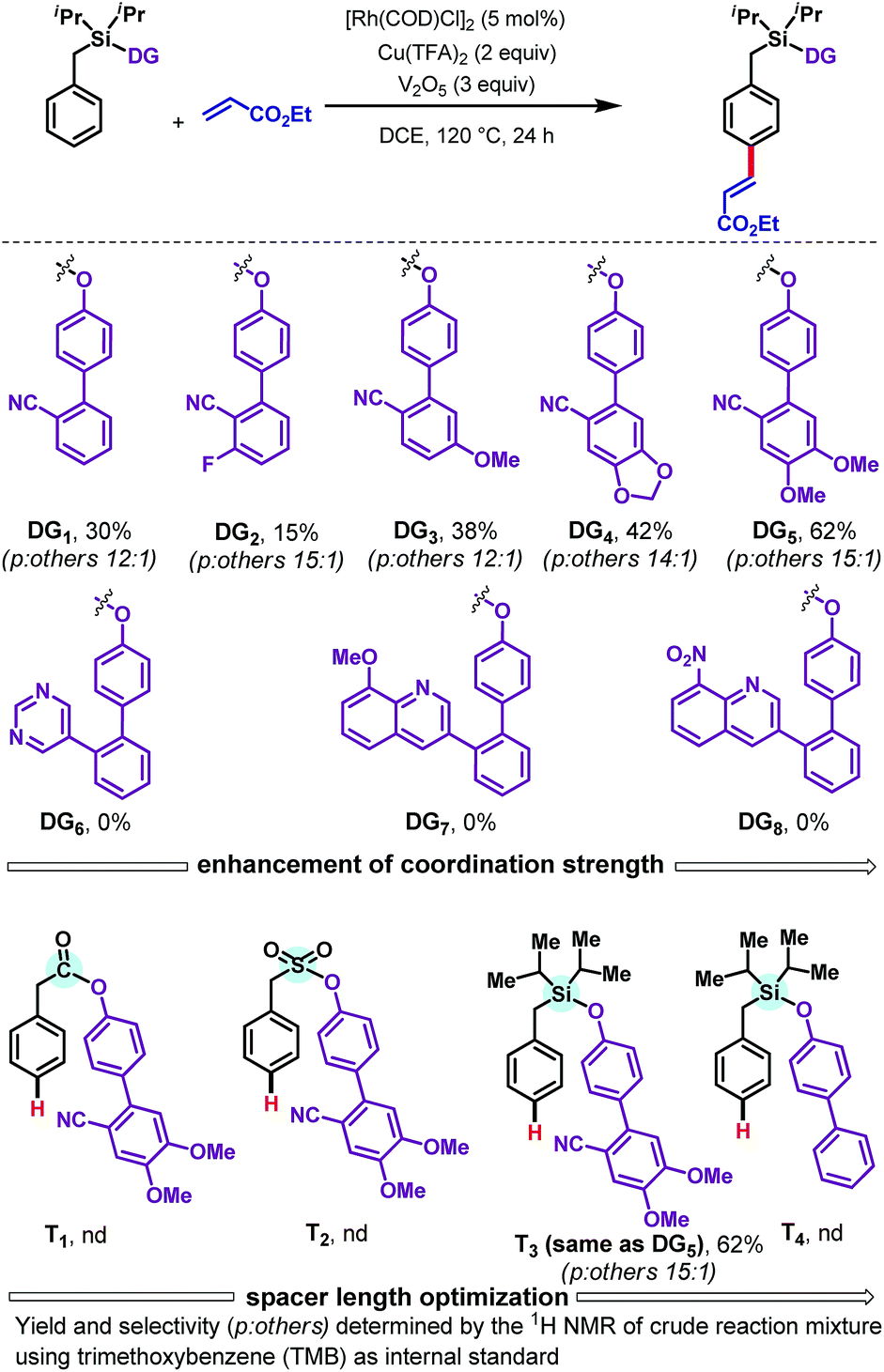 | ||
| Scheme 2 Evaluation of directing groups.11 | ||
Using best-performing directing group DG5, we optimized the reaction with respect to oxidants. A wide variety of silver and copper salts were tested.11 In contrast to Pd-catalyzed olefinations, silver salts were found to be ineffectual in these Rh-catalyzed reactions, delivering the olefinated products in only trace amounts. Use of Cu(TFA)2 as the oxidant in conjunction with V2O5 as a co-oxidant gave a 62% yield of olefinated product with excellent (15:1) para selectivity. Use of CuCl2 provided a lower (30%) yield of product, but a combination of CuCl2, V2O5 and trifluoroacetic acid (TFA) furnished the olefinated product in excellent (85%) yield, with 15:1 para selectivity.11 Interestingly, in the absence of either V2O5 or TFA, the yield was significantly lower (40% and 30%, respectively). Other acidic additives such as acetic acid (AcOH), triflic acid (CF3SO3H) and pivalic acid (Piv–OH) failed to yield the para-olefinated product.11
With optimized conditions in hand, we explored the scope of the reaction with respect to olefin (Table 1), arene (Tables 2 and 3), and benzylic substituents (Table 4). With respect to the olefin coupling partner (Table 1), a range of acrylates reacted efficiently, including alkyl acrylates 2a–2d, cyclohexyl acrylate 2e, and trifluoroethyl acrylate 2f. The olefinated products were obtained in excellent yields with synthetically useful para-selectivities ranging from 7:1 to 15:1. Apart from acrylates, vinyl sulfones including methyl vinyl sulfone (2g) and phenyl vinyl sulfone (2h) also gave the olefinated products, in 48% and 62% yields, respectively.
| a Ratio of para: others determined by the 1H NMR of crude reaction mixture. |
|---|
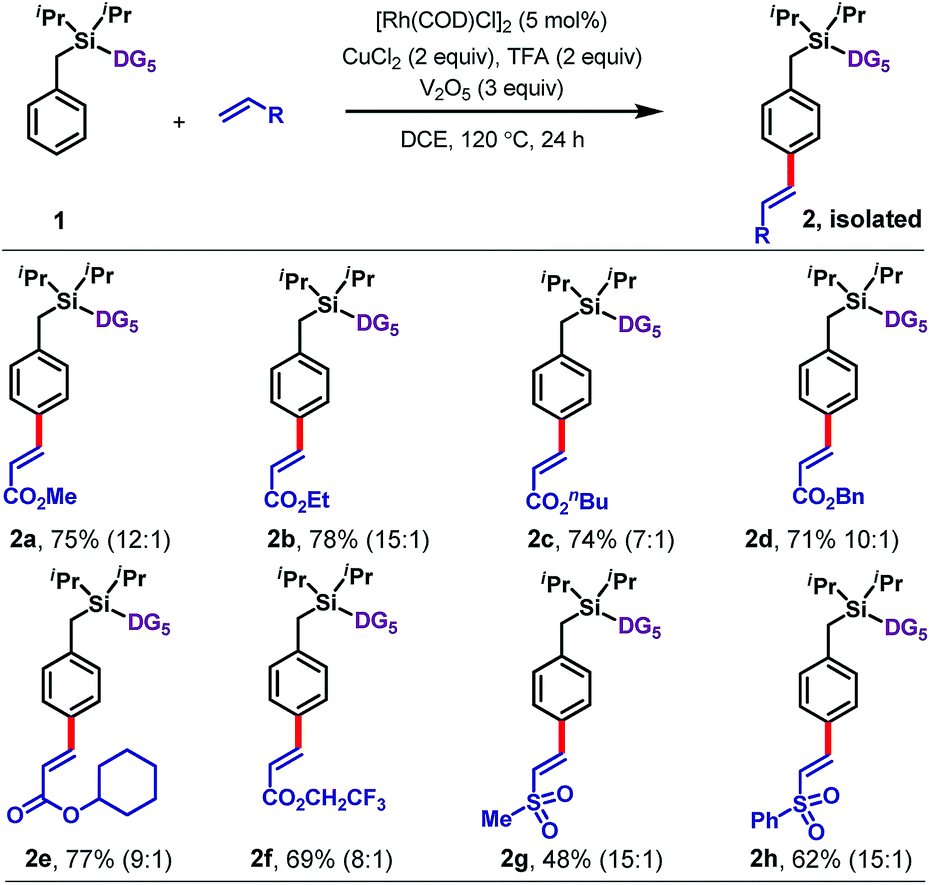
|
| a Ratio of para: others determined by the 1H NMR of crude reaction mixture. |
|---|
|
| a Ratio of para: others determined by the 1H NMR of crude reaction mixture. |
|---|
|
| a Ratio of para: others determined by the 1H NMR of crude reaction mixture. |
|---|
|
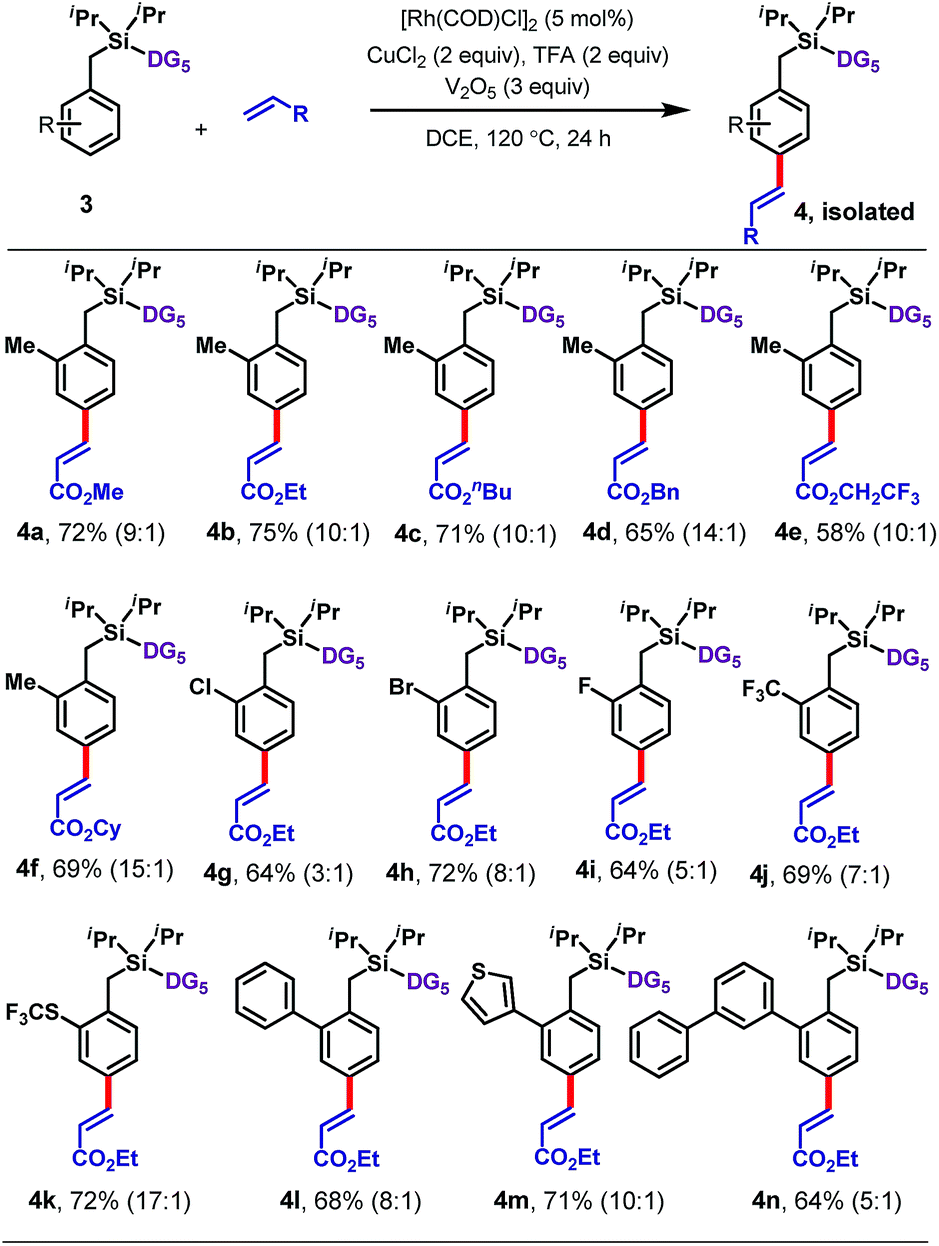
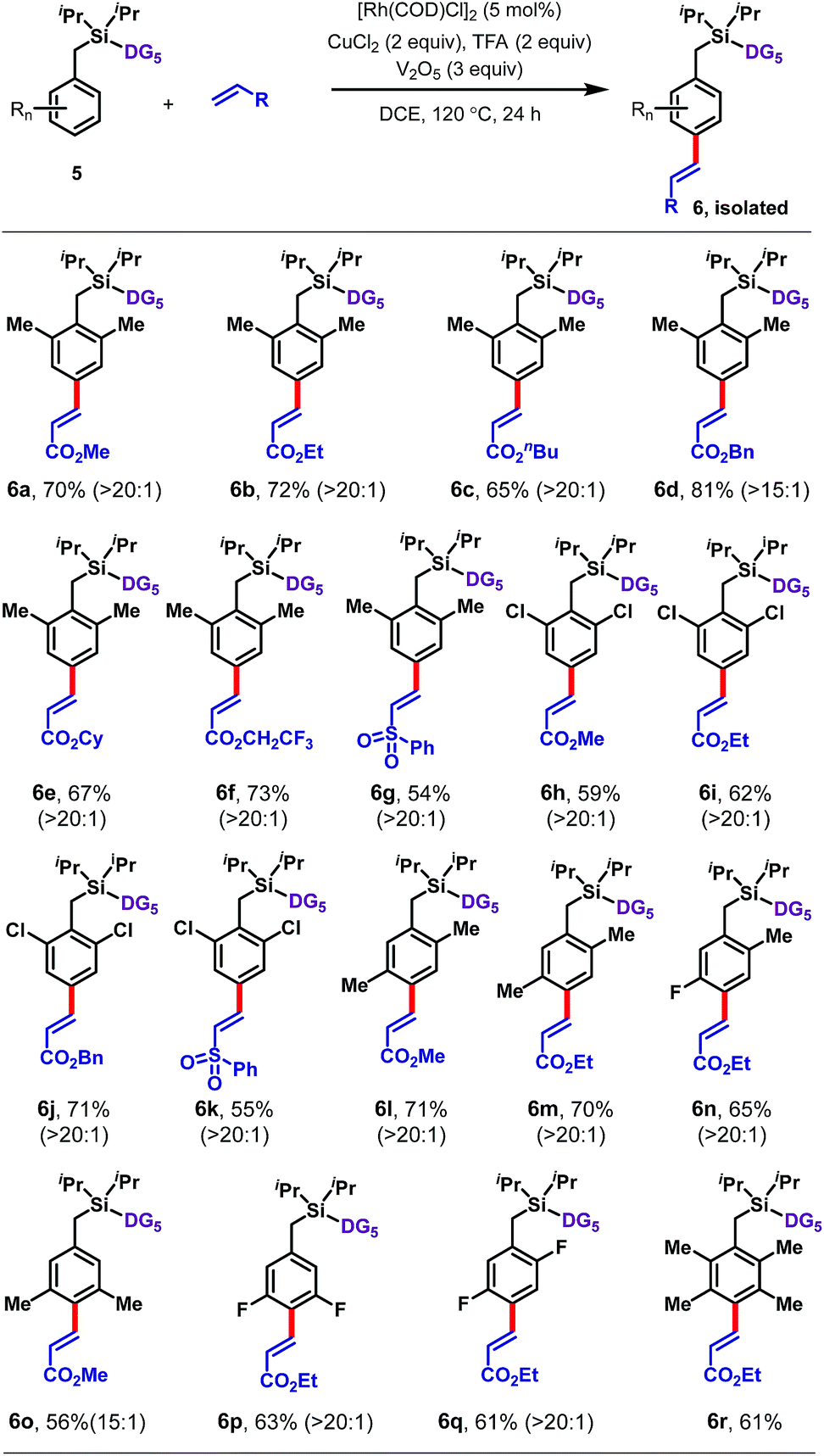
Next an array of substituted arenes was examined (Tables 2 and 3). For monosubstituted arenes, excellent yields and selectivities were obtained irrespective of the electronic nature of the substituent (Table 2). Both electron-rich and electron-deficient arenes were well tolerated, providing yields of up to 75% with upto 17:1 para selectivity.
Disubstituted arenes were also extremely well tolerated (Table 3). The reaction was successfully applied to a range of 2,2, 2,5, 3,5 and 2,6-disubstituted toluenes containing methyl, fluoro, and/or chloro substituents (6a–6q). The selectivities of these reactions were generally higher than those observed for monosubstituted arenes, with all ≥15:1 para selective. Even a tetramethyl-substituted arene was tolerated, reacting with ethyl acrylate to give 6r in 61% yield.
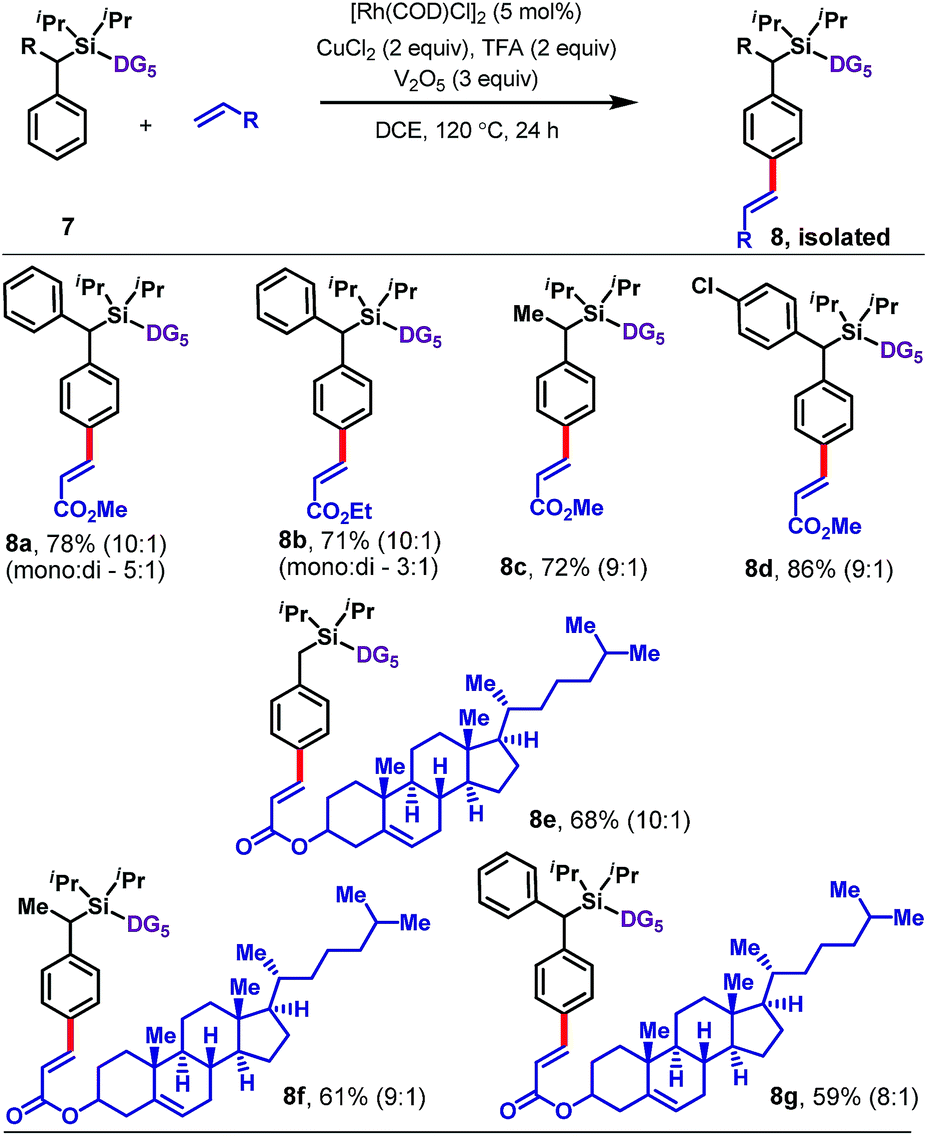
The protocol is also applicable to α-substituted toluene derivatives (Table 4). Substrates bearing methyl, phenyl, or substituted phenyl substituents at the benzylic position reacted with methyl or ethyl acrylate to afford para-olefinated products 8a–8d. The reaction also worked well with a more complex olefin coupling partner, namely, the acrylate derived from cholesterol, which furnished 8e–8g in 59–68% yield.
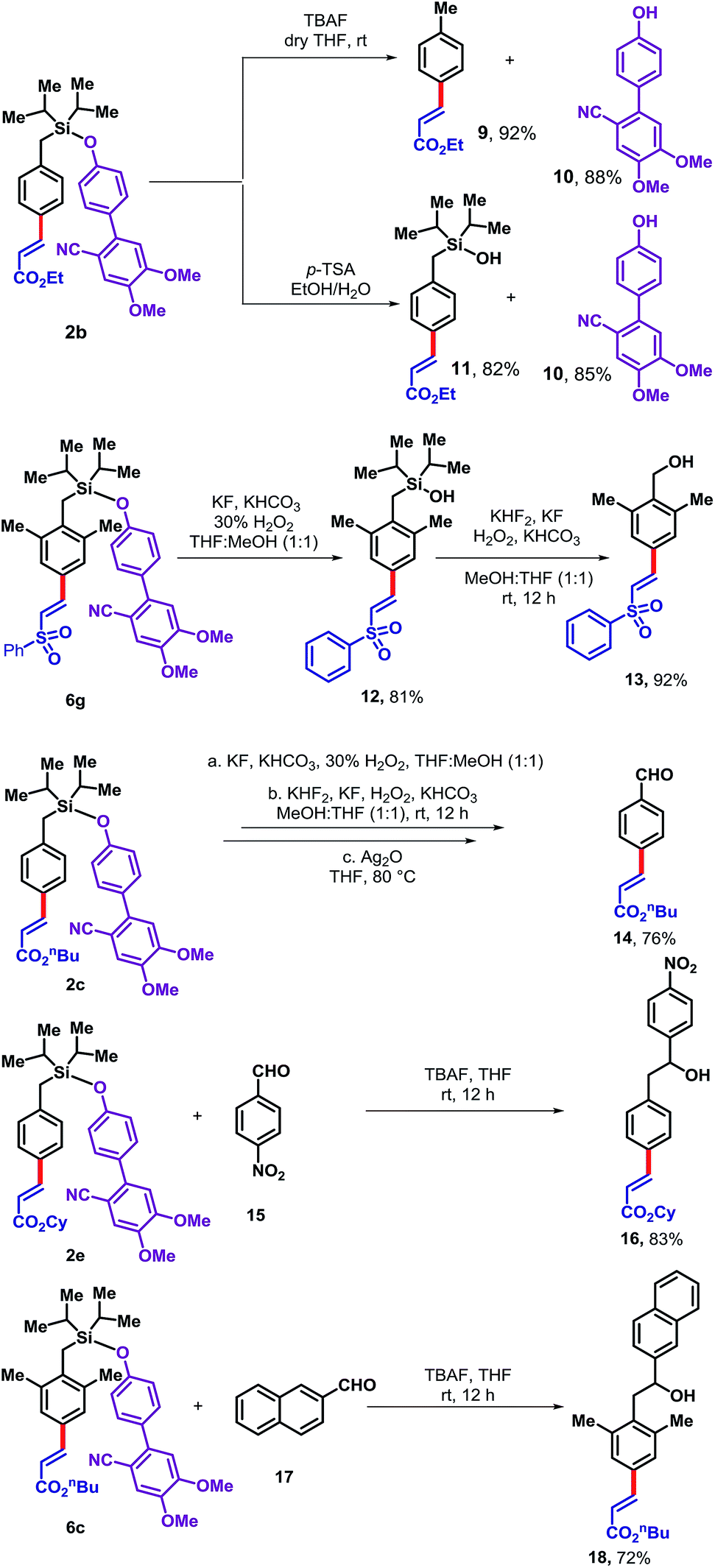
The DG5 directing group can be readily removed from the olefinated product in several ways (Scheme 3). Treatment of 2b with TBAF furnished the desilylated product 9 in 92% yield and allowed the DG5 alcohol 10 to be recovered in 88% yield for reutilization. Alternatively, treatment of 2b with p-TSA generated the corresponding silanol derivative 11 in 82% yield along with an 85% recovery of the DG5 alcohol. In principle, silanol 11 could be further used as a directing group for ortho functionalization. Therefore, the silyl-linked DG5 represents a traceless directing group enabling access to multi-functionalized arenes. While the para-olefinated product 6g has been treated with KF, KHCO3 and H2O2, it produced the corresponding silanol (12). The silanol derivative was then employed under modified Tamao's oxidation condition to produce corresponding benzyl alcohol (13). Another derivative 2c was treated under similar condition to provide the benzyl alcohol which subsequently oxidized to the corresponding benzaldehyde derivative (14) in 76% yield. The silyl based template can act as a nucleophile in presence of TBAF. To demonstrate that, 4-nitrobenzaldehyde (15) and 2-naphthaldehyde (17) was treated with para-olefinated product 2e and 6c, respectively to produce corresponding benzyl alcohols (16 and 18 in 83% and 72%, respectively).
 | ||
| Scheme 3 Removal of the directing group and diversification of the para-olefinated products.11 | ||
 | ||
| Scheme 4 Experiments with a deuterium-labeled substrate.11 | ||
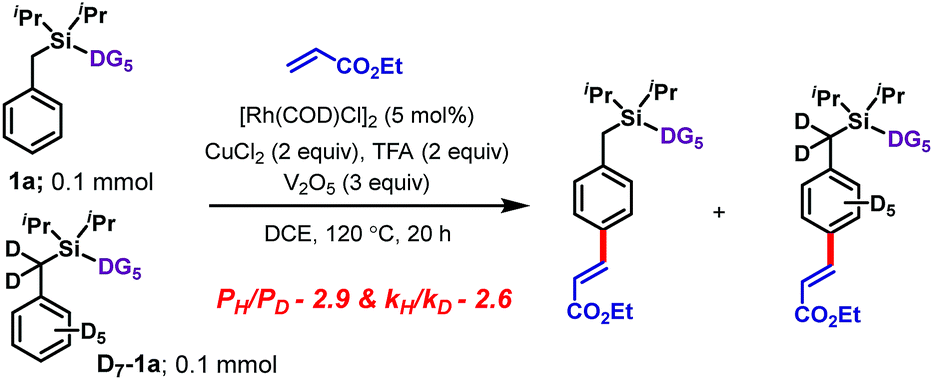
Isotope labeling experiments were conducted involving an intermolecular competition using substrate 1a and its deuterated analogue D7-1a and a PH/PD value of 2.9 and kH/kD value of 2.6 were obtained (Scheme 4).11 Furthermore, a detailed kinetic study indicated that the reaction was first order with respect to the substrate and zero order with respect to the olefin.11 Together, these results suggest that the C–H bond activation is likely to be the rate-determining step of the catalytic cycle. A plausible catalytic cycle for the para-olefination is shown in Scheme 5. In this mechanism, the Rh(I) catalyst precursor is first oxidized to Rh(III). The main steps in the cycle consist of C–H activation, migratory insertion, β-hydride elimination, and reductive elimination.11
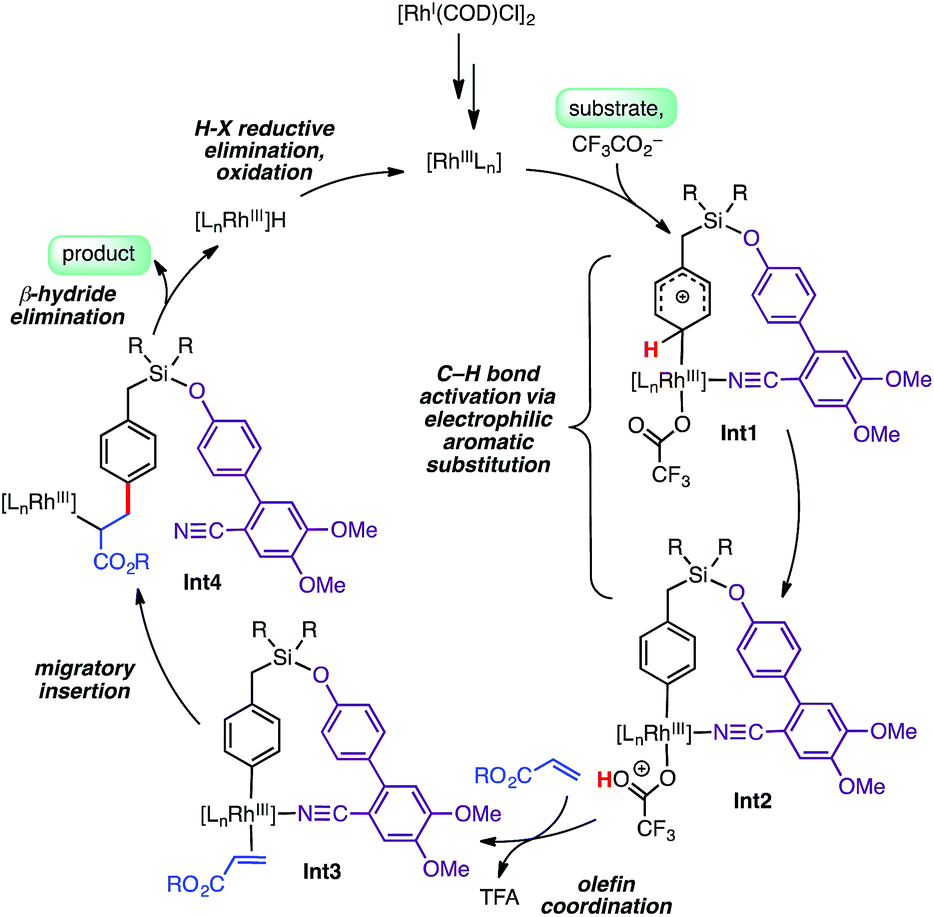 | ||
| Scheme 5 Possible catalytic cycle for para-selective Rh-catalyzed olefination. | ||
We explored the C–H activation process using density functional theory (DFT) (Fig. 1). Computations with the M06 functional using a model of DG1 with trifluoroacetate anion as the base predicted that the C–H bond activation follows an electrophilic aromatic substitution pathway, with a distinct intermediate Int1, rather than a concerted metalation–deprotonation pathway.10p,12 Transition structures for C–H bond breaking at the para and meta positions are shown in Fig. 1.
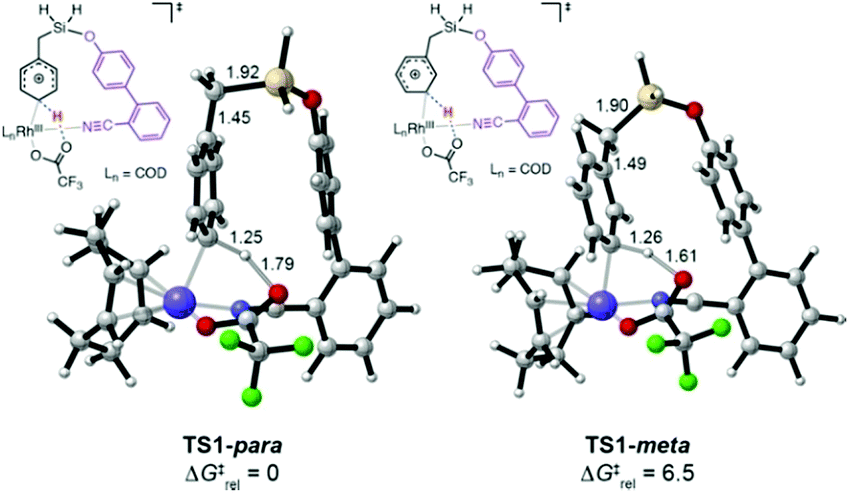 | ||
| Fig. 1 Transition states for Rh(III)-mediated para-C–H and meta-C–H bond activation, computed with M06/6-311+G(d,p)-SDD//M06/6-31G(d,p)-LANL2DZ in SMD dichloroethane. Distances in Å, ΔG‡rel in kcal mol−1. | ||
The para transition state, TS1-para, is 6.5 kcal mol−1 lower in energy than the meta transition state TS1-meta. A fragment-based analysis of the TSs12 reveals that the preference for para-C–H activation is due to a β-silicon effect. The interaction of the arene with Rh(III) endows it with arenium cation character, and this interaction is strengthened in TS1-para because the C–Si bond (which lies perpendicular to the ring) stabilizes the positive charge through hyperconjugation. Computations also revealed the roles of the DG methoxy and nitrile substituents.12 Incorporation of two methoxy groups on the DG activates the substrate toward C–H bond breaking, lowering the barrier by 1.6 kcal mol−1 relative to TS1-para. A TS in which the nitrile is not bound to Rh was computed to be 23 kcal mol−1 higher in energy than TS1-para, indicating that the coordination of the nitrile to Rh strongly stabilizes the C–H activation transition state.
Conclusions
In summary, herein we have reported the first example of a Rh-catalyzed distal para-C–H functionalization reaction. The Rh-catalyzed olefination of toluenes using the Si-linked DG5 directing group displays broad substrate tolerance. Electron-rich and electron-deficient arenes are coupled with electron-deficient olefins in high yield and selectivity. Mechanistic studies are consistent with a catalytic cycle in which the C–H bond activation is rate-determining. This work reveals the potential of Rh catalysis to diversify the scope of functionalizations in the realm of remote para-C–H activation.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This activity is funded by CSIR, India. Financial support received from IITB-Monash Research Academy (fellowship to UD and SP), UGC-India (fellowship to SM), Australian Research Council (DP180103047 to E. H. K.), and the University of Queensland (fellowship to E. H. K.) is gratefully acknowledged. High-performance computing resources were provided by the Australian National Computational Infrastructure and the UQ Research Computing Centre. Helpful discussions with Dr Romain Lepage (UQ) are gratefully acknowledged.Notes and references
- (a) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068–5083 RSC; (b) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633–639 CrossRef CAS PubMed; (c) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (e) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (f) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed; (g) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed; (h) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (i) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- (a) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed; (d) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (e) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed; (f) R. Giri, S. Thapa and A. Kafle, Adv. Synth. Catal., 2014, 356, 1395–1411 CrossRef CAS; (g) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC; (h) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (i) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (j) M. Pichette Drapeau and L. J. Gooßen, Chem.–Eur. J., 2016, 22, 18654–18677 CrossRef CAS PubMed; (k) A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (l) T. Satoh and M. Miura, Synthesis, 2011, 3395–3409, DOI:10.1055/s-0030-1258225; (m) G. Shi and Y. Zhang, Adv. Synth. Catal., 2014, 356, 1419–1442 CrossRef CAS; (n) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70–76 RSC; (o) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2013, 31–41 Search PubMed; (p) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (q) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC; (r) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (s) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC; (t) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 2–28 CrossRef PubMed; (u) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (v) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826–834 CrossRef CAS PubMed; (w) Y. Ogiwara, M. Tamura, T. Kochi, Y. Matsuura, N. Chatani and F. Kakiuchi, Organometallics, 2014, 33, 402–420 CrossRef CAS; (x) F. Kakiuchi, T. Kochi, E. Mizushima and S. Murai, J. Am. Chem. Soc., 2010, 132, 17741–17750 CrossRef CAS PubMed.
- (a) H. Shi, P. Wang, S. Suzuki, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14876–14879 CrossRef CAS PubMed; (b) H.-J. Xu, Y. Lu, M. E. Farmer, H.-W. Wang, D. Zhao, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200–2203 CrossRef CAS PubMed; (c) H.-J. Xu, Y.-S. Kang, H. Shi, P. Zhang, Y.-K. Chen, B. Zhang, Z.-Q. Liu, J. Zhao, W.-Y. Sun, J.-Q. Yu and Y. Lu, J. Am. Chem. Soc., 2019, 141, 76–79 CrossRef CAS PubMed; (d) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed; (e) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443–10450 CrossRef PubMed; (f) S. Li, H. Ji, L. Cai and G. Li, Chem. Sci., 2015, 6, 5595–5600 RSC; (g) R.-J. Mi, Y.-Z. Sun, J.-Y. Wang, J. Sun, Z. Xu and M.-D. Zhou, Org. Lett., 2018, 20, 5126–5129 CrossRef CAS PubMed; (h) L. Zhang, C. Zhao, Y. Liu, J. Xu, X. Xu and Z. Jin, Angew. Chem., Int. Ed., 2017, 56, 12245–12249 CrossRef CAS PubMed; (i) S. Bag, R. Jayarajan, U. Dutta, R. Chowdhury, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 12538–12542 CrossRef CAS PubMed; (j) M. Bera, S. Agasti, R. Chowdhury, R. Mondal, D. Pal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 5272–5276 CrossRef CAS PubMed; (k) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed; (l) U. Dutta, A. Modak, B. Bhaskararao, M. Bera, S. Bag, A. Mondal, D. W. Lupton, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 3162–3168 CrossRef CAS; (m) R. Jayarajan, J. Das, S. Bag, R. Chowdhury and D. Maiti, Angew. Chem., Int. Ed., 2018, 57, 7659–7663 CrossRef CAS PubMed; (n) A. Modak, T. Patra, R. Chowdhury, S. Raul and D. Maiti, Organometallics, 2017, 36, 2418–2423 CrossRef CAS; (o) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed; (p) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed; (q) L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J.-Q. Yu, ACS Cent. Sci., 2015, 1, 394–399 CrossRef CAS PubMed; (r) Z. Jin, L. Chu, Y.-Q. Chen and J.-Q. Yu, Org. Lett., 2018, 20, 425–428 CrossRef CAS PubMed; (s) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed; (t) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463–466 CrossRef CAS PubMed; (u) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef CAS; (v) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed; (w) N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed; (x) J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed; (y) F. Fumagalli, S. Warratz, S.-K. Zhang, T. Rogge, C. Zhu, A. C. Stückl and L. Ackermann, Chem.–Eur. J., 2018, 24, 3984–3988 CrossRef CAS PubMed; (z) M. T. Mihai, H. J. Davis, G. R. Genov and R. J. Phipps, ACS Catal., 2018, 8, 3764–3769 CrossRef CAS; (a a) M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149–171 RSC; (a b) R. Ferraccioli, Synthesis, 2013, 45, 581–591 CrossRef CAS; (a c) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed., 1997, 36, 119–122 CrossRef CAS; (a d) F. Faccini, E. Motti and M. Catellani, J. Am. Chem. Soc., 2004, 126, 78–79 CrossRef CAS PubMed; (a e) Z. Dong and G. Dong, J. Am. Chem. Soc., 2013, 135, 18350–18353 CrossRef CAS PubMed; (a f) Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed; (a g) Z. Dong, J. Wang, Z. Ren and G. Dong, Angew. Chem., Int. Ed., 2015, 54, 12664–12668 CrossRef CAS PubMed; (a h) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed; (a i) G.-C. Li, P. Wang, M. E. Farmer and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 6874–6877 CrossRef CAS PubMed; (a j) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (a k) R. Bisht and B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84–87 CrossRef CAS PubMed; (a l) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (a m) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (a n) D. W. Robbins and J. F. Hartwig, Angew. Chem., Int. Ed., 2013, 52, 933–937 CrossRef CAS PubMed.
- (a) B. E. Haines, Y. Saito, Y. Segawa, K. Itami and D. G. Musaev, ACS Catal., 2016, 6, 7536–7546 CrossRef CAS; (b) M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef CAS PubMed; (c) J. A. Leitch, C. L. McMullin, A. J. Paterson, M. F. Mahon, Y. Bhonoah and C. G. Frost, Angew. Chem., Int. Ed., 2017, 56, 15131–15135 CrossRef CAS PubMed; (d) S. Okumura and Y. Nakao, Org. Lett., 2017, 19, 584–587 CrossRef CAS PubMed; (e) L. Yang, K. Semba and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 4853–4857 CrossRef CAS PubMed; (f) C. Tian, X. Yao, W. Ji, Q. Wang, G. An and G. Li, Eur. J. Org. Chem., 2018, 2018, 5972–5979 CrossRef CAS; (g) L. Zhu, X. Qi, Y. Li, M. Duan, L. Zou, R. Bai and Y. Lan, Organometallics, 2017, 36, 2107–2115 CrossRef CAS.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS PubMed.
- T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS PubMed.
- A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 14903–14907 CrossRef CAS PubMed.
- A. Maji, A. Dahiya, G. Lu, T. Bhattacharya, M. Brochetta, G. Zanoni, P. Liu and D. Maiti, Nat. Commun., 2018, 9, 3582 CrossRef PubMed.
- (a) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed; (b) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458–462 CrossRef CAS PubMed; (c) B. Berzina, I. Sokolovs and E. Suna, ACS Catal., 2015, 5, 7008–7014 CrossRef CAS; (d) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904–6907 CrossRef CAS PubMed.
- (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed; (b) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (c) S. Cui, Y. Zhang and Q. Wu, Chem. Sci., 2013, 4, 3421–3426 RSC; (d) C. Feng, D. Feng and T.-P. Loh, Org. Lett., 2013, 15, 3670–3673 CrossRef CAS PubMed; (e) T. K. Hyster, L. Knörr, T. R. Ward and T. Rovis, Science, 2012, 338, 500–503 CrossRef CAS PubMed; (f) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443–1460 CrossRef CAS; (g) B. Li, J. Ma, W. Xie, H. Song, S. Xu and B. Wang, Chem.–Eur. J., 2013, 19, 11863–11868 CrossRef CAS PubMed; (h) G. Li, Z. Ding and B. Xu, Org. Lett., 2012, 14, 5338–5341 CrossRef CAS PubMed; (i) X. Li and M. Zhao, J. Org. Chem., 2011, 76, 8530–8536 CrossRef CAS PubMed; (j) B. Liu, Y. Fan, Y. Gao, C. Sun, C. Xu and J. Zhu, J. Am. Chem. Soc., 2013, 135, 468–473 CrossRef CAS PubMed; (k) S. H. Park, J. Y. Kim and S. Chang, Org. Lett., 2011, 13, 2372–2375 CrossRef CAS PubMed; (l) N. K. Mishra, J. Park, S. Sharma, S. Han, M. Kim, Y. Shin, J. Jang, J. H. Kwak, Y. H. Jung and I. S. Kim, Chem. Commun., 2014, 50, 2350–2352 RSC; (m) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188–1191 CrossRef CAS PubMed; (n) F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982–9983 CrossRef CAS PubMed; (o) M. Presset, D. Oehlrich, F. Rombouts and G. A. Molander, Org. Lett., 2013, 15, 1528–1531 CrossRef CAS PubMed; (p) X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799–2808 CrossRef CAS PubMed; (q) Y. Shen, G. Liu, Z. Zhou and X. Lu, Org. Lett., 2013, 15, 3366–3369 CrossRef CAS PubMed; (r) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540–542 CrossRef CAS PubMed; (s) F. Wang, G. Song and X. Li, Org. Lett., 2010, 12, 5430–5433 CrossRef CAS PubMed; (t) X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem.–Eur. J., 2013, 19, 11898–11903 CrossRef CAS PubMed; (u) J. Zhou, B. Li, F. Hu and B.-F. Shi, Org. Lett., 2013, 15, 3460–3463 CrossRef CAS PubMed; (v) C. Zhu and J. R. Falck, Chem. Commun., 2012, 48, 1674–1676 RSC; (w) K. Shibata, S. Natsui and N. Chatani, Org. Lett., 2017, 19, 2234–2237 CrossRef CAS PubMed; (x) Y. Kita, M. Tobisu and N. Chatani, Org. Lett., 2010, 12, 1864–1867 CrossRef CAS PubMed.
- See the ESI† for detailed descriptions.
- We also considered several other mechanisms in which the CH bond cleavage step is mediated by either Rh(III) or Rh(I), details are provided in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01824g |
| This journal is © The Royal Society of Chemistry 2019 |