
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA universal method for sensitive and cell-free detection of CRISPR-associated nucleases†
Kurt J.
Cox‡
 abc,
Hari K. K.
Subramanian‡
d,
Christian Cuba
Samaniego
d,
Elisa
Franco
*d and
Amit
Choudhary
*abc
abc,
Hari K. K.
Subramanian‡
d,
Christian Cuba
Samaniego
d,
Elisa
Franco
*d and
Amit
Choudhary
*abc
aChemical Biology and Therapeutics Science, Broad Institute of MIT and Harvard, 415 Main Street, Rm 3012, Cambridge, MA 02142, USA. E-mail: achoudhary@bwh.harvard.edu; Fax: +1 617 715 8969; Tel: +1 617 714 7445
bDepartment of Medicine, Harvard Medical School, Boston, MA 02115, USA
cDivisions of Renal Medicine and Engineering, Brigham and Women's Hospital, Boston, MA 02115, USA
dDepartment of Mechanical Engineering, University of California – Riverside, Riverside, CA – 92521, USA. E-mail: efranco@engr.ucr.edu; Tel: +1 951 827 2442
First published on 16th January 2019
Abstract
A multitude of biological applications for CRISPR-associated (Cas) nucleases have propelled the development of robust cell-based methods for quantitation of on- and off-target activities of these nucleases. However, emerging applications of these nucleases require cell-free methods that are simple, sensitive, cost effective, high throughput, multiplexable, and generalizable to all classes of Cas nucleases. Current methods for cell-free detection are cumbersome, expensive, or require sophisticated sequencing technologies, hindering their widespread application beyond the field of life sciences. Developing such cell-free assays is challenging for multiple reasons, including that Cas nucleases are single-turnover enzymes that must be present in large excess over their substrate and that different classes of Cas nucleases exhibit wildly different operating mechanisms. Here, we report the development of a cell-free method wherein Cas nuclease activity is amplified via an in vitro transcription reaction that produces a fluorescent RNA:small-molecule adduct. We demonstrate that our method is sensitive, detecting activity from low nanomolar concentrations of several families of Cas nucleases, and can be conducted in a high-throughput microplate fashion with a simple fluorescent-based readout. We provide a mathematical framework for quantifying the activities of these nucleases and demonstrate two applications of our method, namely the development of a logic circuit and the characterization of an anti-CRISPR protein. We anticipate our method will be valuable to those studying Cas nucleases and will allow the application of Cas nuclease beyond the field of life sciences.
Introduction
CRISPR-associated (Cas) nucleases are furnishing transformative technologies for genome editing and functional genomics. The commonly employed Cas nucleases that cleave DNA include Cas9 and Cpf1 (or Cas12).1 These nucleases recognize their substrate sequence via a Protospacer Adjacent Motif (PAM) sequence and base-pairing of the target sequence by a guide RNA (gRNA) borne by the nuclease. Upon target recognition, Cas nucleases induce a double-strand break, following which the cell's repair machinery can be co-opted to alter the genomic sequence. Catalytically inactive or impaired Cas-nuclease-bearing effector domains allow loci-specific genome manipulation.2–5 For example, a fusion of catalytically impaired Cas9 to base-modifying enzymes has produced “base-editors” that allow base conversion (e.g., C → T) at specific genomic sites, while a fusion of catalytically-inactive Cas nucleases to transcriptional activators or repressors has enabled gene transcription and repression.6There are a slew of sensitive, orthogonal, and high-throughput methods that can quantify the on- and off-target activities of these nucleases in cellular and even organismal settings.7,8 Paradoxically, the development of general and high-throughput cell-free assays for Cas nucleases has lagged despite several obvious applications.9 For example, while the most-studied Cas9 is from Streptococcus pyogenes (SpCas9), an active search is ongoing for next-generation nucleases as well as anti-CRISPR molecules to control their activity, and these pursuits will benefit from such assays. Chemically modified gRNAs are becoming preferred reagents over natural gRNAs as they provide higher specificity, stability, and lower immunogenicity.10,11 Cell-free assays could be used to screen synthetic gRNAs to identify ideal candidates for further cell-based studies, as such screens cannot be directly performed in cell-based assays because of the large amount of input material required and high associated costs. The availability of low-cost and efficient assays will also impact several areas of synthetic biology involving the development of synthetic nucleic acid circuits and diagnostics. Circuits using nucleic-acid elements can perform complex logical computations,12–14 exhibit dynamic behavior,15 or potentially create biological controllers16 by leveraging the ability of catalytically impaired SpCas9 to interfere with or regulate transcription. Finally, the availability of cell-free assays will guide the mechanistic understanding of extant and emerging nucleases.
An ideal cell-free assay for Cas nucleases should meet the following criteria. First, the assay should be sensitive enough to continuously detect low nanomolar amounts of nuclease and, ideally, be implementable in a microplate format with an easy readout. This is challenging as Cas nucleases are single-turnover enzymes that tightly bind to their DNA substrates and products,17,18 and a large excess of enzyme relative to the substrate (typically >10-fold) is needed for adequate detection of activity. Second, the assay must be modular and adaptable to accommodate the complex and diverse attributes of Cas nucleases, such as their enormous diversity of PAM sequences and their relative binding orientation – for example, Cas9 recognizes a 3′-PAM, while Cpf1 recognizes a 5′-PAM. Third, the assay should work well in a broad range of temperatures, as the activity of many Cas nucleases is temperature dependent,19 and genome editing may be performed in organisms with varying body temperatures. Fourth, the assay should allow multiplexed and simultaneous quantitation of several nucleases for the standardized measurement and direct comparison of novel nucleases, allowing one to benchmark and directly compare nuclease activities under several reaction conditions. Finally, such assay should be cost-effective and not require specialized instruments or data-analysis methods.
Current in vitro methods for nuclease-activity detection, including gel-based DNA cleavage assays, PCR and isothermal amplification reactions,20–22 next-generation sequencing methods,23 and cell-free transcription–translation assays,24 do not meet the aforementioned criteria. The use of radiolabeled nucleotides in standard gel-cleavage assays can increase the nuclease detection limit, but such approaches require specific radiation protocols, specialized imaging equipment, and are tedious and time-consuming. Furthermore, continuous kinetic monitoring of reaction rates is challenging using gel-based workflows. Sensitivity can be boosted using the products of nuclease cleavage as templates for DNA-polymerase-based exponential amplification reactions, whereby increasing the amplification cycle time increases the detection limit.20–22 However, these assays involve multiple liquid-handling steps, including the necessity for heating and denaturing the Cas nuclease before amplification. Furthermore, these assays involve endpoint measurements and preclude real-time monitoring of cleavage, prohibiting their use in a continuous CRISPR-based circuit. Electrochemiluminescent assays, while highly sensitive, also suffer from these two drawbacks.25 Next-generation sequencing approaches are expensive and require specialized equipment and knowledge not easily accessible to many laboratories. Cell-free translation/transcription strategies meet some of the criteria, but they are expensive, require multiple components and steps, and are operational in a narrow temperature range.24 Recent work using fluorophore/quencher pairs to assay Cas9-mediated DNA-cleavage has resulted in powerful high-throughput screening platforms, although these assays still required high concentrations of enzyme (∼200 nM).26 The FRET-based “Cas beacon assay” also satisfies many of these criteria, but it has been optimized to detect DNA-binding interactions with the nuclease/gRNA complex and not cleavage events.27
Towards this end, we developed a sensitive, quantifiable, and continuous cell-free assay that can be generalized to numerous Cas nucleases. We hypothesized that coupling the nuclease activity to an in vitro RNA transcription readout would yield an assay with the aforementioned attributes. Because transcription yields multiple RNA transcripts, the signal stemming from the single-turnover activity of Cas nuclease will be amplified. We report the use of the GFP-mimetic Spinach RNA28,29 as a fluorescent transcription reporter,30 which allowed us to perform the assay in a microplate format using easily accessible plate readers. Furthermore, the fluorescent readout made it possible to measure the activities of multiple Cas nucleases at nanomolar concentrations. This effective amplification of the signal with the Spinach RNA reporter allowed the dynamic range of the assay to be tuned according to experimental needs, a feature that is absent in gel-based assays. To enable benchmarking of Cas nucleases, we present a mathematical model to quantify the activities of different nucleases. While our Spinach assay reads Cas nuclease activity in a “signal-off” fashion, the assay detects nuclease inhibition (e.g., by anti-CRISPR proteins) in a “signal-on” fashion and can facilitate the discovery and validation of anti-CRISPR molecules. To demonstrate this utility, we used the Spinach assay to detect SpCas9 inhibition by an anti-CRISPR protein. Finally, we demonstrated a simple conditional switch using SpCas9 activity as a proof-of-concept for logic and dynamic circuits. Such conditional switches are key elements in many nucleic-acid-based synthetic circuits and can potentially turn a gene ON or OFF using internal transcriptional signals.
Results and discussion
Development of a cell-free assay for measuring the activity of Cas nucleases
To enhance the detection limit of Cas nuclease activity, we coupled the output of this activity to an in vitro transcription (IVT) reaction that produces the RNA aptamer Spinach (Fig. 1a). We used a synthetic gene-like construct (a ‘genelet’) consisting of a bacteriophage T7 RNA Polymerase (T7 RNAP) promoter upstream of the region that codes for the Spinach RNA. Upon binding to the small molecule 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), a stable fluorescent complex is produced only in the presence of intact RNA.28 Our goal was to identify optimal assay conditions to minimize the production of fluorescent transcripts upon disruption of the genelet by the Cas nuclease, making it possible to easily detect Cas nuclease activity by comparing fluorescence measurements.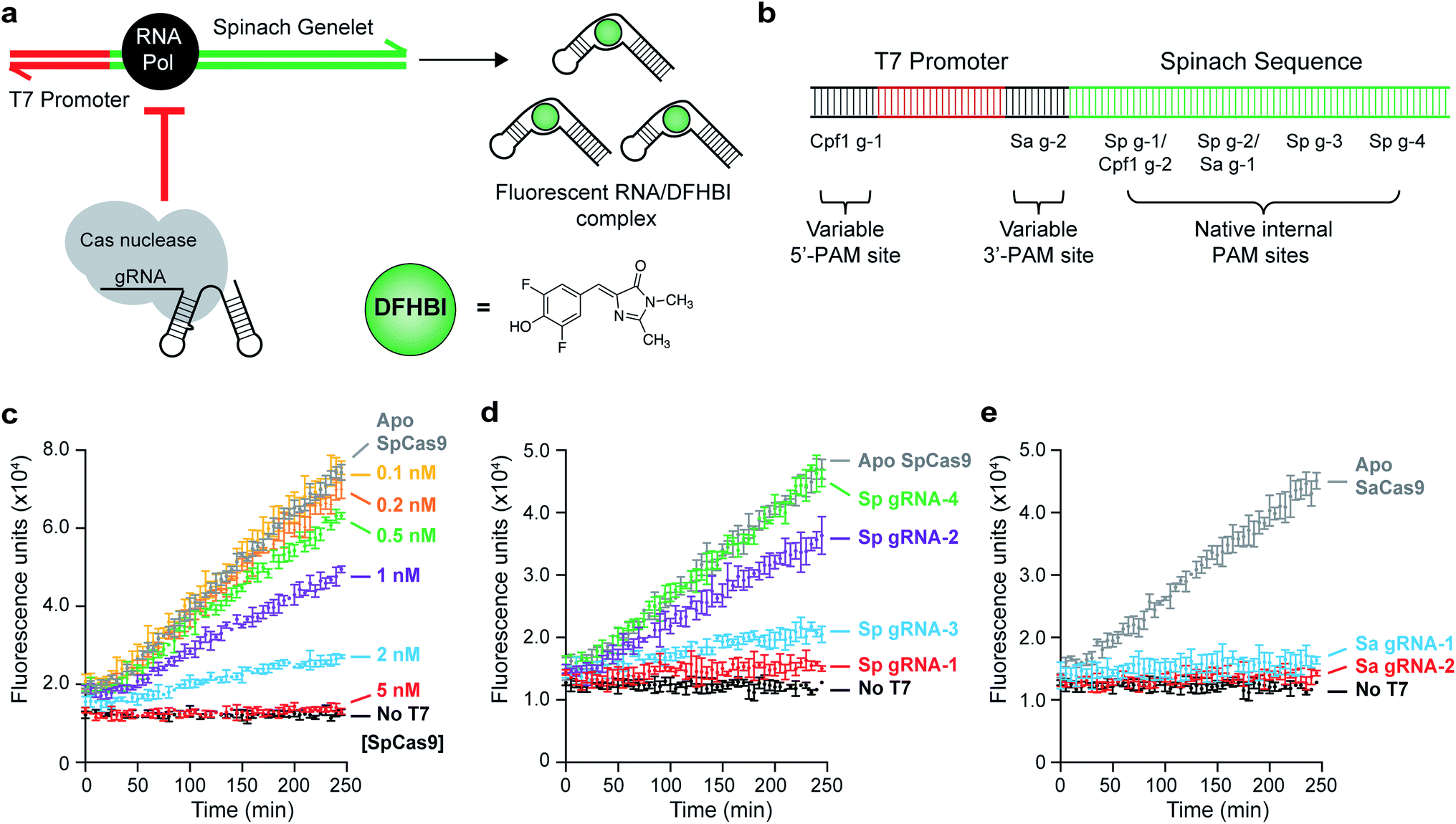 | ||
| Fig. 1 Proof of principle of a Spinach assay for detecting Cas nuclease activity. (a) Schematic of a Spinach-based assay for monitoring the activity of Cas nucleases. In the absence of nuclease, T7 RNA polymerase is recruited to a T7-promoter-containing DNA template to transcribe the Spinach RNA aptamer, which can bind to the fluorogenic molecule 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI). DNA binding or cleavage by the nuclease results in the complete termination of transcription or production of unproductive RNA, resulting in the loss of fluorescence. Cas nucleases can recognize PAM sites native to the T7 and Spinach sequences or variable PAMs that are proximal and distal to the T7 promoter. (b) Schematic of the DNA template detailing gRNA sites, both engineered and native. (c) SpCas9-gRNA targeting site Sp g-1 causes dose-dependent loss of Spinach fluorescence. ApoSpCas9 at 5 nM did not result in cleavage, indicating that this loss is due to cleavage of the Spinach DNA template. Error bars represent the standard deviation from n = 3 technical replicates. (d) SpCas9-gRNA-mediated fluorescence loss is dependent on the position of the gRNA, with PAM sites closer to the T7 promoter (in order: Sp g-1, g-2, g-3, and g-4) being more efficient. ApoSpCas9 at 2 nM did not result in cleavage. Error bars represent the standard deviation from n = 3 technical replicates. (e) Generalization of Cas nuclease-mediated inhibition of IVT to SaCas9. Active SaCas9-gRNA (5 nM) can be used at both an endogenous PAM site (Sa g-1) and an installed GGGT proximal PAM site (Sa g-2). ApoSaCas9 (5 nM) did not result in cleavage. Error bars represent the standard deviation from n = 3 technical replicates. | ||
We initiated our studies by designing gRNAs that bind to and/or cleave PAM-containing sites within the Spinach DNA template (Fig. 1b), allowing preliminary optimization of the assay with SpCas9. Indeed, we were able to titer the amount of DNA template used (0.1 nM, Fig. S1†) to detect nanomolar levels of SpCas9 activity using a gRNA-targeting site Sp g-1 (Fig. 1c). This activity was dependent on both the SpCas9 concentration and on the cleavage site. Scanning the length of the Spinach sequence with four different gRNAs (Sp g-1 through Sp g-4) revealed that binding events 5′ to the region coding for the DFHBI-binding L12 loop31 resulted in fluorescence loss, while binding after this loop still produced capable fluorogenic RNA products (Fig. 1d).
These results indicated that no modifications would be needed to assess this assay in the context of SpCas9, although it did not guarantee assay generalizability to Cas nucleases with more complex PAM recognitions. Indeed, the Spinach gene contained only one NNGGGT and TTTN site each, which are the PAM recognition sequences for SaCas9 (ref. 32) and AsCpf1/LbCpf1,33 respectively. To overcome this limitation, we inserted additional sequences in our reporter gene that could accommodate arbitrary PAM sites – one between the T7 promoter and the Spinach gene (3′-PAM site, intended for 3′-PAM-binding Cas nucleases), and one upstream of the T7 promoter (5′-PAM site, intended for 5′-PAM-binding Cas enzymes such as Cpf1). The 3′-PAM site contained a TAGGGT SaCas9 PAM, and the 5′-PAM site contained a TTTC Cpf1 PAM (Fig. 1b). Because early termination of Spinach transcription resulted in optimal fluorescence loss, we reasoned that these sites would allow direct targeting of the T7 promoter to completely abolish transcription. When comparing the activity of SaCas9 with a gRNA targeting an internal Spinach site (Sa g-1) and the 3′-PAM variable site (Sa g-2), we observed a comparable loss of DFHBI fluorescence with nanomolar levels of SaCas9 (Fig. 1e).
To assess the generalizability of our assay, we tested the activities of three different Cpf1 orthologs: AsCpf1, LbCpf1, and FnCpf1. In general, the Cpf1 orthologs had a lower activity compared to the Cas9 nucleases, as was previously reported,34 although nanomolar detection was still observable (Fig. 2). Of the orthologs tested, FnCpf1 exhibited the widest dose response range with a similar activity for the two target sites tested. While successful inhibition of fluorescence was observed for both AsCpf1 and LbCpf1, their efficiencies were lower than FnCpf1 and more dependent on the location of the gRNA site (Fig. 2a, b). The installed 5′-PAM site was generally active toward all Cpf1 orthologs tested (Fig. 2a). Interestingly, ApoAsCpf1 at 50 nM exhibited markedly higher activity compared to other ApoCpf1 orthologs, which is likely due to nonspecific collateral DNase activity that has recently been described in both AsCpf1 (ref. 35) and LbCpf1.36 To our knowledge, this is the first observation of this behavior in the absence of a bound gRNA, which was described as a prerequisite for this activity.36
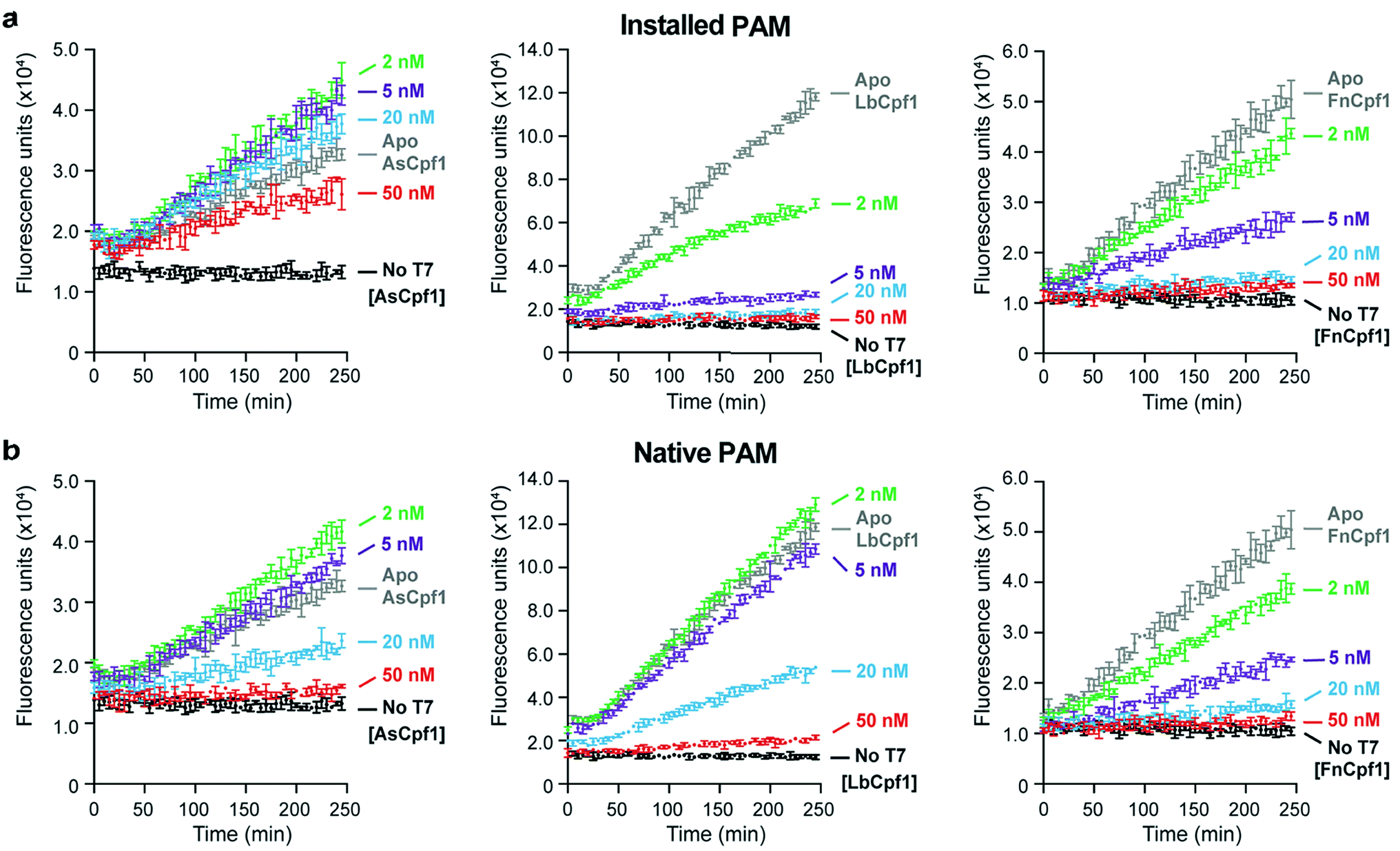 | ||
| Fig. 2 Comparison of Cpf1 activities using the Spinach assay. (a) Generalization of Cas-nuclease-mediated inhibition of IVT to AsCpf1 (left), LbCpf1 (middle), and FnCpf1 (right). Active Cpf1-gRNA binds to and/or cleaves the installed 5′-TTTC PAM site (Cpf1 g-1) in a dose-dependent manner. Error bars represent the standard deviation from n = 3 technical replicates. (b) The same assay as in (a) using the native TTTC PAM site. | ||
Mathematical modeling and data fitting
We developed a simple mathematical model (described in the Experimental section) for tracking the transcription of Spinach aptamer. The model can estimate the normalized transcription rate of the fluorescent reporter from the experimental data given in Fig. 1 and 2. The slopes of the kinetic traces in these figures are proportional to the RNA reporter transcription rate. The transcription rate, in turn, is inversely proportional to the activity of the Cas nuclease. This model divides the slope measured in each experiment by the slope of a control transcription experiment lacking the Cas nuclease to obtain a normalized slope that reflects a transcription rate unaffected by the nuclease and is therefore lower than the rate when transcription is reduced by the nuclease. The normalized slope of the kinetic traces provides a quantitative measure of the Cas9 and Cpf1 activities under different conditions. Tables S2–S11† show all the parameters obtained by fitting the model, and the fits are shown in Fig. S4–S13.† Given that the experimental conditions are the same, the activity of different enzymes can be easily compared using the calculated slope. As an example, we compared the activity of different Cpf1 orthologs presented as ‘normalized slope’ (Fig. 3). These results confirm quantitatively that, as noted earlier, the AsCpf1 prefers an internal PAM site while LbCpf1 prefers the distal PAM site, particularly at enzyme concentrations at or above 20 nM.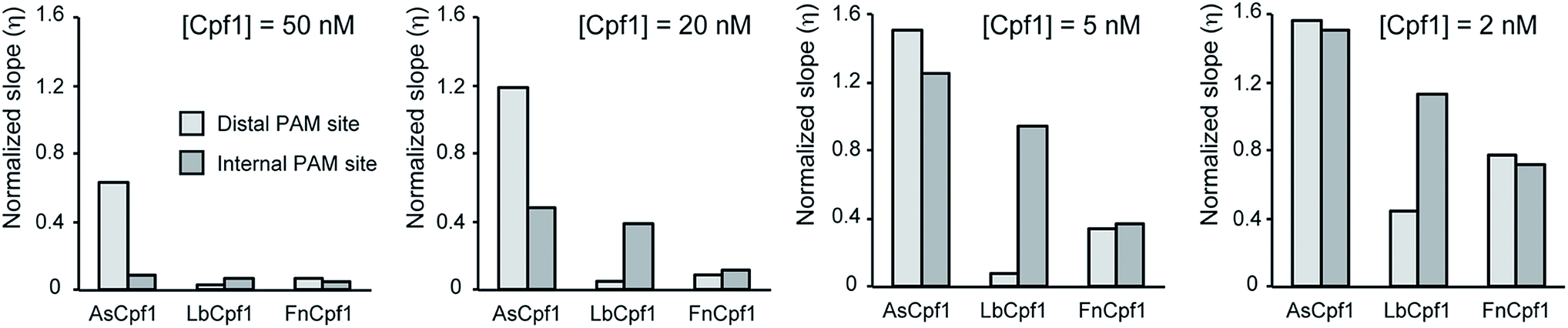 | ||
| Fig. 3 Modeling of Cpf1 activities in the Spinach assay. Comparison between the normalized slopes calculated by fitting the mathematical model to the experimental results for various Cpf1 enzymes. A higher value of η implies a higher Spinach transcription, which means a lower activity of the Cpf1 enzyme. Cpf1 enzymes behave differently when different PAM site locations are used, especially at concentrations of 20 nM and 50 nM. AsCpf1 prefers an internal PAM site while LbCpf1 prefers the distal PAM site. | ||
Monitoring anti-CRISPR inhibition of SpCas9 using the Spinach assay
Given the interest in discovering inhibitors of Cas nucleases,37,38 it is crucial to have assays that can sensitively screen for such molecules with sensitive detection of Cas nuclease activity. To that end, we surveyed the inhibitory effects of the DNA-mimetic anti-CRISPR protein AcrIIA4 (ref. 39) on SpCas9 using both a traditional gel-based DNA cleavage assay and our Spinach assay. We verified that AcrIIA4 could inhibit SpCas9 in a dose-dependent manner using a DNA cleavage assay using 100 nM of SpCas9 (Fig. 4a), recapitulating previously observed results.39 Interestingly, we were able to titrate AcrIIA4 using only 5 nM of SpCas9 in our Spinach assay, corresponding to a 20-fold increase in sensitivity (Fig. 4b). The observed findings from our Spinach assay are similar to the results obtained by the DNA cleavage assay with the added benefit of a fluorescent readout that easily quantifies the inhibition (IC50 = 9.1 nM) and directly compares the potencies of potential inhibitors (Fig. 4c). Furthermore, as we started monitoring the reaction immediately following the addition of the DNA substrate, we demonstrated that our assay allows the real-time observation of Cas9 activity in the presence of an inhibitor.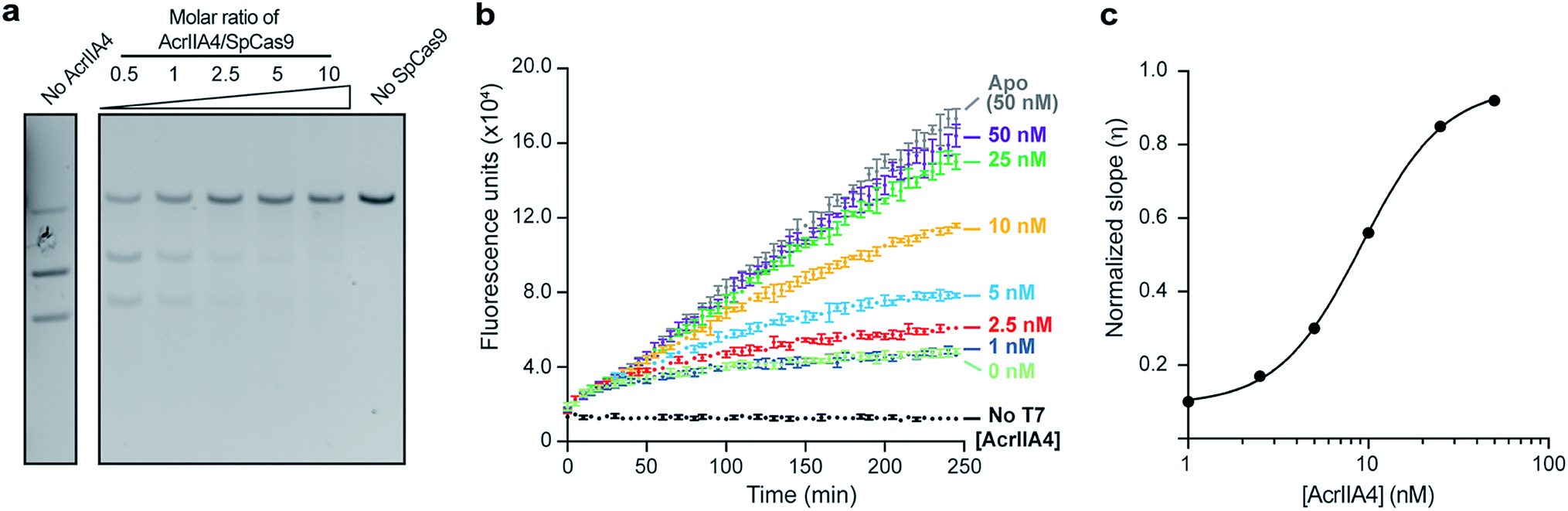 | ||
| Fig. 4 Validating AcrIIA4-mediated inhibition of SpCas9 using the Spinach assay. (a) Gel-cleavage assay showing the dose-dependent inhibition of SpCas9 (100 nM) by variable amounts of AcrIIA4. (b) Spinach assay applied to dose-dependent inhibition of SpCas9 (5 nM) by AcrIIA4. Our Spinach assay increases the detection limit of SpCas9 by 20-fold compared to the gel-cleavage assay. As SpCas9/AcrIIA4 constructs were directly added to DNA and T7 polymerase, this represents real-time monitoring of SpCas9 activity. Error bars represent the standard deviation from n = 3 technical replicates. (c) Dose curve of AcrIIA4 inhibition of SpCas9 from a model fitting the data shown in Fig. 4B. The fits are shown in Fig. S13,† and the normalized slopes are shown in Table S11.† A higher value of η implies a higher Spinach transcription and SpCas9 inhibition. The IC50 calculated from this data is 9.1 nM. | ||
Quantifying conditional activation of SpCas9 with the Spinach assay
To demonstrate further applications of this assay, we designed a conditionally activated system using SpCas9. Methods to rationally control the activity of SpCas9 or catalytically inactive SpCas9 (dSpCas9) are of interest because these enzymes can be used to regulate the activity of different genes in a cell40 or an in vitro system. Conditional activation is intended as a regulatory mechanism by which inactive SpCas9 is re-activated only in the presence of a relevant signal. A natural way to achieve this is to exploit the programmability of the gRNA molecules that activate Cas nucleases, so with this in mind, we designed a system in which toehold-mediated strand displacement controls the functionality of gRNA.41 This concept is similar to the light-activated system demonstrated by Jain et al., which uses a photocleavable gRNA locking strand to activate SpCas9 target binding and cleavage.42 In our case, we used RNA as a trigger signal to uncage the gRNA, allowing the input of the conditional switch to be the output of another gene (such as an RNA transcript) to conditionally activate SpCas9 only when the certain trigger gene is active.The gRNA was incubated with a strand complementary to the target-binding region, called the lock strand (Fig. 5a). Upon binding with the lock strand, the gRNA is inactivated. The lock strand is designed with an 8-base toehold, which enables displacement of the lock strand and activation of the gRNA using a trigger RNA that is fully complementary to the lock strand (Fig. 5a). Once the gRNA is activated, SpCas9 proceeds to bind the target DNA. We were able to show conditional activation of a T7 RNAP-driven genelet that produces Spinach RNA (Fig. 5b). In the absence of the trigger RNA, the locked gRNA could not engage with the target genelet, leading to an increase in fluorescence upon Spinach transcription (green trace). In the presence of the trigger RNA, the gRNA was activated, and the formation of the Spinach transcript was blocked, leading to no fluorescence (blue trace).
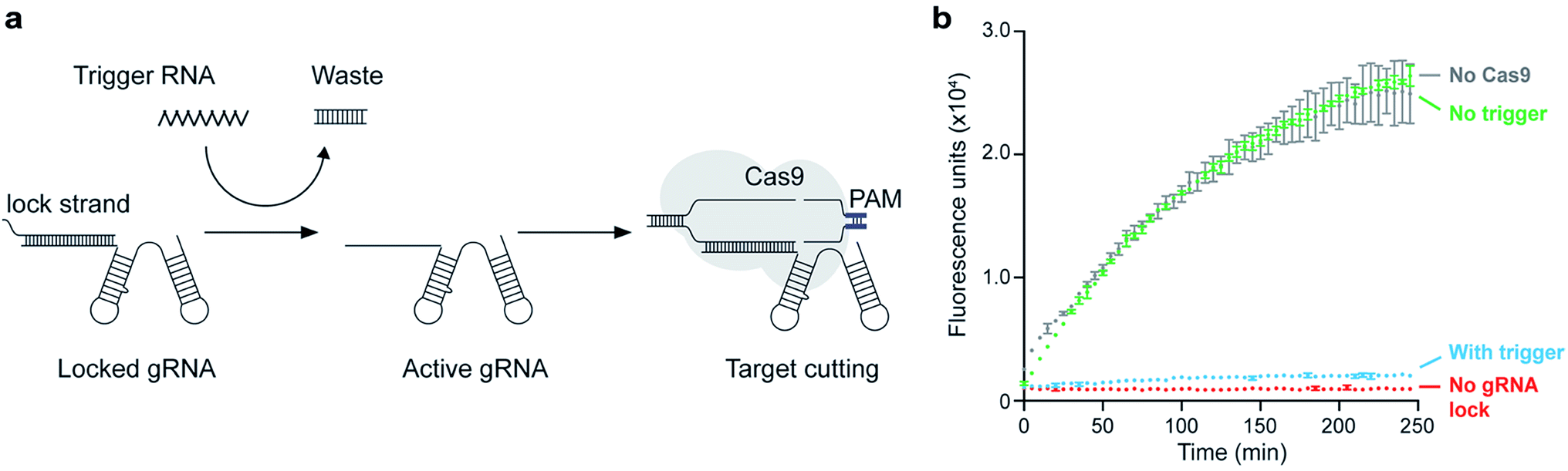 | ||
| Fig. 5 Application of Spinach assay to detect conditional activation of SpCas9. (a) Schematic diagram detailing the steps for conditional activation of SpCas9. The gRNA is pre-annealed with a complementary strand (the lock strand) that binds to the target-binding region. This inactivates the gRNA. An RNA trigger can then displace the lock strand through toehold-mediated strand displacement, thereby activating the gRNA and leading to target cleavage. (b) Demonstration of conditional activation of SpCas9 using the Spinach cleavage assay. The grey trace shows the build-up of fluorescence over time in the presence of the Spinach genelet and T7 RNAP with NTPs. The red trace shows a positive control for cleavage where gRNA and SpCas9 were added to the transcription solution (the mix was incubated at 37 °C for 16 hours prior to measuring); since the genelet is fully cleaved, there is no fluorescence. For the green trace, the gRNA was pre-annealed with a locking strand and added to transcription mix along with SpCas9; the production of Spinach aptamer is not much affected in this case, as evidenced by the fluorescence. The blue trace is a similar setup to the red trace except that there is also a trigger RNA present. The trigger unlocks the gRNA, and the genelet is fully cleaved, thereby inhibiting Spinach production. The error bars for each trace represent the standard deviation from n = 3 technical replicates. | ||
Conclusions
We have presented a cell-free assay to characterize Cas nuclease activity using the in vitro transcription of synthetic DNA templates (genelets) that produce the RNA Spinach aptamer, a well-known fluorescent reporter. This assay is simple and can be executed in any standard plate reader. As the number of available fluorescent RNA aptamer/fluorophore pairs are increasing (such as DFHBI-1T,43 Broccoli,29 Corn,44 Mango,45 Malachite Green,46 and Hoechst47), the assay could be made even more sensitive and be expanded to simultaneously track multiple Cas nuclease reactions with multiple reporters.Since Spinach transcription amplifies the template into multiple Spinach transcripts, it is possible to detect Cas9/Cpf1 activity from a very low quantity of the target template (0.1 nM). By identifying the best target locations on the Spinach genelet for optimal activity, we were able to install arbitrary PAM sites to study any conceivable Cas nuclease. The ease in the design of the genelet to suit any type of Cas nuclease makes this a versatile assay, which we demonstrate by detecting the activities of five Cas nucleases from two different CRISPR-types. For SpCas9 and SaCas9, we showed detection of up to low nanomolar levels of the enzyme, allowing for more biologically relevant characterizations of activity without the need for radiolabeled nucleotides. Despite the Cpf1 nucleases having lower activities than their Cas9 counterparts, we were also able to profile their activity in the nanomolar range. Interestingly, we reveal different activities in terms of target site preference and cleavage efficiency between the different Cpf1 orthologs. Our simple mathematical model to fit the experimental results has enabled us to quantitatively compare activities of Cas9 and Cpf1 enzymes across experiments, which is particularly important in high-throughput experiments where activities of various enzyme inhibitors and enhancers are being probed. Furthermore, we show that this assay can be easily used to titrate anti-CRISPR molecules and quantitatively access their inhibitory effects on SpCas9. Due to the low concentrations of SpCas9 needed, this assay would be a useful starting point to screen for potential inhibitors. Thus, the Spinach assay provides a general, quantitative, sensitive, and moderate-throughput means to assess Cas nuclease activity in a general fashion.
As a proof-of-concept application of our Spinach assay, we demonstrated a simple conditional switch to activate SpCas9. Such conditional switches are critical components in logic and dynamic regulatory modules using Cas nucleases, including oscillators and bistable systems.48 The Cas9-based conditional switch proposed here could be a useful addition to the toolbox of conditional switches used in synthetic biology. To be practical for in vivo applications, the trigger sequence should be independent of the target. In our current design, this is not the case, but such independence can be achieved using transduction gates that use toehold-mediated strand displacement to decouple the input signal from the target.12,49
Given the impact Cas nucleases have had in a wide array of fields, tools to systematically characterize their activity and kinetic behavior in a variety of conditions are extremely valuable. While crystallography and other such techniques can yield structural insights into the action of Cas nucleases, studying their kinetic and dynamic mechanisms require high-throughput assays conducted in a well-controlled environment. In vitro cell-free methods like the one described herein are well suited for this purpose. Another advantage of our approach is that it continuously monitors the operation of the gene targeted by the Cas nuclease; this suggests that we will also be able to use our in vitro assays to quantify how small molecules or other enzymes interact with or kinetically regulate the activity of Cas9 nucleases. For example, it was recently reported that the procession of RNA polymerase could dislodge Cas9 from bound and cleaved DNA and that this effect is enhanced if Cas9 has annealed to the template strand.50 Furthermore, this release of Cas9 allows multiple turnovers to take place. As our Spinach assay directly assesses Cas nuclease activity as a function of RNA polymerase transcription and provides a method for continuous detection, we can directly study the effects of active transcription for SpCas9 as well as a variety of different Cas nucleases. Thus, we foresee that our assay will enable a variety of novel studies on the complex mechanics of Cas nuclease-mediated genome editing in a controlled environment.
Experimental
Cas9/Cpf1 and AcrIIA4 protein expression and purification
Streptococcus pyogenes Cas9 (SpCas9), Streptococcus aureus Cas9 (SaCas9), Acidaminococcus sp. Cpf1 (AsCpf1), Lachnospiraceae bacterium ND2006 Cpf1 (LbCpf1), Francisella tularensis subsp. novicida Cpf1 (FnCpf1), and AcrIIA4 from Listeria monocytogenes were expressed in E. coli BL21 (DE3) Rosetta cells and purified as previously described (Fig. S2†).23,33,37IVT DNA oligonucleotides and gRNA synthesis
All oligonucleotides were purchased from IDT DNA (Coralville, IA) and were desalted for in vitro transcription experiments. The oligonucleotides used in the conditional activation experiments were purchased from IDT with PAGE purification. Oligo-annealing solutions were prepared by mixing complementary strands (10 μM final concentration) together in 1× Cas9 assay buffer (20 mM of Tris–HCl, pH = 7.5; 150 mM of KCl; 1 mM of EDTA; 50 mM of MgCl2). The oligonucleotides were annealed by heating to 95 °C for five minutes followed by slow cooling to 25 °C at a rate of 0.1 °C s−1 to produce a double-stranded oligonucleotide. The gRNA templates were generated by annealing a sequence-specific oligo with either a “universal” oligo containing the Cas-nuclease-specific tracrRNA backbone (SpCas9 and SaCas9) or a T7 oligo to yield a double-stranded T7 promoter (all Cpf1 nucleases). See Table S1† for a list of all sequences. RNA was transcribed using HiScribe T7 Quick High Yield RNA Synthesis Kits (NEB, E2050S). To assess gRNA quality, 200 ng of purified RNA was heated to 95 °C in loading buffer (ThermoFisher, LC6876) and 50 mM of EDTA for 10 minutes and run on a 15% TBE-Urea gel (ThermoFisher, EC68855) for 70 minutes at 200 V. The gels were stained with Sybr Green (ThermoFisher) and imaged by UV (Fig. S3†).Fluorescence data acquisition and gel imaging
For in vitro transcription assays, fluorescence was continuously monitored at 37 °C using an i3x SpectraMax (Molecular Devices; Spinach-DFHBI – Ex 468/Em 502 nm; black, 384-well plate with plastic lid and clear bottom; bottom read; Corning, 8794BC). For conditional Cas9 experiments, fluorescence was measured on an H1m plate reader (Biotek Instruments; Spinach-DFHBI – Ex 469/Em 501 nm; fluorescein – Ex 495/Em 520; 96-well V-bottom plates; Corning Costar 3357). The 96-well plates were sealed during measurement using Costar 3080 microplate storage mats. Gel images were acquired with a C600 (Azure Biosystems).Plasmid cleavage and AcrIIA4 inhibition assays
In a typical DNA-cleavage assay, the Cas9:gRNA complex was formed by mixing each component at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 (Cas9:gRNA) molar ratio and incubating at room temperature for 5–15 minutes. Cas9:gRNA (100 nM) and AcrIIA4 protein (variable concentration) were mixed together in 1× assay buffer for five minutes at room temperature prior to the addition of 5 nM (100 ng per 20 μL) of linearized plasmid and incubated at 37 °C for 30 min. A T7-promoter containing the Spinach sequence cloned into pUC57-Kan and linearized with AsiS1 was used as the plasmid substrate. Loading buffer (6×, NEB) was directly added to reactions and run on a 1.4% agarose gel containing 0.01% ethidium bromide and imaged by UV. The IC50 was determined using GraphPad with fitting to the Hill equation.
:1.2 (Cas9:gRNA) molar ratio and incubating at room temperature for 5–15 minutes. Cas9:gRNA (100 nM) and AcrIIA4 protein (variable concentration) were mixed together in 1× assay buffer for five minutes at room temperature prior to the addition of 5 nM (100 ng per 20 μL) of linearized plasmid and incubated at 37 °C for 30 min. A T7-promoter containing the Spinach sequence cloned into pUC57-Kan and linearized with AsiS1 was used as the plasmid substrate. Loading buffer (6×, NEB) was directly added to reactions and run on a 1.4% agarose gel containing 0.01% ethidium bromide and imaged by UV. The IC50 was determined using GraphPad with fitting to the Hill equation.
Cas-nuclease-binding in vitro transcription Spinach assay
HiScribe T7 High Yield RNA in vitro transcription kits were purchased from NEB (E2040S). The Spinach aptamer template and non-template oligonucleotides were annealed as described above. In a typical assay, a Cas9/Cpf1:gRNA complex was first formed as described above. Cas9/Cpf1 without gRNA (ApoCas9/Cpf1) was treated similarly. A typical assay was performed by mixing the following components together from 10× stocks to get the indicated final concentrations: NTPs (6.7 mM), 10× T7 reaction buffer (0.67×), murine RNase inhibitor (1.3 U; NEB, M0314L), DFHBI (1 μM; Lucerna), DNA template (0.1 nM), and water to a final volume of 25 μL. ApoCas9/Cpf1 or Cas9/Cpf1:gRNA complexes (10×) were added to initiate cleavage and were incubated at 37 °C for 30 minutes. Transcription was initiated by adding 2 μL of T7 RNA polymerase or was omitted to assess the background fluorescence. Reactions (27 μL) were transferred to a 384-well plate and the fluorescence was monitored at 37 °C for up to four hours. For AcrIIA4 inhibition experiments, the Cas9:gRNA complexes were incubated with AcrIIA4 for five minutes in the presence of NTPs, reaction buffer, murine RNase inhibitor, and DFHBI at room temperature. The DNA template was added followed immediately by T7 RNA polymerase and incubation at 37 °C.RNA synthesis and purification for conditional activation experiments
The trigger RNA and gRNA used for the conditional activation experiments were synthesized from gene templates and purified using polyacrylamide gel electrophoresis (PAGE). For gRNA, the dsDNA template was ordered from IDT DNA (Coralville, IA) using the ‘gblocks’ option. For trigger RNA, the individual template and non-template strand were ordered form IDT DNA with PAGE purification and annealed in 1× NEB transcription buffer (40 mM of Tris–HCl, 6 mM of MgCl2, 2 mM of spermidine, 1 mM of dithiothreitol, pH 7.9 at 25 °C) prior to transcription. Both gene templates were designed with a T7 RNAP promoter. RNA strands were individually transcribed in vitro using the AmpliScribe T7-Flash transcription kit (#ASF3507, Epicenter, Inc.) from the corresponding DNA templates (Table S1†). RNA strands were gel extracted using 10% denaturing polyacrylamide gel electrophoresis (PAGE). Next, the RNA was eluted using 0.3 M of sodium acetate at pH 5.3. Finally, the RNA was precipitated using ethanol and glycogen. The samples were then resuspended in 10–15 μL of Ambion nuclease-free water. The concentration was measured by absorbance at 260 nm using a Nanodrop 2000c Spectrophotometer.Spinach assay demonstrating conditional activation of Cas9
First, the locking strand, pre-annealed Spinach gene, and the gRNA were incubated at room temperature for 15 minutes (the locking strand was at 3.75× excess of the gRNA and 15× excess of the Spinach gene) in the presence of 1× NEB transcription buffer (40 mM of Tris–HCl, 6 mM of MgCl2, 2 mM of spermidine, 1 mM of dithiothreitol, pH 7.9 at 25 °C). The mixture was then incubated at 37 °C with the trigger RNA strand (at 1.3× excess of the locking strand) and Cas9 (at a final concentration of 0.3 μM; NEB catalog #M0386T) for 10 minutes. To this mixture, NTPs (5 mM each final concentration), DFHBI (5 mM each final concentration), and MgCl2 (14 mM each final concentration) were added. The final concentrations of the nucleic acid strands in the solution were: Spinach gene – 0.15 μM; locking strand – 2.25 μM, gRNA – 0.6 μM; trigger RNA – 3 μM. For controls lacking a specific ingredient, the mixture was prepared by skipping the appropriate ingredient and making up the missing volume with water. This reaction mixture was incubated at 37 °C for 15 hours. After incubation, 2.4 μL of T7 RNAP (200 U μL−1; Cellscript, Catalog # C-T7300K) was added to 37.6 μL of the Cas9 reaction mixture for a final volume of 40 μL. Fluorescence measurements were made with 12 μL of the mixture in one well of the 96-well plate. To average out the variation between measurements in different wells, three 12 μL wells were measured per sample.Mathematical model and data fitting
We developed a simple model to quantify the relative changes in transcription rate of our template variants after Cas9 incubation. We denoted with e(t) the concentration of unbound (free) T7 RNAP and with etot the total concentration of RNAP; g indicated the concentration of unbound (free) Spinach reporter template, and gtot indicated the total concentration of template.We assumed that the reactions, including template, gRNA, and Cas9, reach a steady state prior to the addition of T7 RNAP. Therefore, when transcription starts, the concentration of template that has been cleaved by Cas9 is at steady state. The goal of the model is to quantify how pre-incubation with Cas9 affects the transcription rate of the template sample.
Typically, one would model the RNA transcription velocity as
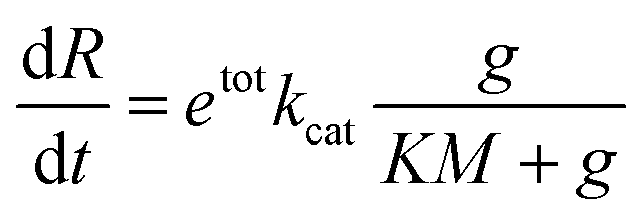
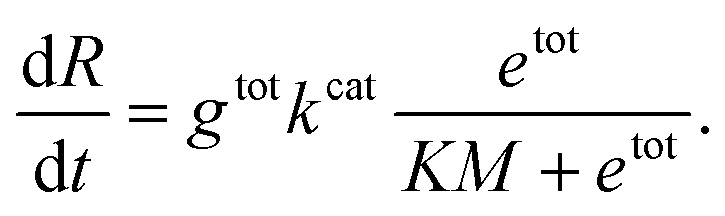
In this expression, kcat and KM are determined by the sequence and length of the promoter and the surrounding template domains; therefore, these parameters are influenced by the pre-incubation reaction with Cas9 and by the location of the PAM sequence. Further, the concentration of template gtot that is a viable substrate for T7 depends on the efficiency of Cas9 activity. Finally, during the first hours of transcription during which the enzyme activity does not fluctuate significantly, the term
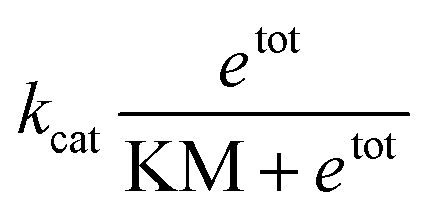
Instead of estimating all the parameters of our simple model, we quantified how the transcription rate varied in distinct assays by normalizing their outcome in relation to a control experiment in which the template was not pre-incubated with Cas9. The Spinach assays provide a fluorescence signal representing the RNA concentration over time; the slope of this fluorescence signal is proportional to the transcription rate. However, the RNA concentration and transcription rate can be directly estimated only via calibration experiments mapping Spinach RNA concentrations to the corresponding fluorescence levels.51 In contrast, the relative change in the slope can be quantified without a fluorescence calibration by simply dividing the slope of each transcription experiment by the slope of the corresponding control experiment (in which the template was not pre-incubated with Cas9).
The normalized slope is
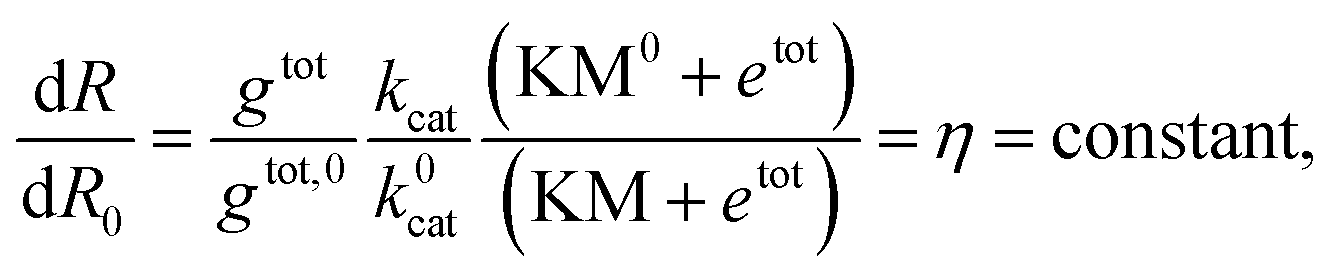
| (F = S × t + b) |
Author contributions
K. J. C. and H. K. K. S. contributed equally to this work.Conflicts of interest
Broad Institute has filed PCT/US2017/59365 and PCT/US2018/058466 that claim inventions relating to assays in this manuscript.Acknowledgements
This work was supported by the Burroughs Wellcome Fund (Career Award at the Scientific Interface to A. C.), DARPA (Brdi N66001-17-2-4055 to A. C.), NIH (1R21AI126239-01 to A. C.), Army Research Office award W911NF1610586 (to A. C.), and by the Department of Energy through grant DE-SC001 0426 to E. F., which supported the salary of H. K. K. S. This work is dedicated to Professor Ronald T. Raines on the occasion of his 60th birthday.References
- E. V. Koonin, K. S. Makarova and F. Zhang, Curr. Opin. Microbiol., 2017, 37, 67–78 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef PubMed.
- J. D. Sander and J. K. Joung, Nat. Biotechnol., 2014, 32, 347–355 CrossRef CAS PubMed.
- A. C. Komor, A. H. Badran and D. R. Liu, Cell, 2017, 168, 20–36 CrossRef CAS PubMed.
- A. C. Komor, A. H. Badran and D. R. Liu, ACS Chem. Biol., 2018, 13, 383–388 CrossRef CAS PubMed.
- B. P. Kleinstiver, M. S. Prew, S. Q. Tsai, V. V. Topkar, N. T. Nguyen, Z. Zheng, A. P. Gonzales, Z. Li, R. T. Peterson, J. R. Yeh, M. J. Aryee and J. K. Joung, Nature, 2015, 523, 481–485 CrossRef PubMed.
- H. K. Kim, M. Song, J. Lee, A. V. Menon, S. Jung, Y. M. Kang, J. W. Choi, E. Woo, H. C. Koh, J. W. Nam and H. Kim, Nat. Methods, 2017, 14, 153–159 CrossRef CAS PubMed.
- T. R. deBoer, N. Wauford, J. Y. Chung, M. S. Torres Perez and N. Murthy, ACS Chem. Biol., 2018, 13, 461–466 CrossRef CAS PubMed.
- A. Hendel, R. O. Bak, J. T. Clark, A. B. Kennedy, D. E. Ryan, S. Roy, I. Steinfeld, B. D. Lunstad, R. J. Kaiser, A. B. Wilkens, R. Bacchetta, A. Tsalenko, D. Dellinger, L. Bruhn and M. H. Porteus, Nat. Biotechnol., 2015, 33, 985–989 CrossRef CAS PubMed.
- D. E. Ryan, D. Taussig, I. Steinfeld, S. M. Phadnis, B. D. Lunstad, M. Singh, X. Vuong, K. D. Okochi, R. McCaffrey, M. Olesiak, S. Roy, C. W. Yung, B. Curry, J. R. Sampson, L. Bruhn and D. J. Dellinger, Nucleic Acids Res., 2018, 46, 792–803 CrossRef CAS PubMed.
- N. Srinivas, J. Parkin, G. Seelig, E. Winfree and D. Soloveichik, Science, 2017, 358 Search PubMed.
- D. Y. Zhang, A. J. Turberfield, B. Yurke and E. Winfree, Science, 2007, 318, 1121–1125 CrossRef CAS PubMed.
- L. Qian and E. Winfree, Science, 2011, 332, 1196–1201 CrossRef CAS PubMed.
- E. Franco, E. Friedrichs, J. Kim, R. Jungmann, R. Murray, E. Winfree and F. C. Simmel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E784–E793 CrossRef CAS PubMed.
- C. Cuba Samaniego and E. Franco, An ultrasensitive motif for robust closed loop control of biomolecular systems, 2017 IEEE 56th Annual Conference on Decision and Control (CDC), Melbourne, VIC, 2017, pp. 5334–5340 Search PubMed.
- A. T. Raper, A. A. Stephenson and Z. Suo, J. Am. Chem. Soc., 2018, 140, 2971–2984 CrossRef CAS PubMed.
- C. D. Richardson, G. J. Ray, M. A. DeWitt, G. L. Curie and J. E. Corn, Nat. Biotechnol., 2016, 34, 339–344 CrossRef CAS PubMed.
- M. A. Moreno-Mateos, J. P. Fernandez, R. Rouet, C. E. Vejnar, M. A. Lane, E. Mis, M. K. Khokha, J. A. Doudna and A. J. Giraldez, Nat. Commun., 2017, 8, 2024 CrossRef PubMed.
- K. Zhang, R. Deng, Y. Li, L. Zhang and J. Li, Chem. Sci., 2016, 7, 4951–4957 RSC.
- M. Huang, X. Zhou, H. Wang and D. Xing, Anal. Chem., 2018, 90, 2193–2200 CrossRef CAS PubMed.
- C. C. Bell, G. W. Magor, K. R. Gillinder and A. C. Perkins, BMC Genomics, 2014, 15, 1002 CrossRef PubMed.
- V. Pattanayak, S. Lin, J. P. Guilinger, E. Ma, J. A. Doudna and D. R. Liu, Nat. Biotechnol., 2013, 31, 839–843 CrossRef CAS PubMed.
- R. Marshall, C. S. Maxwell, S. P. Collins, T. Jacobsen, M. L. Luo, M. B. Begemann, B. N. Gray, E. January, A. Singer, Y. He, C. L. Beisel and V. Noireaux, Mol. Cell, 2018, 69, 146–157 CrossRef CAS PubMed.
- W. Liu, H. Yu, X. Zhou and D. Xing, Anal. Chem., 2016, 88, 8369–8374 CrossRef CAS PubMed.
- K. J. Seamon, Y. K. Light, E. A. Saada, J. S. Schoeniger and B. Harmon, Anal. Chem., 2018, 90, 6913–6921 CrossRef CAS PubMed.
- V. Mekler, L. Minakhin, E. Semenova, K. Kuznedelov and K. Severinov, Nucleic Acids Res., 2016, 44, 2837–2845 CrossRef PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed.
- K. Hofer, L. V. Langejurgen and A. Jaschke, J. Am. Chem. Soc., 2013, 135, 13692–13694 CrossRef PubMed.
- H. Huang, N. B. Suslov, N. S. Li, S. A. Shelke, M. E. Evans, Y. Koldobskaya, P. A. Rice and J. A. Piccirilli, Nat. Chem. Biol., 2014, 10, 686–691 CrossRef CAS PubMed.
- A. E. Friedland, R. Baral, P. Singhal, K. Loveluck, S. Shen, M. Sanchez, E. Marco, G. M. Gotta, M. L. Maeder, E. M. Kennedy, A. V. Kornepati, A. Sousa, M. A. Collins, H. Jayaram, B. R. Cullen and D. Bumcrot, Genome Biol., 2015, 16, 257 CrossRef PubMed.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed.
- D. Kim, J. Kim, J. K. Hur, K. W. Been, S. H. Yoon and J. S. Kim, Nat. Biotechnol., 2016, 34, 863–868 CrossRef CAS PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang, Science, 2018, 360, 439–444 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- L. B. Harrington, K. W. Doxzen, E. Ma, J. J. Liu, G. J. Knott, A. Edraki, B. Garcia, N. Amrani, J. S. Chen, J. C. Cofsky, P. J. Kranzusch, E. J. Sontheimer, A. R. Davidson, K. L. Maxwell and J. A. Doudna, Cell, 2017, 170, 1224–1233 CrossRef CAS PubMed.
- J. Shin, F. Jiang, J. J. Liu, N. L. Bray, B. J. Rauch, S. H. Baik, E. Nogales, J. Bondy-Denomy, J. E. Corn and J. A. Doudna, Sci. Adv., 2017, 3, e1701620 CrossRef PubMed.
- H. Yang and D. J. Patel, Mol. Cell, 2017, 67, 117–127 CrossRef CAS PubMed.
- M. Deaner and H. S. Alper, Metab. Eng., 2017, 40, 14–22 CrossRef CAS PubMed.
- B. Yurke and A. P. Mills, Genet. Program Evolvable Mach., 2003, vol. 4, pp. 111–122 Search PubMed.
- P. K. Jain, V. Ramanan, A. G. Schepers, N. S. Dalvie, A. Panda, H. E. Fleming and S. N. Bhatia, Angew. Chem., Int. Ed. Engl., 2016, 55, 12440–12444 CrossRef CAS PubMed.
- W. Song, R. L. Strack, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 1198–1201 CrossRef CAS PubMed.
- W. Song, G. S. Filonov, H. Kim, M. Hirsch, X. Li, J. D. Moon and S. R. Jaffrey, Nat. Chem. Biol., 2017, 13, 1187–1194 CrossRef CAS PubMed.
- E. V. Dolgosheina, S. C. Jeng, S. S. Panchapakesan, R. Cojocaru, P. S. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, ACS Chem. Biol., 2014, 9, 2412–2420 CrossRef CAS PubMed.
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2005, 127, 12442–12443 CrossRef CAS PubMed.
- S. Sando, A. Narita, M. Hayami and Y. Aoyama, Chem. Commun., 2008, 33, 3858–3860 RSC.
- C. Cuba Samaniego, H. K. K. Subramanian and E. Franco, Design of a bistable network using the CRISPR/Cas system, 2017 IEEE Conference on Control Technology and Applications (CCTA), Mauna Lani, HI, 2017, pp. 973–978 Search PubMed.
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS PubMed.
- R. Clarke, R. Heler, M. S. MacDougall, N. C. Yeo, A. Chavez, M. Regan, L. Hanakahi, G. M. Church, L. A. Marraffini and B. J. Merrill, Mol. Cell, 2018, 71, 42–55 CrossRef CAS PubMed.
- J. Lloyd, C. H. Tran, K. Wadhwani, C. Cuba Samaniego, H. K. K. Subramanian and E. Franco, ACS Synth. Biol., 2018, 7, 30–37 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03426e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |