
Organelle-specific analysis of labile Fe(II) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes†
Tasuku
Hirayama
 *,
Ayaji
Miki
and
Hideko
Nagasawa
*,
Ayaji
Miki
and
Hideko
Nagasawa
Laboratory of Pharmaceutical and Medicinal Chemistry, Gifu Pharmaceutical University, Japan. E-mail: hirayamat@gifu-pu.ac.jp
First published on 6th September 2018
Abstract
Ferroptosis is an emerging type of cell death mode that is dependent on iron. Unfortunately, the detailed analysis of the function of organelle labile Fe(II) in oxidative damage and lethality of the cells has not been demonstrated so far, mainly due to the lack of efficient methods to visualize labile Fe(II) at the targeted organelles. We have recently reported a series of Fe(II)-selective fluorescent probes, i.e., Ac-MtFluNox, Lyso-RhoNox, and ER-SiRhoNox, which can detect Fe(II) specifically in the mitochondria, lysosomes, and endoplasmic reticulum (ER), respectively. These probes demonstrate similar reaction rates and off/on contrasts with various colours and intracellular distributions, enabling simultaneous multi-colour imaging that allows the monitoring of labile Fe(II) levels at each targeted organelle. In this paper, by using a cocktail of these probes, we successfully visualised the aberrant elevation of labile Fe(II) in the lysosomes and ER prior to HT1080 cell death induced by erastin, which is an inducer of ferroptosis.
Significance to metallomicsWe established a useful imaging method to monitor organelle-specific labile Fe(II) simultaneously at mitochondria, lysosomes, and endoplasmic reticulum (ER). This imaging method enabled the first organelle-specific analysis of the alteration of labile Fe(II) levels during ferroptosis, which is an emerging cell death mode involving iron as the critical factor. Since organelle-specific detection of labile Fe(II) in living cells is an essential method to elucidate the biological functions of iron, the methodology herein described, which led to the discovery of an increase in labile Fe(II) in lysosomes and the ER during ferroptosis, contributes to the understanding of the role of Fe(II) in ferroptosis. |
Introduction
Iron is the most abundant transition metal species in the human body and plays a number of essential roles with respect to physiological functions such as oxygen transport, energy production, and metabolic enzymes1–3 and pathological functions including carcinogenesis4,5 and other pathogenesis.6–8 The concentration of iron in our body cells is appropriately maintained by a sophisticated homeostatic system. Iron homeostasis is strictly controlled even at the cell level through a complicated network of iron sensing/transport mechanisms among the organelles,9 and the dysfunction of this iron-regulating machinery often results in severe cell oxidative damage through aberrant production of highly reactive oxygen species (hROS) such as hydroxyl radicals via Fenton chemistry, where Fe(II) ions act as a generator of hROS.10–13Ferroptosis was reported by Stockwell and Dixon et al. as a new cell death mode different from apoptosis and necrosis, which was first described as erastin-induced cell death of oncogenic Ras-expressing cells such as HT1080 fibrosarcoma cells.14 Since then, several characteristics of ferroptosis have been reported, including the shrinkage of mitochondria and aberrant lipid peroxidation. The most characteristic feature of ferroptosis is the inhibition effect of deferoxamine (DFO), a clinically-used iron chelator, which demonstrates the requirement of iron for the cell death process.14,15 Much effort to gain a deeper understanding of ferroptosis has been made, revealing that the attenuation of cellular antioxidant capacity through inhibition of glutathione peroxidase 4 (GPx4) and/or xCT, which causes the depletion of glutathione, is the primary factor governing such a phenomenon.14–16 The dysfunction of the cellular antioxidant system causes accumulation of lipid peroxides (R-OOH),17 which are readily converted to radical species (RO˙) upon reaction with Fe(II). Very recently, the elucidation of the intracellular location of lethal lipid peroxides has attracted particular attention.18,19 Although the iron-dependency of ferroptosis is known, there is still limited knowledge on the alteration in intracellular iron homeostasis during the process of ferroptosis20 because of the lack of useful methods to monitor labile iron, which is involved in the oxidative damage in living cells.21–23
Although iron is mainly present in the human body in protein-bound form, labile iron, which is non-protein bound or weakly protein-bound iron species, is also found in living systems.24,25 Motivated by the idea that labile Fe(II) rather than Fe(III) is likely to contribute to the oxidative damage of cells through the intracellular generation of radical species, we were interested in monitoring the Fe(II) species in the process of ferroptosis.26 We developed several fluorescent probes that exhibit turn-on fluorescence responses selective to Fe(II),27–30 in particular, to labile Fe(II) species involved in oxidative stress.31–34 Besides, we have recently reported a mitochondria-targeted fluorescent probe, the MtFluNox/Ac-MtFluNox system, which was shown to detect mitochondrial Fe(II) in living cells.35 To date, we have developed Fe(II)-selective fluorescent probes that work in the endoplasmic reticulum (ER),29 mitochondria,35 lysosomes,28 and plasma membranes.30 Among them, we selected Ac-MtFluNox, Lyso-RhoNox,28 and ER-SiRhoNox29 (the last two referred to as HMRhoNox-M and SiRhoNox-1, respectively, in our previous work) for the present work because they exhibit similar reaction rates and off/on contrast in response to Fe(II) while displaying independent colour regions, which would enable the simultaneous detection of Fe(II) at their targeted organelles in living cells by multi-colour imaging. We have anticipated that this technique could be a useful tool to elucidate how the lethal lipid peroxidation is induced in cells during ferroptosis. Herein, we demonstrate a multi-colour imaging method to monitor intracellular labile Fe(II) simultaneously at mitochondria, lysosomes, and ER with a cocktail of fluorescent probes, Ac-MtFluNox, Lyso-RhoNox, and ER-SiRhoNox. Furthermore, this imaging method allowed us to detect aberrant elevations of labile Fe(II) in the lysosomes and ER prior to ferroptotic cell death.
