
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi(II)-catalyzed mono-selective ortho-arylation of unactivated aryl C–H bonds utilizing amino acids as a directing group†
Zhanqing
Cong‡
abc,
Feng
Gao‡
ab and
Hong
Liu
 *abc
*abc
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zu Chong Zhi Road, Shanghai, 201203, China. E-mail: hliu@simm.ac.cn
bUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, China
cSchool of Life Science and Technology, ShanghaiTech University, 100 Haike Road, Shanghai 201210, China
First published on 8th April 2019
Abstract
The nickel(II)-catalyzed ortho-arylation of unactivated C–H bonds utilizing amino acids as directing groups with aryl iodides or bromides as coupling electrophiles is described. This protocol features excellent mono-selectivity, good regioselectivity, and wide functional group tolerance. Additionally, the obtained products bearing a biaryl motif and an amino acid represent bioactive molecules with wide bioactivities.
Introduction
Biaryls are privileged π-conjugated structural units, and are widely found in pharmaceuticals, agrochemicals, and materials science (Fig. 1).1 Despite the significant progress made in achieving biaryl scaffolds via transition metal-catalyzed cross-coupling between aryl halides and organometallic reagents,2 these reliable methods suffer from harsh conditions, prefunctionalization of substrates, and the extra generation of metal halides. Therefore, there remains the need to develop efficient and environmental-friendly synthetic methods to furnish biaryl derivatives.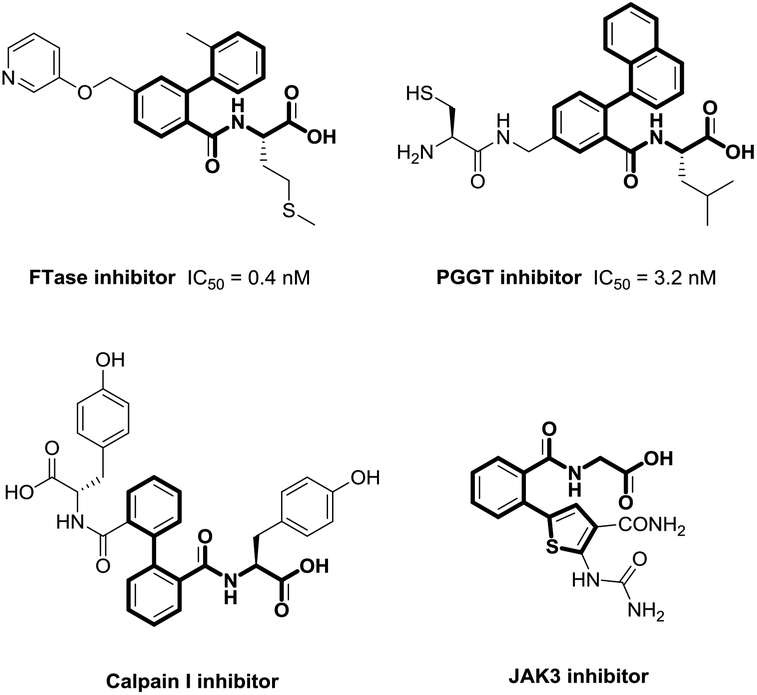 | ||
| Fig. 1 Representative bioactive molecules. | ||
Over the past few decades, transition metal-catalyzed direct C(sp2)–H arylation has been extensively studied as an attractive and complementary access to construct biaryl derivatives.3 In this context, chelation-assisted strategy has been demonstrated as one of the most powerful methods for regioselective transforming C–H bonds.4 A wide variety of monodentate or bidentate directing groups have been evaluated to achieve transition metal-mediated regioselective C–H activation, thus, compatible with broad substrates. However, these methodologies mostly rely on the use of expensive and toxic second- or third-row transition metals, such as palladium, ruthenium, and rhodium (Scheme 1a). Recent attention has been shifted on earth-abundant 3d transition metals.5 Among them, nickel is emerging as a robust and versatile catalyst for C–H activation, owing to its unique activity and low-cost.6 Pioneered by Chatani's work,7 the combination of nickel catalysis and N-heterocyclic bidentate directing groups, specifically referring to 8-aminoquinoline (AQ)8 and (pyridin-2-yl)isopropyl (PIP),9 has shown superior activity for the construction of aromatic C–C bonds. But in most cases, the selectivity for monoarylation versus diarylation was not ideal with the aid of AQ or PIP (Scheme 1b).8h,i,9b,c Very recently, environmental-friendly and inexpensive amino acids have been employed as novel bidentate directing groups, which have been well demonstrated in palladium-catalyzed C–H functionalization and showed extraordinary reactivity.10 In continuation of our recent effort on direct C–H functionalization,11 we proposed the construction of biaryl derivatives via Ni(II)-catalyzed amino acid directed C–H cleavage in a mono-selective manner. It is worth notice that amino acids act as not only directing groups, but also a crucial part of the final products with wide potential bioactivities.12 Herein, we reported the nickel-catalyzed highly mono-selective ortho-arylation of unactivated aryl C–H bonds utilizing amino acid as a directing group (Scheme 1c).
