
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-dependent shotgun proteomics revealed distinct effects of an organoruthenium prodrug and its activation product on colon carcinoma cells
Samuel M.
Meier-Menches
 *a,
Katja
Zappe
a,
Andrea
Bileck
b,
Dominique
Kreutz
a,
Ammar
Tahir†
a,
Margit
Cichna-Markl
a and
Christopher
Gerner
*a
*a,
Katja
Zappe
a,
Andrea
Bileck
b,
Dominique
Kreutz
a,
Ammar
Tahir†
a,
Margit
Cichna-Markl
a and
Christopher
Gerner
*a
aDepartment of Analytical Chemistry, University of Vienna, Vienna, Austria. E-mail: samuel.meier@univie.ac.at; christopher.gerner@univie.ac.at
bDepartment of Nephrology and Hypertension and Department of BioMedical Research, Inselspital, Bern University Hospital, University of Bern, Switzerland
First published on 8th August 2018
Abstract
Activation kinetics of metallo-prodrugs control the types of possible interactions with biomolecules. The intact metallo-prodrug is able to engage with potential targets by purely non-covalent bonding, while the activated metallodrug can form additional coordination bonds. It is hypothesized that the additional coordinative bonding might be favourable with respect to the target selectivity of activated metallodrugs. Thus, a time-dependent shotgun proteomics study was conducted in HCT116 colon carcinoma cells with plecstatins, which are organoruthenium anticancer drug candidates. First, the target selectivity was evaluated in a time-dependent fashion, which accounted for their hydrolysis kinetics. The binding selectivity increased from 50- to 160-fold and the average specificity from 0.72 to 0.86, respectively, from the 2 h to the 4 h target profiling experiment. Target profiling after 19 h did not reveal significant enrichments, possibly due to deactivation of the probe via arene cleavage. Up to 450 interactors were identified in the target profiling experiments. A plecstatin analogue that substituted a hydrogen bond acceptor with a hydrogen bond donor abrogated the target selectivity for plectin in HCT116 whole cell lysates, underlining the necessity of this hydrogen bond acceptor for a strong interaction between plecstatin and plectin. Second, time-dependent response profiling experiments provided evidence that plecstatin-2 induced an integrated stress response (ISR) in HCT116 cell culture. The phosphorylation of eIF2α, a key mediator of the ISR, after 3 h treatment indicated that this perturbation was initiated by the intact plecstatin-2 prodrug, while the effects of plectin-targeting are mediated by activated plecstatin-2.
Significance to metallomicsCell-level investigations of metallodrugs by shotgun proteomics are still scarce despite the evident advantages of these methods. We performed time-dependent target profiling experiments with organoruthenium-based plecstatins in cell lysates of HCT116 colon carcinoma cells. The changes in target selectivity over time correlated with the activation status of the metallo-prodrug. The target engagement of the plecstatins was tested by substituting a hydrogen bond acceptor with a hydrogen bond donor, which abrogated the plectin-targeting ability. Moreover, time-dependent response profiling experiments in HCT116 cell cultures indicated the induction of an integrated stress response by the prodrug before activation. |
Introduction
Bioinorganic medicinal chemistry is a growing field of research that requires the development of methods to characterise effects of metal-based pharmaceuticals (metallodrugs) in the cellular context and identify their cellular targets.1 Romero-Canelón and Sadler suggested the use of systems pharmacology approaches to assess metallodrug effects in 20152 and proteomics3–6 or transcriptomics7,8 approaches are being reported with increasing frequency. Such cell-level investigations are still challenging because metallodrugs are considered to be rather unspecific and the detailed modes of action are indeed elusive in most cases.1 Additionally, metallodrugs may change their binding properties in time due to ligand exchange reactions.9 For example, these compounds may be prodrugs that hydrolyse a labile metal-halido bond. The prodrug may interact with potential targets via non-covalent bonding that may aid in orienting the prodrug with respect to the target site.10 The activated species can additionally coordinate to biological nucleophiles that may increase the binding strength and thus, potentially, the binding selectivity. Evidently, coordination bonds are inherently different from organic covalent bonds. This was illustratively shown for a chlorambucil-functionalized RAPTA derivative, which was exposed to 9-ethylguanine and analysed by mass spectrometry (MS).11 This bi-functional ruthenium(II) compound can potentially coordinate 9-ethylguanine to the metal centre or alkylate 9-ethylguanine by the nitrogen mustard moiety. Under kinetic control, the nucleobase coordinated quickly to the hydrolysed metal centre, but alkylation of 9-ethylguanine via attack on the aziridinium of the nitrogen mustard was the final stable product.MS techniques are nowadays applicable to several research questions, ranging from the analysis of metallodrugs in model systems to the study of their tissue distribution.12,13 This versatility is mainly enabled by the orthogonal types of sample ionization, most commonly inductively coupled plasma (ICP) and electrospray ionization (ESI).12 The former is preferentially employed to quantify metal isotopes in complex biological matrices, while the latter was initially used to study model systems and provides molecular information on the interaction products between metallodrugs and biomolecules. Recently, considerable efforts were undertaken to extend these techniques to the study of metallodrugs in the cellular context by applying MS-based shotgun proteomics, termed response profiling,6 besides other techniques based on gel electrophoresis.14–17 The main advantage of the MS-based shotgun approach relates to the online combination of peptide separation and analysis by LC-MS/MS allowing for the routine identification of five peptides per second and a total of >5000 proteins.6 Response profiling yields an indirect, but global assessment of effects on the cellular proteome upon drug treatment in living cells. It was found that response profiling is applicable across diverse classes of metallodrugs, whose representatives induced specific protein regulation patterns, thus suggesting specific modes of action.6 This method confirmed known and suggested unrecognized drug effects.
Besides elucidating the modes of action of metallodrugs on a cellular level, target identification approaches emerged that frequently make use of MS-based techniques. For example, the protein targets of colloidal bismuth subcitrate in Helicobacter pylori were investigated by a gel-electrophoresis-ICP-MS approach.18 Multiple targets were reported for a cyclometallated gold(III) complex by photo-crosslinking and subsequent 2D-PAGE and MS analysis of the fluorescent spots.19,20 A similar strategy showed the mitochondrial heat shock protein 60 as a target for gold(III) porphyrins.21 Furthermore, putative targets for RAPTA compounds were suggested by a target profiling approach.22 Target profiling experiments are based on affinity purification strategies by immobilizing a drug candidate on a solid support and the subsequent sampling of directly bound protein targets. Although powerful, this approach is usually performed using cell lysates and prone to non-selective interactions. It is thus usually employed as a hypothesis-generating procedure on the path to target identification and validation.
We were recently able to identify the protein target of an organometallic ruthenium(arene) prodrug by an integrated target-response profiling approach.5 Integrating the two dimensions provides a strong hypothesis on probable drug targets by combining the target spectrum of a drug in cell lysates with the effects of the same drug on the proteome of living cells. Accordingly, [chlorido(η6-p-cymene)(N-(4-fluorophenyl)-2-pyridinecarbothioamide)ruthenium(II)] chloride (plecstatin-1, Scheme 1A) was shown to target plectin, a scaffold protein and cytolinker, which was validated in plectin knock-out cells and showed anti-invasive effects in vivo.5 Bioimaging studies then proved that the compound entered tumours in vivo.23
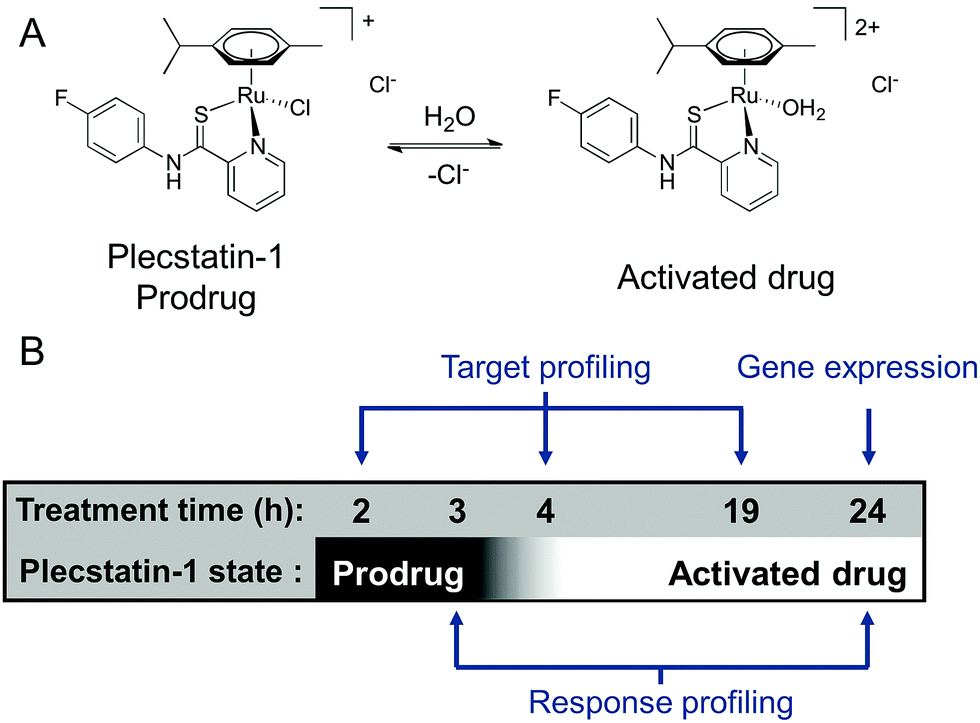 | ||
| Scheme 1 (A) Chemical structures of plecstatin-1 as a prodrug and activated drug. (B) The experimental design of the time-dependent analysis of plecstatins involved three time points for target profiling, two time points for response profiling and one time point for gene expression analysis. | ||
Because ligand exchange reactions were rarely accounted for in cell-level investigations on the proteome, this work aimed to provide insight into the time-dependent target selectivity of plecstatins depending on the activation status of the therapeutic agent (Scheme 1B). Thus, three time points were chosen for target profiling experiments reflecting their hydrolysis rates. In a second step, the target selectivity was further assessed by a plecstatin-analogue based on N-(4-hydroxyphenyl)-2-pyridinecarbothioamide in which a hydrogen bond acceptor was replaced by a hydrogen bond donor. Third, the treatment effects of plecstatin-2 on the HCT116 colon carcinoma cell line were evaluated in detail by time-dependent response profiling as it remained unclear how the reduced mitochondrial membrane potential (MMP) and induced reactive oxygen species (ROS)5 were related to plectin targeting. The findings herein provide unprecedented evidence that simultaneously occurring, but per se unrelated cellular effects result from the speciation of a metallo-prodrug. In other words, specific representatives of the activation cascade of a metallo-prodrug induce distinct drug effects.
Experimental
Synthesis
[Chlorido(η6-toluene)(N-(4-hydroxyphenyl)-2-pyridinecarbothioamide)ruthenium(II)]chloride (Ru-OH-1). A previously published method was largely followed.24 In brief, N-(4-hydroxyphenyl)-2-pyridinecarbothioamide (97 mg, 0.42 mmol) was dissolved in MeOH (10 mL) under argon and was stirred until a clear solution was obtained. [Ru(η6-toluene)Cl2]2 (105 mg, 0.2 mmol) was suspended in MeOH (10 mL) and was added under argon. The reaction mixture was stirred over night at room temperature in the dark. After several minutes, the suspension turned deep red. The reaction mixture was dried, redissolved in dichloromethane (10 mL) and filtered. Hexane (20 mL) was added for precipitation at 4 °C. Red crystals formed that were filtered, washed with hexane and dried. Yield: 186 mg (92%). Elemental analysis found: C, 44.75; H, 3.54; N, 5.22; S, 5.97, calculated for C19H18Cl2ON2RuS·H2O: C, 44.53; H, 3.93; N, 5.47; S, 6.26. MS (ESI+): m/z 423.0101 ([M − HCl − Cl]+, mtheor = 423.0104, 1 ppm). 1H NMR (500.1 MHz, d4-MeOD, 25 °C): δ = 9.71 (d, 3J(H1,H2) = 5 Hz, 1H, H-1), 8.39 (d, 3J(H3,H4) = 6 Hz, 1H, H-4), 8.29 (t, 3J(H2,H3)/(H3,H4) = 6 Hz, 1H, H-3), 7.83 (t, 3J(H1,H2)/(H2,H3) = 6 Hz, 1H, H-2), 7.46 (d, 3J(H8,H9)/(H11,H12) = 9 Hz, 2H, H-9/H-11), 6.96 (d, 3J(H8,H9)/(H11,H12) = 9 Hz, 2H, H-8/H-12), 6.11 (t, 3J(H14,H15)/(H15,H16) = 6 Hz, 1H, H-15), 6.09 (t, 3J(H16,H17)/(H17,H18) = 6 Hz, 1H, H-17), 5.93 (d, 3J(H17,H18) = 6 Hz, 1H, H-18), 5.80 (t, 3J(H15,H16)/(H16,H17) = 6 Hz, 1H, H-16), 5.60 (d, 3J(H14,H15) = 6 Hz, 1H, H-14), 2.28 (s, 3H, H-19) ppm. 13C(1H) NMR (125.8 MHz, d4-MeOD, 25 °C): δ = 193.0 (C-6), 160.3 (C-1), 159.6 (C-10), 155.1 (C-5), 141.1 (C-3), 130.5 (C-2), 130.2 (C-7), 127.7 (C-9/C-11), 124.8 (C-4), 117.2 (C-8/C-12), 107.5 (C-13), 92.6 (C-15), 90.9 (C-17), 88.0 (C-18), 83.8 (C-16), 83.7 (C-14), 19.4 (C-19) ppm.
[Chlorido(η6-benzylammonium)(N-(4-hydroxyphenyl)-2-pyridinecarbothioamide)ruthenium(II)]dichloride (P1). A previously published method was followed.5 In brief, N-(4-hydroxyphenyl)-2-pyridinecarbothioamide (44 mg, 0.19 mmol) was dissolved in DMF (2 mL) under argon and was stirred until a clear solution was obtained. [Ru(η6-benzylammonium)Cl2]2Cl2 (63 mg, 0.1 mmol) was suspended in DMF (1 mL) and was added under argon. The reaction mixture was stirred over night at room temperature in the dark. A clear dark orange solution formed. Chloroform (30 mL) was added and then cooled to 4 °C. The red precipitate was filtered under argon and dried in vacuo. Yield: 98 mg (94%). Elemental analysis found: C, 39.31; H, 3.65; N, 6.73; S, 5.37, calculated for C19H20Cl3ON3RuS·0.4CHCl3: C, 39.25; H, 3.46; N, 7.08; S, 5.40. HR-MS (ESI+): m/z 438.0203 ([M − 2HCl − Cl]+, mtheor = 438.0213, 2.3 ppm). 1H NMR (500.1 MHz, d4-MeOD, 25 °C): δ = 9.74 (d, 3J(H1,H2) = 6 Hz, 1H, H-1), 8.43 (d, 3J(H3,H4) = 7 Hz, 1H, H-4), 8.32 (t, 3J(H2,H3)/(H3,H4) = 7 Hz, 1H, H-3), 7.86 (t, 3J(H1,H2)/(H2,H3) = 6 Hz, 1H, H-2), 7.46 (d, 3J(H8,H9)/(H11,H12) = 9 Hz, 2H, H-9/H-11), 6.96 (d, 3J(H8,H9)/(H11,H12) = 9 Hz, 2H, H-8/H-12), 6.44 (t, 3J(H14,H15)/(H15,H16) = 6 Hz, 1H, H-15), 6.35 (t, 3J(H16,H17)/(H17,H18) = 6 Hz, 1H, H-17), 6.18 (d, 3J(H17,H18) = 6 Hz, 1H, H-18), 5.96 (d, 3J(H14,H15) = 6 Hz, 1H, H-14), 5.94 (t, 3J(H15,H16)/(H16,H17) = 6 Hz, 1H, H-16), 4.14 (q, 3J(H19,NH) = 14 Hz, 2H, H-19) ppm. 13C(1H) NMR (125.8 MHz, d4-MeOD, 25 °C): δ = 160.5 (C-1), 159.7 (C-10), 155.0 (C-5), 141.5 (C-3), 130.8 (C-2), 130.3 (C-7), 127.6 (C-9/C-11), 125.0 (C-4), 117.2 (C-8/C-12), 92.9 (C-14), 92.0 (C-17), 88.6 (C-16), 85.9 (C-15), 83.9 (C-18), 42.6 (C-19) ppm.
[Chlorido(biotin–dPEG(4)-amido-η6-benzyl)(N-(4-hydroxyphenyl)-2-pyridinecarbothioamide)ruthenium(II)]chloride (Ru-OH-2). The precursor P1 (2.99 mg, 0.005 mmol) was dissolved in DMF (0.12 mL) and biotin–dPEG(4)–NHS (2.95 mg, 0.005 mmol) dissolved in DMF (0.12 mL) was added. Triethylamine (10 μL) was added and the flask was purged with argon and stirred for 1 h. The in situ generated Ru-OH-2 was directly immobilized on the streptavidin beads. HR-MS (ESI+): 911.2406 ([M − HCl]+, mtheor = 911.2414, 0.9 ppm).
Stability test
Compound Ru-OH-2 was prepared according to the procedure outlined above and the reaction was monitored over time by taking aliquots after 30, 60 and 240 min. High resolution mass spectra were recorded on a Maxis UHR ESI time-of-flight mass spectrometer (Bruker Daltonics) employing the following parameters: capillary −4.5 kV, nebulizer 0.4 bar, dry gas 8 L min−1, dry temperature 180 °C, 400 Vpp funnel RF, 4 eV quadrupole ion energy and 110 μs transfer time. Aliquots were diluted with MeOH to 1 μM and injected by direct infusion into the mass spectrometer at a flow rate of 3 L min−1. Generally, spectra were recorded in positive ion mode over 0.5 min and averaged. The Data Analysis 4.0 software package (Bruker Daltonics) was used for processing.Cell culture
HCT116 cells (purchased from the ATCC) were cultivated to approximately 108 cells per T75 flask for pull-down experiments using McCoy's 5A modified medium (Gibco, Life Technologies, containing 2 mM L-glutamine), supplemented with penicillin, streptomycin and 10% heat-inactivated fetal-calf serum. The cancer cells were expanded in T25 flasks for response profiling experiments and treated with half the IC50-concentrations of plecstatin-2 (15 μM) or arsenic trioxide (5 μM) for 3 h. The cells were fractionated into cytoplasmic (CYT) and nuclear (NE) fractions before digestion.6 For the PCR experiments, the cells were analogously treated with plecstatin-2 or arsenic trioxide for 24 h. Three biological replicates were used for each condition.Pull-down experiments
HCT116 colon carcinoma cell pellets were suspended in non-denaturing lysis buffer (1.5 mL) and homogenized by passing through a gauge syringe twenty times. The lysis buffer consisted of NaCl (100 mM), Tris HCl (50 mM), MgCl2 (1.5 mM), NaF (25 mM), NP-40 (0.2%), glycerol (5%), DDM (0.2%), TLCK (10 μg mL−1), DTT (1 mM), Na3VO4 (1 mM), PMSF (1 mM) and pepstatin/leupeptin/aprotinin (each 1 μg mL−1), at pH 7.4. The suspension was centrifuged (3500 rpm, 5 min, 4 °C) and the supernatant was collected containing the whole protein cell lysate. In normal pull-downs, the HCT116 whole cell lysate was directly exposed to the immobilized compound, whereas the whole cell lysate was pre-incubated for 1 h with free drug (20 μM in DMF) for competitive pull-downs.Pierce Streptavidin UltraLink resins were washed with lysis buffer. The biotin conjugated metallodrugs were added and incubated for 30 min. After washing with lysis buffer, the HCT116 whole cell lysate (1.5 mL) was added and incubated independently for 2 or 19 h, respectively. The beads were then transferred into spin columns (MobiSpin Column “F”, Mobitec) and washed with lysis buffer and HEPES buffer (pH 8, HEPES–NaOH 50 mM, EDTA 0.5 mM, including PMSF 1 mM, TLCK 10 μg mL−1 and pepstatin/leupeptin/aprotinin (each 1 μg mL−1)). Proteins were eluted with 250 μL elution buffer (formic acid 50%, urea 2 M) and stored at −20 °C.
Protein digestion
Protein samples from response profiling containing 25 μg protein (determined by Bradford assay) were loaded onto molecular weight cut-off filters. The acidic samples of the pull-down experiments containing 30 μg protein were first neutralized with ammonium bicarbonate buffer (pH 8) on the molecular weight cut-off filters. Proteins were then reduced with dithiothreitol at 37 °C and alkylated with iodoacetamide. Then, proteins were digested with trypsin/LysC and the peptides were collected and dried according to a published procedure.6Hyphenated mass spectrometry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 resolution at m/z 200, AGC target 1e6, 20 ms max. accumulation time in the IT) and fragment MS (AGC target 5e4, isolation 1 m/z, HCD 30% NCE, intensity threshold 6.7e2, apex trigger 6–20 s, charge exclusion 1 and >5, dynamic exclusion 30 s, 250 ms fill time). The resolution was 17500 at m/z = 200.
:acetonitrile:formic acid, 98:2:0.1) and B (water:acetonitrile:formic acid, 80:20:0.1). Each dried sample was reconstituted with 10 fmol synthetic peptide standard (in 30% FA, 5 μL) and with eluent A (40 μL) and the injection volume was 5 μL. Chromatographic separations were performed with a Pepmap100 pre-column (2 cm × 75 μm, C18, Thermo Fisher Scientific) and Pepmap100 analytical column (50 cm × 75 μm, C18, Thermo Fisher Scientific). For the target profiling, the chromatographic method consisted of a 38 min stepped gradient from 7–35% B and a 85 min total run time. A gradient from 8–40% B over 120 min was used for the cytoplasmic and nuclear fractions of the response profiling experiments.
000 resolution at m/z 200, AGC target 1e6, 20 ms max. accumulation time in the IT) and fragment MS (AGC target 5e4, isolation 1 m/z, HCD 30% NCE, intensity threshold 6.7e2, apex trigger 6–20 s, charge exclusion 1 and >5, dynamic exclusion 30 s, 250 ms fill time). The resolution was 17500 at m/z = 200.
:acetonitrile:formic acid, 98:2:0.1) and B (water:acetonitrile:formic acid, 80:20:0.1). Each dried sample was reconstituted with 10 fmol synthetic peptide standard (in 30% FA, 5 μL) and with eluent A (40 μL) and the injection volume was 5 μL. Chromatographic separations were performed with a Pepmap100 pre-column (2 cm × 75 μm, C18, Thermo Fisher Scientific) and Pepmap100 analytical column (50 cm × 75 μm, C18, Thermo Fisher Scientific). For the target profiling, the chromatographic method consisted of a 38 min stepped gradient from 7–35% B and a 85 min total run time. A gradient from 8–40% B over 120 min was used for the cytoplasmic and nuclear fractions of the response profiling experiments.
Data evaluation
Proteins were searched with MaxQuant (version 1.5.2.8) including the Andromeda search engine for label-free quantification of cytoplasmic fractions, nuclear fractions and pull-down samples. A maximum of two missed cleavages and at least two unique peptides were required for identifications. The first search peptide tolerance was 20 ppm and the main search tolerance was set to 5 ppm for this Orbitrap instrument type. Protein and peptide false-discovery rate (FDR) was set to 0.01. We included N-terminal acetylation and methionine oxidation as variable modifications and cysteine carbamidomethylation as a fixed modification. For searches of phosphorylated proteins, dynamic modifications on serine, threonine and tyrosine were added. The minimum score was set to 40. Proteins were searched against the human UniProtKB/SwissProt database. Protein isoforms were usually combined into protein groups. Data sets were analyzed with Perseus (Version 1.5.1.6). After log(2) transformation of the LFQ-values, rows were filtered based on valid values with a minimum number of 6 values in at least one group. Additional searches were performed using Proteome Discoverer (1.4.0.288) with Mascot and a Mascot Significance Threshold of 0.01 on the peptide level. Here, all proteins were included with >2 peptides and PSMs (peptide-spectrum matches) and a score of >30. Only proteins with a positive enrichment were considered for each pull-down experiment.The bubble plot contains four dimensions. The y-axis represents the enrichment of a protein and is calculated as the ratio of the LFQ-areas of a given protein (two biological and two technical replicates) in the normal pull-down with respect to the competitive pull-down. The x-axis denotes the confidence of the enrichment and is obtained by a p-value of the mentioned LFQ-areas. The size of the bubbles represents the intensity in the mass spectrometric analysis. Finally, the colour represents the specific binding probability (P) and is a measure of specificity of a given protein to bind to the small molecule probe. It is obtained via the CRAPome27 database and is calculated as follows:
| P = 1 − (CRAPome N° of samples containing the protein)/(CRAPome total N° of samples)·(CRAPome average SC)/(average SC of pull-down) |
Data repository
The raw data of the proteomics experiments can be found at the PRIDE database under the following identifiers: PXD005367–72 (3 h response profiling), PXD005373–6 (24 h response profiling), PXD005377–8 and PXD005386–9 (2 h, 4 h and 19 h target profiling with plecstatins) and PXD010318–9 (4 h target profiling with Ru-OHs).Gene expression experiments
After harvesting cells with RNAprotect® Cell Reagent (Qiagen), RNA was isolated and purified using the AllPrep DNA/RNA Mini Kit (Qiagen), according to the manufacturer's protocol. However, instead of β-mercaptoethanol, dithiothreitol (final concentration 0.04 M) was added to Buffer RLT Plus for cell lysis. RNA concentration was determined with the Qubit™ RNA HS Assay Kit using a Qubit™ 3 Fluorometer (ThermoFisher Scientific). Reverse transcription of 1 μg RNA was performed with the QuantiTect® Reverse Transcription Kit (Qiagen) following the manufacturer's instructions.Gene expression was measured by real-time quantitative polymerase chain reaction (qPCR) using the resulting cDNA, QuantiTect® Primer Assays and Rotor-Gene® SYBR® Green PCR Kit in the Rotor-Gene Q instrument (all from Qiagen). According to proteomics results, GAPDH was chosen as house-keeping gene. QuantiTect® Primer Assays were Hs_GAPDH_1_SG, Hs_TIMM17A_1_SG, Hs_TOMM40_1_SG, Hs_ATF3_1_SG, Hs_DDIT3_1_SG and Hs_CLPP_1_SG. PCR program included an initial activation step of 5 min at 95 °C, followed by a 40× repeated 2-step cycling of 5 s denaturation at 95 °C and 10 s combined annealing and elongation at 60 °C. Specificity of PCR was checked by melting curve analysis.
Relative expression was calculated using the ΔΔCT method and was expressed as 2−ΔΔCT. Two-tailed Student's t-test for independent samples (CI = 0.95) without Welch correction was performed on ΔΔCT values with SPSS® Statistics version 25 (IBM) to test for differences in gene expression by the individual treatment compared to the untreated control. Differences were considered statistically significant at a p value ≤0.05.
Results and discussion
Non-platinum metallodrugs such as those belonging to the ruthenium and osmium classes are widely investigated as anticancer agents because they promise to follow novel modes of action.1 While DNA was revealed as the main target of platinum anticancer agents already in the 1970s,28 it was unclear until recently, whether derivatives of the ruthenium class would target DNA or rather proteins. Indications on the DNA versus protein binding preference were obtained from crystal soaking experiments with the nucleosome core particle, which is the basic chromatin structure consisting of a histone octamer surrounded by double-stranded DNA.29 While oxaliplatin was shown to preferentially interact with the oligonucleotide in this model system,29 the organometallic ruthenium(arenes) turned out to be tuneable with respect to this property, depending on the inert ligands.30 In the same setup, the drug candidate [chlorido(η6-p-cymene)(N-(4-fluorophenyl)-2-pyridinecarbothioamide)ruthenium(II)]chloride (plecstatin-1, Scheme 1A) was shown to exclusively coordinate to histidines 79 and 106 of histone 2B.24 Then, an MS-based shotgun proteomics method was developed to identify potential protein targets of plecstatin-1 by a combination of affinity purification of cell lysates and in vitro treatment of cell cultures.5 Unexpected target selectivity was detected for plecstatins and plectin was validated as the main target.5 Depolarization of the mitochondrial membrane potential (MMP) and induction of reactive oxygen species (ROS) were also observed upon plecstatin treatment, but it remained unclear how these effects were related to plectin targeting.5Plecstatin-1 was found to hydrolyse with a half-life of approximately 50 min in 104 mM sodium chloride, while the osmium analogue did not hydrolyse significantly within 14 h under the same conditions.24 However, a 2 h treatment revealed a virtually identical organ distribution pattern for plecstatin-1 and its isosteric osmium-analogue, suggesting that the compounds remain largely intact in vivo for this time period.23 The compounds are probably stabilized by rapid non-covalent binding to serum proteins,31 which was already observed for the ruthenium(III)-based KP1019.32
In order to investigate the impact of the hydrolysis behaviour on the target selectivity of plecstatins, time-dependent target profiling experiments were performed using cell lysates of HCT116 colon carcinoma cells. Target profiling was carried out as a differential experiment using the biotin-modified plecstatin-3 (Fig. 1), which was immobilized on streptavidin-containing beads. On the one hand, the immobilized plecstatin-3 was exposed to native whole cell lysates of HCT116 cells, termed normal pull-down. On the other hand, the native whole cell lysate was pre-treated with plecstatin-2 in order to saturate selective binding partners before adding to the immobilized plecstatin-3. The second approach was termed competitive pull-down. The normal pull-down identifies selective and non-selective interactors of the immobilized plecstatin-3, while the competitive pull-down identifies only non-selective interactors. A differential analysis of the normal and competitive pull-down may then reveal potential selective protein interactors of the immobilized plecstatin-3. For the time-dependent experiment, the normal and competitive pull-downs were incubated for 2, 45 and 19 h, respectively, to account for hydrolysis kinetics of plecstatins.
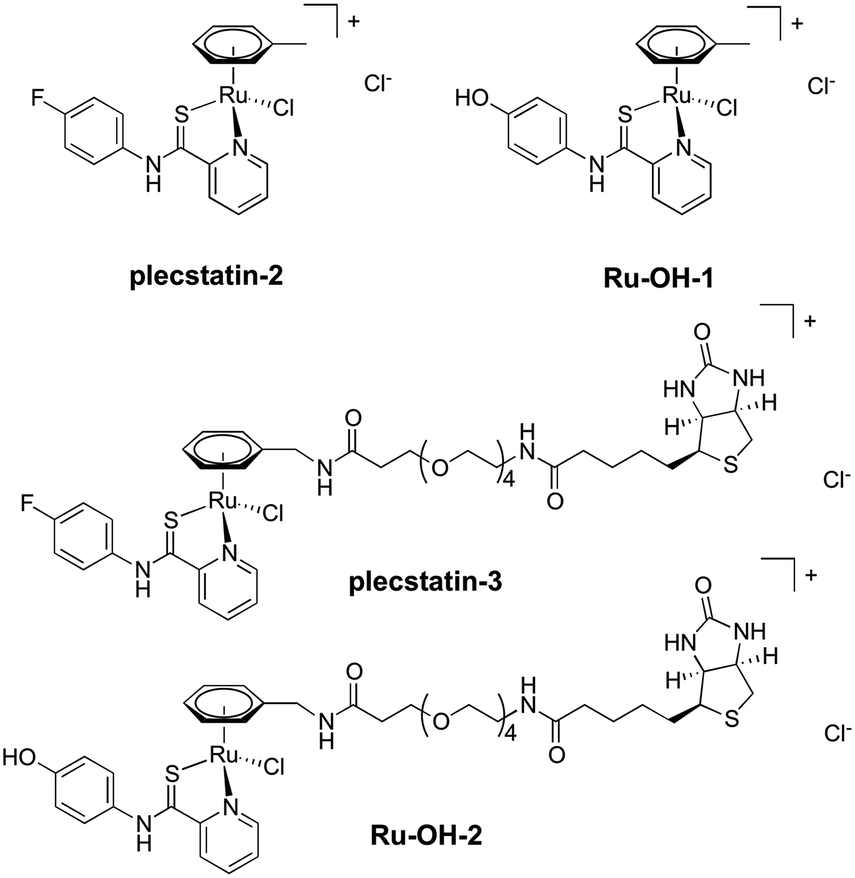 | ||
| Fig. 1 Chemical structures of the compounds investigated in this study. | ||
Approximately 350–450 potential interactors were identified in the pull-down experiments at different time points. The Venn diagram in Fig. 2 depicts the number and overlap of identified interactors at different incubation times. Several potential interactors were identified in all three pull-down experiments among which were most notably five ribosomal proteins (RPS3, RPS3A, RPS9, RSP16, RPL5) and two proteins related to tight junctions (tight junction protein 1 and cingulin).
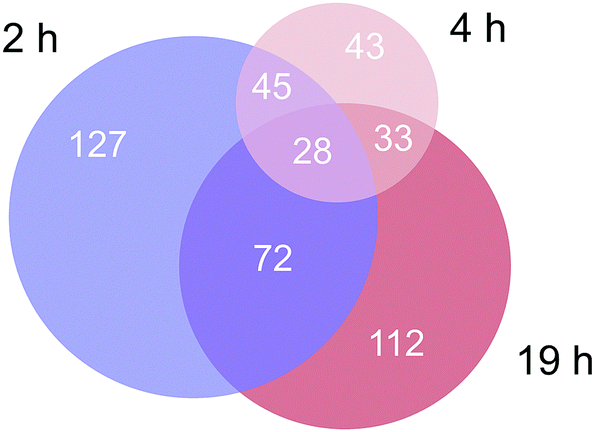 | ||
| Fig. 2 Venn diagram of the number of identified proteins in the three time-dependent pull down experiments. | ||
The 2 h pull-down revealed twelve potential interactors with an enrichment of up to 50-fold (Fig. 3). These interactors are considered fast binders that interact with the intact plecstatin-3 prodrug via non-covalent and electrostatic bonding. Notably, the ribosomal proteins (RPL23, RPL34 and RPS17L) were among the most enriched proteins, besides high mobility group protein A1 and the RNA processing protein MFAP1. The histone HIST1H2BL was also enriched, but showed low binding specificity (i.e. grey coloured). The overall specificity of the potential interactors was low as indicated by the colour code, giving an average specificity of 0.72.
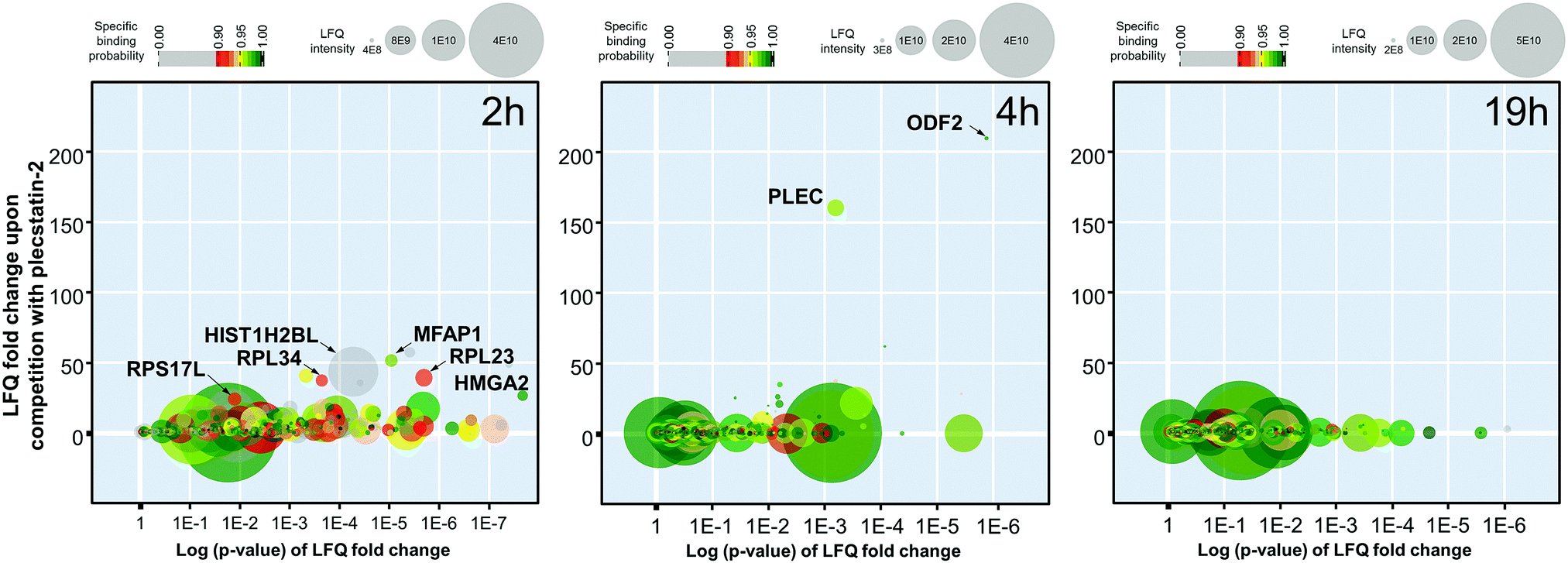 | ||
| Fig. 3 Graphic representation of the time-dependent pull-down experiments of plecstatin-3 using whole cell lysates of HCT116 colon carcinoma cells and plecstatin-2 as the competitor after incubating for 2, 4 or 19 h. | ||
The enrichment increased to >150-fold for two potential interactors after 4 h including plectin (PLEC) as already reported.5 The 4 h pull-down experiment returned the lowest number of potential interactors that might additionally be indicative of an increased overall selectivity. Furthermore, the average specificity increased to 0.86 (i.e. green coloured). This indicated that the hydrolysed plecstatin, which forms additional coordinative bonds, increases the binding selectivity and specificity with respect to the prodrug.
Finally, the enrichment of potential interactors dropped to <6-fold after incubating for 19 h. This loss of enrichment may be attributed either to cleavage of the arene from the ruthenium leading to deactivation of the affinity probe, denaturing processes of the native proteins in the whole cell lysate or a combination thereof.
In an attempt to further understand the target engagement of the plecstatins to plectin, an analogue of plecstatin-2 was synthesized using N-(4-hydroxyphenyl)-2-pyridine-carbothioamide as the chelating ligand. The resulting Ru-OH-1 (Fig. 1) features a hydrogen bond donor (–OH) instead of a hydrogen bond acceptor (–F) in para-position of the phenyl. Moreover, the biotin conjugate Ru-OH-2 was prepared in analogy to plecstatin-3. Ru-OH-2 was stable for at least 4 h and its identity was evidenced by high resolution mass spectrometry (Fig. 4A). Thus, a 4 h pull-down experiment was performed with Ru-OH-2 and using Ru-OH-1 as the competitor. This pull-down experiment using HCT116 cell lysates yielded a total of 581 identified interactors of which 141 showed an enrichment of at least 1.5-fold. It was immediately evident from the pull-down plot that Ru-OH-2 was less selective towards plectin displaying a plectin enrichment of only two-fold (Fig. 4B). Thus, replacing the hydrogen bond acceptor by a hydrogen bond donor completely abrogated the target selectivity of the plecstatins and underlines the necessity of this hydrogen bond acceptor for a strong interaction with plectin. In fact, only 13% of the total identified interactors in the 4 h pull-down experiments were detected with both plecstatins and Ru-OHs, which suggested that the two compounds had distinct target spectra in HCT116 cell lysates despite their similarity.
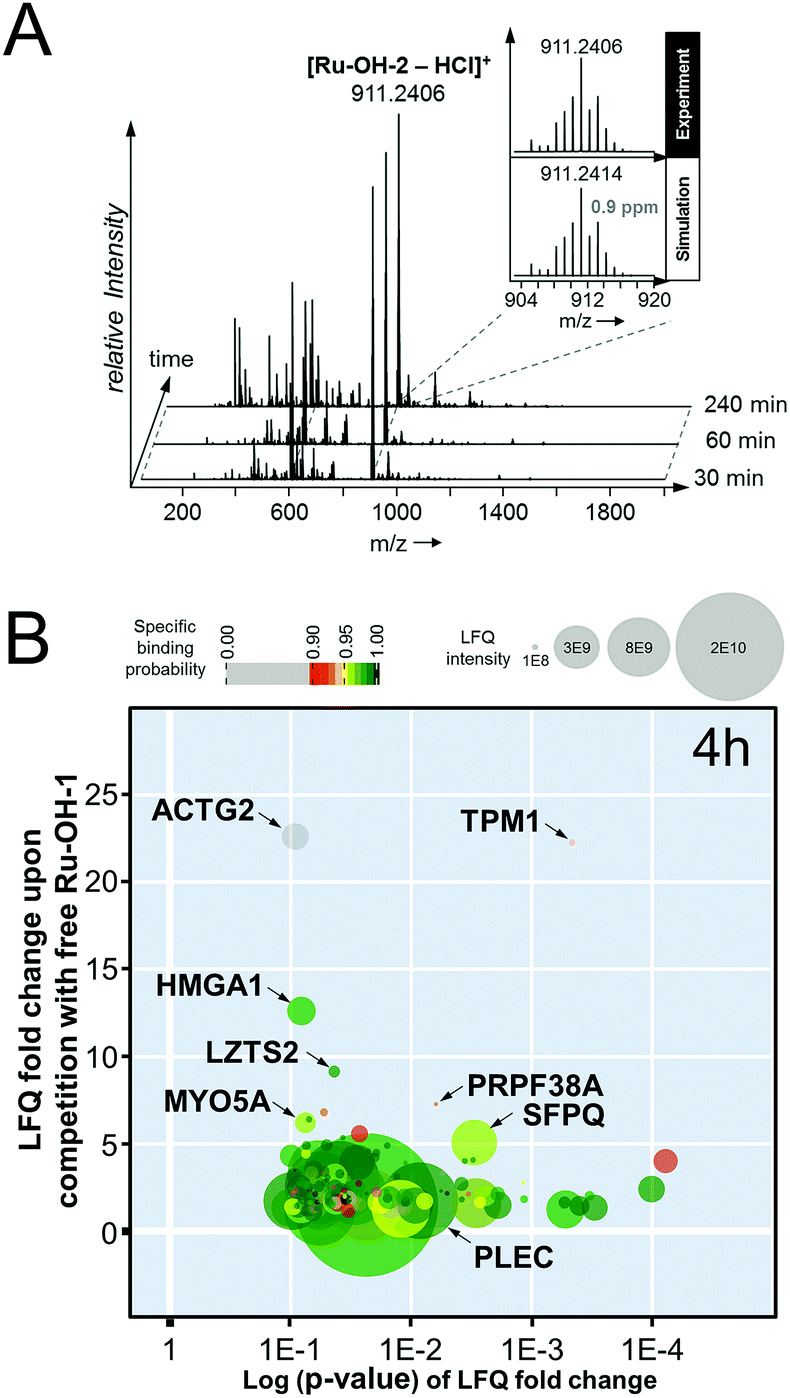 | ||
| Fig. 4 (A) Stability of the biotin-modified Ru-OH-2 over 4 h monitored by high resolution mass spectrometry. (B) Result of the pull-down experiment with Ru-OH-2 and Ru-OH-1 as the competitor using whole cell lysates of HCT116 cancer cells and an incubation time of 4 h. | ||
Based on these findings, the MS-based shotgun proteomics experiment of plecstatin-2 treated HCT116 colon carcinoma cell lines5 was re-evaluated in more detail. In this experiment, cytoplasmic (CYT) and nuclear (NE) fractions were separated and independently analysed.5,6 The 580 significantly regulated proteins were searched for enriched gene ontology (GO) terms against the human gene background and then grouped accordingly. The summed regulations of each group of proteins were then displayed in a two-coordinate plot featuring the total protein regulation in the CYT and NE fractions as the x- and y-axis, respectively (Fig. 5). The CYT-regulated protein groups relating to cell junctions, filaments, actin and microtubules are indicative of plectin targeting because plectin is located in the cytoplasm as a cytolinker of the intermediate filament and present in hemidesmosomes that regulate cell junctions.5,33,34
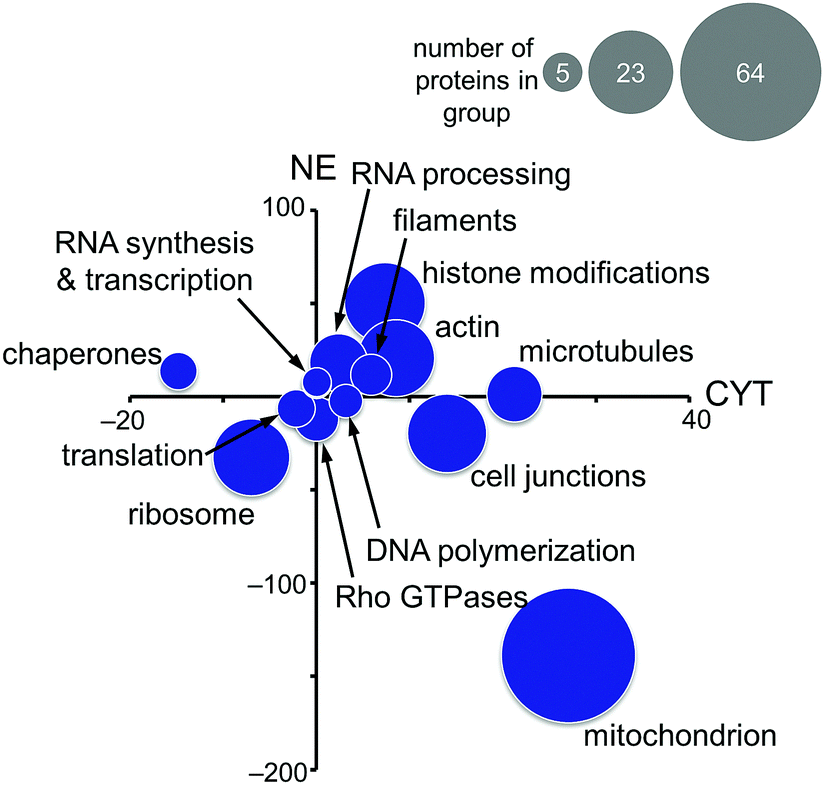 | ||
| Fig. 5 Significantly regulated proteins of HCT116 cancer cells treated for 24 h with half the IC50 concentration of plecstatin-2 were grouped according to gene ontology terms and the summed regulations are depicted. The x-axis represents the fold-changes in the cytoplasmic (CYT) fraction and the y-axis the fold-changes in the nuclear (NE) fraction. The size of the circle represents the number of proteins in each group. | ||
Mitochondrial proteins were found to be strongly regulated and in large number, especially mitochondrial transporters and mitochondrial ribosomes (Fig. 5). Cytoplasmic ribosomes were additionally down-regulated in both the cytoplasmic and nuclear fractions, as well as proteins involved in translation. As shown above, ribosomal proteins were identified as probable interactors of the intact plecstatin prodrug.
Chaperones such as heat shock 70 kDa proteins are known to be particularly induced in cells upon exposure to divalent transition metals35 and constitute a rescue attempt of the cell upon drug insult.24 Specifically, heat shock 70 kDa protein 6 (HSPA6) was down-regulated 15-fold in the cytoplasm and 5-fold up-regulated in the nucleus (Fig. 6A). This translocation was paralleled by an upregulation of heat shock 70 kDa protein 12A (HS12A) in the nuclear fraction. In contrast, the mitochondrial heat shock protein 60 kDa (CH60) was unaffected by the treatment.
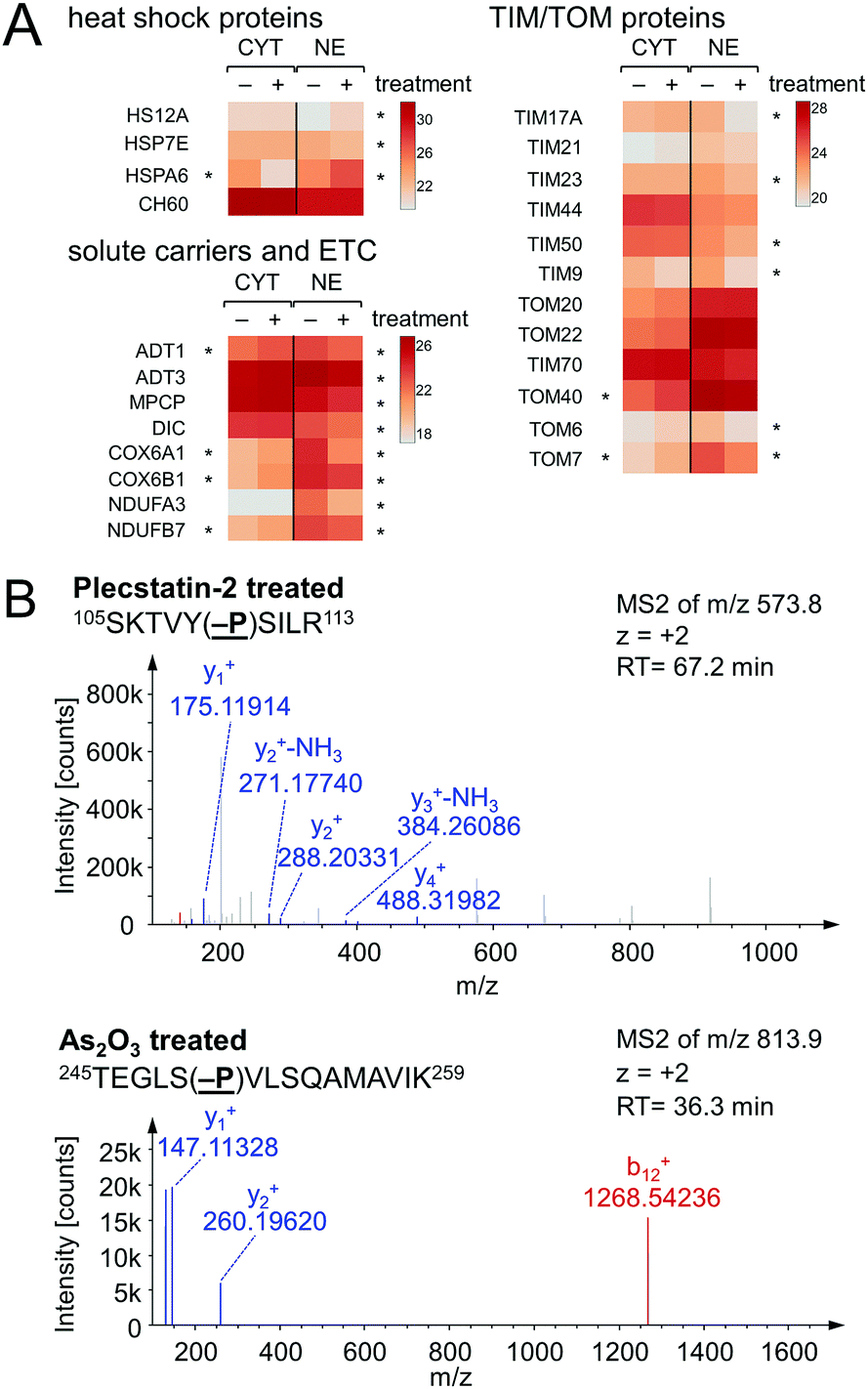 | ||
| Fig. 6 (A) Selected proteins involved in drug effects in the cytoplasmic (CYT) and nuclear (NE) fractions of HCT116 cancer cells treated with half the IC50 concentration of plecstatin-2. An asterisk highlights a multi-parameter significant protein regulation. (B) HCT116 cells were treated with half the IC50 concentration of plecstatin-2 or arsenic trioxide for 3 h. Phosphorylation of eIF2α, the key initiator of the integrated stress-response, was observed in the tandem mass spectrum of the label-free shotgun proteomics approach in both cases. | ||
The observed alterations in heat shock proteins, the increase in ROS5 and the concomitant down-regulation of translation and ribosomal proteins suggested that treatment with plecstatin-2 caused an integrated stress response (ISR) in HCT116 cancer cells.36 Arsenic trioxide is a classical inducer of the ISR, which is mediated by transient phosphorylation of the eukaryotic initiation factor 2α (eIF2α) between 3–8 h after drug perturbation.37 Thus, a response profiling experiment was performed by treating HCT116 cells with sub-cytotoxic concentrations of plecstatin-2 or arsenic trioxide for 3 h. At this time point, the cellular effects are most likely mediated by the intact plecstatin-2 prodrug. Arsenic trioxide was used as positive control and phosphorylation events were determined as recently reported by evaluating phosphorylated serine, tyrosine and threonine residues.38 Selective phosphorylation of eIF2α was determined in the tandem mass spectra after by treating HCT116 cells with plecstatin-2 and arsenic trioxide (Fig. 6B).
An ISR is characterised by reduced cytosolic protein synthesis that reportedly depletes proteins with short half-lives in mitochondria. This leads to a decrease in abundance of TIM17A, a transport protein in the inner mitochondrial membrane.37 The attenuated import of proteins into mitochondria confers stress-resistance against oxidative insult.36 This effect was confirmed by treating HCT116 cells with plecstatin-2 for 24 h, which led to the down-regulation of the transporter of inner mitochondrial membrane 17A (TIM17A) in the NE fraction (Fig. 6A). Because mitochondria are anchored to the cytoskeleton, mitochondrial proteins are expected to be found in the NE fraction. Importantly, the related scaffold proteins TIM21 and TIM44 were unaffected by the treatment. Besides TIM17A, several substrates of these transporters, especially members of the solute carrier protein family SLC25 were down-regulated as well, including the ADP/ATP translocases 1 and 3 (ADT1 and ADT3), the mitochondrial phosphate carrier protein (MPCP) and the mitochondrial dicarboxylate carrier (DIC, Fig. 6A). In some cases, these proteins accumulated in the cytoplasmic fraction further supporting a reduced capability of protein import into mitochondria (Fig. 6A). Similarly, several additional mitochondrial proteins encoded in the nucleus accumulated in the cytoplasmic fraction upon treatment, while being reduced in the nuclear fraction, among which were cytochrome c oxidase subunits 6A1 and 6B1 (COX6A1 and COX6B1) and NADH dehydrogenase 1α subunit 3 and 1β subunit 7 (NDUFA3 and NDUFB7).
The previously observed depolarization of the MMP after plecstatin treatment5 can thus be rationalized by a reduced protein import into mitochondria upon ISR induction. Furthermore, a prolonged depolarized MMP can lead to degradation of TIM23, ultimately inhibiting cell proliferation.39 TIM23 was reported to diminish by 60–90% within 1–2 days after treating with the kinase inhibitor staurosporin.39 Treating HCT116 cells with plecstatin-2 reduced TIM23 by 56% in the nuclear fraction within 24 h (Fig. 6A). We also found TIM50 to be down-regulated, which was reported to be degraded after TIM23.39 An increased cell population in the G0/G1 phase of the cell cycle supports the reduced proliferative activity upon treatment with plecstatin-2.5
Finally, the induction of an ISR was further investigated on the mRNA level by gene expression analysis via real-time quantitative polymerase chain reaction (qPCR). Again, HCT116 colon carcinoma cells were treated with sub-cytotoxic concentrations of plecstatin-2 or arsenic trioxide for 24 h. Arsenic trioxide was used as the positive control for ISR induction. The effects on the Translocase of Inner Mitochondrial Membrane 17A (TIMM17A), Translocase of Outer Mitochondrial Membrane 40 (TIMM40), Activating Transcription Factor 3 (ATF3), DNA Damage Inducible Transcript 3 (DDIT3, also known as CHOP-10) and Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit (CLPP) expression were evaluated (Fig. 7). ATF3 and DDIT3 are members of the stress-response genes upon ISR induction40 and CLPP is a marker for the mitochondrial unfolded protein response (UPRmt).36
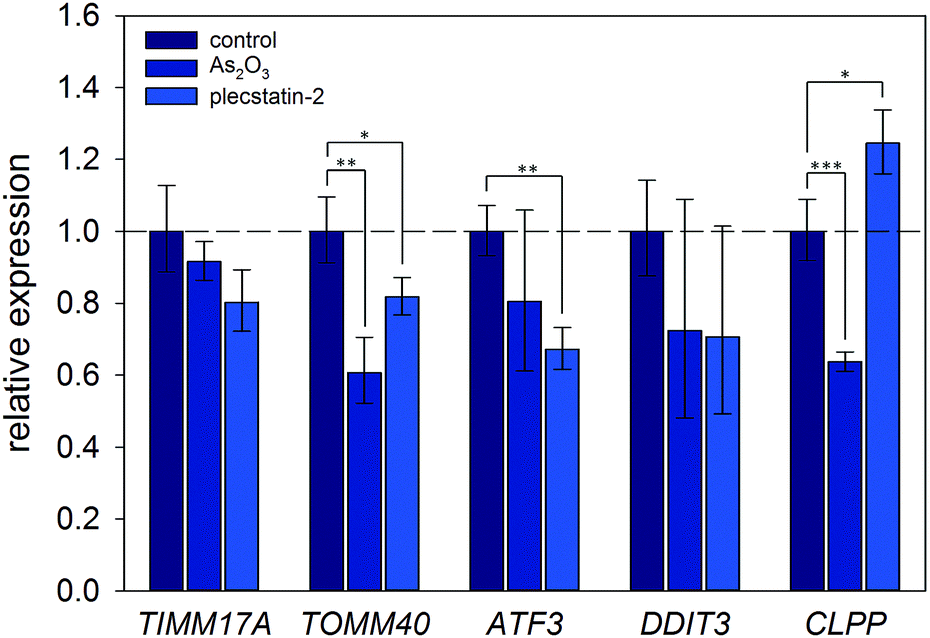 | ||
| Fig. 7 Gene expression analysis of key protein transcripts involved in the ISR and UPRmt. HCT116 cancer cells were treated with half the IC50 concentration of plecstatin-2 or arsenic trioxide for 24 h compared to untreated controls. Data are represented as mean ± SD and significant changes are marked with * for p ≤ 0.05, ** for p ≤ 0.01 and *** for p ≤ 0.001. | ||
Plecstatin-2 was found to decrease the expression of TOMM40 (p ≤ 0.05) and ATF3 (p ≤ 0.01) (Fig. 7). Arsenic trioxide, the positive control, decreased the expression of TOMM40 (p ≤ 0.01) but did not affect the expression of ATF3. Neither plecstatin-2 nor arsenic trioxide significantly altered gene expression of TIMM17A and DDIT3. The UPRmt marker CLPP was up-regulated by plecstatin-2 (p ≤ 0.05), but down-regulated by arsenic trioxide (p ≤ 0.001), indicating effects by plecstatin-2 beyond the ISR. In accordance with literature data, gene expression is considerably less affected by an ISR, than the proteome.41 In sum, our experiments support the induction of mitochondrial stress by plecstatin-2, particularly an ISR, as an additional drug effect besides plectin-targeting.
Conclusions
Time-dependent proteomics-based investigations on the detailed cellular effects of metallo-prodrugs are still rare, possibly due to the challenge to account for their hydrolysis behaviours. Time-dependent target profiling experiments were performed and showed that the activated plecstatins displayed the highest target selectivity and binding specificity in whole cell lysates of HCT116 colon carcinoma cells. A strong target engagement with plectin relies on the hydrogen bond acceptor (–F) of plecstatins. Substituting this hydrogen bond acceptor with a hydrogen bond donor (–OH) abrogated plectin enrichment in the target profiling assay. Treating HCT116 cell cultures with plecstatin-2 induced an integrated stress response (ISR), similarly to arsenic trioxide, and this provided a rationale for the depolarization of the mitochondrial membrane potential and induction of reactive oxygen species, as previously observed after plecstatin-treatment. Phosphorylation of eIF2α, a key mediator of the ISR, after 3 h indicated that plecstatin-2 induced an ISR by the intact prodrug, while the plectin-targeting effects are mediated by the activated metallodrug.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the mass spectrometry core facility of the Faculty of Chemistry, University of Vienna, for access to their instruments. Ricarda Bugl of the NMR core facility is acknowledged for acquiring NMR spectra.References
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Structure-activity relationships for ruthenium and osmium anticancer agents – towards clinical development, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- I. Romero-Canelon and P. J. Sadler, Systems approach to metal-based pharmacology, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4187–4188 CrossRef CAS PubMed.
- R. F. S. Lee, A. Chernobrovkin, D. Rutishauser, C. S. Allardyce, D. Hacker, K. Johnsson, R. A. Zubarev and P. J. Dyson, Expression proteomics study to determine metallodrug targets and optimal drug combinations, Sci. Rep., 2017, 7, 1590 CrossRef PubMed.
- Y. C. Wang, L. G. Hu, F. Xu, Q. Quan, Y. T. Lai, W. Xia, Y. Yang, Y. Y. Chang, X. M. Yang, Z. F. Chai, J. W. Wang, I. K. Chu, H. Y. Li and H. Z. Sun, Integrative approach for the analysis of the proteome-wide response to bismuth drugs in Helicobacter pylori, Chem. Sci., 2017, 8, 4626–4633 RSC.
- S. M. Meier, D. Kreutz, L. Winter, M. H. M. Klose, K. Cseh, T. Weiss, A. Bileck, B. Alte, J. C. Mader, S. Jana, A. Chatterjee, A. Bhattacharyya, M. Hejl, M. A. Jakupec, P. Heffeter, W. Berger, C. G. Hartinger, B. K. Keppler, G. Wiche and C. Gerner, An organoruthenium anticancer agent shows unexpected target selectivity for plectin, Angew. Chem., Int. Ed., 2017, 56, 8267–8271 CrossRef CAS PubMed.
- D. Kreutz, A. Bileck, K. Plessl, D. Wolrab, M. Groessl, B. K. Keppler, S. M. Meier and C. Gerner, Response profiling using shotgun proteomics enables global metallodrug mechanisms of action to be established, Chem. – Eur. J., 2017, 23, 1881–1890 CrossRef CAS PubMed.
- J. M. Hearn, I. Romero-Canelon, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Potent organo-osmium compound shifts metabolism in epithelial ovarian cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3800–E3805 CrossRef CAS PubMed.
- J. M. Hearn, G. M. Hughes, I. Romero-Canelon, A. F. Munro, B. Rubio-Ruiz, Z. Liu, N. O. Carragher and P. J. Sadler, Pharmaco-genomic investigations of organo-iridium anticancer complexes reveal novel mechanism of action, Metallomics, 2018, 10, 93–107 RSC.
- J. Reedijk, Metal-Ligand Exchange Kinetics in Platinum and Ruthenium Complexes, Platinum Met. Rev., 2008, 52, 2–11 CrossRef CAS.
- V. Graziani, A. Marrone, N. Re, C. Coletti, J. A. Platts and A. Casini, A multi-level theoretical study to disclose the binding mechanisms of gold(III)-bipyridyl compounds as selective aquaglyceroporin inhibitors, Chem. – Eur. J., 2017, 23, 13802–13813 CrossRef CAS PubMed.
- A. A. Nazarov, S. M. Meier, O. Zava, Y. N. Nosova, E. R. Milaeva, C. G. Hartinger and P. J. Dyson, Protein ruthenation and DNA alkylation: chlorambucil-functionalized RAPTA complexes and their anticancer activity, Dalton Trans., 2015, 44, 3614–3623 RSC.
- C. G. Hartinger, M. Groessl, S. M. Meier, A. Casini and P. J. Dyson, Application of mass spectrometric techniques to delineate the modes-of-action of anticancer metallodrugs, Chem. Soc. Rev., 2013, 42, 6186–6199 RSC.
- M. Wenzel and A. Casini, Mass spectrometry as a powerful tool to study therapeutic metallodrugs speciation mechanisms: current frontiers and perspectives, Coord. Chem. Rev., 2017, 352, 432–460 CrossRef CAS.
- Y. Wang, Q. Y. He, R. W. Sun, C. M. Che and J. F. Chiu, GoldIII porphyrin 1a induced apoptosis by mitochondrial death pathways related to reactive oxygen species, Cancer Res., 2005, 65, 11553–11564 CrossRef CAS.
- F. Guidi, I. Landini, M. Puglia, F. Magherini, C. Gabbiani, M. A. Cinellu, S. Nobili, T. Fiaschi, L. Bini, E. Mini, L. Messori and A. Modesti, Proteomic analysis of ovarian cancer cell responses to cytotoxic gold compounds, Metallomics, 2012, 4, 307–314 RSC.
- F. Guidi, A. Modesti, I. Landini, S. Nobili, E. Mini, L. Bini, M. Puglia, A. Casini, P. J. Dyson, C. Gabbiani and L. Messori, The molecular mechanisms of antimetastatic ruthenium compounds explored through DIGE proteomics, J. Inorg. Biochem., 2013, 118, 94–99 CrossRef CAS PubMed.
- T. Gamberi, L. Massai, F. Magherini, I. Landini, T. Fiaschi, F. Scaletti, C. Gabbiani, L. Bianchi, L. Bini, S. Nobili, G. Perrone, E. Mini, L. Messori and A. Modesti, Proteomic analysis of A2780/S ovarian cancer cell response to the cytotoxic organogold(III) compound Aubipy(c), J. Proteomics, 2014, 103, 103–120 CrossRef CAS PubMed.
- L. G. Hu, T. F. Cheng, B. He, L. Li, Y. C. Wang, Y. T. Lai, G. B. Jiang and H. Z. Sun, Identification of metal-associated proteins in cells by using continuous-flow gel electrophoresis and inductively coupled plasma mass spectrometry, Angew. Chem., Int. Ed., 2013, 52, 4916–4920 CrossRef CAS.
- S. K. Fung, T. T. Zou, B. Cao, P. Y. Lee, Y. M. E. Fung, D. Hu, C. N. Lok and C. M. Che, Cyclometalated gold(III) complexes containing N-heterocyclic carbene ligands engage multiple anti-cancer molecular targets, Angew. Chem., Int. Ed., 2017, 56, 3892–3896 CrossRef CAS.
- O. Karaca, S. M. Meier-Menches, A. Casini and F. E. Kuhn, On the binding modes of metal NHC complexes with DNA secondary structures: implications for therapy and imaging, Chem. Commun., 2017, 53, 8249–8260 RSC.
- D. Hu, Y. G. Liu, Y. T. Lai, K. C. Tong, Y. M. Fung, C. N. Lok and C. M. Che, Anticancer gold(III) porphyrins target mitochondrial chaperone Hsp60, Angew. Chem., Int. Ed., 2016, 55, 1387–1391 CrossRef CAS PubMed.
- M. V. Babak, S. M. Meier, K. V. M. Huber, J. Reynisson, A. A. Legin, M. A. Jakupec, A. Roller, A. Stukalov, M. Gridling, K. L. Bennett, J. Colinge, W. Berger, P. J. Dyson, G. Superti-Furga, B. K. Keppler and C. G. Hartinger, Target profiling of an antimetastatic RAPTA agent by chemical proteomics: relevance to the mode of action, Chem. Sci., 2015, 6, 2449–2456 RSC.
- M. H. M. Klose, S. Theiner, C. Kornauth, S. M. Meier-Menches, P. Heffeter, W. Berger, G. Koellensperger and B. K. Keppler, Bioimaging of isosteric osmium and ruthenium anticancer agents by LA-ICP-MS, Metallomics, 2018, 10, 388–396 RSC.
- S. M. Meier, M. Hanif, Z. Adhireksan, V. Pichler, M. Novak, E. Jirkovsky, M. A. Jakupec, V. B. Arion, C. A. Davey, B. K. Keppler and C. G. Hartinger, Novel metal(II) arene 2-pyridinecarbothioamides: a rationale to orally active organometallic anticancer agents, Chem. Sci., 2013, 4, 1837–1846 RSC.
- F. K. Cheung, C. Lin, F. Minissi, A. L. Criville, M. A. Graham, D. J. Fox and M. Wills, An investigation into the tether length and substitution pattern of arene-substituted complexes for asymmetric transfer hydrogenation of ketones, Org. Lett., 2007, 9, 4659–4662 CrossRef CAS.
- M. A. Bennett and A. K. Smith, Arene ruthenium(II) complexes formed by dehydrogenation of cyclohexadienes with ruthenium(III) trichloride, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC.
- D. Mellacheruvu, Z. Wright, A. L. Couzens, J. P. Lambert, N. A. St-Denis, T. Li, Y. V. Miteva, S. Hauri, M. E. Sardiu, T. Y. Low, V. A. Halim, R. D. Bagshaw, N. C. Hubner, A. Al-Hakim, A. Bouchard, D. Faubert, D. Fermin, W. H. Dunham, M. Goudreault, Z. Y. Lin, B. G. Badillo, T. Pawson, D. Durocher, B. Coulombe, R. Aebersold, G. Superti-Furga, J. Colinge, A. J. R. Heck, H. Choi, M. Gstaiger, S. Mohammed, I. M. Cristea, K. L. Bennett, M. P. Washburn, B. Raught, R. M. Ewing, A. C. Gingras and A. I. Nesvizhskii, The CRAPome: a contaminant repository for affinity purification-mass spectrometry data, Nat. Methods, 2013, 10, 730–736 CrossRef CAS.
- J. M. Pascoe and J. J. Roberts, Interactions between mammalian-cell DNA and inorganic platinum compounds 1. DNA interstrand crosslinking and cytotoxic properties of platinum(II) compounds, Biochem. Pharmacol., 1974, 23, 1345–1357 CrossRef CAS.
- B. Wu, P. Droge and C. A. Davey, Site selectivity of platinum anticancer therapeutics, Nat. Chem. Biol., 2008, 4, 110–112 CrossRef CAS.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. J. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity, Nat. Commun., 2014, 5, 3462 CrossRef.
- M. H. M. Klose, A. Schöberl, P. Heffeter, W. Berger, C. G. Hartinger, G. Koellensperger, S. M. Meier-Menches and B. K. Keppler, Serum-binding properties of isosteric ruthenium and osmium anticancer agents elucidated by SEC-ICP-MS, Monatsh. Chem., 2018 Search PubMed , accepted.
- N. Cetinbas, M. I. Webb, J. A. Dubland and C. J. Walsby, Serum-protein interactions with anticancer Ru(III) complexes KP1019 and KP418 characterized by EPR, J. Biol. Inorg. Chem., 2010, 15, 131–145 CrossRef CAS.
- L. Winter and G. Wiche, The many faces of plectin and plectinopathies: pathology and mechanisms, Acta Neuropathol., 2013, 125, 77–93 CrossRef CAS.
- G. Wiche, S. Osmanagic-Myers and M. J. Castanon, Networking and anchoring through plectin: a key to IF functionality and mechanotransduction, Curr. Opin. Cell Biol., 2015, 32, 21–29 CrossRef CAS.
- M. Murata, P. Gong, K. Suzuki and S. Koizumi, Differential metal response and regulation of human heavy metal-inducible genes, J. Cell. Physiol., 1999, 180, 105–113 CrossRef CAS.
- P. M. Quiros, A. Mottis and J. Auwerx, Mitonuclear communication in homeostasis and stress, Nat. Rev. Mol. Cell Biol., 2016, 17, 213–226 CrossRef CAS.
- T. K. Rainbolt, N. Atanassova, J. C. Genereux and R. L. Wiseman, Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation, Cell Metab., 2013, 18, 908–919 CrossRef CAS.
- A. Bileck, R. L. Mayer, D. Kreutz, T. Weiss, S. Taschner-Mandl, S. M. Meier, A. Slany and C. Gerner, Evaluation of inflammation-related signaling events covering phosphorylation and nuclear translocation of proteins based on mass spectrometry data, J. Proteomics, 2017, 152, 161–171 CrossRef CAS.
- C. G. Goemans, P. Boya, C. J. Skirrow and A. M. Tolkovsky, Intra-mitochondrial degradation of Tim23 curtails the survival of cells rescued from apoptosis by caspase inhibitors, Cell Death Differ., 2008, 15, 545–554 CrossRef CAS.
- H. Y. Jiang, S. A. Wek, B. C. McGrath, D. Lu, T. W. Hai, H. P. Harding, X. Z. Wang, D. Ron, D. R. Cavener and R. C. Wek, Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response, Mol. Cell. Biol., 2004, 24, 1365–1377 CrossRef CAS.
- P. M. Quiros, M. A. Prado, N. Zamboni, D. D'Amico, R. W. Williams, D. Finley, S. P. Gygi and J. Auwerx, Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals, J. Cell Biol., 2017, 216, 2027–2045 CrossRef CAS PubMed.
Footnote |
| † Current address: Department of Pharmacognosy, Faculty of Life Sciences, University of Vienna, Althanstraße 14, 1090 Vienna, Austria. |
| This journal is © The Royal Society of Chemistry 2019 |