Methods
Chemicals
All the chemicals used in this study except for the fluorescent probes for Fe(II) were commercial products. The fluorescent probes, Ac-MtFluNox/MtFluNox, Lyso-RhoNox, ER-SiRhoNox, Ac-MtRhodol, Lyso-Rhodamine, and ER-SiRhodamine were synthesised according to our previous reports.28,29,35Steady-state fluorescence measurements
Fluorescence spectra were recorded using a JASCO FP6600 instrument with a slit width of 5 nm and 6 nm for excitation and emission, respectively. The path length was 1 cm with a cell volume of 3.0 mL. Excitation was provided at 510 nm (MtFluNox), 550 nm (Lyso-RhoNox), and 630 nm (ER-SiRhoNox). For all the fluorescence measurements, the probes were used at a final concentration of 2 μM (from 1 mM stock solution in DMSO) in 50 mM HEPES buffer (pH 7.4).Cell culture experiments
Human fibrosarcoma (HT1080) cells or human breast adenocarcinoma (MCF-7) cells were cultured in modified essential medium (MEM, Gibco) containing 10% fetal bovine serum (FBS, Gibco), 1% antibiotic–antimycotic solution (Gibco), and 2 mM glutamate in a 5% CO2/95% air incubator at 37 °C. Two days before use, the cells (8 × 104 HT1080 cells per well or 5 × 104 MCF-7 cells per well) were seeded on glass-bottomed dishes (Advanced TC CELLVIEW™ Greiner). For dissolving the probes and erastin, dimethyl sulfoxide with high purity (DMSO infinity pure, FUJIFILM Wako Pure Chemical Corporation) was used.Confocal fluorescence imaging experiments
Confocal microscopic fluorescence images were acquired with an Olympus IX83 microscope equipped with a 130 W mercury lamp, an EMCCD camera (Hamamatsu Photonics, ImagEM) and a disk scan confocal unit (DSU). Images were obtained with appropriate filter sets for each dye as follows.Ac-MtFluNox: FITC filter set (ex: 465–500 nm, em: 516–556 nm, dichroic: 495 nm);
Lyso-RhoNox: rhodamine filter set (ex: 532–554 nm, em: 572–642 nm, dichroic: 562 nm);
ER-SiRhoNox: Cy5 filter set (ex: 608–648 nm, em: 672–712 nm, dichroic: 660 nm)
For all imaging experiments, Hank's Balanced Salt Solution (HBSS, Gibco) containing calcium chloride and magnesium chloride without phenol red was used.
For Fe(II) uptake experiments, HT1080 cells were stained with a cocktail or each of Ac-MtFluNox (1 μM from 1 mM stock in DMSO), Lyso-RhoNox (1 μM from 1 mM stock in DMSO), and ER-SiRhoNox (5 μM from 1 mM stock in DMSO) in HBSS at 37 °C for 30 min and then washed with MEM without FBS. Then, the cells were incubated in MEM without FBS in the presence or absence of 100 μM ferrous ammonium sulphate hexahydrate (FAS, Fe(NH4)2(SO4)2·6H2O, from 10 mM stock solution in water) at 37 °C for 30 min. After washing the cells with HBSS (×2), the cells were imaged by the corresponding filter set as described above.
For imaging the ferroptosis process, HT1080 cells or MCF-7 cells were treated with erastin14 (10 μM from 2 mM stock in DMSO) in the presence or absence of DFO (100 μM from 10 mM stock in water) in the complete medium at 37 °C for 30 min for the indicated times (2, 3, and 5 h). Then, the cells were washed with HBSS and stained by the cocktail of probes as described above. After incubation at 37 °C for 1 h, the cells were washed with HBSS and then imaged. For all the control experiments, the same amounts of the vehicles were added to the incubation medium.
For the imaging study using Ac-MtRhodol,35 Lyso-Rhodamine,28 and ER-SiRhodamine,29 HT1080 cells were treated with erastin (10 μM) for 3 h as described above and stained with a cocktail of Ac-MtRhodol (1 μM from 1 mM stock in DMSO), Lyso-Rhodamine (1 μM from 1 mM stock in DMSO), and ER-SiRhodamine (1 μM from 1 mM stock in DMSO) in HBSS at 37 °C for 1 h. The cells were washed with HBSS and then imaged.
For the imaging of lipid peroxidation, HT1080 cells were treated with erastin (10 μM) in the presence or absence of DFO (100 μM) for 3 h as mentioned above and stained with C11-BODIPY (1 μM from 1 mM stock in DMSO, Thermo Fisher Scientific, Inc.) in HBSS at 37 °C for 30 min. The cells were washed with HBSS and then imaged.
The regions of interest (ROI) were selected by setting thresholds with the Default program for dark background in ImageJ. Average fluorescence intensities of the ROI inside cells (190 cells on average) were measured for each image, and a total of 3 or 5 images were analyzed for each condition by using ImageJ. Statistical analyses were performed using Student's t-test. Pearson's correlation values (Rcoloc) were calculated by the Coloc2 program of Fiji.36 The Rcoloc values were obtained for each field of view, and the averages of the Rcoloc values for each condition were calculated.
Cell viability assay (MTT assay)
HT1080 cells (5 × 104 cells per well) or MCF-7 cells (5 × 104 cells per well) were seeded on a 96-well plastic dish (TPP) and cultured in complete medium at 37 °C for 24 h in a 5% CO2/95% air incubator. Then, the cells were treated with erastin (10 μM) with or without DFO (100 μM) in complete medium for the indicated times. Next, the cells were treated with thiazolyl blue tetrazolium bromide (MTT reagent, Sigma Aldrich, final: 0.5 mg mL−1 (form 5 mg mL−1 stock)) at room temperature for 4 h, and then the medium was removed. DMSO (100 μL) was added to each well, and absorption at 560 nm was measured to calculate cell viability.Quantification of total intracellular iron by atomic absorption spectrometry
HT1080 cells (1 × 106 cells per well) were seeded on 10 cm dishes two days prior to use. The cells were treated with erastin (10 μM) at 37 °C for 3 h in complete medium. After removal of the medium, the cells were washed with phosphate-buffered saline (PBS) twice. The cells were then washed with cold PBS (6 mL × 3). The cells were removed from the dishes by using a scraper, and then the suspension was centrifuged (1000 rpm, 5 min). The supernatant was carefully removed, and the cells were re-suspended in conc. HNO3 (100 μL). The suspension was heated at 90 °C for 4 h to dissolve the cell bodies. The lysate was diluted to 2 mL with distilled water. The concentrations of iron in the samples were measured by furnace atomic absorption spectroscopy with a Shimadzu AA-7000 atomic absorption spectrometer. The obtained values (ng mL−1) were normalised with the cell numbers (per 106 cells). A total of four dishes were prepared for each experiment; three dishes were used for iron quantification, and the fourth dish was used to calculate the number of cells. To determine the cell numbers, the cells were collected by trypsinisation at the same time point with the other three dishes, and the number of cells was counted by using a hematocytometer.Results and discussion
Multi-colour imaging of labile Fe(II) in HT1080 cells
Recently, we have reported a series of Fe(II)-specific fluorescent probes that localise to mitochondria (Ac-MtFluNox, Pearson's correlation value (Rcoloc) with MitoTracker® DeepRed FM = 0.81 ± 0.03),35 lysosomes (Lyso-RhoNox, Rcoloc with LysoTracker® Green DND-26 = 0.80 ± 0.02),28 and the endoplasmic reticulum (ER) (ER-SiRhoNox, Rcoloc with ER-Tracker™ Green = 0.80 ± 0.02)29 with green, orange, and deep-red fluorescence, respectively (Fig. 1a). All of these probes are based on N-oxide chemistry, which we previously established as a Fe(II)-specific chemical switch, and exhibit turn-on fluorescence response to Fe(II) with oxidation state selectivity. The probes show highly selective turn-on fluorescence response toward Fe(II) and are stable against biologically relevant reactive oxygen species, reactive sulphur species, and reactive nitrogen species (Fig. S1, ESI†).28,29,35 We envisaged that these probes could be applied simultaneously for the monitoring of labile Fe(II) in living cells because their off/on contrasts and reaction kinetics are similar while exhibiting non-overlapped fluorescence spectra and excitation wavelengths and different intracellular localisations. Since the excitation wavelengths and fluorescence spectra of the probes are not significantly overlapped, their fluorescence signals can be detected separately by using the appropriate filter set for microscopic observation (Fig. 1a and b). The apparent reaction rates of MtFluNox, Lyso-RhoNox, and ER-SiRhoNox (2 μM probe) with Fe(II) (20 μM) in an aqueous buffer (50 mM HEPES, pH 7.4) were calculated to be 2.1 × 10−3 s−1, 2.2 × 10−3 s−1, and 1.7 × 10−3 s−1, respectively. Thus, the small difference in the responsiveness regarding the off/on ratio and the kinetics between the probes indicates the feasibility of the simultaneous imaging of labile Fe(II) at the three different organelles, i.e., mitochondria, lysosomes, and ER, in living cells.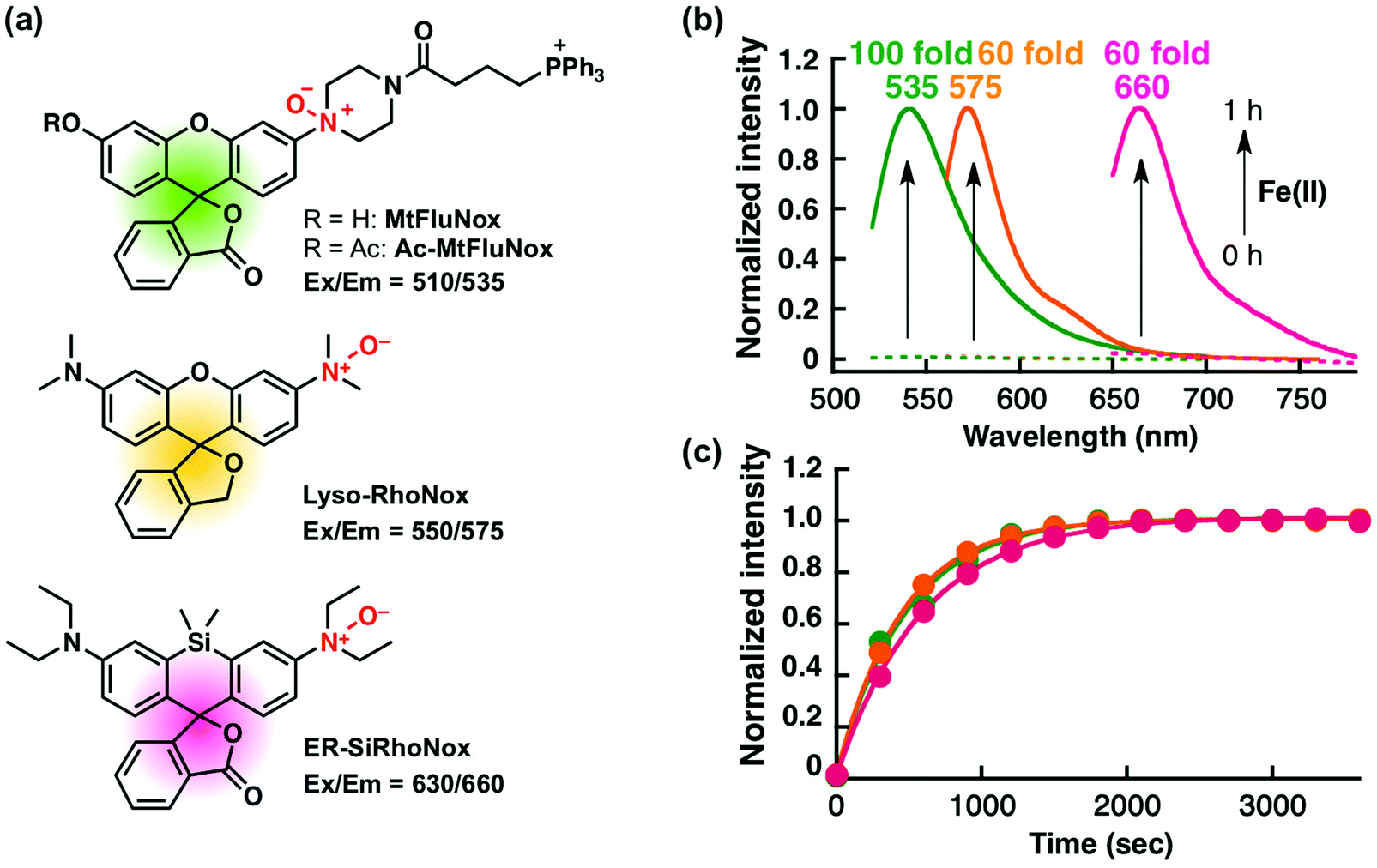 | ||
| Fig. 1 (a) Structures and excitation/emission wavelengths of the fluorescent probes used in this study. Ac-MtFluNox is converted to MtFluNox intracellularly by esterase. (b) Normalised fluorescence spectral changes of the probes (2 μM): MtFluNox (green), Lyso-RhoNox (orange), and ER-SiRhoNox (magenta). Dashed lines and solid lines indicate before and after the reaction with Fe(II) (20 μM, supplemented as FeSO4) for 1 h in 50 mM HEPES buffer (pH 7.4). (c) Plots of normalised fluorescence intensities at 535 nm (green, MtFluNox), 575 nm (orange, Lyso-RhoNox), and 660 nm (magenta, ER-SiRhoNox) against time. Excitation is provided at 510 nm, 550 nm, and 630 nm for MtFluNox, Lyso-RhoNox, and ER-SiRhoNox, respectively. | ||
We first explored the performance of a cocktail of Ac-MtFluNox, Lyso-RhoNox, and ER-SiRhoNox to monitor exogenously supplemented Fe(II) by using our multi-colour imaging approach. HT1080 cells were treated with the three probes at the same time, and Fe(II) ions were then supplemented as ferrous ammonium sulphate. Significant increases in the fluorescence signals were observed with the corresponding organelle-specific pattern (Fig. 2a–d). As can be seen, Ac-MtFluNox, Lyso-RhoNox, and ER-SiRhoNox exhibit fluorescence increase in their corresponding colour regions and the typical staining patterns for mitochondria, lysosomes, and ER, respectively, which is consistent with our previous reports.28,29,35 The fluorescence signals from each probe could be detected only in the corresponding channels exclusively, which was confirmed by single-staining with each of the probes (Fig. S2, ESI†). The merged image shows little overlap (indicated as white colour) and provides low Pearson's correlation values (Rcoloc(MtFluNox vs. Lyso-RhoNox) = −0.20 ± 0.07, Rcoloc(MtFluNox vs. ER-SiRhoNox) = 0.44 ± 0.06, Rcoloc(Lyso-RhoNox vs. ER-SiRhoNox) = 0.29 ± 0.04, (n = 3)), which suggests that the probes were effective at their targeted organelles.
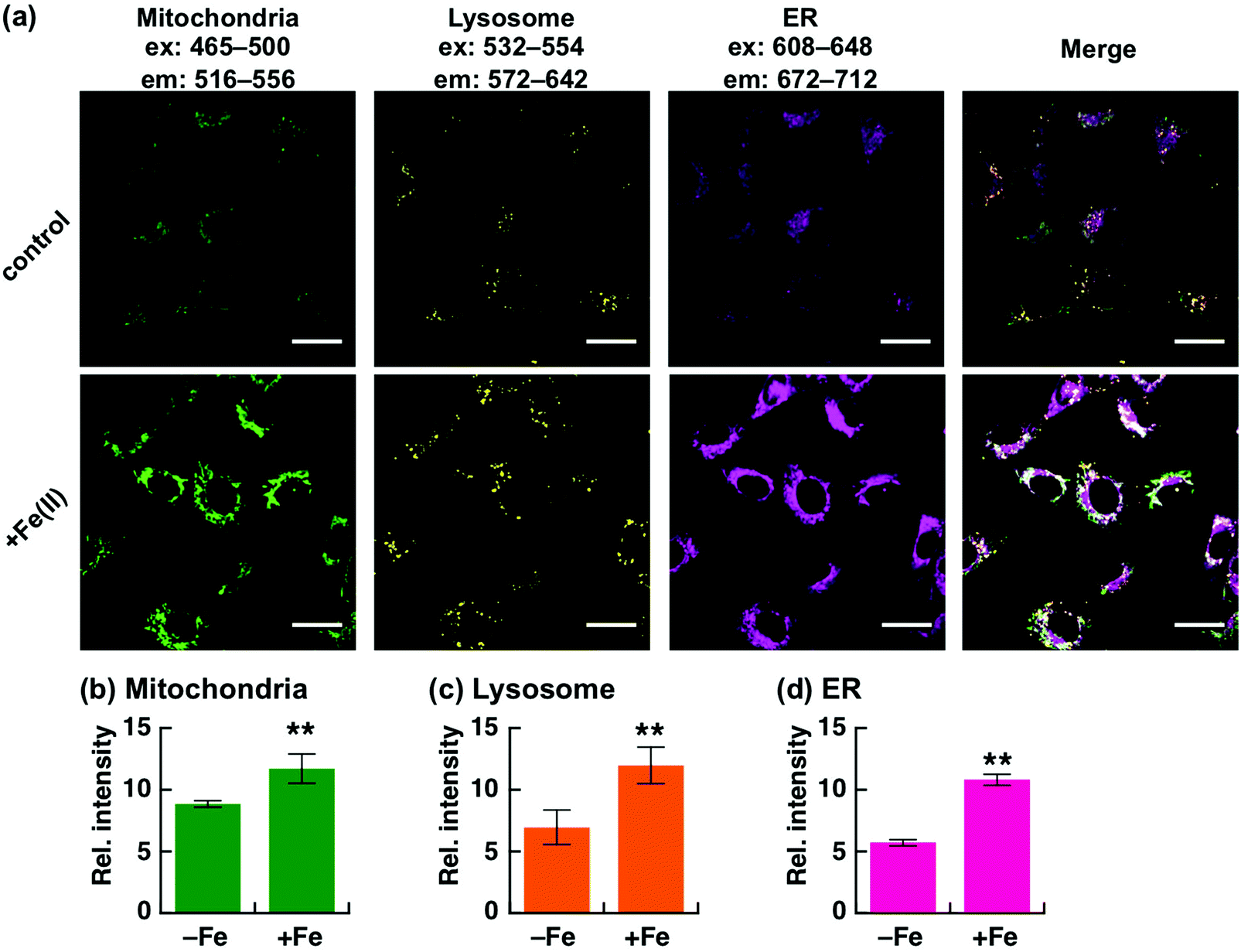 | ||
| Fig. 2 Fluorescence microscopic analysis for multi-colour detection of Fe(II) at each targeted organelle. (a) Representative images of HT1080 cells after treatment without (top) and with Fe(II) (supplemented as ferrous ammonium sulphate hexahydrate) at 37 °C for 30 min (bottom). Left to right: images acquired by using filter sets to detect Fe(II) in mitochondria (green), lysosomes (yellow), and ER (magenta). The rightmost images indicate the merged images of the same field of view of the three images on the left. (b–d) Quantification of the images of each channel acquired in each condition. Error bars indicate ±S.E.M. Statistical analysis was performed using Student's t-test. **P < 0.05 (n = 5). Scale bars indicate 25 μm. | ||
Analysis of labile Fe(II) level during ferroptosis
Although Chang et al. had previously reported an increase in cellular labile Fe(II) level prior to ferroptotic cell death, which was detected by using a ratiometric fluorescent probe,20 a detailed organelle-specific analysis has not been provided to date. To this aim, we selected HT1080 cells because they are widely known to undergo ferroptotic cell death induced by erastin.14,15 In order to design the imaging analysis, we first evaluated the time course of the cell death induced by erastin treatment (10 μM). Since a significant occurrence of cell death (less than 50% viability) was detected from 6 h following erastin treatment (Fig. S3a, ESI†), which is consistent with the previous study by Stockwell et al.,14 we performed the imaging study after 2, 3, and 5 h of treatment to investigate the labile Fe(II) levels before cell death. The erastin-induced cell death of HT1080 cells was cancelled by co-treatment with erastin (10 μM) and DFO (100 μM) (Fig. S3b, ESI†). In this context, we performed multi-colour imaging of labile Fe(II) in HT1080 cells treated with erastin (10 μM) by using the cocktail of Ac-MtFluNox, Lyso-RhoNox, and ER-SiRhoNox as the organelle-specific indicators of labile Fe(II) and DFO as the ferroptosis inhibitor (Fig. 3).14 No significant change in the fluorescence signals was observed in the cells after 2 h of erastin treatment, whereas significant decreases were found in the lysosomes and ER of the cells incubated with DFO (Fig. 3a and b). This result indicates that DFO effectively captures the basal labile Fe(II), which is converted to an inactive form against our probes in the cells. In contrast, treatment of the cells with erastin for 3 h caused an increase in the fluorescence signals in all the organelles monitored (Fig. 3c and d). The fluorescence increase was cancelled by the supplementation of DFO in the lysosomes and ER, but that in mitochondria was not affected. A similar trend was observed after 5 h of treatment (Fig. 3e and f). Considering the critical role of DFO as an inhibitor of ferroptosis as previously reported, the fluorescence increase observed in the lysosomes and ER can be attributed to the elevation of labile Fe(II). In contrast, the mitochondrial fluorescence signals remain practically unchanged after DFO treatment during ferroptosis. It is known that shrinkage of mitochondria occurs during the erastin-induced ferroptotic cell death of HT1080 cells.14 This would result in an enhancement of the local concentration of the mitochondria-targeted probe in the shrunk mitochondria, which would be accompanied by an increment in the fluorescence signal regardless of the labile Fe(II) levels. To confirm this hypothesis, we performed a multi-colour imaging study of erastin-treated HT1080 cells using Ac-MtRhodol,35 Lyso-Rhodamine,28 and ER-SiRhodamine,29 which are the fluorescent counterparts of the probes without N-oxides (Fig. S4a, ESI†). Among the three organelles monitored, only mitochondria exhibited a fluorescence increase (Fig. S4b–d, ESI†), suggesting that the shrinkage of mitochondria and the concomitant increase in the concentration of the mitochondria-targeted fluorescent molecule (MtRhodol in this case), rather than labile Fe(II), were responsible for the increase in the mitochondrial fluorescence signal. On the other hand, the fluorescence levels in lysosomes and ER were significantly increased by erastin treatment and suppressed by co-incubation with DFO. Besides, the absence of significant changes in the accumulation levels of the dyes (Fig. S4c and d, ESI†) indicates that the fluorescence enhancements were due to the elevation of the labile Fe(II) levels in the lysosomes and ER. Torii et al. reported that ferritin degradation in lysosomes plays a critical role during ferroptosis in HT1080 cells37,38 since the degradation of ferritin liberates a large amount of labile Fe(II) and induces lipid peroxidation in the perinuclear regions, which is a characteristic feature of ferroptotic cell death.38 Our results are consistent with this observation (Fig. 3c and d). In that work, it was proposed that overproduction of labile Fe(II) may be involved in the perinuclear oxidative damage during ferroptosis,26,38 which could be invoked to explain the observed elevation of labile Fe(II) in the ER (Fig. 3c–f) in the present work. The abnormally increased Fe(II) level in the ER and lysosomes readily triggers lipid peroxidation in the perinuclear regions through Fe(II)-induced Fenton chemistry, whereas DFO-treatment removed Fenton chemistry-active labile Fe(II).11 We performed fluorescence microscopic analysis of lipid peroxidation levels using C11-BODIPY (Fig. S5, ESI†),39 to find that lipid peroxidation was significantly enhanced in the HT1080 cells upon treatment with erastin for 3 h and cancelled by DFO treatment. We observed no significant increase in the total iron content in the erastin-treated cells (Fig. S6, ESI†) indicating that not upregulation of iron influx, but the alteration of endogenous homeostasis of labile iron contributed to the cell death process. Taken together, our results suggest that ferroptotic cell death is triggered by intracellular elevation of the labile Fe(II) level in the lysosomes and ER, but not in the mitochondria, which is consistent with the results of previous studies about the intracellular location of ROS generation during ferroptosis.18,19,38 In contrast, the multi-colour imaging study conducted on MCF-7 cells, which are not sensitive to erastin-induced ferroptosis, revealed that the labile Fe(II) level does not change upon the treatment with erastin (10 μM). As in the case of HT1080 cells, MCF-7 cell viability was assayed, resulting in no significant cell death upon the addition of erastin (10 μM) up to 12 h (Fig. S7, ESI†), at which time point HT1080 cells were completely dead in the presence of the same concentration of erastin. In contrast to HT1080, incubation of MCF-7 cells in the presence of erastin did not cause any significant change in the fluorescence signals from the mitochondria, lysosomes, or ER (Fig. S8, ESI†), indicating that the labile Fe(II) level was not altered by erastin treatment in MCF-7 cells. Therefore, it seems reasonable to conclude that the increase in the labile Fe(II) in the lysosomes and ER is specific to erastin-induced cell death, i.e., ferroptosis, in HT1080 cells.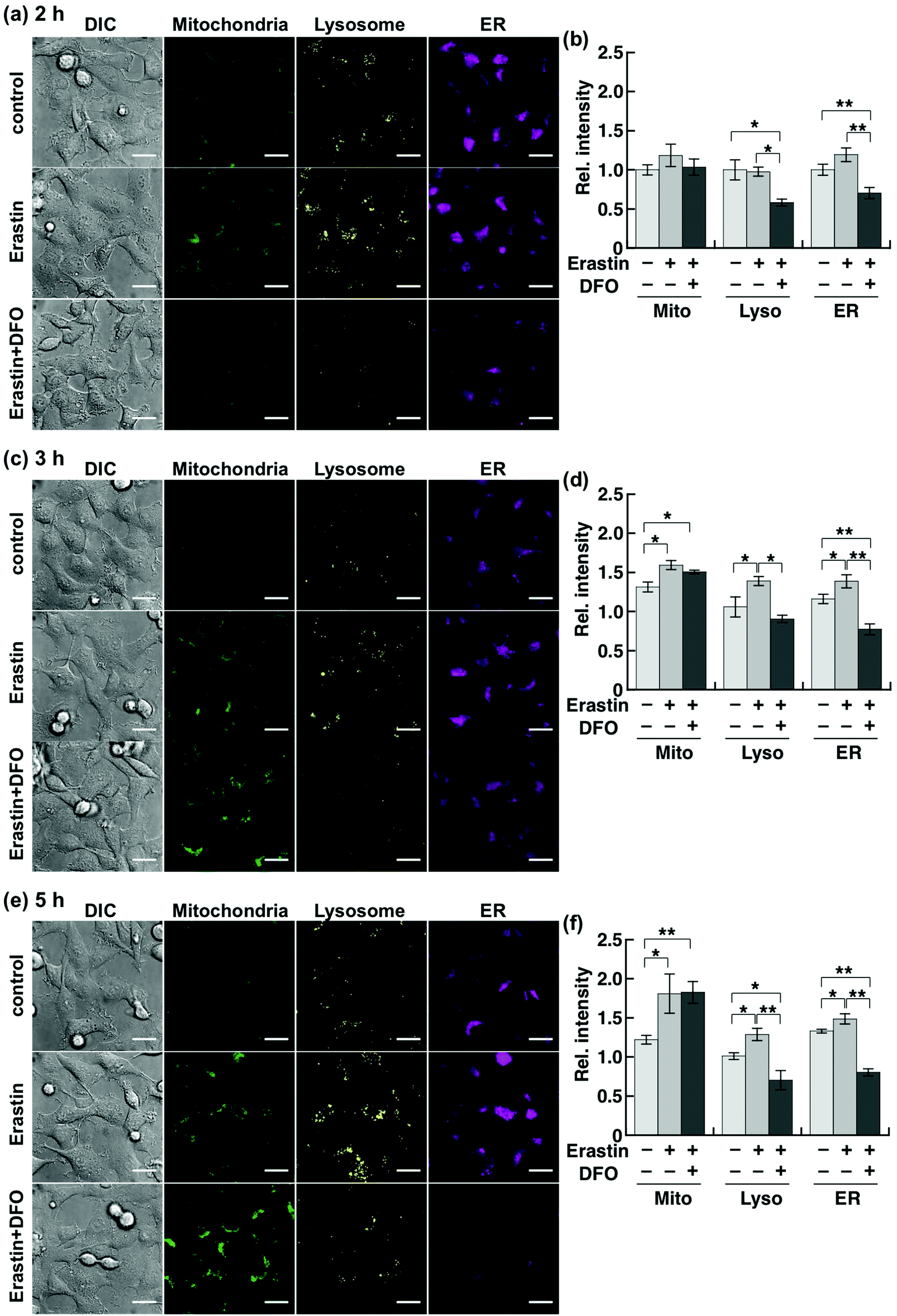 | ||
| Fig. 3 Multi-colour fluorescence microscopic analyses of labile Fe(II) in mitochondria (green), lysosomes (yellow), and ER (magenta) in HT1080 cells. The cells were treated with erastin (10 μM) in the presence or absence of DFO (100 μM) and then stained by the cocktail of the probes at 37 °C for 1 h. (a), (c) and (e) Representative images of the control cells (top) and the cells treated with erastin in the absence (middle) and the presence (bottom) of DFO at 37 °C for (a) 2 h, (c) 3 h, and (e) 5 h. Left to right: images acquired by differential interference contrast (DIC) and by using filter sets to detect Fe(II) in the mitochondria (green), lysosome (yellow), and ER (magenta). (b), (d) and (f) Quantification of images of each channel in (a), (c), and (e), respectively. Light grey: control. Grey: erastin-treated cells. Dark grey: erastin-treated cells in the presence of DFO. Error bars indicate ±S.E.M. Statistical analysis was performed using Student's t-test. *P < 0.05, **P < 0.01 (n = 5). Scale bars indicate 25 μm. | ||
Conclusions
We have presented a new method for the simultaneous monitoring of intracellular labile Fe(II) in the mitochondria, lysosomes, and ER by using a cocktail of various colour organelle-targeted fluorescent probes, i.e., Ac-MtFluNox (mitochondria), Lyso-RhoNox (lysosomes), and ER-SiRhoNox (ER). We had previously developed these probes by exploiting N-oxide chemistry, and a kinetic study had revealed that all the probes reacted to Fe(II) ions with similar off/on contrast and reaction rates, thereby enabling the simultaneous imaging study of labile Fe(II) in living cells. We applied this cocktail of probes to analyse the alteration of local labile Fe(II) levels during ferroptosis, which is an emerging iron-dependent cell death mode. A series of imaging studies revealed that the labile Fe(II) levels were elevated in the lysosomes and ER during erastin-induced cell death, which confirms the involvement of the labile Fe(II) present in the lysosomes and ER in the lethality. In contrast, although mitochondrial fluorescence signals increased in mitochondria prior to cell death, this fluorescence change was not primarily due to the elevation of the Fe(II) level but due to mitochondria shrinking, according to the imaging results obtained using the fluorescent counterpart MtRhodol and the ineffectiveness of DFO supplementation. In addition, an imaging study conducted on MCF-7 cells, which are insensitive to erastin-induced cell death (ferroptosis), revealed that the elevation of labile Fe(II) in the lysosomes and ER is characteristic of ferroptosis. To the best of our knowledge, this is the first report that describes the monitoring of labile Fe(II) in cellular organelles. We are certain that our cocktail of probes will potentially contribute to the elucidation of the organelle-specific behaviour of labile Fe(II) in living cells.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI (Grant-in-aid for Young Scientist (A), No. 25702050 for T. H.), The Naito Foundation (for T. H.), and The Kato Memorial Bioscience Foundation (for T. H.). This work was supported by JSPS A3 Foresight Program. We thank Ms Emi Inaba and Mr Shusaku Hirosawa for their technical assistance.References
- S. J. Lippard and J. M. Berg, Principles of Bioinorganic Chemistry, University Science Books, 1997 Search PubMed.
- T. Ganz, Physiol. Rev., 2013, 93, 1721–1741 CrossRef CAS PubMed.
- E. C. Theil and D. J. Goss, Chem. Rev., 2009, 109, 4568–4579 CrossRef CAS PubMed.
- S. V. Torti and F. M. Torti, Nat. Rev. Cancer, 2013, 13, 342–355 CrossRef CAS.
- S. Toyokuni, Cancer Sci., 2009, 100, 9–16 CrossRef CAS PubMed.
- K. V. Kowdley, Gastroenterology, 2004, 127, S79–S86 CrossRef CAS.
- M. C. Kruer, Int. Rev. Neurobiol., 2013, 110, 165–194 CrossRef CAS.
- L. Zecca, M. B. H. Youdim, P. Riederer, J. R. Connor and R. R. Crichton, Nat. Rev. Neurosci., 2004, 5, 863–873 CrossRef CAS.
- M. W. Hentze, M. U. Muckenthaler, B. Galy and C. Camaschella, Cell, 2010, 142, 24–38 CrossRef CAS.
- J. A. Imlay, S. M. Chin and S. Linn, Science, 1988, 240, 640–642 CrossRef CAS.
- M. J. Burkitt and R. P. Mason, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8440–8444 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridgeb, FEBS Lett., 1992, 307, 108–112 CrossRef CAS.
- S. Enami, Y. Sakamoto and A. J. Colussi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 623–628 CrossRef CAS PubMed.
- S. J. Dixon, K. M. Lemberg, M. R. Lamprecht, R. Skouta, E. M. Zaitsev, C. E. Gleason, D. N. Patel, A. J. Bauer, A. M. Cantley, W. S. Yang, B. Morrison and B. R. Stockwell, Cell, 2012, 149, 1060–1072 CrossRef CAS PubMed.
- B. R. Stockwell, J. P. Friedmann Angeli, H. Bayir, A. I. Bush, M. Conrad, S. J. Dixon, S. Fulda, S. Gascón, S. K. Hatzios, V. E. Kagan, K. Noel, X. Jiang, A. Linkermann, M. E. Murphy, M. Overholtzer, A. Oyagi, G. C. Pagnussat, J. Park, Q. Ran, C. S. Rosenfeld, K. Salnikow, D. Tang, F. M. Torti, S. V. Torti, S. Toyokuni, K. A. Woerpel and D. D. Zhang, Cell, 2017, 171, 273–285 CrossRef CAS.
- W. S. Yang, R. Sriramaratnam, M. E. Welsch, K. Shimada, R. Skouta, V. S. Viswanathan, J. H. Cheah, P. A. Clemons, A. F. Shamji, C. B. Clish, L. M. Brown, A. W. Girotti, V. W. Cornish, S. L. Schreiber and B. R. Stockwell, Cell, 2014, 156, 317–331 CrossRef CAS PubMed.
- R. Shintoku, Y. Takigawa, K. Yamada, C. Kubota, Y. Yoshimoto, T. Takeuchi, I. Koshiishi and S. Torii, Cancer Sci., 2017, 108, 2187–2194 CrossRef CAS PubMed.
- H. Feng and B. R. Stockwell, PLoS Biol., 2018, 16, e2006203 CrossRef PubMed.
- M. M. Gaschler, F. Hu, H. Feng, A. Linkermann, W. Min and B. R. Stockwell, ACS Chem. Biol., 2018, 13, 1013–1020 CrossRef CAS.
- A. T. Aron, M. O. Loehr, J. Bogena and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 14338–14346 CrossRef CAS.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS.
- T. Hirayama and H. Nagasawa, J. Clin. Biochem. Nutr., 2017, 60, 39–48 CrossRef CAS.
- A. T. Aron, A. G. Reeves and C. J. Chang, Curr. Opin. Chem. Biol., 2018, 43, 113–118 CrossRef CAS.
- O. Kakhlon and Z. I. Cabantchik, Free Radical Biol. Med., 2002, 33, 1037–1046 CrossRef CAS.
- Z. I. Cabantchik, Front. Pharmacol., 2014, 5, 1–11 CAS.
- S. J. Dixon and B. R. Stockwell, Nat. Chem. Biol., 2014, 10, 9–17 CrossRef CAS PubMed.
- T. Hirayama, K. Okuda and H. Nagasawa, Chem. Sci., 2013, 4, 1250–1256 RSC.
- M. Niwa, T. Hirayama, K. Okuda and H. Nagasawa, Org. Biomol. Chem., 2014, 12, 6590–6597 RSC.
- T. Hirayama, H. Tsuboi, M. Niwa, A. Miki, S. Kadota, Y. Ikeshita, K. Okuda and H. Nagasawa, Chem. Sci., 2017, 8, 4858–4866 RSC.
- M. Niwa, T. Hirayama, I. Oomoto, D. O. Wang and H. Nagasawa, ACS Chem. Biol., 2018, 13, 1853–1861 CrossRef CAS.
- T. Hirayama, Free Radical Biol. Med., 2018 DOI:10.1016/j.freeradbiomed.2018.07.004.
- T. Mukaide, Y. Hattori, N. Misawa, S. Funahashi, L. Jiang, T. Hirayama, H. Nagasawa and S. Toyokuni, Free Radical Res., 2014, 48, 990–995 CrossRef CAS.
- T. Imamura, T. Hirayama, K. Tsuruma, M. Shimazawa, H. Nagasawa and H. Hara, Exp. Eye Res., 2014, 129, 24–30 CrossRef CAS.
- T. Adachi, S. Nonomura, M. Horiba, T. Hirayama, T. Kamiya, H. Nagasawa and H. Hara, Sci. Rep., 2016, 6, 20928 CrossRef CAS.
- T. Hirayama, S. Kadota, M. Niwa and H. Nagasawa, Metallomics, 2018, 10, 794–801 RSC.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676 CrossRef CAS.
- W. Hou, Y. Xie, X. Song, X. Sun, M. T. Lotze, H. J. Zeh, R. Kang and D. Tang, Autophagy, 2016, 12, 1425–1428 CrossRef CAS.
- S. Torii, R. Shintoku, C. Kubota, M. Yaegashi, R. Torii, M. Sasaki, T. Suzuki, M. Mori, Y. Yoshimoto, T. Takeuchi and K. Yamada, Biochem. J., 2016, 473, 769–777 CrossRef CAS PubMed.
- G. P. C. Drummen, L. C. M. van Liebergen, J. A. F. Op den Kamp and J. A. Post, Free Radical Biol. Med., 2002, 33, 473–490 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on cell culture and cytotoxicity assays. See DOI: 10.1039/c8mt00212f |
| This journal is © The Royal Society of Chemistry 2019 |