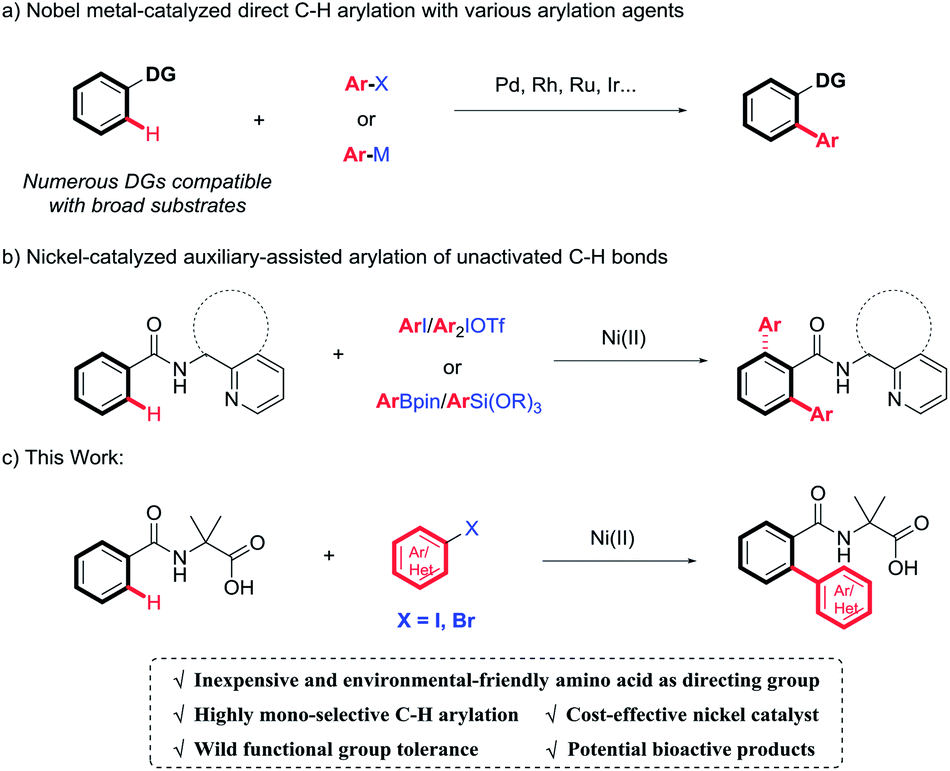 | ||
| Scheme 1 Transition metal-catalyzed arylation of unactivated C–H Bonds. | ||
Results and discussion
We initiated our studies by investigating the reaction parameters of the coupling between N-benzoyl α-amino acid 1a with 1-iodo-4-methylbenzene 2a using commercially available nickel(II) salts (Table 1). All of the nickel complexes showed catalytic activity and Ni(OTf)2 was proved to be the most effective catalyst, affording the desired mono-arylation product 3a in 82% yield (entries 1–4). The efficiency of the reaction was significantly affected by different solvents with DMSO being identified as optimal and no desired product was obtained in nonpolar solvent (entries 5–8). Further optimization of bases revealed that Na2CO3 was crucial for the conversion (entries 9–11). Additionally, the yield was decreased without MesCOOH (entry 12). The absence of TBAI also led to a slight decrease of yield (entry 13). By contrast, the reaction gave no conversion when nickel(II) catalyst was omitted (entry 14).|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | [Ni] | Base | Solvent | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), catalyst (10 mol%), MesCOOH (20 mol%), base (0.6 mmol), TBAI (0.2 mmol), solvent (2 mL), 140 °C, air, 16 h. b NMR yield. Values in parentheses are the isolated yields of 3a. c In absence of MesCOOH. d In absence of TBAI. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Ni(acac)2 | Na2CO3 | DMF | 13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | NiCl2 | Na2CO3 | DMF | 21 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | NiI2 | Na2CO3 | DMF | 12 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | Ni(OTf)2 | Na2CO3 | DMF | 82 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Ni(OTf)2 | Na2CO3 | NMP | 25 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Ni(OTf)2 | Na2CO3 | DMSO | 88 (81) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Ni(OTf)2 | Na2CO3 | Toluene | N.R. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | Ni(OTf)2 | Na2CO3 | Dioxane | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | Ni(OTf)2 | KOAc | DMSO | N.R. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | Ni(OTf)2 | K3PO4 | DMSO | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | Ni(OTf)2 | NaHCO3 | DMSO | Trace | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12c | Ni(OTf)2 | Na2CO3 | DMSO | 73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13d | Ni(OTf)2 | Na2CO3 | DMSO | 62 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | — | Na2CO3 | DMSO | N.R. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
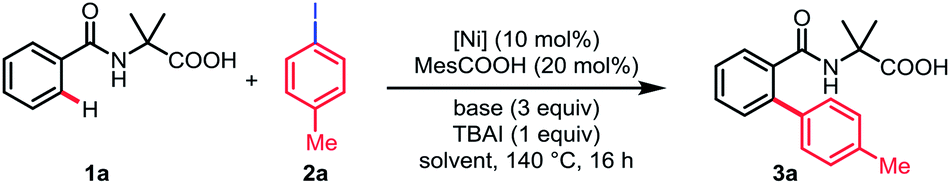
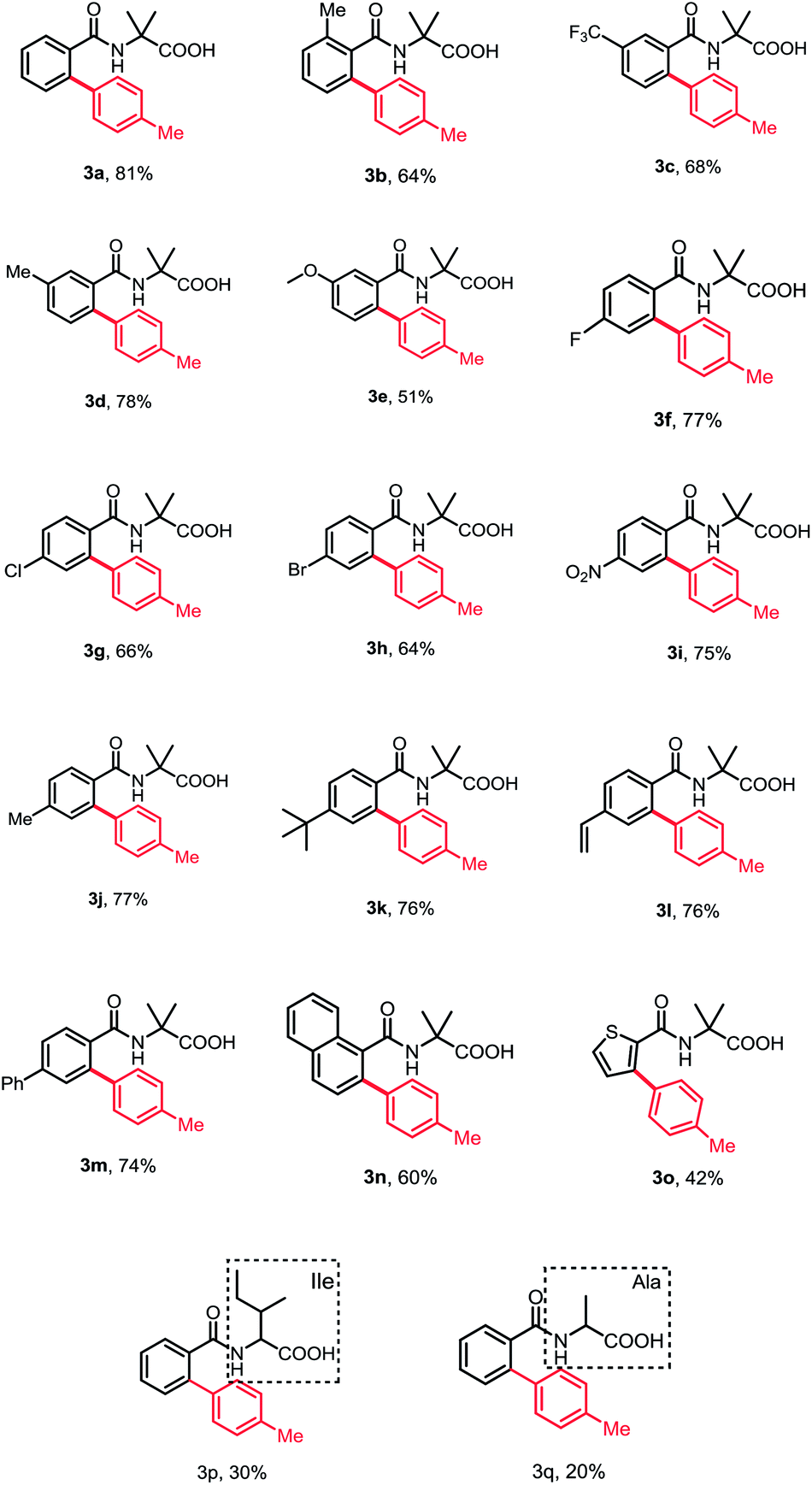
With the optimized reaction conditions in hand, we investigated the substrate scope of N-benzoyl α-amino acid derivatives. Generally, various substituents on the aromatic ring were tolerated in this reaction and generated the corresponding products in moderate to high yields without diarylation products being detected (Table 2). The aromatic ring bearing a substituent at ortho-position, such as methyl, could afford the desired products in moderate yield, probably due to the steric hindrance (3b). When meta-substituted substrates were employed, the C–H bond arylation took place at the less sterically hindered position in good yields (3c–3e). To our delight, the substrates bearing either electron-donating or -withdrawing groups at para-position furnished the mono-arylation products selectively in satisfactory yields, irrespective of the electronic nature of the substituents (3f–3m). Halogens were also well tolerated under standard conditions, revealing the protocol may have more potential applications (3f–3h). Furthermore, the naphthyl and thienyl substrates afforded the desired products in moderate yields (3n and 3o). The other natural amino acid directing groups have showed less activity in this reaction, probably due to the absence of the Thorpe–Ingold effect (3p and 3q).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1 (0.2 mmol), 2a (0.6 mmol), Ni(OTf)2 (10 mol%), MesCOOH (20 mol%), Na2CO3 (0.6 mmol), and TBAI (0.2 mmol) in DMSO (2 mL) at 140 °C for 16 h. All listed yields are isolated ones. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
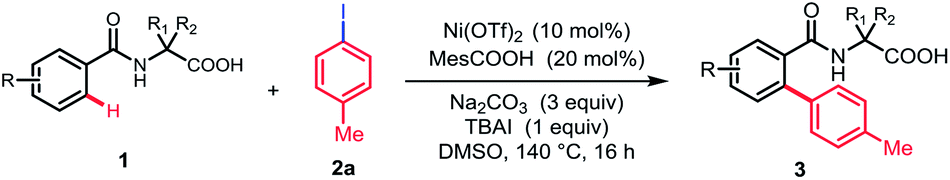
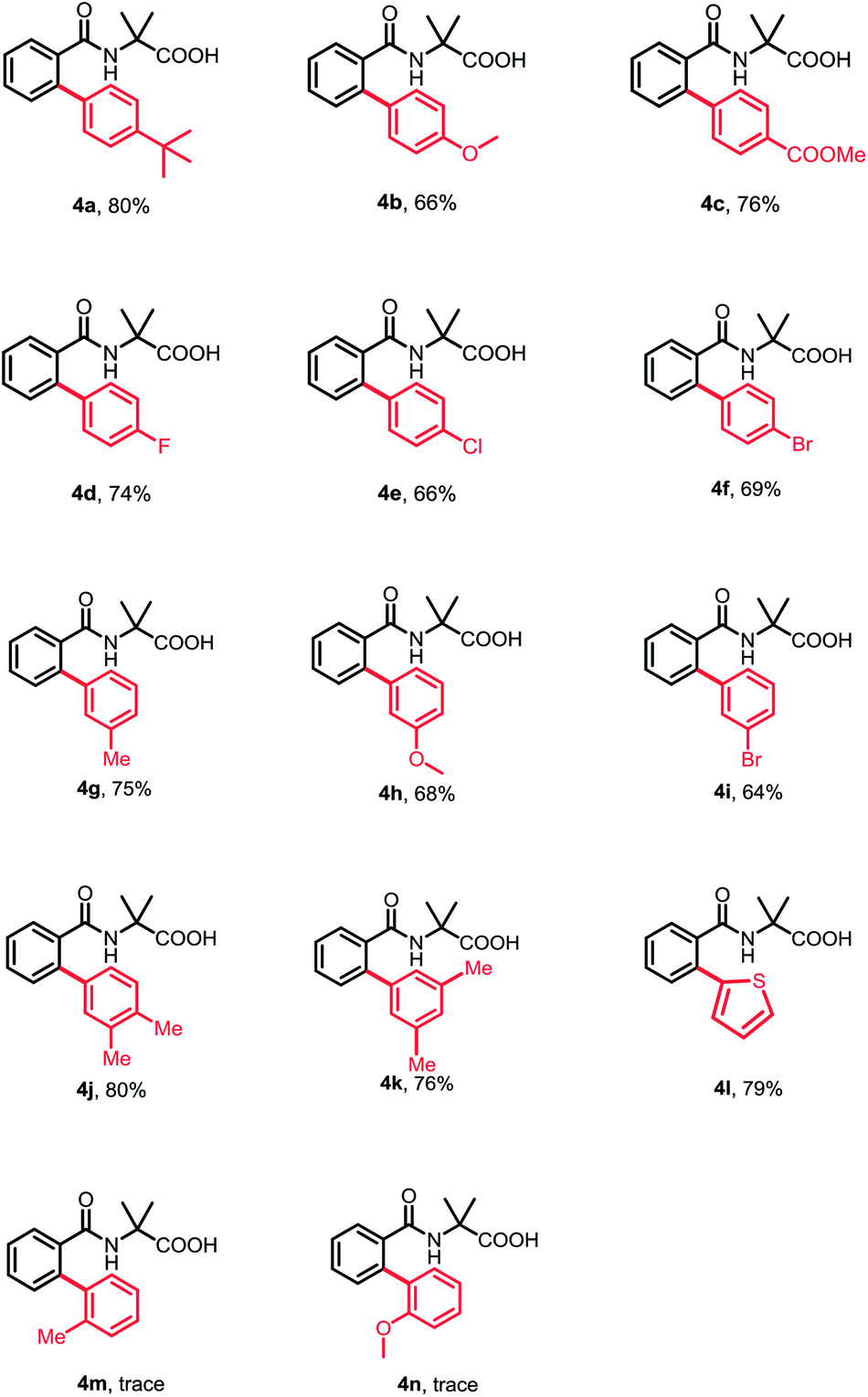
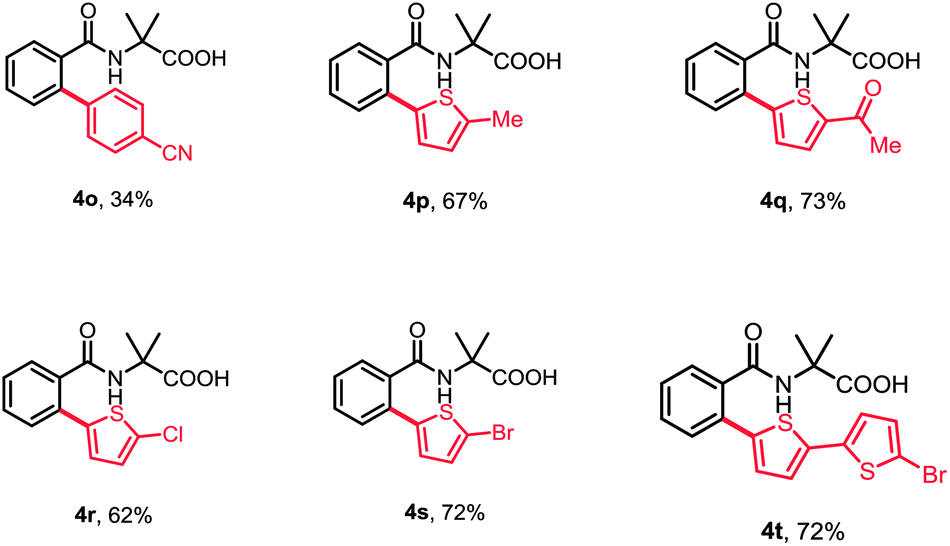
Subsequently, a wide range of aryl halides were examined under standard conditions. As illustrated in Table 3, various aryl iodides could be tolerated in this reaction and gave mono-arylation products in moderate to high yields. The aryl iodides bearing either electron-donating or -withdrawing groups at para- and meta-position proceeded smoothly to afford the corresponding arylation products in good to high yields (4a–4i). Additionally, multisubstituted aryl iodides and heterocyclic iodide provided the desired products in good yields (4j–4k). The introduction of a substituent at ortho-position of aryl iodides afforded no desired products, probably because of interference with the oxidative addition process (4m and 4n). To broaden the usability and enhance the practicality of this method, we tried to apply this reaction with less reactive aryl bromides. Fortunately, 4-bromobenzonitrile was found feasible in the reaction (4o). Furthermore, the thiophene bromides showed great compatibility in this reaction and various functional groups were well tolerated under standard conditions, such as halides and carbonyl, guaranteeing further transformation (4p–4t).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1a (0.2 mmol), 2 (0.6 mmol), Ni(OTf)2 (10 mol%), MesCOOH (20 mol%), Na2CO3 (0.6 mmol), and TBAI (0.2 mmol) in DMSO (2 mL) at 140 °C for 16 h. All listed yields are isolated ones. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| X = I | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| X = Br | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
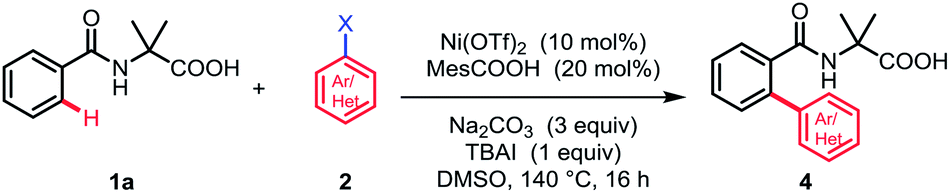
Importantly, the ortho-arylation could be carried out on a gram scale to afford 3a in 80% yield. Moreover, the amino acid group could be easily removed and afforded the corresponding ester 5 in nearly quantitative yield (Scheme 2).
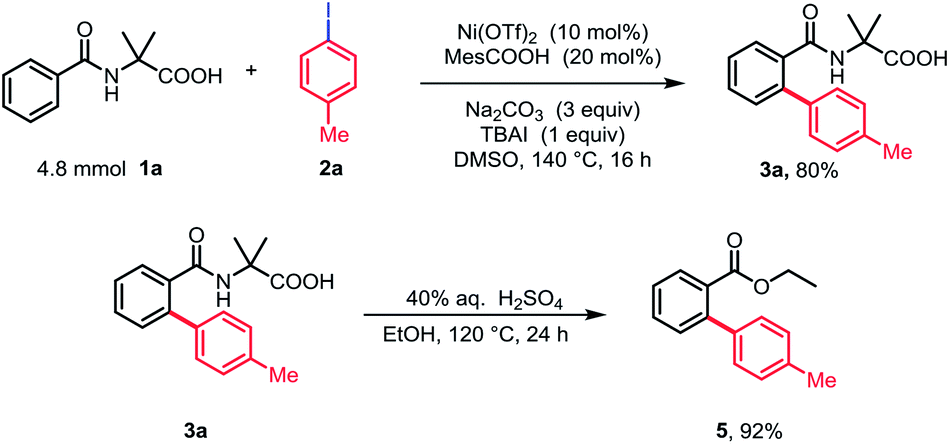 | ||
| Scheme 2 Gram-scale synthesis and removal of the directing group. | ||
To understand the reaction mechanism, a series of mechanistic experiments were carried out. The H/D exchange experiment demonstrated that the cleavage of the C–H bond was an irreversible process (Scheme 3A). Furthermore, the radical scavenger experiments were performed by the addition of 2,2,6,6-tetramethyl-1-piperidinoxyl (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT). The reaction efficiency was not affected, indicating that a single-electron transfer (SET) process was probably not involved (Scheme 3B). By employing 1a-d5 as the substrate, the kinetic isotope effect (KIE) was observed to be 1.2, suggesting that the C–H bond cleavage was not the rate-limiting step.13
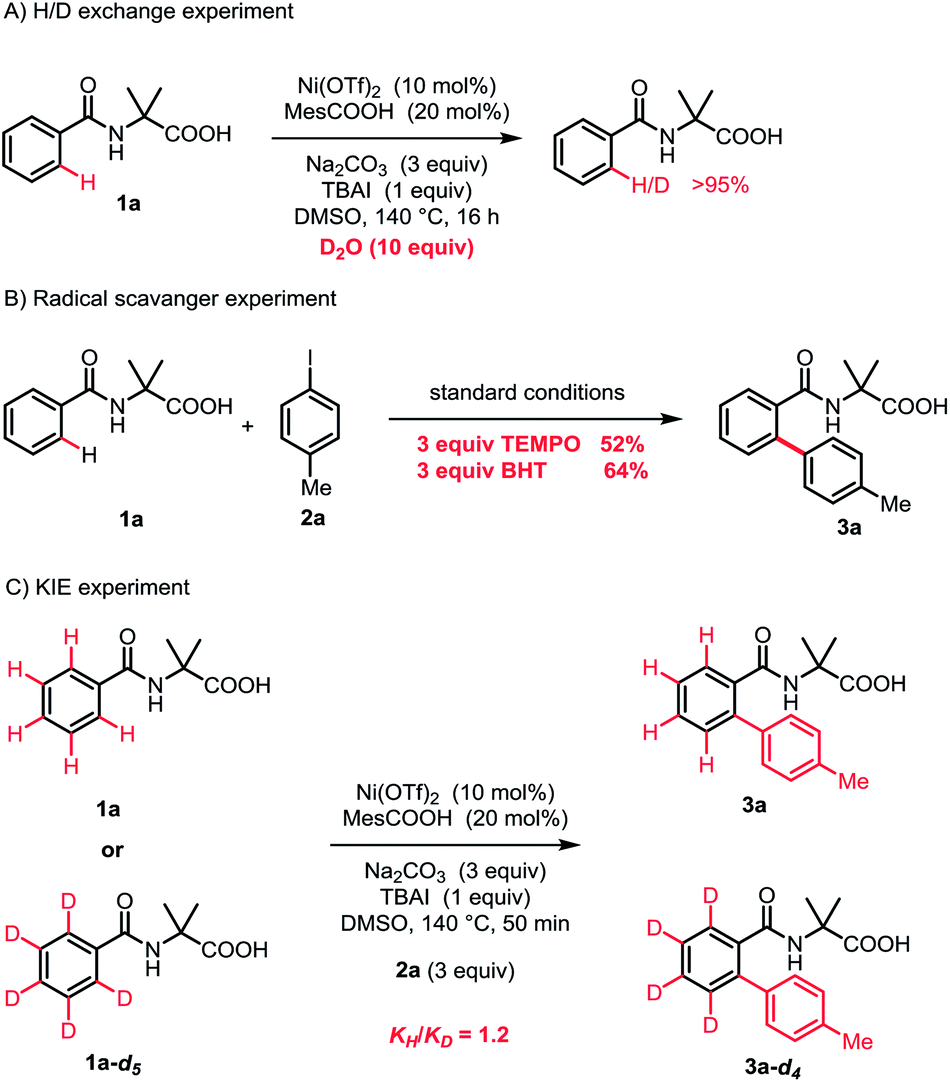 | ||
| Scheme 3 Mechanistic investigations. | ||
Based on the preliminary results and reported literatures,8b,8d,8f,8g a plausible mechanism was proposed. First, the coordination of 1a to the nickel catalyst generated a nickel intermediate A, followed by a concerted metalation–deprotonation (CMD) process14 to produce the nickel complexes B irreversibly. Then the oxidative addition of PhI to intermediate B led to a high-valent Ni intermediate C, which underwent reductive elimination to release the catalyst and give the final product 3a (Scheme 4).
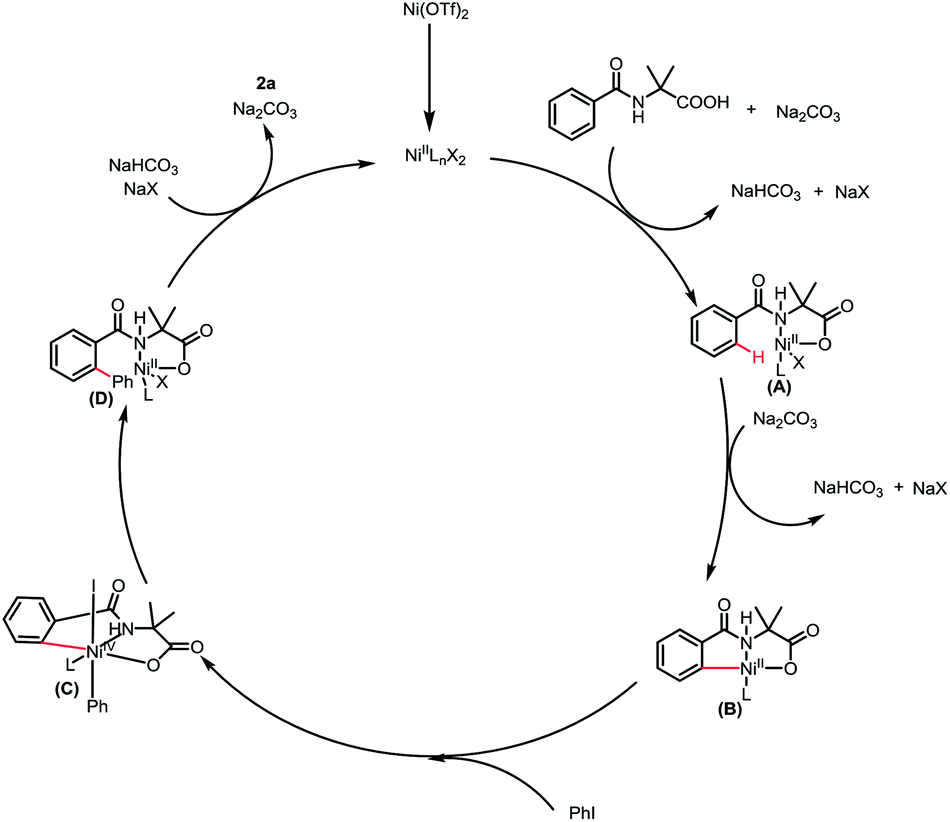 | ||
| Scheme 4 Plausible mechanism. | ||
Conclusions
In conclusion, we developed a nickel(II)-catalyzed ortho-arylation of unactivated aryl C–H bonds utilizing inexpensive amino acid as a directing group. This protocol features excellent mono-selectivity, good regioselectivity, and wide functional tolerance. Moreover, the resulted productions bearing a biaryl motif and an amino acid represent bioactive molecules with wide bioactivities, especially the natural amino acid molecules, which will be of great importance to medicinal chemists.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (81620108027, 21632008 and 81602975), the Major Project of Chinese National Programs for Fundamental Research and Development (2015CB910304).Notes and references
- (a) I. Cepanec, Synthesis of Biaryls, Elsevier, New York, 2004 Search PubMed; (b) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS; (c) Y. Deng, Y. W. Chin, H. Chai, W. J. Keller and A. D. Kinghorn, J. Nat. Prod., 2007, 70, 2049–2052 CrossRef CAS; (d) Y. J. Wu, J. Guernon, J. Shi, L. Marcin, M. Higgins, R. Rajamani, J. Muckelbauer, H. Lewis, C. Chang, D. Camac, J. H. Toyn, M. K. Ahlijanian, C. F. Albright, J. E. Macor and L. A. Thompson, J. Med. Chem., 2016, 59, 8593–8600 CrossRef CAS.
- (a) P. Leowanawat, N. Zhang, A. M. Resmerita, B. M. Rosen and V. Percec, J. Org. Chem., 2011, 76, 9946–9955 CrossRef CAS; (b) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636–15639 CrossRef CAS; (c) I. Hussain, J. Capricho and M. A. Yawer, Adv. Synth. Catal., 2016, 358, 3320–3349 CrossRef CAS; (d) F. Lv and Z.-J. Yao, Sci. China: Chem., 2017, 60, 701–720 CrossRef CAS; (e) S. Mao, Z. Chen, L. Wang, D. B. Khadka, M. Xin, P. Li and S. Q. Zhang, J. Org. Chem., 2019, 84, 463–471 CrossRef CAS.
- (a) M. E. D. A. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef; (b) I. V. Sereginn and V. Gevorgya, Chem. Soc. Rev., 2007, 36, 1173–1193 RSC; (c) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949–957 CAS; (d) J.-W. Park and C.-H. Jun, ChemCatChem, 2009, 1, 69–71 CrossRef CAS; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (f) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (g) P. Y. Choy, S. M. Wong, A. Kapdi and F. Y. Kwong, Org. Chem. Front., 2018, 5, 288–321 RSC; (h) R. Das and M. Kapur, Asian J. Org. Chem., 2018, 7, 1217–1235 CrossRef CAS.
- (a) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450–2494 CrossRef CAS; (b) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS; (d) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906–6919 RSC.
- (a) A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087–4109 CAS; (b) S. Bezzenine-Lafollee, R. Gil, D. Prim and J. Hannedouche, Molecules, 2017, 22, 1901/1–1901/29 CrossRef CAS PubMed; (c) J. Chen and Z. Lu, Org. Chem. Front., 2018, 5, 260–272 RSC; (d) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS.
- (a) Y. Tamaru, Modern Organonickel Chemistry, Wiley-VCH, 2005 CrossRef; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS; (c) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef CAS; (d) J.-P. Wan, Y. Li and Y. Liu, Org. Chem. Front., 2016, 3, 768–772 RSC.
- (a) H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 14952–14955 CrossRef CAS; (b) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef CAS.
- (a) M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef CAS PubMed; (b) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509–15512 CrossRef CAS PubMed; (c) M. Iyanaga, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11933–11939 CrossRef CAS PubMed; (d) L. C. M. Castro and N. Chatani, Chem. Lett., 2015, 44, 410–421 CrossRef CAS; (e) N. Lv, Z. Chen, Y. Liu, Z. Liu and Y. Zhang, Org. Lett., 2018, 20, 5845–5848 CrossRef CAS PubMed; (f) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed; (g) A. P. Honeycutt and J. M. Hoover, ACS Catal., 2017, 7, 4597–4601 CrossRef CAS; (h) Y. Cheng, Y. Wu, G. Tan and J. You, Angew. Chem., Int. Ed., 2016, 55, 12275–12279 CrossRef CAS PubMed; (i) A. Yokota, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11922–11932 CrossRef CAS PubMed.
- (a) F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187–4192 RSC; (b) Y. J. Liu, Y. H. Liu, S. Y. Yan and B. F. Shi, Chem. Commun., 2015, 51, 6388–6391 RSC; (c) S. Y. Yan, Y. J. Liu, B. Liu, Y. H. Liu and B. F. Shi, Chem. Commun., 2015, 51, 4069–4072 RSC.
- (a) X. M. Zhou, Q. Wang, W. H. Zhao, S. S. Xu, W. Zhang and J. M. Chen, Tetrahedron Lett., 2015, 56, 851–855 CrossRef CAS; (b) G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Millers, M. A. Pos, P. M. Scola, K. S. Yeung and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 3338–3351 CrossRef CAS PubMed; (c) L. Wei, Y. W. Dong, L. Z. Yong, Y. Fei and M. C. Jun, Adv. Synth. Catal., 2016, 358, 1968–1974 CrossRef; (d) L. C. M. Castro and N. Chatani, Chem.–Eur. J., 2014, 20, 4548–4553 CrossRef PubMed; (e) J. Kim, M. Sim, N. Kim and S. Hong, Chem. Sci., 2015, 6, 3611–3616 RSC; (f) T. Toba, Y. Hu, A. T. Tran and J. Q. Yu, Org. Lett., 2015, 17, 5966–5969 CrossRef CAS PubMed; (g) C. Wang, C.-P. Chen, J.-Y. Zhang, J. Han, Q. Wang, K. Guo, P. Liu, M.-Y. Guan, Y.-M. Yao and Y.-S. Zhao, Angew. Chem., Int. Ed., 2014, 53, 9884 CrossRef CAS PubMed; (h) M. Guan, Y. Pang, J. Zhang and Y. Zhao, Chem. Commun., 2016, 52, 7043 RSC; (i) J. Han, P. Liu, C. Wang, Q. Wang, J. Zhang, Y. Zhao, D. Shi, Z. Huang and Y. Zhao, Org. Lett., 2014, 16, 5682 CrossRef CAS PubMed.
- (a) W. Zhu, D. Zhang, N. Yang and H. Liu, Chem. Commun., 2014, 50, 10634–10636 RSC; (b) F. Gao, W. Zhu, D. Zhang, S. Li, J. Wan and H. Liu, J. Org. Chem., 2016, 81, 9122–9130 CrossRef CAS PubMed; (c) S. Li, W. Zhu, F. Gao, C. Li, J. Wang and H. Liu, J. Org. Chem., 2017, 82, 126–134 CrossRef CAS PubMed; (d) X. W. Wu, B. Wang, S. B. Zhou, Y. Zhou and H. Liu, ACS Catal., 2017, 7, 2494–2499 CrossRef CAS.
- (a) J. D. Ochocki and M. D. Distefano, MedChemComm, 2013, 4, 476–492 RSC; (b) A. Montero, M. Alonso, E. Benito, A. Chana, E. Mann, J. M. Navas and B. Herradon, Bioorg. Med. Chem. Lett., 2004, 14, 2753–2757 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- (a) G. Ercolano, F. Farina, S. Cavaliere, D. Jones and J. Roziere, Nanomaterials, 2016, 6, 236/1–236/12 CrossRef CAS PubMed; (b) S. J. Freakley, J. Ruiz-Esquius and D. J. Morgan, Surf. Interface Anal., 2017, 49, 794–799 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. See DOI: 10.1039/c9ra00749k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |