
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFine-tuning of wettability in a single metal–organic framework via postcoordination modification and its reduced graphene oxide aerogel for oil–water separation†
Sunhwi
Eom
,
Dong Won
Kang
,
Minjung
Kang
,
Jong Hyeak
Choe
,
Hyojin
Kim
,
Dae Won
Kim
and
Chang Seop
Hong
 *
*
Department of Chemistry, Korea University, Seoul 02841, Republic of Korea. E-mail: cshong@korea.ac.kr
First published on 16th January 2019
Abstract
Elaborate control of wettability in a single platform is essential for materials’ applications towards oil–water separation, but it still remains challenging. Herein, we performed postcoordination modification of Mg2(dobpdc) with monoamines of various alkyl chain lengths, enabling both long-term hydrolytic stability and facile fine-tuning of wettability. An efficient separation of oil–water mixtures was achieved by using the octylamine-appended framework (OctA). We also prepared an OctA/reduced graphene oxide aerogel that showed exceptional absorption capacities towards organic solvents and oil as well as superb recyclability with maintained absorbency.
Introduction
Water is severely polluted by oil spills, causing major impact on the environment and human health. The water pollution arising from oil leakage must be immediately removed before it damages the surrounding environment.1 Water remediation methods are based on conventional artificial technologies such as separation, filtration, centrifugation, floatation, and electrochemistry.2,3 The representative hydrophobic/oleophilic materials for oil removal include fabrics, sponges, metallic meshes, and carbon and its derivatives.4–9 Despite the notable absorption performance of some of these materials, the fine-tuning of wettability is a big challenge because their structures are not systematically designable and controllable.Metal–organic frameworks (MOFs) are a new class of crystalline solids with a large surface area and tunable pore properties. Most MOFs show hydrolytic instability and are unsuitable for oil–water separation. Therefore, the design and synthesis of hydrophobic and water-stable MOFs are imperative for their potential application in oil–water separation. The construction of such frameworks was attempted by introducing hydrophobic moieties into the backbone. When long alkyl and/or fluorine-containing organic linkers were utilized, MOFs became hydrophobic.10–14 In addition, the incorporation of polymers into the frameworks or exposure to corrugated aromatic hydrocarbon surfaces induced hydrophobicity of MOFs.15–17 Nonetheless, these approaches associated with ligand modification by hydrophobic side groups may require expensive ligand components or multistep synthetic procedures. For practical application in oil–water separation, facile synthesis of a water-stable and hydrophobic framework is ultimately desirable.
Recently, the combination of MOFs with hydrophobic polymeric materials was explored to create MOF-based hybrid composites. Only a few hybrid MOF composites with hydrophobicity have been reported to date, exhibiting diverse functions such as photocatalytic ability, removal of toxic substances, and oil–water and hydrocarbon separation.12,18–21 ZIF-8 has been exclusively utilized as a pillar component for the fabrication of such hybrid composites. However, the reported ZIF-8-based composite materials have shown very low absorption capacities for organic solvents or oil in comparison with other hydrophobic materials.22
The expanded version of MOF-74, Mg2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) with a high density of open metal sites, has been investigated mostly to capture CO2 from flue gas emissions after diamine functionalization.23–29 Herein, we report a facile yet unique method for postsynthetically inducing hydrophobicity of the hydrophilic Mg2(dobpdc) framework. The platform is readily transformable from hydrophilic to hydrophobic by appending monoamines with various alkyl chain lengths to the open metal sites, which has not been demonstrated before. This simply modified hydrophobic framework with long-term hydrolytic stability enables effective separation of oil (hexadecane) and water, leading to successful oil removal. Moreover, the composite MOF/reduced graphene oxide aerogel (rGA) provides significant absorption capacities for organic liquids, which are much greater than those provided by MOF-based composites and comparable to those of top-performing materials,12,18–20,22 and excellent reusability without capacity loss.
Results and discussion
The open metal sites of Mg2(dobpdc) were postcoordinated by monoamines H–(CH2)n–NH2 with different numbers of carbons in the alkyl group from n = 3 to 16 to produce amine-appended MOFs (PA, n = 3; BA, n = 4; PenA, n = 5; HexA, n = 6; OctA, n = 8; DcA, n = 10; TdA, n = 14; HdA, n = 16). The elemental analysis revealed that the open metal sites were partially filled by the monoamines. The occupancy of the amine groups tended to decrease from 88% for PA to 55% for HdA, because the amines with shorter alkyl groups diffuse into the channels more easily than the longer amines. The structure of Mg2(dobpdc) was maintained even after the postcoordination modifications, as confirmed by the powder X-ray diffraction (PXRD) profiles shown in Fig. 1a. The characteristic N–H stretching peaks in the infrared (IR) spectra of the samples were centered at 3282 and 3341 cm−1, indicating the existence of the amine groups in the Mg2(dobpdc) framework (Fig. S1†). We conducted X-ray photoelectron spectroscopy (XPS) to determine the binding energies of Mg 2p before and after amine grafting (Fig. S2†). The binding energy of Mg 2p for Mg2(dobpdc) was estimated to be 50.0 eV, while the binding energies for amine-functionalized samples were centered at 49.5 eV. The coordination of amine groups to the exposed Mg sites accounts for the reduced binding energy. Thermogravimetric analysis (TGA) data showed that the thermal collapse of the grafted amines occurred over a broad temperature range, and their decomposition temperatures increased as the chain length increased (Fig. S3†).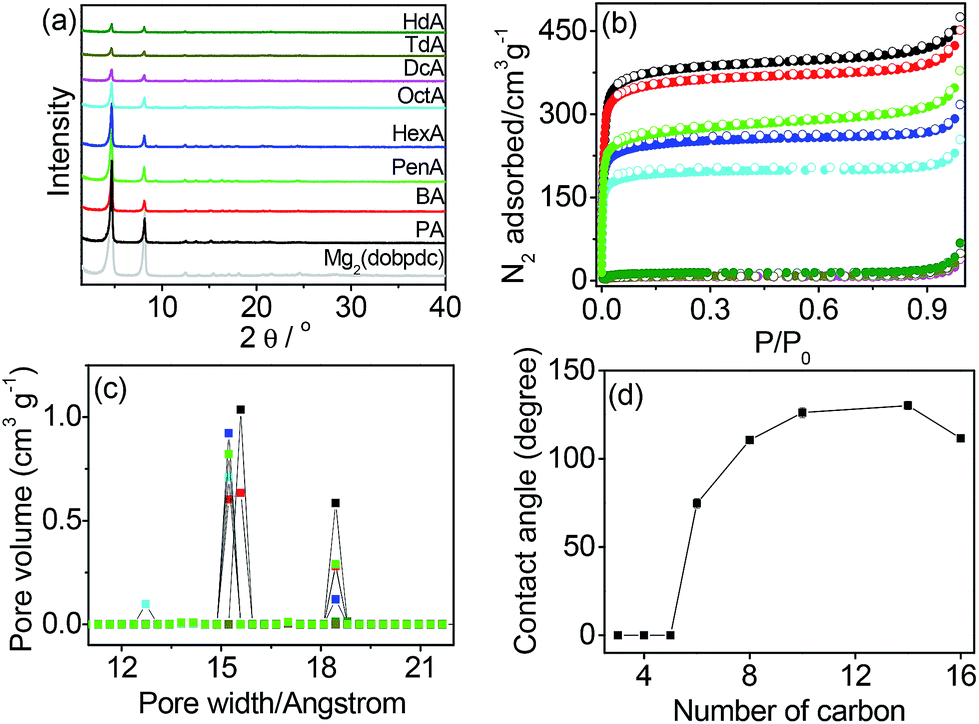 | ||
| Fig. 1 (a) PXRD profiles, (b) N2 isotherms at 77 K, and (c) pore size distributions of monoamine-appended Mg2(dobpdc). Color codes in (a)–(c) represent the same compounds. (d) Contact angles of the frameworks as a function of the number of carbons in the monoamine. | ||
The N2 isotherms at 77 K were measured to determine the BET surface areas (Fig. 1b). The surface areas gradually decreased from 1476 m2 g−1 for PA to 772 m2 g−1 for OctA and then abruptly decreased to 28–53 m2 g−1 for DcA, TdA, and HdA. The values are much smaller than that (3280 m2 g−1) of the non-modified framework, which is attributed to the amines grafted onto the open metal sites of the Mg2(dobpdc) framework. In the pore size distributions (Fig. 1c), pores were observed at 18.4 and 15.6 Å for PA. It is interesting to note that the pores at 15.2 Å emerged from n = 4 (BA) and remained until n = 8 (OctA). This result indicates that the alkyl chain lengths ranging from n = 4 to 8 do not affect the smaller pore size of the framework. No pores existed with n larger than 10 because all pores were blocked by the amines with long alkyl chains. The scanning electron microscopy (SEM) images of monoamine-functionalized Mg2(dobpdc) were similar to that of Mg2(dobpdc) (Fig. S4†).
To examine the hydrophobicity with amines appended onto the open metal sites of the framework, we measured the contact angles of water droplets dropped on the solid surface of each sample (Fig. 1d and S5†). No contact angle was observed until n = 5, implying that the surface of the solids remained hydrophilic. The modified framework became hydrophobic at n = 8, and the contact angle was larger than 110°. Hydrophobicity was maintained until n = 16. After the contact angles were saturated, they slightly decreased with increasing n. This is likely due to the grafting of few monoamines onto the open metal sites as the chain length increases (refer to elemental analysis results in the Experimental section). We further investigated the hydrophobicity by measuring water adsorption isotherms (Fig. S6†). For PA, water vapor was adsorbed above P/P0 = 0.4 and desorbed with hysteresis, suggesting hydrophilicity. In comparison, negligible water adsorption occurred in OctA and DcA, which is consistent with the hydrophobicity confirmed by the contact angle measurements. The structural integrity was checked by PXRD before and after water adsorption (Fig. S7†). We also tested the long-term stability in water, and found that the solids with n ≤ 6 were unstable after 24 h immersion, whereas the samples with n ≥ 8 were stable even after immersion in water for 7 d (Fig. S8†). The incorporation of longer amines into the pores of the framework promotes both hydrophobicity and water stability. This result indicates that wettability and water stability are readily controllable by such simple modification of the pore surface using the open metal sites, which has been demonstrated for the first time in this study.30,31
The hydrophobicity was further investigated by immersing the pelletized samples in a mixed solvent system with a water layer (bottom) and an oil layer (top) composed of hexadecane colored with a yellowish Sudan I dye (Fig. S9a†). The hydrophilic sample pellets were dispersed over the water layer. In contrast, the hydrophobic sample pellets floated on the water layer and moved into the oil layer, indicating that organic pollutants can be readily extracted. We carried out a sorption test of hexadecane using an OctA pellet. After the pellet was placed on an oil droplet, the yellow oil diffused into it (Fig. S9b†).
Oil–water separation was conducted using the hydrophobic MOF OctA. To visually distinguish between oil (hexadecane) and water, we stained the water with Acid Orange dye (Fig. 2). The separation membrane was fabricated by sandwiching MOF powder between two gauze layers. A mixture of oil and water was poured into a syringe. The syringe was slightly tilted so that the oil part touched the membrane and selectively penetrated through the film. It is obvious that only oil was filtered down, while the orange water part remained in the syringe, suggesting that this membrane made of OctA provides effective oil–water separation. The other hydrophobic materials also showed similar separation behaviors (Fig. S10–S12†). Moreover, we attempted to separate water-in-toluene emulsions using this framework. The opaque emulsions indicate the existence of colloidal water (Fig. S13†).32 After the separation of the emulsions by an identical membrane, we obtained a clean solution only containing toluene. Thus, the hydrophobic OctA enables the successful separation of oil–water mixtures. Furthermore, we carried out the time-dependent separation process to obtain the permeation flux and separation efficiency of the membrane (Fig. S14†).33,34
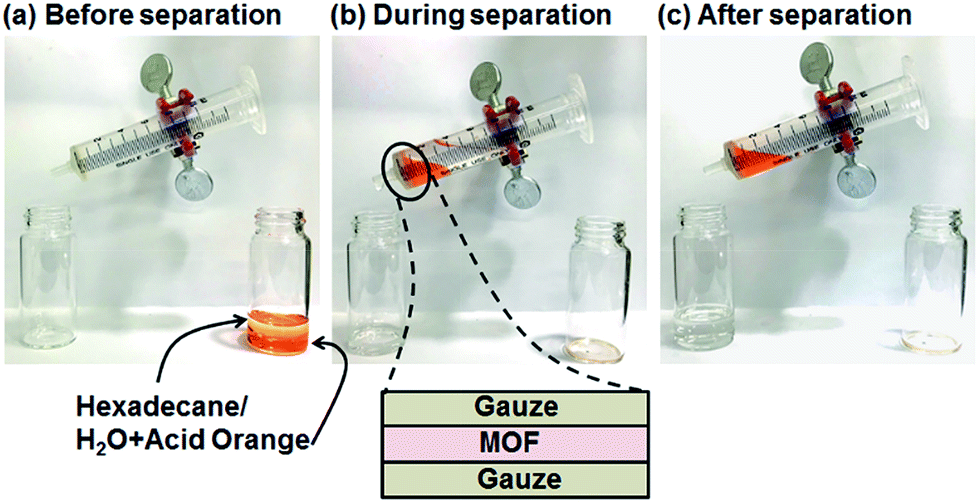 | ||
| Fig. 2 Photos taken (a) before oil–water separation of OctA, (b) during separation, and (c) after separation. The water layer was colored with Acid Orange dye. The separation membrane was composed of three layers of gauze/MOF/gauze as shown in the figure. | ||
A MOF/rGA composite was synthesized by a one-pot reaction of graphene oxide (GO) and a MOF in the presence of hydrazine hydrate (Fig. 3a). To prepare proper aerogels, the MOF should be well dispersed over water. The dispersion tests showed that OctA was the most homogeneously dispersed, but the other hydrophobic MOFs were partly dispersed or rapidly precipitated (Fig. S15†). After the one-pot reaction, OctA only produced a well-formed aerogel with a uniform distribution of OctA over rGA (Fig. S16†). Therefore, we concentrated on the properties of the OctA/rGA aerogel. According to a previous report, GO contains hydroxyl and epoxide groups located above and below the GO plane, and carboxylic groups at the edge.4 These groups could be reduced by hydrazine hydrate.35 To examine the compositional changes after the reduction, the IR spectra were measured. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1723 cm−1 was visible in GO but disappeared in the OctA/rGA aerogel, which indicates that reduction of the corresponding functional groups occurred (Fig. S17†). As seen in the X-ray photoelectron spectroscopy (XPS) results, the intensity ratio of C/O for rGA was smaller than that for GO, further indicating the reduction of CO groups by the reducing agent (Fig. 3b). Binding energies (50 and 88 eV) were present in the XPS spectrum of OctA, which are assignable to Mg 2p and 2s peaks, respectively. These peaks were also seen in the XPS spectrum of the OctA/rGA composite, which indicates that OctA was impregnated in rGA. Notably, the N 1s peak (351 eV) emerged in the XPS data of both OctA and OctA/rGA. In the narrow scan of the XPS spectra of the core-level C 1s peak, the CO peak in GO was absent in rGA due to the reduction, while the CO and C–N peaks appeared in OctA/rGA, confirming the presence of the dobpdc4− ligand and octylamine group in the composite (Fig. S18†). Given that N atoms only originate from octylamines grafted onto the open metal sites of the Mg2(dobpdc) platform, this result implies that the amine groups appended to the platform remained intact even in the composite formed by one-pot synthesis.
O stretching at 1723 cm−1 was visible in GO but disappeared in the OctA/rGA aerogel, which indicates that reduction of the corresponding functional groups occurred (Fig. S17†). As seen in the X-ray photoelectron spectroscopy (XPS) results, the intensity ratio of C/O for rGA was smaller than that for GO, further indicating the reduction of CO groups by the reducing agent (Fig. 3b). Binding energies (50 and 88 eV) were present in the XPS spectrum of OctA, which are assignable to Mg 2p and 2s peaks, respectively. These peaks were also seen in the XPS spectrum of the OctA/rGA composite, which indicates that OctA was impregnated in rGA. Notably, the N 1s peak (351 eV) emerged in the XPS data of both OctA and OctA/rGA. In the narrow scan of the XPS spectra of the core-level C 1s peak, the CO peak in GO was absent in rGA due to the reduction, while the CO and C–N peaks appeared in OctA/rGA, confirming the presence of the dobpdc4− ligand and octylamine group in the composite (Fig. S18†). Given that N atoms only originate from octylamines grafted onto the open metal sites of the Mg2(dobpdc) platform, this result implies that the amine groups appended to the platform remained intact even in the composite formed by one-pot synthesis.
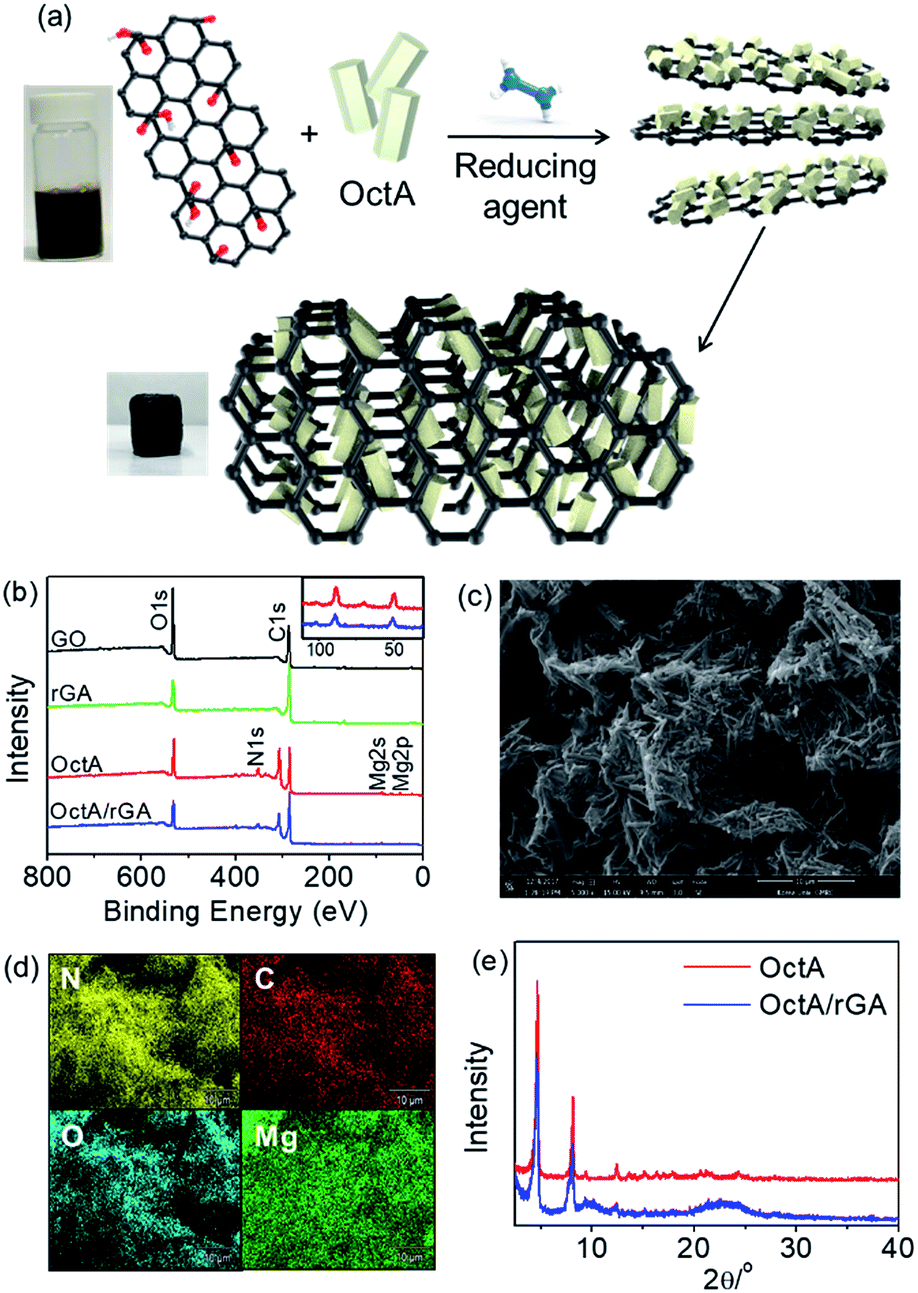 | ||
| Fig. 3 (a) Schematic illustration of the proposed self-assembly process of rGO and OctA for the synthesis of OctA/rGA. (b) XPS of GO, rGA, OctA, and OctA/rGA. The inset shows the magnification of the binding energy region of Mg. (c) SEM image of OctA/rGA. (d) EDS elemental mapping of OctA/rGA. (e) PXRD of OctA and OctA/rGA. | ||
The existence of OctA in the aerogel was additionally verified by examination of the surface morphology by SEM; the image of OctA/rGA shows that rod-shaped OctA particles were well incorporated into the rGA (Fig. 3c). Moreover, the homogeneous distribution of OctA over the aerogel was confirmed by energy dispersive spectroscopy (EDS) mapping (Fig. 3d). The PXRD pattern of OctA was discernible in OctA/rGA, suggesting that the phase of the MOF remained in the composite gel (Fig. 3e). The OctA loading, determined by TGA, was consistent with the nominal values, indicating that all MOFs added to the suspension were incorporated into the aerogel (Fig. S19 and Table S1†).36 The porosity was checked using the N2 isotherm of OctA/rGA at 77 K (Fig. S20†). For GO, there was no water contact angle because of the hydrophilic character arising from hydroxyl, epoxide, and carboxylic groups. The contact angle (25°) of rGA increased slightly with respect to GO, which is associated with the reduction of the functional groups (Fig. S21†). The enhanced hydrophobicity allows the aggregation of rGO sheets. When Mg2+ was used as a cross-linker, the gel Mg2+/rGA showed an increased contact angle of 69°. It is interesting to note that the inclusion of OctA facilitated the rapid gel formation of OctA/rGA and a high contact angle of 134°, which is greater than that (110°) for OctA. The hydrophobic OctA particles with functional groups could favor extensive interaction with reduced GO sheets, leading to enhancement of the water contact angle. These results demonstrate that the reduction of some hydrophilic functional groups in GO leads to the effective crosslinking of rGO with OctA and the subsequent self-assembly of the reduced GO sheets, resulting in the formation of the consequent hydrophobic hydrogel.
To investigate the composite aerogel's absorption behavior with various organic liquids, we stained hexadecane with Sudan I (Fig. 4a–c). The yellow hexadecane was rapidly absorbed by the aerogel when the liquid was touched by the OctA/rGA composite. The absorption capacity was estimated using the formula 100(Wwet − Wdry)/Wdry, where Wwet and Wdry denote the weights of wet and dry samples, respectively. Fig. 4d presents the weight gain upon solvent absorption by the composite aerogel. The absorption capacity of OctA ranged from 423 to 1012 wt%, while Mg2+/rGA exhibited a capacity in the range of 1900 to 4600 wt%. Notably, the absorption capacity of the OctA/rGA aerogel was much greater than that of OctA and Mg2+/rGA, and ranged from 4700 to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 122 wt%. We also investigated the absorption capacities of DcA/rGA, TdA/rGA, and HdA/rGA, which were smaller than that of OctA/rGA (Fig. S22†). The distribution of MOF particles was probed using SEM images of the composites (Fig. S23†). Contact angle measurements revealed that hydrophobicity was maintained (Fig. S24†). These results demonstrated that the reduced capacities could be due to the poor formation of aerogels, as shown in Fig. S16.†
122 wt%. We also investigated the absorption capacities of DcA/rGA, TdA/rGA, and HdA/rGA, which were smaller than that of OctA/rGA (Fig. S22†). The distribution of MOF particles was probed using SEM images of the composites (Fig. S23†). Contact angle measurements revealed that hydrophobicity was maintained (Fig. S24†). These results demonstrated that the reduced capacities could be due to the poor formation of aerogels, as shown in Fig. S16.†
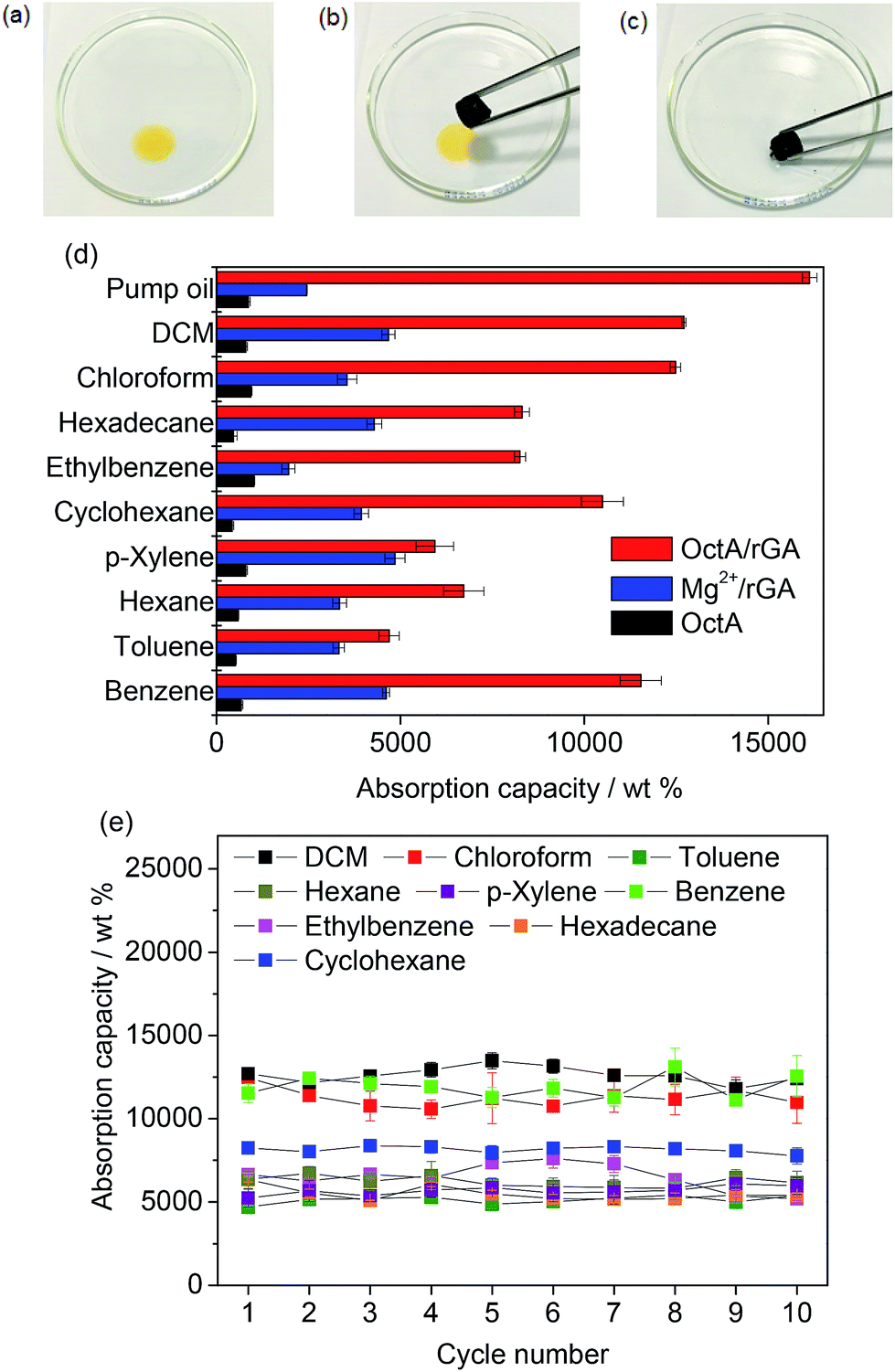 | ||
| Fig. 4 Absorption process of hexadecane colored with Sudan I by OctA/rGA: (a and b) before absorption and (c) after absorption. (d) Absorption capacities of OctA/rGA with various organic liquids. (e) Recyclability tests of absorption capacities of OctA/rGA for organic liquids. | ||
The different densities and viscosities of the corresponding organic substances contribute to different absorption capacities. There was no swelling in the size of OctA/rGA even after absorbing the pump oil with the highest capacity value, suggesting that the oil diffuses into the pores of the aerogel (Fig. S25†).
The capacity values of OctA/rGA exceed those of conventional resins, sponges, and foams as well as MOF-incorporated composites, and are comparable to those of top-performing porous materials.12,19,37–40 The reusability of the aerogel was assessed through recycling tests on the organic solvents (Fig. 4e). We ran 10 cycles of absorption–desorption processes. The absorbed samples were activated for 30 min at the boiling temperature of each solvent except for hexadecane which was desorbed at 100 °C for 24 h. For benzene, the adsorption capacity remained significant between 11500 and 13100 wt% during the repeated cycles. The other organic liquids also maintained their capacities within narrow variation ranges. The compositional distributions in the aerogel were examined by SEM-EDS elemental mapping analyses (Fig. S26–S32†). Mg elements were observed in the images together with N atoms from monoamines, highlighting the strong binding affinity of the OctA particles to the rGA platform during absorption–desorption cycles. The contact angle measurements confirmed that the hydrophobicity was well maintained throughout the cycles, and the IR and PXRD data of the samples corroborated the structural integrity after the cycling tests (Fig. S33–S35†).
Conclusions
In summary, we prepared amine-Mg2(dobpdc) frameworks appended with monoamines with various alkyl chain lengths. Postcoordination modification of open metal sites in the hydrophilic framework with alkyl amines of various chain lengths allows fine tuning of wettability, which constitutes a facile and unique strategy for the realization of hydrophobic materials. Excellent oil–water separation by OctA was demonstrated, which indicates that this material is very promising for oil cleaning applications. Furthermore, the OctA/rGA composite material shows superior absorption capacity for organic liquids and substantial recyclability.Experimental
Preparation
All chemicals and solvents used in the synthesis were obtained from commercial companies and utilized without further purification. H4dobpdc was prepared according to the literature.23Synthesis of [Mg2(dobpdc)(DMF)2] [Mg2(dobpdc)]
The solid was synthesized according to a slightly modified literature procedure.41 H4dobpdc (128 mg, 0.467 mmol), MgBr2·6H2O (409.2 mg, 1.400 mmol), and 16 mL of solvent (DMF:EtOH = 1:1, v/v) were loaded in a 35 mL cell and sealed with a PTFE cap. The mixture was reacted in a microwave reactor (CEM Discover) at 130 °C, 150 psi, and 50 W. After 20 min, the white powder was collected via filtration and washed with DMF several times. The product was dried under vacuum. Mg2(dobpdc) was immersed in MeOH for 3 days and treated under vacuum (<10−2 kPa) at 390 °C for 90 min to yield the fully activated Mg2(dobpdc).
Synthesis of [Mg2(dobpdc)(C3H9N)1.76(H2O)0.24]·1.06(H2O) (PA)
PA was prepared via a slightly modified literature procedure.24 Fully activated Mg2(dobpdc) (100 mg, 0.313 mmol) at 390 °C was transferred to a 250 mL Schlenk flask in a glove box. Before adding propylamine, the Schlenk flask containing Mg2(dobpdc) was heated to 250 °C for 30 min under vacuum to remove water again and cooled to room temperature. After 100 mL of dry toluene was transferred to a 250 mL-Schlenk flask containing the activated Mg2(dobpdc) using a cannula, propylamine was added to the mixture. The mixture was sonicated for 12 h at 60 °C. The solid was separated by centrifugation and washed with dry toluene and hexane five times in order. The product was dried under Ar flow. Yield: 92.26%. Anal. calcd for C20H24Mg2N2O6: C, 51.71; H, 5.52; N, 5.52. Found: C, 51.71; H, 5.33; N, 5.52.Synthesis of [Mg2(dobpdc)(C4H11N)1.6(H2O)0.4]·1.2(H2O) (BA)
BA was synthesized by a procedure identical to the preparation of PA except for the use of butylamine. Yield: 98.13%. Anal. calcd for C22H28Mg2N2O6: C, 52.73; H, 5.73; N, 4.82. Found: C, 52.72; H, 5.77; N, 4.55.Synthesis of [Mg2(dobpdc)(C5H13N)1.51(H2O)0.39]·0.91(H2O) (PenA)
PenA was synthesized by a procedure identical to the preparation of PA except for the use of pentylamine. Yield: 92.14%. Anal. calcd for C24H32Mg2N2O6: C, 54.42; H, 6.02; N, 4.45. Found: C, 54.66; H, 6.11; N, 4.40.Synthesis of [Mg2(dobpdc)(C6H15N)1.5(H2O)0.5]·1.1(H2O) (HexA)
HexA was synthesized by a procedure identical to the preparation of PA except for the use of hexylamine. Yield: 97.45%. Anal. calcd for C26H36Mg2N2O6: C, 55.31; H, 6.40; N, 4.21. Found: C, 55.41; H, 6.52; N, 4.45.Synthesis of [Mg2(dobpdc)(C8H19N)1.5(H2O)0.5]·0.3(H2O) (OctA)
OctA was synthesized by a procedure identical to the preparation of PA except for the use of octylamine. Yield: 97.64%. Anal. calcd for C30H44Mg2N2O6: C, 59.25; H, 6.90; N, 3.99. Found: C, 59.04; H, 7.03; N, 3.89.Synthesis of [Mg2(dobpdc)(C10H23N)1.32(H2O)0.68]·1.2(H2O) (DcA)
DcA was synthesized by a procedure identical to the preparation of PA except for the use of decylamine. Yield: 84.63%. Anal. calcd for C34H52Mg2N2O6: C, 60.61; H, 7.06; N, 3.43. Found: C, 52.72; H, 5.77; N, 4.55.Synthesis of [Mg2(dobpdc)(C13H29N)1.1(H2O)0.9]·2.5(H2O) (TdA)
TdA was synthesized by a procedure identical to the preparation of PA except for the use of tetradecylamine. Yield: 81.63%. Anal. calcd for C40H64Mg2N2O6: C, 61.92; H, 7.06; N, 3.43. Found: C, 62.17; H, 7.86; N, 2.87.Synthesis of [Mg2(dobpdc)(C16H35N)0.9(H2O)1.1]·2.2(H2O) (HdA)
HdA was synthesized by a procedure identical to the preparation of PA except for the use of hexadecylamine. Yield: 70.49%. Anal. calcd for C46H76Mg2N2O6: C, 60.63; H, 7.66; N, 2.68. Found: C, 60.81; H, 7.77; N, 2.39.Preparation of GO sheets
GO sheets were prepared through a modified Hummers' method.19 Graphite powder (2 g) was dispersed in 46 mL of concentrated sulfuric acid, followed by the slow addition of 6 g KMnO4. The mixture was stirred in an ice bath and then was kept at 35 °C for 5 h. Deionized water (100 mL) was added dropwise to the mixture and then the resulting slurry was heated at 90 °C for 10 min. The dark brown slurry was filtered and washed several times with HCl, water, and ethanol to obtain the final product.Synthesis of OctA/rGA
OctA (12 mg) was dispersed in 6 mL of distilled water by sonication for 1 h, which was added to a mixture of 6 mL of 2 mg mL−1 GO aqueous solution and 12 μL of hydrazine hydrate. The resulting mixture was sealed tightly and placed in an electrical oven at 95 °C for 1 h to produce a hydrogel. The hydrogel was washed with distilled water and hexane several times, and then freeze-dried into an aerogel.Synthesis of Mg2+/rGA
Mg2+/rGA was synthesized by a procedure identical to that of OctA/rGA except for the use of MgBr2 instead of OctA.Synthesis of rGA
6 mL of distilled water was added to a mixture of 6 mL of 2 mg mL−1 GO aqueous solution and 12 μL of hydrazine hydrate. The mixture was sealed tightly and placed in an electrical oven at 95 °C for 2 h to produce a hydrogel.Physical measurements
IR spectra were obtained with an ATR module using a Nicolet iS10 FT-IR spectrometer. TGA was carried out under a N2 (99.999%) atmosphere (flow rate = 50 mL min−1) in the temperature range of 25–800 °C (heating rate = 10 °C min−1) using a Scinco TGA-N 1000 instrument. Powder XRD patterns were recorded using Cu Kα radiation (λ = 1.5406) on a Rigaku Ultima III diffractometer with a scan speed of 2° min−1 and a step size of 0.02°. Elemental analyses for C, H, and N were performed at the Elemental Analysis Service Center of Sogang University. XPS data were collected using X-tool, and EDX-SEM analysis was performed using a Quanta 250 FEG instrument at the Semiconductor & Display Green Manufacturing Research Center at Korea University. Contact angle measurements were conducted using a contact angle analyzer (Phoenix 10).Gas sorption measurements
Before sorption analysis, the samples were degassed at 120 °C under vacuum for 2 h. All gases used in the measurements were highly pure (99.999%). N2 gas sorption measurements at 77 K were carried out up to 1 atm using a Micromeritics 3flex instrument. Water vapor isotherms at 298 K were obtained using a Micromeritics ASAP2020 instrument equipped with a vapor source.Absorption measurement
OctA/rGA was soaked in several organic solvents at room temperature until equilibrium was reached. The resultant samples were then removed from the solvents and weights were measured quickly to avoid solvent evaporation from the samples.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by a Korea CCS R&D Center (KCRC) grant funded by the Korean government (the Ministry of Science, ICT, & Future Planning (MSIP)) (NRF-2012-0008901) and by the Basic Science Research Program (NRF-2018R1A2A1A05079297).Notes and references
- B. Dubansky, A. Whitehead, J. T. Miller, C. D. Rice and F. Galvez, Environ. Sci. Technol., 2013, 47, 5074–5082 CrossRef PubMed.
- A. B. Nordvik, J. L. Simmos, K. R. Bitting, A. Lewis and T. Storm-Kristiansen, Spill Sci. Technol. Bull., 1996, 3, 107–122 CrossRef.
- K. Gaaseidnes and J. Turbeville, Pure Appl. Chem., 1999, 71, 95–101 Search PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef.
- L. Feng, Z. Zhang, Z. Mai, Y. Ma, B. Liu, L. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 43, 2012–2014 CrossRef PubMed.
- Z. Xue, Y. Cao, N. Liu, L. Feng and L. Jiang, J. Mater. Chem. A, 2014, 2, 2445–2460 RSC.
- L. Wang, Y. Zhao, Y. Tian and L. Jiang, Angew. Chem., Int. Ed., 2015, 54, 14732–14737 CrossRef PubMed.
- A. M. Rather, N. Jana, S. Begum, H. K. Srivastava and U. Manna, Green Chem., 2017, 19, 4527–4532 RSC.
- J. Zhang, H. Liu and L. Jiang, Adv. Funct. Mater., 2017, 27, 1606544 CrossRef.
- M. Zhang, X. Xin, Z. Xiao, R. Wang, L. Zhanga and D. Sun, J. Mater. Sci. Eng. A, 2017, 5, 1168–1175 Search PubMed.
- S. T. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560–4561 CrossRef PubMed.
- K. Jayaramulu, K. K. Datta, C. Rosler, M. Petr, M. Otyepka, R. Zboril and R. A. Fischer, Angew. Chem., Int. Ed., 2016, 55, 1178–1182 CrossRef PubMed.
- C. Yang, U. Kaipa, Q. Z. Mather, X. Wang, V. Nesterov, A. F. Venero and M. A. Omary, J. Am. Chem. Soc., 2011, 133, 18094–18097 CrossRef PubMed.
- L. Li, Q. Yang, S. Chen, X. Hou, B. Liu, J. Lu and H. L. Jiang, Chem. Commun., 2017, 53, 10026–10029 RSC.
- Z. Zhang, H. T. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920–925 CrossRef PubMed.
- K. P. Rao, M. Higuchi, K. Sumida, S. Furukawa, J. Duan and S. Kitagawa, Angew. Chem., Int. Ed., 2014, 53, 8225–8230 CrossRef PubMed.
- G. Huang, Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef PubMed.
- W. Zhang, Y. Hu, J. Ge, H. L. Jiang and S. H. Yu, J. Am. Chem. Soc., 2014, 136, 16978 CrossRef PubMed.
- J. Mao, M. Ge, J. Huang, Y. Lai, C. Lin, K. Zhang, K. Meng and Y. Tang, J. Mater. Sci. Eng. A, 2017, 5, 11873–11881 Search PubMed.
- P. Jing, S.-Y. Zhang, W. Chen, L. Wang, W. Shi and P. Cheng, Chem.–Eur. J., 2018, 24, 3754–3759 CrossRef PubMed.
- Z.-R. Jiang, J. Ge, Y.-X. Zhou, Z. U. Wang, D. Chen, S.-H. Yu and H.-L. Jiang, NPG Asia Mater., 2016, 8, e253 CrossRef.
- B. Wang, W. Liang, Z. Guo and W. Liub, Chem. Soc. Rev., 2015, 44, 336–361 RSC.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef PubMed.
- W. R. Lee, S. Y. Hwang, D. W. Ryu, K. S. Lim, S. S. Han, D. Moon, J. Choi and C. S. Hong, Energy Environ. Sci., 2014, 7, 744–751 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef.
- W. R. Lee, H. Jo, L.-M. Yang, H. Lee, D. W. Ryu, K. S. Lim, J. H. Song, D. Y. Min, S. S. Han, J. G. Seo, Y. K. Park, D. Moon and C. S. Hong, Chem. Sci., 2015, 6, 3697 RSC.
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runcevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef.
- P. J. Milner, J. D. Martell, R. L. Siegelman, D. Gygi, S. C. Weston and J. R. Long, Chem. Sci., 2018, 9, 160–174 RSC.
- P. Deria, Y. G. Chung, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Sci., 2015, 6, 5172–5176 RSC.
- J. G. Nguyen and S. M. Cohen, J. Am. Chem. Soc., 2010, 132, 4560–4561 CrossRef.
- G. Wang, B. Yu, S. Chen and H. Uyama, Sci. Rep., 2017, 7, 6534 CrossRef.
- S. Ye, Q. Cao, Q. Wang, T. Wang and Q. Peng, Sci. Rep., 2016, 6, 37591 CrossRef.
- J. P. Chaudhary, S. K. Nataraj, A. Gogda and R. Meena, Green Chem., 2014, 16, 4552–4558 RSC.
- P. G. Ren, D. X. Yan, X. Ji, T. Chen and Z. M. Li, Nanotechnology, 2011, 22, 055705 CrossRef.
- H. Zhu, X. Yang, E. D. Cranston and S. Zhu, Adv. Mater., 2016, 28, 7652–7657 CrossRef PubMed.
- G.-R. Shan, P.-Y. Xu, Z.-X. Weng and Z.-M. Huang, J. Appl. Polym. Sci., 2003, 89, 3309–3314 CrossRef.
- C. Liu, J. Yang, Y. Tang, L. Yin, H. Tang and C. Li, Colloids Surf., A, 2015, 468, 10–16 CrossRef.
- B. Ge, X. Men, X. Zhu and Z. Zhang, J. Mater. Sci., 2015, 50, 2365–2369 CrossRef.
- S. Yang, L. Chen, L. Mu and P. C. Ma, J. Colloid Interface Sci., 2014, 430, 337–344 CrossRef PubMed.
- W. R. Lee, J. E. Kim, S. J. Lee, M. Kang, D. W. Kang, H. Y. Lee, V. Hiremath, J. G. Seo, H. Jin, D. Moon, M. Cho, Y. Jung and C. S. Hong, ChemSusChem, 2018, 11, 1694–1707 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04581j |
| This journal is © The Royal Society of Chemistry 2019 |