
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase-free conversion of glycerol to methyl lactate using a multifunctional catalytic system consisting of Au–Pd nanoparticles on carbon nanotubes and Sn-MCM-41-XS†
Zhenchen
Tang
a,
Dina G.
Boer
a,
Ali
Syariati
 b,
Mihaela
Enache
b,
Petra
Rudolf
b,
Hero J.
Heeres
a and
Paolo P.
Pescarmona
*a
b,
Mihaela
Enache
b,
Petra
Rudolf
b,
Hero J.
Heeres
a and
Paolo P.
Pescarmona
*a
aChemical Engineering Group, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.p.pescarmona@rug.nl
bZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands
First published on 4th July 2019
Abstract
Multifunctional catalytic systems consisting of physical mixtures of (i) bimetallic Au–Pd nanoparticles (average size of 3–5 nm) supported on functionalised carbon nanotubes (CNTs) and (ii) Sn-MCM-41 nanoparticles (50–120 nm), were synthesised and investigated for the base-free, selective conversion of glycerol to methyl lactate in a batch reactor. The catalysts were characterised by means of transmission electron microscopy, N2-physisorption, energy-dispersive X-ray spectroscopy, X-ray photoelectron spectroscopy and by Boehm titration. The catalyst based on bimetallic AuPd/CNTs showed much higher activity than the monometallic Au or Pd counterparts, thus indicating synergetic effects. Functionalisation of the CNTs by oxidative treatments had a positive effect on catalyst performance, which was correlated to the observed increase in surface acidity and hydrophilicity. The highest yield of methyl lactate achieved in this work was 85% at 96% glycerol conversion (140 °C, 10 h at 30 bar air), which is the highest yield ever reported in the literature so far. Insights in the reaction pathway were obtained by monitoring the conversion-time profiles for intermediates and their possible role as inhibitors. Batch recycling experiments demonstrated the excellent reusability of the catalyst.
Introduction
Biomass conversion into biofuels and biobased chemicals represents a sustainable alternative to the use of fossil resources like oil, gas and coal.1–3 Considering the finite nature of the latter, growing research efforts are being dedicated to the development of efficient catalytic routes for biomass valorisation. The catalysts employed in large scale refinery processes are generally not suitable for the conversion of biomass, which consists of compounds with a much higher content of oxygenated groups compared to fossil resources. Therefore, new catalytic methodologies need to be developed. For the upgrading of biobased compounds into valuable chemicals, multistep syntheses are often required and as a consequence there is a strong need for developing multifunctional catalysts, which ideally should be able to promote the different steps in a one-pot reaction.3 When implementing this in the design of a heterogeneous catalytic system, the challenge is to combine two or more kinds of functional sites, such as acid and metallic sites, and to make them work efficiently and cooperatively. In this work, we present the design, synthesis, characterisation and testing of a novel, highly efficient multifunctional catalytic system for the conversion of glycerol to methyl lactate. Glycerol is considered a very attractive biobased platform molecule with high potential for conversion into a broad scope of interesting bulk and fine chemicals.4–6 It is obtained in large quantities as side-product from the biodiesel industry by transesterification of triglycerides with methanol (≈1.2 × 106 t glycerol was produced by the biodiesel industry in 2016).7,8 Therefore, the transformation of glycerol into valuable chemicals has attracted a lot of interest from both academia and industry.9 Among the most promising platform molecules that can be produced from glycerol are lactic acid and the related esters (alkyl lactates).7,10 Lactic acid can be used for the production of poly-lactic acid, a biodegradable bio-polymer with various applications in the food, pharmaceutical and packaging industry.10 Alkyl lactates find application as green solvents and as precursors of lactide. Currently, lactic acid is commercially produced by a fermentation process using carbohydrates as the feed. This process has a relatively low volumetric production rate and generates large amounts of salts during the product work-up.11,12 Therefore, there is an incentive to develop efficient chemo-catalytic methodologies to obtain lactic acid or alkyl lactates from biobased glycerol in a one-pot reaction. This route involves two main consecutive steps: (1) the selective oxidation of glycerol to a triose (dihydroxyacetone and/or glyceraldehyde) and (2) the rearrangement of the trioses into lactic acid (aqueous medium) or alkyl lactate (alcoholic medium) (Scheme 1).13–15 Initial research was typically conducted in strongly basic medium, supposedly essential to promote the rate of the oxidation in the first step of the reaction, and to increase the rate of the rearrangement in the second step by neutralising the formed lactic acid.16–18 A major disadvantage of using a base is the cost required to convert the obtained metal lactate back to lactic acid by an acidification step with a mineral acid, producing large amounts of salt waste. In order to develop a greener and more cost-effective process, it is crucial to design catalytic systems that do not require the use of a homogeneous base for achieving high activity and selectivity in this reaction. This can be achieved by employing noble metals for catalysing the partial oxidation of glycerol (step A in Scheme 1), and a solid acid to catalyse the successive rearrangement of the trioses into lactate/lactic acid (steps B and C in Scheme 1). Au nanoparticles supported on ultra-stable Y (USY) zeolite was the first catalyst reported for the conversion of glycerol to methyl lactate (ML) in methanol, without the need of a base (Table S1, entry 1†).13 Higher yields of methyl lactate (79%) were obtained by incorporating Sn in a related Au/USY zeolite system, though oxygen instead of air was used as the oxidant (Table S1, entry 2†).14 A recent study form our group demonstrated that physical mixtures of Au/CuO and Sn-MCM-41-XS (i.e. Sn-MCM-41 with 50–120 nm particle size) also efficiently convert glycerol into methyl lactate, achieving up to 63% yield at 95% glycerol conversion (Table S1, entry 3†).15 The use of a solid acid like Sn-MCM-41-XS, which contains a combination of mild Brønsted acid sites and Lewis acid sites, is preferable in terms of selectivity compared to zeolites, which display stronger Brønsted acid sites.19,20 An additional advantage of this catalytic system is that supporting the metal nanoparticles on a different material from that providing the Lewis acid sites offers the possibility of modifying and tuning the nature of the support towards enhanced catalytic performance of the whole system.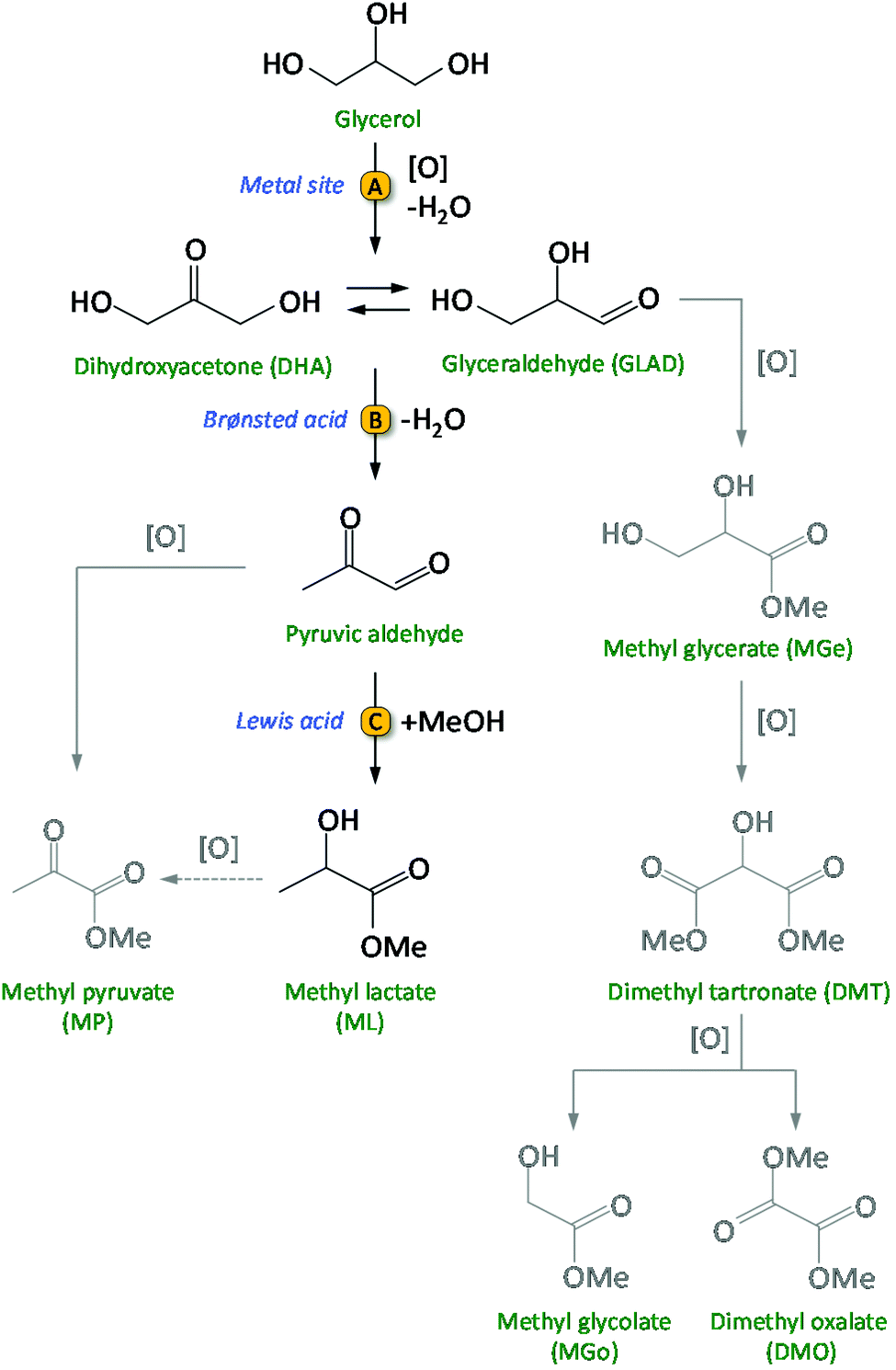 | ||
| Scheme 1 Catalytic route from glycerol to methyl lactate (ML) and, in grey, the possible side-products. | ||
In order to promote the cascade reaction in Scheme 1 in a base-free reaction medium, two kinds of catalytic species are needed: (i) (supported) noble metal nanoparticles (NPs) for the partial oxidation of glycerol; (ii) a solid acid catalyst with Lewis and Brønsted acid sites for the rearrangement step.13,14,20,21 Here, we report for the first time the use of bimetallic Au–Pd nanoparticles supported on (functionalised) carbon nanotubes as catalysts for the oxidation step of glycerol to glyceraldehyde or dihydroxyacetone. Previous work demonstrated that this first step of the reaction is the one on which research should be mainly focussed to increase the overall activity and selectivity of the catalytic system.15 Therefore, we focussed our efforts on designing an enhanced catalyst for this initial step, while employing the optimum solid acid catalyst identified in previous work (i.e. Sn-MCM-41-XS)15,20,22 for promoting the rearrangement of trioses into lactates. We reasoned that the catalyst for the partial oxidation of glycerol could be improved by employing bimetallic nanoparticles, as the presence of a second metal can allow tuning the electronic properties, morphology and stability of the nanoparticles towards increased activity and selectivity.23,24 Therefore, we chose to combine Au with another, relatively less expensive noble metal, Pd, which has proven to lead to increased catalytic performance when used in an alloy with Au in several partial oxidation reactions.3,8,23–28 A second factor that plays an important role for the catalytic behaviour of (bi)metallic nanoparticles is the nature of the support.29,30 In this work, functionalised multi-walled carbon nanotubes (CNTs) were selected as support for the bimetallic nanoparticles. Carbon nanotubes display a large, accessible external surface area and high chemical and thermal stability, which make them attractive supports for metal nanoparticles.31–34 Additionally, carbon nanotubes can be modified by oxidative methods to tune their surface properties like acidity, hydrophilicity and dispersibility.30,35–37 Such functionalisation of the CNTs surfaces was shown to have a positive effect on catalyst performance due to enhanced interactions between the metallic nanoparticles as well as with the substrates.30–32,38,39 However, there is no report in the literature on the use of mono- or bimetallic Au-based nanoparticles supported on functionalised CNTs for the base-free glycerol oxidation to lactic acid/alkyl lactates. Based on these considerations, our strategy involved the synthesis, characterisation and testing of multifunctional heterogeneous catalytic systems for the conversion of glycerol to methyl lactate, consisting of physical mixtures of (i) bimetallic Au–Pd nanoparticles supported on different functionalised CNTs and (ii) Sn-MCM-41-XS. The performance of the bimetallic nanoparticles was compared to the monometallic (Au or Pd) counterparts. The choice of using a physical mixture of the supported nanoparticles and Sn-MCM-41-XS rather than supporting the metal nanoparticles directly on Sn-MCM-41-XS was based on the previously observed infeasibility of achieving high loading of well dispersed nanoparticles without causing the deterioration of this mesoporous material.15 In addition, the use of two materials allows higher flexibility in terms of adjustment of the Sn to noble metal ratios. The batch reactions were carried out in methanol (which has been identified as a “recommended” solvent in the context of green chemistry40) without the addition of an external base and using air as the oxidant at relatively mild conditions for this reaction (see Table S1†).
Results and discussion
Synthesis, characterisation and catalytic performance of monometallic Au, Pd and bimetallic Au–Pd catalysts on unmodified CNTs
A series of catalysts consisting of monometallic Au, monometallic Pd or bimetallic Au–Pd nanoparticles (with a Au/Pd molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1 or 1:3) supported on unmodified CNTs were synthesised by a colloid immobilisation (CI) method. The actual loading of the metals on the CNTs supports, as well as the molar ratios between Au and Pd, were determined by inductively coupled plasma optical emission spectrometry (ICP-OES) (Table 1). The total metal loading ranged from 0.9% to 1.2%, which indicates only slight deviations from the theoretical loading of 1.0 wt%. The higher loading for the materials with higher relative gold content suggests that the observed deviations might be caused by the inaccuracy of the Au content (45–55%) in the commercial Au precursor (HAuCl4·xH2O). As a consequence, the molar ratio between Au and Pd is slightly higher than the theoretical values. The low metal loading of these materials implies that the textural properties of the carbon nanotubes do not get significantly affected by the deposition of the nanoparticles, as shown by the only slightly lower values of surface area and pore volume of AuPd/CNTs compared to the parent CNTs (entries 1 and 6 in Table 2).
:1, 1:1 or 1:3) supported on unmodified CNTs were synthesised by a colloid immobilisation (CI) method. The actual loading of the metals on the CNTs supports, as well as the molar ratios between Au and Pd, were determined by inductively coupled plasma optical emission spectrometry (ICP-OES) (Table 1). The total metal loading ranged from 0.9% to 1.2%, which indicates only slight deviations from the theoretical loading of 1.0 wt%. The higher loading for the materials with higher relative gold content suggests that the observed deviations might be caused by the inaccuracy of the Au content (45–55%) in the commercial Au precursor (HAuCl4·xH2O). As a consequence, the molar ratio between Au and Pd is slightly higher than the theoretical values. The low metal loading of these materials implies that the textural properties of the carbon nanotubes do not get significantly affected by the deposition of the nanoparticles, as shown by the only slightly lower values of surface area and pore volume of AuPd/CNTs compared to the parent CNTs (entries 1 and 6 in Table 2).
| Entry | Catalyst | Au + Pd loading (wt%) | Molar ratio of Au/Pd | |
|---|---|---|---|---|
| Theoretical | Experimental | |||
| 1 | Au/CNTs | 1.2 | — | — |
| 2 | Au3Pd/CNTs | 1.0 | 3 | 3.1 |
| 3 | AuPd/CNTs | 1.0 | 1 | 1.2 |
| 4 | AuPd3/CNTs | 1.0 | 0.33 | 0.4 |
| 5 | Pd/CNTs | 0.9 | — | — |
| Entry | Sample | Specific surface areaa (m2 g−1) | Pore volumeb (cm3 g−1) | C atomic%c | O atomic%c | S atomic%c | Total acid sitesd (mmol g−1) | Stronger acid sitesd (mmol g−1) |
|---|---|---|---|---|---|---|---|---|
| a BET surface area. b BJH cumulative volume of pores, between 17 and 3000 Å width, based on the desorption isotherm. c C, O, S atomic content (mol%), as determined by EDX. d The amount of total and stronger acid sites was determined by Boehm titration of NaOH and NaHCO3, respectively. CNTs-O, CNTs-N, CNTs-S, CNTs-NS denote the CNTs treated by H2O2, HNO3, H2SO4 and H2SO4–HNO3, respectively. n.d. = not determined. | ||||||||
| 1 | CNTs | 258 | 2.1 | 99.2 | 0.8 | — | 0.022 | 0.002 |
| 2 | CNTs-O | 313 | 2.0 | 98.7 | 1.3 | — | 0.90 | 0.42 |
| 3 | CNTs-N | 306 | 2.0 | 98.5 | 1.5 | — | 1.3 | 0.88 |
| 4 | CNTs-S | 259 | 2.1 | 98.8 | 1.1 | 0.11 | 0.46 | 0.23 |
| 5 | CNTs-NS | 274 | 1.6 | 97.3 | 2.6 | 0.10 | 2.4 | 1.3 |
| 6 | AuPd/CNTs | 241 | 2.0 | n.d. | n.d. | n.d. | n.d. | n.d. |
TEM images of the bimetallic catalysts demonstrate the successful synthesis of metal nanoparticles with an average size of around 6 nm and a relatively broad particle size distribution from 1 to 16 nm (Fig. 1A). The relatively larger nanoparticles consist of aggregated and partially coalesced smaller nanoparticles. This is probably due to the weak interaction between the CNTs surface and the nanoparticles during the colloid immobilisation process. As a reference, bimetallic Au–Pd nanoparticles supported on unmodified activated carbon (AC) using the same synthesis protocol also showed (Fig. 1B) coalesced nanoparticles with a large average size (9.9 nm) and a large particle size distribution (1–24 nm), thus supporting the hypothesis that unfunctionalised carbon surfaces tend to lead to relatively large nanoparticles.
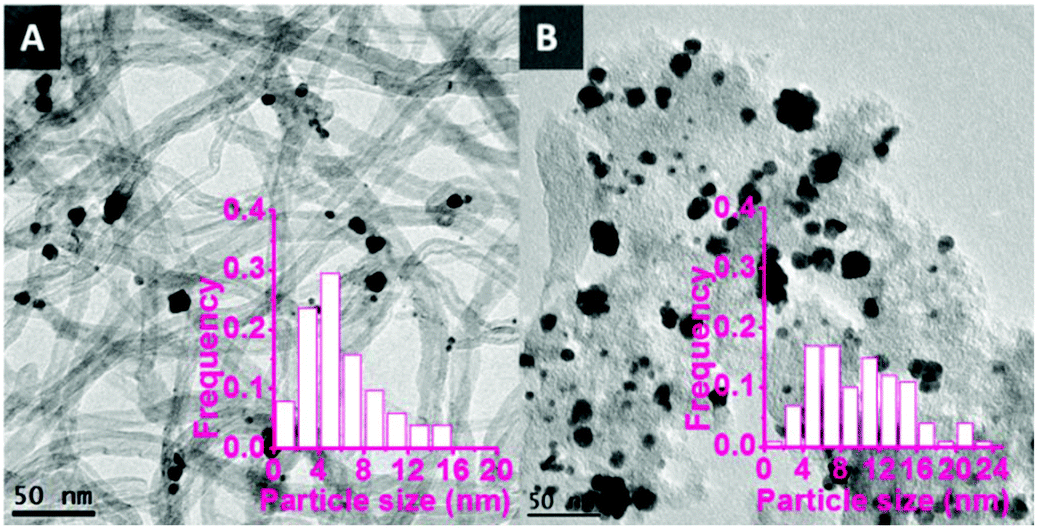 | ||
| Fig. 1 TEM pictures of bimetallic Au–Pd nanoparticles supported on unmodified CNTs and AC. A: AuPd/CNTs, average size = 6.0 nm; B: AuPd/AC, average size = 9.9 nm. | ||
The monometallic Au/CNTs, Pd/CNTs and bimetallic AuPd/CNTs catalysts were characterised by XPS to determine the surface composition and, in the case of the bimetallic catalyst, to investigate whether the metals are actually forming an alloy, or are present as isolated Au and Pd nanoparticles, or as a combination of the two. Based on thermodynamic considerations, the formation of an Au–Pd alloy is expected to be favoured as negative enthalpies of formation have been observed for the Au–Pd system.41 XPS confirms the presence of the metals employed in the synthesis in all samples (Au/CNTs, Pd/CNTs and AuPd/CNTs, Fig. 2). The analysis of the chemical states of Au and Pd was based on the Au 4f7/2 and Pd 3d3/2 signals, which are devoid of the overlap observed between Au 4d5/2 and Pd 3d5/2 or between Au 4f5/2 and Pd 4s.42,43 The Au 4f7/2 peak was found at 84.2 eV in the monometallic Au/CNTs catalyst (Fig. 2a), while it was shifted by about 0.5 eV towards lower binding energy in the bimetallic AuPd/CNTs (Fig. 2b). The same trend was found for the Pd 3d3/2 peak, which was shifted by 0.8 eV towards lower binding energy in the bimetallic AuPd/CNTs sample (Fig. 2d) compared to the monometallic Pd/CNTs one (Fig. 2c). The observed decrease in binding energy of both Au 4f7/2 and Pd 3d3/2 compared to the monometallic counterparts is in line with what has been previously observed in other Au–Pd systems and has been attributed to the formation of an Au–Pd alloy.44,45
 | ||
| Fig. 2 XPS signals of the Au 4f core level region for (a) Au/CNTs, (b) AuPd/CNTs, and of the Pd 3d core level region for (c) Pd/CNTs, (d) AuPd/CNTs. | ||
XRD analysis of the AuPd/CNT materials (Fig. 3) confirmed the formation of the Au–Pd alloy. After loading the Au–Pd nanoparticles on the CNTs, only one new, broad peak (due to the small size of the particles) appears in the XRD pattern at 39.36°. Its position is in between that of the (111) peak of monometallic Au (38.27°) and of the (111) peak of monometallic Pd (40.42°), in agreement with the formation of an Au–Pd alloy.
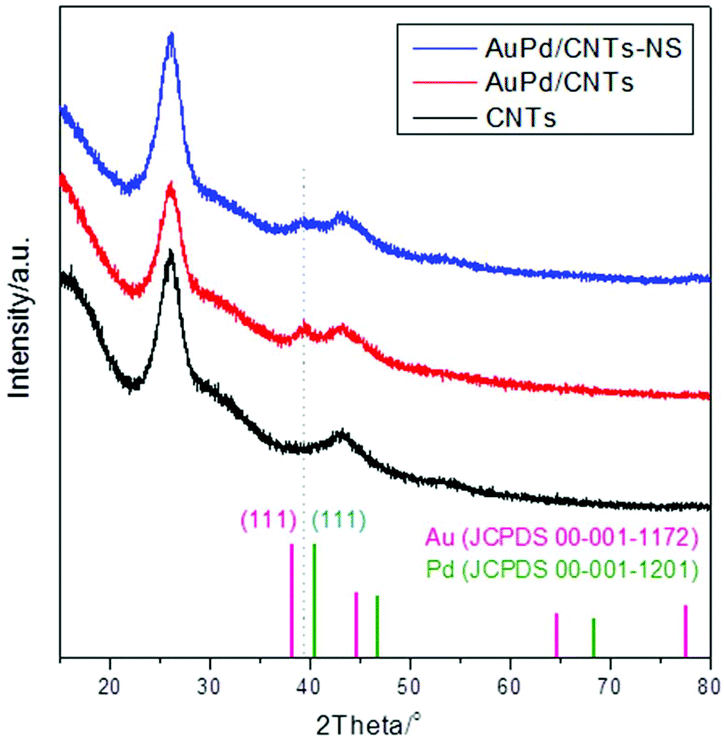 | ||
| Fig. 3 XRD patterns of CNTs before and after loading Au–Pd nanoparticles. | ||
The two monometallic and the bimetallic Au–Pd catalysts on unmodified CNTs were investigated for the conversion of glycerol into methyl lactate in combination with Sn-MCM-41-XS in a batch set-up (140 °C, 30 bar air pressure, Fig. 4). The monometallic Au/CNTs catalyst showed 25% conversion of glycerol and 93% selectivity towards methyl lactate. By gradually increasing the relative Pd content of the catalyst (nominal Au/Pd molar ratio of 3/1; 1; and 1/3), the conversion first increased from 41% to 47% and then decreased to 41% (Fig. 4). The conversion further decreased to 35% when the monometallic Pd/CNTs was used. Conversely, the selectivity towards methyl lactate remained nearly constant at around 90% regardless of the Au/Pd molar ratio.46 The role of the (bi)metallic nanoparticles is to catalyse the first step of glycerol oxidation (i.e. the initial activation of oxygen and/or glycerol, step A in Scheme 1).15,20 The optimum synergistic effect was found for a nominal Au/Pd molar ratio of 1 (actual ratio = 1.2, see Table 1), which matches our anticipation that alloying Au with Pd would lead to enhanced activity. This trend is in agreement with the increase in the rate of the oxidation of alcohol groups of other organic compounds (e.g. benzyl alcohol and 5-hydroxymethylfurfural) that was observed by using bimetallic Au–Pd instead of monometallic Au catalysts.30,47
 | ||
| Fig. 4 Performance of the catalytic system consisting of mono- or bimetallic Au–Pd on unmodified CNTs (with different Au/Pd values) and Sn-MCM-41-XS. Reaction conditions: 0.25 M glycerol in 20 ml methanol; 0.1 g of AuPd/CNTs; 0.2 g of Sn-MCM-41-XS; 140 °C; 30 bar air pressure; 4.5 h batch time. | ||
Synthesis, characterisation and catalytic performance of bimetallic Au–Pd catalysts supported on functionalised CNTs
The study on the catalysts based on Au–Pd nanoparticles supported on CNTs demonstrated the advantage of the bimetallic nanoparticles compared to their monometallic counterparts, but also indicated that the carbon nanotubes are not an optimum support compared to previously reported metal oxides.15 With the purpose of improving the interaction with the support and thus decrease the nanoparticle size, we decided to modify the CNTs used as supports by oxidative treatments using a number of reagents, viz. H2O2, HNO3, H2SO4 and the combination of HNO3 and H2SO4. These treatments are known to largely enhance the density of functional groups (e.g. –OH, –CHO and –COOH) on the CNTs surface.31,39 Additionally, the specific surface area of the CNTs was increased by the oxidative treatment with H2O2, HNO3 and H2SO4–HNO3 (compare entries 1 to 2, 3 and 5 in Table 2), though it was not affected by the treatment with H2SO4 (entry 4, Table 2). Such increase in specific surface area is generally ascribed to loss of structural integrity and size reduction of the CNTs.31,48 The pore volume did not change significantly upon treatment with H2O2, HNO3 and H2SO4, but it decreased to 1.6 cm3 g−1 with the harsher H2SO4–HNO3 treatment.Elemental analysis of the CNTs by EDX allowed determining the C, O and S content at the surface, which provides information on the degree of functionalisation of the CNTs. In the untreated CNTs, the oxygen amount is only 0.8%, whereas all the treatments largely enhanced the oxygen content of the CNTs. The highest value (2.6%, entry 5, Table 2) was obtained upon treatment with a H2SO4–HNO3 mixture. The increased O content indicates the formation of considerable amounts of oxygenated groups (e.g. –OH, –CHO, –COOH) on the surface of the CNTs.48,49 The presence of such oxygenated groups leads to a higher surface acidity, but also significantly improves the hydrophilicity of the CNTs surface. Both features can be important for the catalytic performance of the supported (bi)metallic nanoparticles. The higher surface acidity can promote the acid-catalysed dehydration step of the conversion of glycerol into methyl lactate (i.e. step B in Scheme 1). The higher hydrophilicity can: (i) enhance the interaction with the nanoparticles during their preparation by colloidal immobilisation; (ii) affect the reaction rates by changing the adsorption constants of substrates and products on the surface; (iii) promote the dispersion of the CNTs in a polar reaction medium (which is an asset both for the preparation of the supported nanoparticles as well as for the catalytic reaction). The treatment of CNTs with H2SO4 (entries 4 and 5) is known to lead to the formation of sulphonic acid groups (–SO3H) on the CNTs surface, which is confirmed by the presence of significant amounts of sulphur in these materials (0.1%, see Table 2). The number and strength of the acid sites of the functionalised CNTs were evaluated by Boehm titration with an aqueous solution of NaOH, for the total number of acid sites, and with an aqueous solution of NaHCO3 for the determination of the stronger acid sites (i.e. carboxylic and sulphonic groups).50,51 The untreated CNTs material has a low amount of acid sites (0.022 mmol g−1 in total, entry 1, Table 2). All treatments resulted in an increase in total acid sites. The highest values in the concentration of both strong (1.3 mmol g−1) and total (2.4 mmol g−1) acid sites were obtained upon treatment of the CNTs with H2SO4–HNO3. The total amount of acid sites correlates well with the oxygen content after treatment, Fig. S1.†49 The anticipated effect of the functionalisation of the carbon nanotubes on their ability to form a stable dispersion in a polar medium was estimated by sonicating the CNTs for 1 h in water, followed by storage without agitation. After one week in static conditions, the parent CNTs and CNTs-S had separated at the bottom of the vial, whereas the CNTs treated by H2O2, HNO3 and H2SO4–HNO3 (i.e. those with the highest degree of functionalisation, see Table 2) were still dispersed in the water phase (Fig. S2†). Placing a droplet of each of these suspensions on a filter paper indicated a different degree of aggregation of each type of functionalised CNTs (Fig. S2†). Particularly, the larger, uniform, dark grey spot generated by CNTs-NS showed that this material has the highest tendency to from a homogenous dispersion in water. This result correlates well with the higher oxygen content and total acid amount of these functionalised CNTs, which cause the material to be more hydrophilic.29 The partial cleavage of CNTs into smaller fragments that has been reported to be caused by the harsh treatment with H2SO4–HNO3 probably also contributes to the enhanced dispersibility of CNTs-NS.48,52
The functionalised CNTs were used as support for a range of bimetallic Au–Pd catalysts prepared using a similar CI method to the one employed with unmodified CNTs (vide supra). As anticipated, the functionalisation method had a large effect on the average size of the Au–Pd NPs on the support,48,52 as evidenced by TEM analysis (Fig. 5). The average particle size and size distribution show a correlation with the degree of functionalisation of the CNTs surface,41,45 with the Au–Pd nanoparticles supported on CNTs prepared in the presence of HNO3 (CNTs-N and CNTs-NS, i.e. those with the highest concentration of functional groups, see Table 2) having the smallest average size (∼3.5 nm), with a narrow particle size distribution. The smaller average size of the Au–Pd nanoparticles observed by TEM in AuPd/CNTs-NS (3.5 nm, Fig. 5D) compared to those in AuPd/CNTs (6.0 nm, Fig. 1A) correlates well with the less intense and broader peak of the Au–Pd alloy observed in the XRD pattern of the former material (Fig. 3).
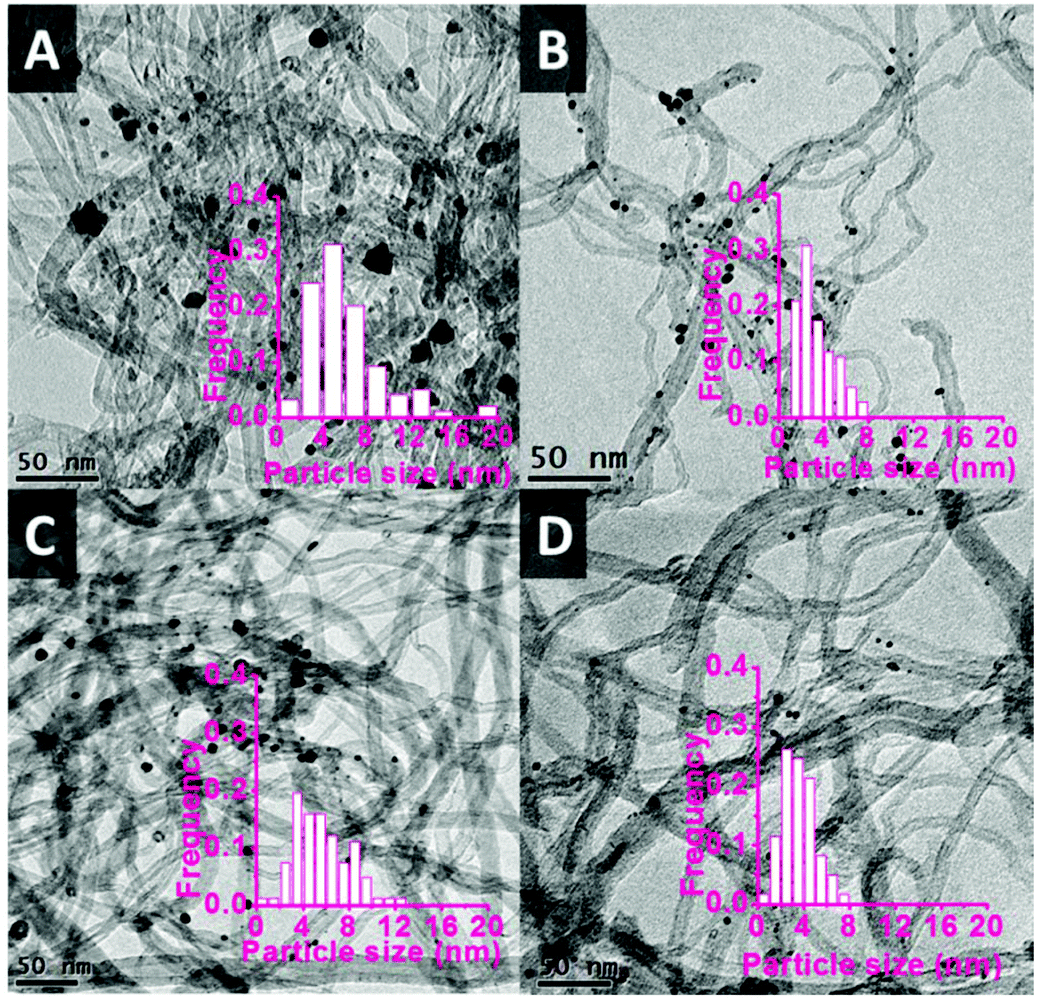 | ||
| Fig. 5 TEM pictures of Au–Pd alloy nanoparticles supported on various functionalised CNTs. A: AuPd/CNTs-O, average size = 6.2 nm; B: AuPd/CNTs-N, average size = 3.3 nm; C: AuPd/CNTs-S, average size = 5.2 nm; D: AuPd/CNTs-NS, average size = 3.5 nm. | ||
The bimetallic Au–Pd nanoparticles supported on functionalised CNTs were investigated in combination with Sn-MCM-41-XS as heterogeneous catalytic system for the oxidative conversion of glycerol into methyl lactate (Table 3). We expected these catalytic systems to be superior to the counterpart based on Au–Pd nanoparticles supported on the unfunctionalised CNTs because: (i) the well-dispersed Au–Pd nanoparticles have smaller average size, (ii) the functionalised CNTs disperse better in the polar reaction medium and (iii) provide additional surface Brønsted acid sites for promoting the dehydration step from trioses to pyruvic aldehyde.13,15,20 Indeed, the rate of glycerol conversion was higher with the functionalised CNTs, the only exception being the CNTs treated with H2O2, which performed as the untreated CNTs (Table 3, entries 1 to 5). The impact of the nature of the support on the performance of the catalytic system was highlighted by the dramatic increase in the conversion of glycerol passing from the untreated CNTs (54% conversion) to the CNTs that underwent the treatment with HNO3–H2SO4 and which display the largest concentration of acid groups, including strong sulphonic groups (81% conversion with 70% yield of ML). With prolonged reaction time (9 h), the yield of methyl lactate reached 85% at 96% glycerol conversion (Table 3, entry 6), which to the best of our knowledge is the highest yield ever reported for the conversion of glycerol to methyl lactate using a heterogeneous catalytic system (Table S1†). The main by-products identified by gas chromatography (GC) were methyl pyruvate (MP), methyl glycolate (MGo), dimethyl oxalate (DMO), dimethyl tartronate (DMT) and methyl glycerate (MGe), see Scheme 1. Carbon balances for all runs exceeded 98%, indicating that the formation of products that would not be detected by GC (e.g. higher molecular weight components formed by condensation reactions) is negligible.
| Entry | Catalyst | Conv. (%) | Y of ML (%) | Selectivity (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| ML | MP | MGo | DMO | DMT | MGe | ||||
| a Reaction conditions: 0.25 M glycerol in 20 ml methanol; 0.1 g of AuPd/CNTs; 0.2 g of Sn-MCM-41-XS; 140 °C; 30 bar air; 4.5 h. For all reactions, small amounts of pyruvic aldehyde dimethyl acetal were observed, with selectivity below 0.1%. b Reaction time: 9 h at 140 °C (heating time 0.5 h). c Run without Sn-MCM-41-XS. | |||||||||
| 1 | AuPd/CNTs + Sn-MCM-41-XS | 54 | 47 | 90 | 1.5 | 2.2 | 0.4 | 0.8 | 4.9 |
| 2 | AuPd/CNTs-O + Sn-MCM-41-XS | 54 | 46 | 88 | 2.3 | 3.0 | 0.6 | 0.6 | 5.0 |
| 3 | AuPd/CNTs-N + Sn-MCM-41-XS | 63 | 54 | 88 | 2.5 | 3.4 | 0.5 | 0.9 | 4.4 |
| 4 | AuPd/CNTs-S + Sn-MCM-41-XS | 71 | 61 | 87 | 1.9 | 1.8 | 0.4 | 1.3 | 7.2 |
| 5 | AuPd/CNTs-NS + Sn-MCM-41-XS | 81 | 70 | 87 | 5.6 | 1.6 | 0.2 | 0.3 | 5.1 |
| 6 | AuPd/CNTs-NS + Sn-MCM-41-XSb | 96 | 85 | 88 | 2.7 | 3.5 | 0.6 | 0.6 | 3.2 |
| 7 | AuPd/AC + Sn-MCM-41-XS | 9.3 | 7.5 | 94 | 0.4 | 1.6 | 0.5 | 0.8 | 2.8 |
| 8 | AuPd/CNTsc | 6.8 | 0.4 | 5.7 | 2.1 | 13 | 5.1 | 1.6 | 17 |
The decrease of the Au–Pd particle size when using the unfunctionalised carbon nanotubes as support, which implies that a higher fraction of metal sites is exposed on the surface and thus available as catalytic sites, is most likely the main factor that led to the dramatic increase in activity (from 54% with AuPd/CNTs to 81% with AuPd/CNTs-NS, see Table 3). However, the acidity of the functionalised CNTs also contributed to the observed increase in catalytic activity. This is demonstrated by the higher activity achieved with AuPd/CNTs-S compared to Au–Pd/CNTs (71% vs. 54%): both have similar particle size (∼6 nm), but the former has a large population of acid sites (Table 2). Additionally, the strength of these acid sites also plays a role: AuPd/CNTs-S has a larger particle size and lower amount of acid sites (0.46 mmol g−1) compared to AuPd/CNTs-N (particle size 3.3 nm, acid amount 1.29 mmol g−1) but showed higher activity (71% vs. 63%). This is most likely due to the presence in AuPd/CNTs-S of the sulphonic groups generated by the treatment with H2SO4, as indicated by the presence of sulphur in this material (Table 2). Though both sulphonic and carboxyl groups are labelled as strong acids on the basis of Boehm titration, the sulphonic groups are much stronger acid sites compared to the carbon-based acid groups in AuPd/CNTs-N and, therefore, promote more efficiently the dehydration step of trioses to pyruvic aldehyde.53 A similar reasoning based on the number and strength of the acid sites also allows explaining the significantly higher activity of AuPd/CNTs-NS compared to AuPd/CNTs-N (Table 3), notwithstanding their similar particle size (Fig. 5B and D). In previous work, it was reported that too strong Brønsted acid sites are detrimental as they promote undesired acetalisation reactions of the intermediate pyruvic aldehyde.53 This was not the case with the catalytic system presented here, most likely because the Sn-MCM-41-XS used in combination with the Au–Pd catalysts provides enough Lewis acid sites to rapidly convert the pyruvic aldehyde formed after dehydration into the desired methyl lactate product (step C in Scheme 1).
In order to further prove the importance of using modified CNTs as support for the Au–Pd nanoparticles, the material consisting of Au–Pd nanoparticles supported on activated carbon using a similar CI method (average particle size = 9.9 nm, see Fig. 1B) was tested in combination with Sn-MCM-41-XS as catalyst for the conversion of glycerol to methyl lactate. A much lower activity was observed (9.3% glycerol conversion, see Table 3, entry 7), confirming the prominent role of the use of (functionalised) CNTs on the catalytic activity in this reaction. This lower activity can be ascribed to the larger size of the Au–Pd particles when AC is used as support (Fig. 1), which implies that a smaller fraction of the metal atoms are exposed on the surface and, therefore, are available as active sites. Moreover, the CNTs have different textural properties compared to AC. CNTs have fully open and thus more accessible external surface, whereas AC has a porous structure with large amount of micropores, which can cause mass transfer limitations of glycerol as well as of the reaction intermediates.
AuPd/CNTs was also tested without the addition of Sn-MCM-41-XS, leading to a much lower conversion of glycerol (Table 3, entry 8) and to poor selectivity towards methyl lactate (5.7%). The main by-products were MGo, DMO and MGe, all arising from over-oxidation of the initially formed trioses. This result indicates that Sn-MCM-41-XS plays an essential role by enhancing the rate of the rearrangement reactions of the trioses (and/or pyruvic aldehyde) and thus preventing over-oxidation of the trioses (Scheme 1), which can lead to the formation of inhibitors (e.g. glyceric acid, vide infra).15
The effect of the air pressure used during the catalytic test was investigated with the catalytic system consisting of AuPd/CNTs-S and Sn-MCM-41-XS (Fig. S3†). An almost identical catalytic performance (glycerol conversion around 71%) was observed with air pressure at 16 or 30 bar, whereas the conversion of glycerol dropped to 47% when the pressure was lowered to 10 bar. Based on the reaction stoichiometry, it was calculated that the minimum air pressure required for this reaction is around 8 bar. This indicates that when using 10 bar the reaction kinetics depends on air pressure, whereas it becomes 0th order when the pressure is 16 bar or higher. To get a deeper insight in the kinetics of this reaction, the conversion of glycerol and selectivity to the major intermediates/products was determined as a function of the batch time for the catalytic system consisting of AuPd/CNTs-S and Sn-MCM-41-XS (Fig. 6). The conversion of glycerol increased almost linearly in the first 4.5 h and reached 85% after 15.5 h. The selectivity to methyl lactate was nearly constant at around 90% between 0 and 15.5 h. The main by-product was MGe with a selectivity of around 6%, while the selectivity towards other products were all below 2%. The initial conversion of glycerol (0–4.5 h) was analysed by plotting the natural logarithm of the concentration of glycerol vs. reaction time. The linear fitting gave a R2 value of 0.994, which suggests a first order reaction with respect to glycerol concentration. This fact, in combination with the constant selectivity towards methyl lactate throughout the reaction, points to the initial partial oxidation of glycerol as the rate determining step under the employed reaction conditions. Compared to the initial stage of the reaction, the conversion of glycerol at the second half stage (4.5–15.5 h) only had a slow increase from 68% to 84%, whereas the selectivity to methyl lactate stayed identical (Fig. 6). For this later stage of the reaction, the slope of the natural logarithm of the concentration of glycerol vs. the reaction time is much lower than for the initial stage (Fig. S4†). This indicates that the reaction is no more first order with respect to glycerol concentration and suggests that partial inhibition of the catalyst by reaction intermediates or products might occur. To investigate this potential inhibition of the catalytic activity, a number of tests were performed by including possible reaction intermediates (methyl esters and their corresponding carboxylic acids) in the initial reaction mixture, using a molar ratio of 9 to 1 between glycerol and additional compound (Fig. S5†). The tests carried out in the presence of 10% of methyl esters (glycolate, oxalate or formate) gave similar results to the reaction without any additional compound. However, when performing the experiments in the presence of 10% of the corresponding carboxylic acids (glycolic acid, oxalic acid or formic acid), a clear reduction of the glycerol conversion was observed (Fig. S5†). The effect was even more marked when 10% glyceric acid was employed in the system, leading to a decrease in the conversion of glycerol from 54% to 12% with a dramatic drop in the selectivity towards methyl lactate from 90% to 28%. The inhibition effect by glyceric acid was similar even when only a small amount (2% relative to glycerol) was added to the reaction mixture. On the other hand, the activity only slightly decreased upon the addition of 2% lactic acid. The effect of adding a strong inorganic acid such as H2SO4 (0.05 mM) was also studied: the activity decreased from 54% to 22% but the selectivity towards methyl lactate was not affected. These results prove that the methyl esters of the by-products do not affect significantly the catalysts, whereas the corresponding carboxylic acids, and particularly glyceric acid, negatively affect the catalytic system. Since small amounts of H2O were generated from the oxidation of alcohols and the dehydration of pyruvic aldehyde (Scheme 1), the methyl esters in the reaction mixture may partially hydrolyse to form carboxylic acids. These carboxylic acids probably compete with glycerol in the adsorption on the catalytic surface and thus inhibit the catalytic activity.15 This phenomenon is reversible, as proven by the high reusability of the catalytic system consisting of AuPd/CNTs-NS and Sn-MCM-41-XS.
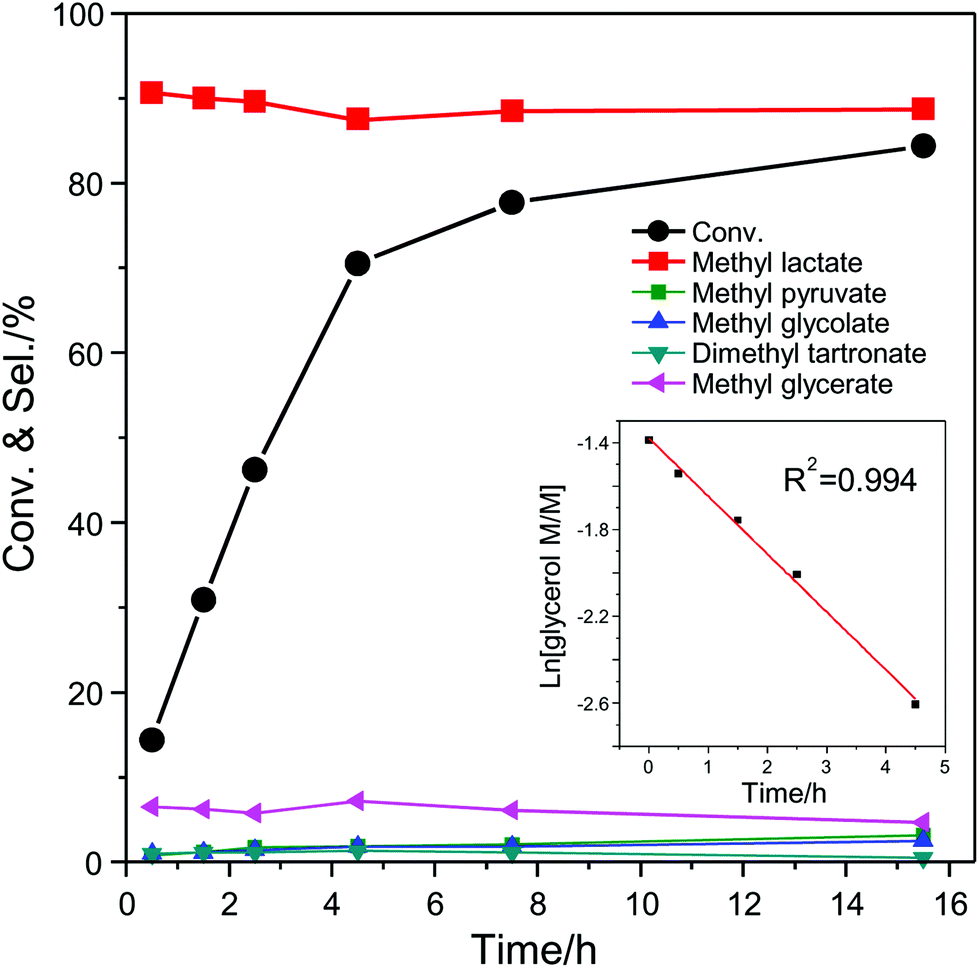 | ||
| Fig. 6 Glycerol conversion and product selectivity as a function of batch time for a catalyst system consisting of AuPd/CNTs-S and Sn-MCM-41-XS. Reaction conditions: 0.25 M glycerol in 20 ml methanol; 0.1 g AuPd/CNTs-S; 0.2 g of Sn-MCM-41-XS; 140 °C; 30 bar air. | ||
For the recycling experiments, the catalyst was separated from the reaction mixture, washed and dried and subsequently re-used for another run. The results show that the activity and selectivity are virtually constant in 5 consecutive runs (Fig. 7), demonstrating excellent catalyst reusability under the operating conditions. The elemental composition of the catalysts was analysed by ICP-OES after 5 runs and did not reveal any changes in the Au/Pd molar ratio in AuPd/CNTs-NS or in the Si/Sn molar ratio in Sn-MCM-41-XS (Table S2†). Moreover, the catalyst reused in 5 consecutive runs was analysed by TEM (Fig. S6†) and showed a very similar average particle size (3.8 nm) compared to the fresh catalyst (3.5 nm, Fig. 5D). The TEM image also revealed that AuPd/CNTs-NS and Sn-MCM-41-XS were intimately mixed, the distance between them being at the nanoscale, which is beneficial for the mass transfer of reactants and intermediates during the reaction. These results indicate that there was negligible change in the catalyst after 5 runs, which correlates well with the observed reusability with no activity loss.
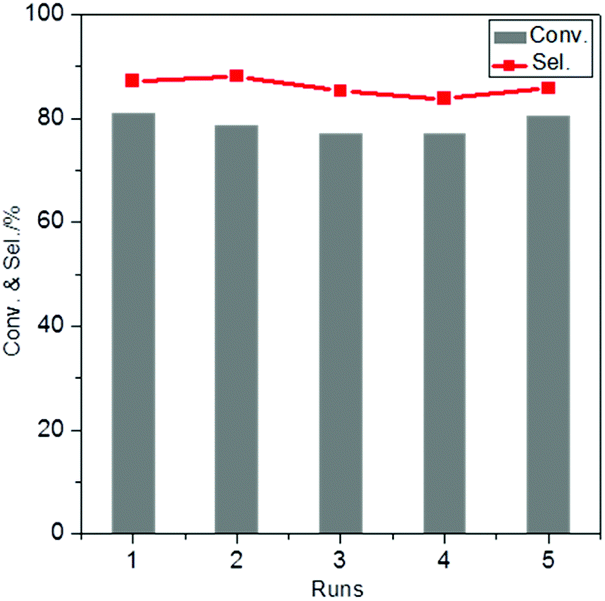 | ||
| Fig. 7 Catalyst recycling experiments in a batch set-up using a catalytic system consisting of AuPd/CNTs-NS and Sn-MCM-41-XS. Reaction conditions: 0.25 M glycerol in 20 ml methanol; 0.1 g of AuPd/CNTs-NS; 0.2 g of Sn-MCM-41-XS; 140 °C; 30 bar air; 4.5 h. | ||
Conclusions
Multifunctional heterogeneous catalytic systems consisting of (i) bimetallic Au–Pd nanoparticles supported on (functionalised) CNTs and (ii) Sn-MCM-41-XS were reported here for the first time for the conversion of glycerol into methyl lactate in a base-free one-pot reaction. Among these materials, the catalytic system employing CNTs functionalised through a HNO3/H2SO4 treatment showed the best performance and gave 85% yield of methyl lactate at 96% conversion of glycerol after 9 h at 140 °C, which is the best catalytic performance reported so far in the literature for base-free systems. This result represents a green advance also considering that the reaction is conducted in methanol, which is a recommended solvent in the context of green chemistry, and at relatively mild conditions compared to those generally employed for the conversion of glycerol. The use of bimetallic Au–Pd nanoparticles was shown to lead to higher activity compared to the monometallic systems (either Au or Pd alone), proving the synergy between Au and Pd. The observed catalytic trends could be rationalised on the basis of a characterisation study, which highlighted the interaction between Au and Pd (XPS), the small particle size (TEM) and the high degree of functionalisation with acidic groups of the CNTs modified by a HNO3/H2SO4 treatment (EDX and Boehm titration). The latter feature is considered crucial for the final catalytic activity, as it led to: (i) increased interaction between the bimetallic nanoparticles and the CNTs surface, with consequent small particle size (average size = 3.5 nm); (ii) presence of surface Brønsted acid sites that promote the dehydration of the trioses intermediates to pyruvic aldehyde; (iii) enhanced dispersion of the AuPd/CNTs-NS material in the polar reaction medium. A kinetic study revealed that the reaction is first order with respect to glycerol, thus identifying the initial oxidation of our starting compound as the rate-determining step. Moreover, the role of the carboxylic acid and ester side-products as possible inhibitors of the active sites was investigated, showing that the former negatively affect the catalytic activity. The reusability of the catalytic system consisting of AuPd/CNTs-NS and Sn-MCM-41-XS was excellent, as proven by the retention of the original activity in 5 consecutive batch recycle experiments. Our strategy of combining a material bearing the catalyst for the oxidation of glycerol (AuPd/CNTs-NS) with one providing the active sites for the consecutive rearrangement of the triose intermediates into the desired lactate product (Sn-MCM-41-XS) proved successful in achieving unsurpassed catalytic performance. This concept of catalyst design is highly flexible and allows careful tuning of each component. Therefore, it has the potential to be applicable for other multi-step reactions necessitating various types of catalytically active sites, as required for the upgrading of other bio-based compounds.Experimental
Reactants and materials
Glycerol (99%), 1,3-dihydroxyacetone dimer (97%), glyceraldehyde (90%), methyl glycolate (98%), methyl lactate (98%), pyruvic aldehyde (40 wt% in H2O), tartronic acid (97%), Au(III) chloride hydrate (99.999%), Pd(II) chloride (99.999%), hydrogen peroxide (30 wt% in H2O), polyvinyl alcohol (PVA, MW 9000–10000, 80% hydrolysed), sodium borohydride (99%), cetyltrimethylammonium bromide (CTAB, 99%), tetraethyl orthosilicate (TEOS, 98%), tin chloride pentahydrate (SnCl4·5H2O, 98%) and multi-walled carbon nanotubes (CNTs, >95% purity, O.D. (outside diameter) × L (length) 6–9 nm × 5 μm) were all purchased from Sigma Aldrich. Active carbon Norit SX1G was purchased from Cabot. Glyceric acid (20 wt% in H2O) was purchased from TCI Chemicals. The H2O used in this work was always of MilliQ grade. All chemicals were used without further purification.
Synthesis of the catalysts
To remove possible metallic residues in the multi-walled carbon nanotubes (e.g. cobalt and molybdenum), the CNTs were washed with a HCl solution prior to use. CNTs (6 g) were suspended in an aqueous HCl solution (5 wt%, 600 mL) and the mixture was refluxed for 2 h at 50 °C. The suspension was filtered (on a Büchner funnel) and washed with MilliQ water until the filtrate was neutral. The resulting solid was dried overnight at 100 °C and used as support for metal nanoparticles or for further functionalisation reactions.The multi-walled carbon nanotubes were functionalised by oxidation using H2O2, HNO3, H2SO4 or HNO3–H2SO4. A representative experimental procedure for the CNTs treatment by H2O2 (CNTs-O) is as follows: CNTs (2.5 g) were suspended in an aqueous H2O2 solution (30 wt%, 25 mL) and the mixture was sonicated for 30 min. The mixture was then refluxed at 65 °C for 6 days while adding a fresh aqueous H2O2 solution (30 wt%, 10 mL) every 12 h (120 mL in total). Next, the suspension was filtered and the solid residue was washed with MilliQ water until the filtrate was neutral. The obtained solid was dried at 100 °C overnight. A representative procedure for CNTs treatment by HNO3 (CNTs-N) is as follows: CNTs (2.5 g) were suspended in an aqueous solution of HNO3 (30 wt%, 50 mL). The mixture was sonicated for 30 min, after which it was refluxed for 2 h by setting the temperature of the oil bath at 140 °C. The suspension was filtered and the solid residue was washed with MilliQ water until the filtrate was neutral. The obtained solid was dried overnight at 100 °C. A representative procedure for CNTs treatment by H2SO4 (CNTs-S) is as follows: CNTs (2.5 g) were suspended in an aqueous solution of H2SO4 (98%, 25 mL) and the mixture was sonicated for 30 min. The mixture was then refluxed by setting the temperature of the oil bath at 150 °C for 15 h. The reaction mixture was diluted with MilliQ water, after which it was filtered and washed with MilliQ water. The resulting solid was dried overnight at 100 °C. A representative procedure for CNTs treatment by HNO3–H2SO4 (CNTs-NS) is as follows: CNTs (2.5 g) were suspended in a mixture of aqueous HNO3 (30 wt%, 25 mL) and H2SO4 (98%, 25 mL) and the slurry was sonicated for 30 min. The mixture was then maintained at 60 °C for 15 h. Next, the reaction mixture was diluted with MilliQ water, after which it was filtered and washed with MilliQ water. The resulting solid was dried overnight at 100 °C.
Mono- and bimetallic Au–Pd catalysts supported on CNTs were prepared using a colloidal immobilisation (CI) method aiming at a theoretical metal loading of 1 wt%.30 The preparation of Au–Pd NPs (1 wt%) supported on the chosen type of CNTs with a molar ratio of Au-to-Pd of 1 was as follows: an aqueous HAuCl4 solution (3.5 g Au per L, 1.85 mL), an aqueous PdCl2·2HCl solution (3.5 g Pd per L, 1.00 mL) and 2 wt% of an aqueous PVA solution (1.2 mL) were added to MilliQ H2O (100 mL) and the mixture was stirred for 30 min at room temperature. A freshly prepared aqueous solution of NaBH4 (0.1 M, 2.54 mL) was added dropwise to the mixture, which was then stirred for 30 min. The pH was set to 2 by the addition of an aqueous H2SO4 solution (1 M). Subsequently, CNTs (1 g) were added to the solution. The CNTs were sonicated (30 min) in MilliQ water (50 mL) prior to use, to achieve a good dispersion. The suspension was stirred for 2 h and subsequently filtered. The residue was washed with H2O (2 L) to neutralise the mixture and to remove PVA. Finally, the solid was dried overnight at 80 °C.
Sn-MCM-41-XS was synthesised according to a previously reported method.20 In short, an aqueous solution of NaOH (2 M, 3.6 mL) was added to a solution of CTAB (1.0 g) in H2O (480 mL), after which the mixture was stirred vigorously at room temperature for 30 min. Next, TEOS (2.29 g) was added dropwise to this solution. Then, a solution of SnCl4·5H2O in ethanol (0.3 M, 1 mL) was added dropwise, after which more TEOS (2.29 g) was added dropwise. After stirring the mixture at room temperature for 2 h, the white gel was filtered and washed three times with MilliQ H2O (10 mL) and then with ethanol (10 mL). The obtained white solid was dried at 70 °C overnight and the resulting powder was calcined at 550 °C in air for 5 h, with a heating rate of 3 °C min−1. The following molar ratio was used: 1 TEOS:0.0135 SnCl4·5H2O:0.33 NaOH:0.125 CTAB:0.9 EtOH:1247 H2O. See Fig. S7† for selected characterisation data for Sn-MCM-41-XS.
Catalytic experiments
The catalytic experiments were carried out in a 100 mL Parr stainless steel autoclave reactor equipped with a Teflon liner and an overhead stirrer. In a typical experiment test, a solution of glycerol in methanol (0.25 M, 20 mL) was loaded into the reactor together with a predetermined amount of the noble metal catalyst and of Sn-MCM-41-XS. The reaction was performed using air as the oxidant (30 bar) for 4.5 h at 140 °C (heating time 0.5 h) at a stirring speed of 800 rpm. Then, the reactor was depressurised and the reaction content was collected and filtered to separate the catalyst. The liquid phase was analysed by gas chromatography using a Thermo Trace GC equipped with a Restek Stabilwax-DA column (30 m × 0.32 mm × 1 μm) and a FID detector. Each compound (reactant or product) was calibrated using solutions of the pure component at 4 different concentrations.The glycerol conversion (Conv./%) is defined by eqn (1):
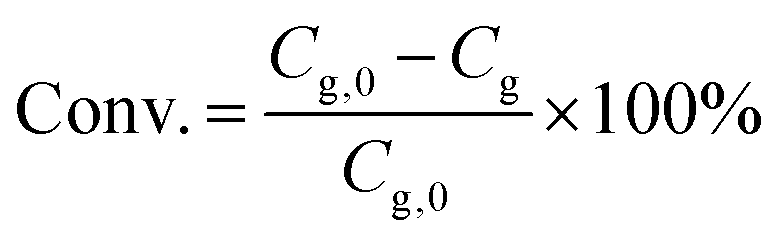 | (1) |
The product selectivity for a compound P is defined by eqn (2):
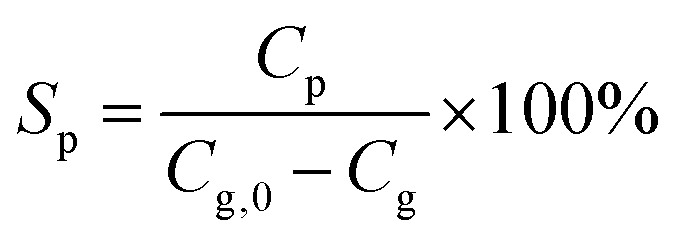 | (2) |
The carbon balance (C/%) was calculated as the sum of the concentration of unreacted glycerol and of the yields of all products detected by GC.
For the catalysts recycling tests, after a small amount of the reaction mixture was collected for analysis at the end of the catalytic test, the remaining mixture was filtered and the catalysts were recovered. The catalysts were washed first with MilliQ H2O (20 mL), then with ethanol (20 mL), and this procedure was repeated 3 times, after which the solid was dried overnight at 100 °C. This solid was used for another batch experiment.
Characterisation of the catalysts
Transmission electron microscopy (TEM) images were obtained using a CM12 (Philips) electron microscope operating at 120 keV. The samples were prepared by ultra-sonication in ethanol, after which a droplet of the suspension was added to a carbon coated 400 mesh copper grid. Images were taken with a slow scanning CCD camera.Nitrogen physisorption isotherms were measured at −196 °C using a Micromeritics ASAP 2420 apparatus. The Brunauer–Emmett–Teller (BET) method was employed to calculate the specific surface area. The Barrett–Joyner–Halenda (BJH) method was used to calculate the pore volume.
Inductively-coupled plasma optical emission spectrometry (ICP-OES) was performed using a PerkinElmer Optima 7000 DV instrument in order to obtain the actual Au and Pd loadings on the supports, as well as the actual Si/Sn ratio in Sn-MCM-41-XS.
Energy-dispersive X-ray spectroscopy (EDX) measurements were performed on a FEI Tecnai T20 electron microscope operating at 200 keV with an Oxford Xmax 80T detector. The samples were prepared by ultra-sonication in ethanol followed by drop-casting of the material on a copper grid.
X-ray Photoelectron Spectroscopy (XPS) was measured by mounting the catalysts on a conductive tape attached to the XPS sample holder. No further treatment was carried out prior to the XPS measurement. The measurements were performed using a Surface Science SSX-100 ESCA instrument equipped with a monochromatic Al Kα X-ray source (hν = 1486.6 eV) and operating at a pressure below 2 × 10−9 mbar. The analysed spot size was 600 μm; an electron flood gun was employed to compensate for charging effects. Spectral analysis included Shirley background subtraction54 and peak deconvolution using the Winspec software package developed by LISE laboratory, University of Namur, Belgium. Binding energies were referenced to the C 1s peak set at 284.5 ± 0.2 eV.
The amount of acid sites per gram of CNTs was measured by Boehm titration.50 For this test, a CNTs sample (0.2 g) was added to an aqueous basic solution of NaHCO3 (0.05 M, 25 mL) to determine the stronger acid sites, such as carboxylic and sulphonic acid groups; or to an aqueous solution of NaOH (0.1 M, 25 mL) to determine the total amount of acid sites. The suspensions were stirred for 24 h at room temperature and filtered before titration. Aliquots (10 mL) of the filtered solutions were titrated with 0.1 M HCl.
The dispersibility of the functionalised CNTs was evaluated by suspending the CNTs (20 mg) in water (20 mL) using ultra-sonication for 30 min. The mixture was stored without agitation for 1 week. Then, one drop of the suspension was taken and dropped on a filter paper to visually determine the state of aggregation of the functionalised CNTs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Zhenchen Tang acknowledges financial support from the China Scholarship Council for his Ph.D. grant. All authors thank Leon Rohrbach, Jan Henk Marsman, Erwin Wilbers, Anne Appeldoorn and Marcel de Vries for technical support, Dr Marc Stuart for support with TEM-EDX and Johannes van der Velde for support with ICP-OES.Notes and references
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS.
- A. Dibenedetto, A. Angelini, M. Aresta, J. Ethiraj, C. Fragale and F. Nocito, Tetrahedron, 2011, 67, 1308–1313 CrossRef CAS.
- Y.-C. Lin, Int. J. Hydrogen Energy, 2013, 38, 2678–2700 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS.
- C. H. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- K. L. Wasewar, A. A. Yawalkar, J. A. Moulijn and V. G. Pangarkar, Ind. Eng. Chem. Res., 2004, 43, 5969–5982 CrossRef CAS.
- Y. Wang, Y. Tashiro and K. Sonomoto, J. Biosci. Bioeng., 2015, 119, 10–18 CrossRef CAS.
- R. K. Pazhavelikkakath Purushothaman, J. van Haveren, I. Melian-Cabrera, E. R. van Eck and H. J. Heeres, ChemSusChem, 2014, 7, 1140–1147 CrossRef CAS.
- T. Lu, X. Fu, L. Zhou, Y. Su, X. Yang, L. Han, J. Wang and C. Song, ACS Catal., 2017, 7, 7274–7284 CrossRef CAS.
- Z. Tang, S. L. Fiorilli, H. J. Heeres and P. P. Pescarmona, ACS Sustainable Chem. Eng., 2018, 6, 10923–10933 CrossRef CAS.
- Y. Shen, S. Zhang, H. Li, Y. Ren and H. Liu, Chem. – Eur. J., 2010, 16, 7368–7371 CrossRef CAS.
- R. K. P. Purushothaman, J. van Haveren, D. S. van Es, I. Melián-Cabrera, J. D. Meeldijk and H. J. Heeres, Appl. Catal., B, 2014, 147, 92–100 CrossRef CAS.
- H. Kishida, F. Jin, Z. Zhou, T. Moriya and H. Enomoto, Chem. Lett., 2005, 34, 1560–1561 CrossRef CAS.
- P. P. Pescarmona, K. P. F. Janssen, C. Delaet, C. Stroobants, K. Houthoofd, A. Philippaerts, C. De Jonghe, J. S. Paul, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1083–1089 RSC.
- L. Li, X. Collard, A. Bertrand, B. F. Sels, P. P. Pescarmona and C. Aprile, J. Catal., 2014, 314, 56–65 CrossRef CAS.
- H. J. Cho, C.-C. Chang and W. Fan, Green Chem., 2014, 16, 3428–3433 RSC.
- L. Li, C. Stroobants, K. Lin, P. A. Jacobs, B. F. Sels and P. P. Pescarmona, Green Chem., 2011, 13, 1175–1181 RSC.
- L. Kesavan, R. Tiruvalam, M. H. A. Rahim, M. I. Bin Saiman, D. I. Enache, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, S. H. Taylor, D. W. Knight, C. J. Kiely and G. J. Hutchings, Science, 2011, 331, 195–199 CrossRef CAS.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS.
- S.-S. Liu, K.-Q. Sun and B.-Q. Xu, ACS Catal., 2014, 4, 2226–2230 CrossRef CAS.
- S. A. Kondrat, P. J. Miedziak, M. Douthwaite, G. L. Brett, T. E. Davies, D. J. Morgan, J. K. Edwards, D. W. Knight, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ChemSusChem, 2014, 7, 1326–1334 CrossRef CAS.
- H.-L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21, 13705–13725 RSC.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105–1136 CrossRef CAS.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CrossRef CAS.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS.
- E. G. Rodrigues, S. A. C. Carabineiro, J. J. Delgado, X. Chen, M. F. R. Pereira and J. J. M. Órfão, J. Catal., 2012, 285, 83–91 CrossRef CAS.
- J. J. Adjizian, P. De Marco, I. Suarez-Martinez, A. A. El Mel, R. Snyders, R. Y. N. Gengler, P. Rudolf, X. Ke, G. Van Tendeloo, C. Bittencourt and C. P. Ewels, Chem. Phys. Lett., 2013, 571, 44–48 CrossRef CAS.
- Z. Zhang, L. Xin, J. Qi, Z. Wang and W. Li, Green Chem., 2012, 14, 2150–2152 RSC.
- S. Wang, Z. Xin, X. Huang, W. Yu, S. Niu and L. Shao, Phys. Chem. Chem. Phys., 2017, 19, 6164–6168 RSC.
- W. Deng, J. Chen, J. Kang, Q. Zhang and Y. Wang, Chem. Commun., 2016, 52, 6805–6808 RSC.
- K. A. Wepasnick, B. A. Smith, J. L. Bitter and D. Howard Fairbrother, Anal. Bioanal. Chem., 2010, 396, 1003–1014 CrossRef CAS.
- H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116–8121 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- S. V. Barabash, V. Blum, S. Müller and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035108 CrossRef.
- Y. Ding, F. Fan, Z. Tian and Z. L. Wang, J. Am. Chem. Soc., 2010, 132, 12480–12486 CrossRef CAS PubMed.
- J. Xu, T. White, P. Li, C. He, J. Yu, W. Yuan and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 10398–10406 CrossRef CAS.
- J. Wang, P. Zhang, Y. Xiahou, D. Wang, H. Xia and H. Möhwald, ACS Appl. Mater. Interfaces, 2018, 10, 602–613 CrossRef CAS.
- P. A. P. Nascente, S. G. C. de Castro, R. Landers and G. G. Kleiman, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 4659–4666 CrossRef CAS.
- In principle, the selectivity over different catalysts should be compared at the same conversion value, because for complex reaction networks the selectivity is often dependent on the conversion degree. Nevertheless, for the conversion of glycerol to methyl lactate a nearly constant selectivity is observed at different conversion values (see ref. 15 or Fig. 6 in this work). Therefore, this analysis of the selectivity over the different catalysts is pertinent notwithstanding the different conversion of glycerol obtained on each of them.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- Y. Yan, J. Miao, Z. Yang, F.-X. Xiao, H. B. Yang, B. Liu and Y. Yang, Chem. Soc. Rev., 2015, 44, 3295–3346 RSC.
- S. Kundu, Y. Wang, W. Xia and M. Muhler, J. Phys. Chem. C, 2008, 112, 16869–16878 CrossRef CAS.
- R. Rodrigues, M. Gonçalves, D. Mandelli, P. P. Pescarmona and W. A. Carvalho, Catal. Sci. Technol., 2014, 4, 2293–2301 RSC.
- H. P. Boehm, Carbon, 1994, 32, 759–769 Search PubMed.
- E. G. Rodrigues, J. J. Delgado, X. Chen, M. F. R. Pereira and J. J. M. Órfão, Ind. Eng. Chem. Res., 2012, 51, 15884–15894 CrossRef CAS.
- F. de Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef CAS.
- D. A. Shirley, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison with previously reported heterogeneous catalysts, correlation between oxygen content and number of total acid sites, dispersion of functionalised carbon nanotubes, conversion of glycerol at different reaction pressures, plot of the concentration of glycerol as a function of the reaction time, conversion and selectivity as a function of the addition of different compounds, TEM pictures of the used catalysts, elemental analysis of the used catalysts, TEM picture of Sn-MCM-41-XS. See DOI: 10.1039/c9gc01521c |
| This journal is © The Royal Society of Chemistry 2019 |