
Membrane-separated electrochemical latrine wastewater treatment†
Yang
Yang
 a,
Lin
Lin
b,
Leda Katebian
Tse
a,
Heng
Dong
c,
Shaokun
Yu
d and
Michael R.
Hoffmann
*a
a,
Lin
Lin
b,
Leda Katebian
Tse
a,
Heng
Dong
c,
Shaokun
Yu
d and
Michael R.
Hoffmann
*a
aDivision of Engineering and Applied Science, Linde-Robinson Laboratory, California Institute of Technology, Pasadena, California 91125, USA. E-mail: mrh@caltech.edu
bEnvironmental Engineering Research Centre, Department of Civil Engineering, The University of Hong Kong, Hong Kong, China
cState Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing 100084, PR China
dDepartment of Civil & Environmental Engineering, University of California, Davis, California 95616, USA
First published on 20th November 2018
Abstract
Electrolysis is demonstrated to be useful for onsite latrine wastewater treatment. Improved wastewater treatment efficiencies are achieved through the use of a combination of cation and anion exchange membranes (CEM/AEM) in electrochemical reactors. Compared to a membrane-free electrolysis cell, the separation of anodic and cathodic chambers using a CEM separator is shown to reduce energy consumption by 51% for COD removal and 87% for NH4+ removal. Furthermore, 51% of the initial [NH4+]0 is recovered via electrodialysis. CEM-separated electrolysis is shown to produce 39% less ClO3− and 92% less chlorinated organic by-products than in a membrane-free reactor. Helminth (Ascaris suum) eggs, which yield parasitic worms, are very resistant to conventional disinfection methods. Membrane-free electrolysis only inactivates 15% of the dosed helminth eggs (100 eggs per mL). In CEM-separated electrolysis, a combination of low pH and in situ chlorine production in the anodic chamber results in 85% inactivation of helminth eggs. In addition, H2, which is produced in the cathodic chamber of the CEM-separated reactor, is directly converted to electricity using a hydrogen–air fuel cell. The hydrogen energy produced during electrolysis is estimated to reduce the overall energy cost of operation by 20%. Recovery of 85% of the initial [PO43−]0 and pH neutralization are achieved by treating the acidic effluent of a CEM-separated electrolysis cell in an AEM-separated electrolysis cell. A one-month continuous operation demonstrates the potential of using both the CEM and AEM during electrolysis to achieve more efficient wastewater treatment while, at the same time, recovering NH4+ and PO43−, for eventual use as agricultural fertilizers.
Water impactWe show that multiple benefits can be accrued by integrating cation and anion exchange membranes in an electrolysis cell for wastewater treatment. The benefits include enhanced pollutant removal, inactivation of helminth eggs, efficient recovery of N and P nutrients, and production of pure H2. Membrane electrolysis is shown to be durable over one month of continuous operation. |
Introduction
Electrochemical oxidation (EO) has been proven to be an efficient alternative treatment technology for use in small-scale onsite wastewater treatment operations.1 Electrolysis is particularly well suited for the decentralized onsite treatment of latrine wastewater that has higher COD and BOD loadings coupled with substantially lower volumetric flows compared to centralized domestic wastewater treatment plants. Previous studies have demonstrated that a combination of biological pretreatment along with electrolysis can totally eliminate the chemical oxygen demand (COD) in a wastewater influent, remove ammonia via breakpoint chlorination, oxidize trace organic contaminants such as personal care products and pharmaceuticals, and disinfect the effluent within several hours.2–6To further improve the treatment efficiency and lower the energy cost of the electrochemical oxidation process, recent studies have been focused primarily on the development of alternative electrode materials.6,7 However, improvements in reactor design and use of alternative reactor configurations may increase the overall efficiency of onsite wastewater treatment. Typical electrolysis reactors are either single cells or membrane or porous disk separated cells. Divided cells with the anode and cathode separated by ion-exchange membranes are normally used in studies to exclude side reactions that occur in the presence of a counter electrode. In addition, the membrane separated cells tend to minimize the loss of electrochemically-generated oxidants such as ClO− and H2O2, which are susceptible to further anodic oxidation (H2O2 → HO2˙ + H+ + e−) or cathodic reduction (ClO− + H+ + e− → Cl− + OH−) in a single cell configuration.8 However, in spite of the potential advantages of using membrane separated cells, single cell reactors have been preferred due to the ease of their construction and their lower operational costs.1,8,9 Ion-exchange membranes are normally considered to be undesired components in electrolysis cells that are designed for water treatment. For example, the addition of a membrane between the anodic and cathodic compartments raises the overall cell potential due to an increase of internal resistance.10,11 Another potential limiting factor is the resulting acidification of the anolyte and alkalization of the catholyte; this normally requires the addition of chemical reagents to adjust the final pH of the membrane-separated electrochemical reactor effluents.1,12–15 Membrane fouling is a major challenge in the long-term operation of bio-electrochemical devices.16 It could also be a potential threat to the stability of abiotic electrochemical systems.
If nutrient recovery during wastewater treatment is a second objective, then membrane-separated electrolysis is feasible in practice. Based on the electrodialysis mechanism, ions can be extracted from wastewater through ion-exchange membranes driven by an electric field applied between the anode and cathode. Cation exchange membrane (CEM) divided cell electrolytic reactors have been reported to separate and concentrate NH4+ from urine17,18 and anaerobic digestate.19 Furthermore, phosphate recovery during electrochemical treatment is achieved with the use of anion exchange membranes (AEMs) in divided cell reactors.14,20,21 Ammonium and phosphate ion recoveries up to 90% have been demonstrated.
Molecular hydrogen produced during wastewater electrolysis has the potential to be used in a hydrogen–oxygen fuel cell.22–24 However, hydrogen gas evolved during single cell electrochemical wastewater treatment is invariably mixed with oxygen and other volatile compounds. Thus, the head-space gases (H2 + O2 in a typical mole ratio of 6 H2 to 1 O2)22,23,25 cannot be utilized directly by introduction into a hydrogen fuel cell without first removing oxygen from the gaseous effluent stream. Moreover, the mixture of hydrogen and oxygen has the potential to be explosive. In contrast, using a divided cell membrane-separated electrolyzer, high purity hydrogen gas (>99.99% as a dry gas) produced in the cathodic chamber can be introduced directly into a hydrogen fuel cell.10 The resulting electrical energy produced by the fuel cell can then be used directly to offset the overall energy cost of electrochemical wastewater treatment.
We have previously demonstrated that membrane-free electrolysis in the presence of chloride ions (e.g., oxidation to produce HOCl/ClO−) effectively inactivates model microorganisms and viruses (e.g., E. coli, Enterococcus, Coliphage MS2, and rAd5).3,5 However, helminth eggs, which yield parasitic worms, are very resistant to conventional disinfection methods (i.e., strong acids and bases, most oxidants and reductants, and surface-active agents).26,27 The impermeable inner lipoprotein layer of the eggshells protects helminth eggs from the effects of chemical reagents. The established method to inactivate helminth eggs requires heating to temperatures above 60 °C.28 It is important to determine if helminth eggs can be inactivated using membrane-free and membrane separated electrolysis.
An overview of electrochemical wastewater treatment is provided in Table S1.† It can be found that the use of ionic exchange membranes during electrolysis is not commonly explored. Herein, we now report on the overall impact of ion-exchange membranes on the onsite electrochemical treatment of latrine wastewater, while examining by-product formation, helminth egg inactivation, nutrient recovery, and hydrogen production and utilization.
Materials and methods
Electrodes
A SnO2/Co–TiO2/Ir0.7Ta0.3Ox (STI) triple layer electrode was prepared by the spray pyrolysis method as described previously with slight modifications of the precursor solutions.2 The initial mixed-metal oxide layer of Ir0.7Ta0.3O2 was deposited onto a flat Ti plate, followed by successive layers of cobalt-doped TiO2 (Co–TiO2) and SnO2. In this study, the STI electrode was prepared without antimony as a selective dopant in the SnO2 layer. We determined that antimony could be excluded from the original formulation without impacting the chlorine evolution reaction efficiency. An accelerated lifetime test of the STI anode was performed in the previous study.2 The estimated lifetime at the maximum current density (25 mA cm−2) tested in this study is ten years (89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 380 h). A boron doped diamond (BDD) electrode (Neocoat) was used as an anode for benchmarking the process efficiency.
380 h). A boron doped diamond (BDD) electrode (Neocoat) was used as an anode for benchmarking the process efficiency.
Electrolysis
STI or BDD anodes with geometric surface areas of 6 cm2 each were coupled with stainless steel cathodes (6 cm2) in a single cell configuration. The reactor configuration for a 25 mL volume membrane-free electrolysis cell is shown in Fig. 1a, while a divided cell reactor consists of one anodic chamber (V = 25 mL) and one cathodic chamber (V = 25 mL) separated by a cation exchange membrane (CEM, Nafion 117) or an anion exchange membrane (AEM, AMI-7001 Membrane International, Inc.) as illustrated in Fig. 1b. In the case of CEM-separated electrolysis, the anodic chamber was filled with wastewater while the cathodic chamber was filled with 30 mM Na2SO4. In the case of AEM electrolysis, the anolyte and catholyte consisted of 30 mM Na2SO4 and wastewater, respectively. The catholyte of the CEM cell and the anolyte of the AEM cell are denoted as recovery solution A (RS-A) and B (RS-B), respectively.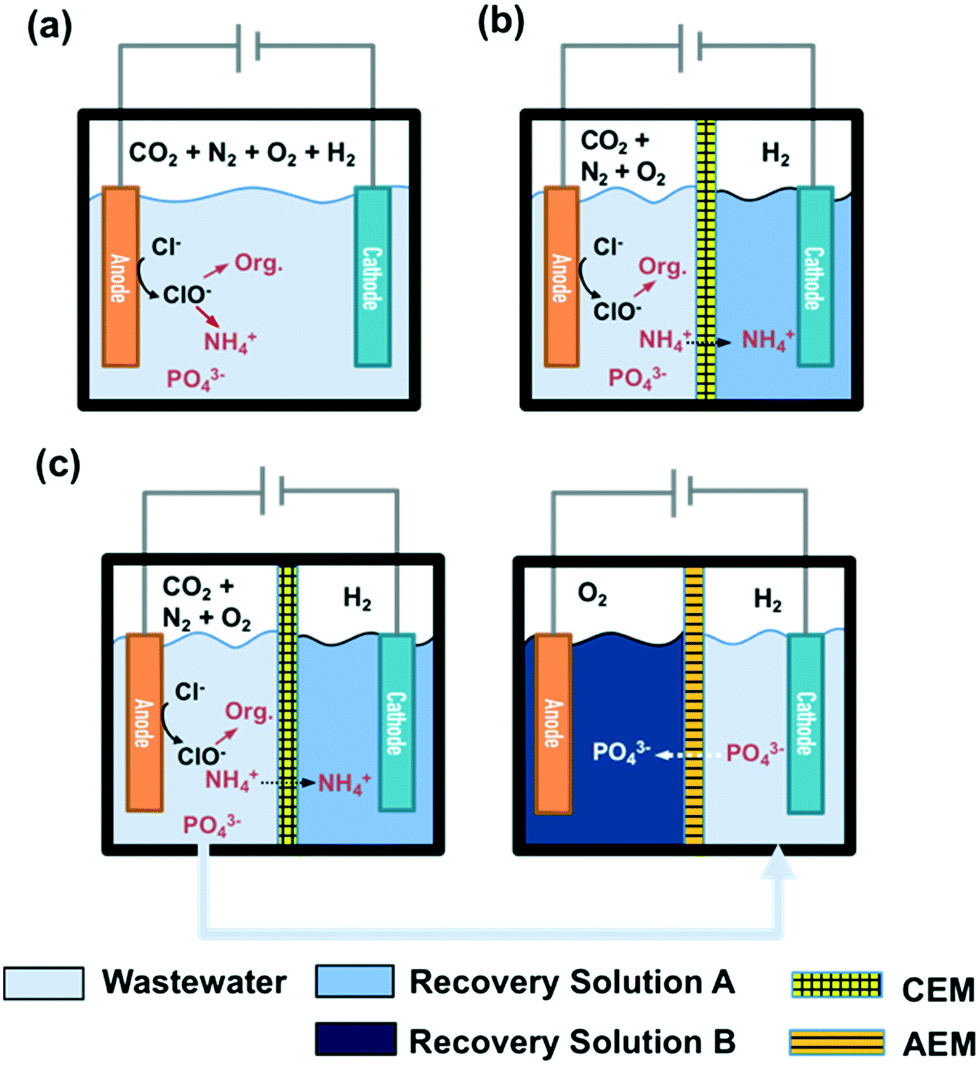 | ||
| Fig. 1 Configurations of a) membrane-free and b) membrane-separated electrolysis reactors. c) Reactor configuration for a two-step membrane-separated electrolysis. | ||
During electrolysis, gas samples were collected in a graduated burette to determine the volumetric gas evolution rate. The volumetric fraction of H2 in the collected gas was determined by GC-TCD (Hewlett-Packard, USA), using 5 and 100 V/V% H2 gas standards for calibration. Prior to gas sample collection, the evolved gas from the cathodic chamber during CEM electrolysis was passed through a 100 mL 0.5 M H2SO4 acid trap to capture the volatilized NH3. Note: no NH4+ was detected in the sulfuric acid trap during 4 h of batch-mode CEM electrolysis; however, volatilization of NH3 becomes important after 24 h of continuous electrolysis (vide infra).
Latrine wastewaters were collected from a prototype recycling toilet and coupled electrochemical system located on the Caltech campus (Pasadena, CA). The collected toilet wastewater contained a mixture of feces, urine and recycled flush water. The COD of the wastewater was determined by dichromate digestion (Hach Method 8000). Cations and anions were quantified by ion chromatography (ICS 2000, Dionex). The latrine wastewater contains 550 mg L−1 COD, 28 mM NH4+, and 50 mM Cl−. The detailed composition is shown in Table S2.†
During electrolysis, free and total chlorine were measured using the DPD (N,N-diethyl-p-phenylenediamine) reagent (Hach Method 8021 and 8167). Water samples were diluted with a 100 mM potassium phosphate buffer (pH = 7) solution prior to DPD analyses to ensure the complete hydrolysis of Cl2(aq) potentially produced during electrolysis. Probe molecule degradation tests were performed using 2 mM benzoic acid (BA) and 2 mM phenol. They were measured using an HPLC (Agilent 1100) equipped with a Zorbax XDB column. Methods for the analyses and inactivation of Ascaris suum eggs can be found in Text S1.† Results obtained from duplicate electrolysis tests are reported as means ± standard deviation.
Results and discussion
Removal of COD and NH4+
Electrolysis of latrine wastewater was investigated first in a single cell reactor using both the STI and BDD anodes in separate experiments. The complete removal of COD and NH4+ required 6 h of electrolysis at a current density of 25 mA cm−2 when using an STI anode in a membrane-less configuration. The evolution of total chlorine is significant only if more than 80% of NH4+ and COD is removed (Fig. S1†).When the STI anode is operated at a lower current density of 5 mA cm−2, only 15% of COD and 7% of NH4+ are removed after 4 h of electrolysis (Fig. 2a and c). In comparison with the STI anode, a BDD anode is more efficient for both COD and NH4+ removal during membrane-free electrolysis. During membrane-free electrolysis when using both the STI and the BDD anode, 0.15–0.2 mM total chlorine was detected (Fig. 2b).
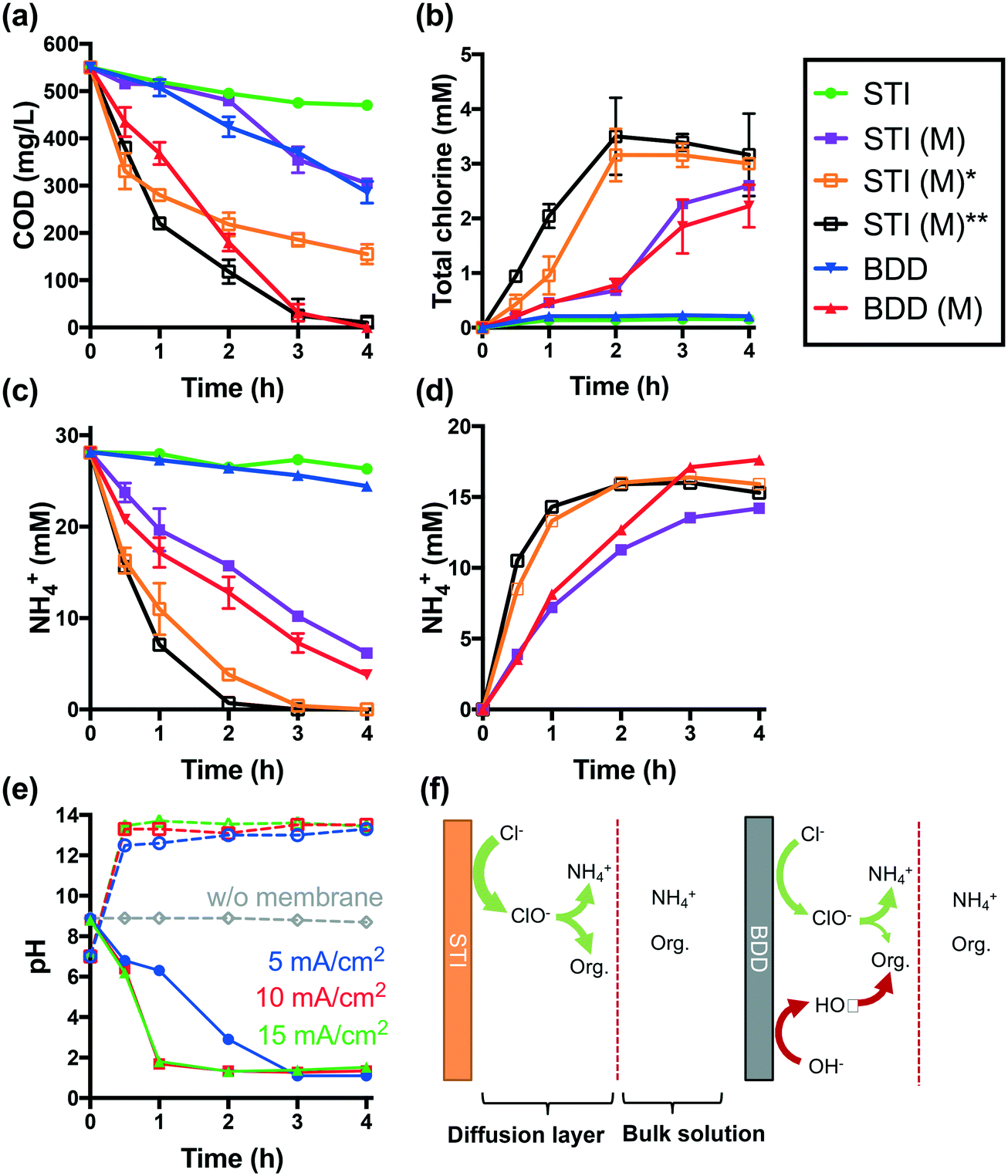 | ||
| Fig. 2 a) COD reduction and b) profiles of total chlorine concentration as a function of electrolysis time. c) NH4+ oxidation in wastewater during electrolysis and d) the recovery of NH4+ in RS-A. Tests marked with “M” were performed in CEM separated electrolysis. All the electrolysis tests were conducted at 5 mA cm−2, except for STI (M)* and STI (M)** which were operated at 10 and 15 mA cm−2, respectively. e) Variation of pH in anodic and cathodic chambers in CEM electrolysis in the STI anode. f) A schematic illustration of electrolysis mechanisms on STI and BDD anodes. | ||
The performance of STI and BDD anodes is determined by their properties. The STI anode is active for chlorine evolution but inefficient for hydroxyl radical production.2 As illustrated in Fig. 2f, the organic components in latrine wastewater can be readily oxidized by electrochemically produced chlorine. In addition to reacting with organics, chlorine reacts with NH4+ to yield combined chlorine (i.e., mono- and di-chloramine), which is converted to N2 upon further chlorination.4,29,30 Electrochemically generated chlorine is rapidly consumed by reactions with both organics and NH4+ within the diffusion layer of the anode before entering the bulk solution. Therefore, low concentrations of total chlorine are detected in the bulk electrolyte solution (Fig. 2b). Given that the chlorine generated on the surface of the STI anode is transformed quickly into less reactive chloramines,4 the COD removal efficiency should be low. However, in the case of the BDD anode, which readily produces hydroxyl and chlorine radical species (e.g., HO˙, Cl˙, and Cl2˙−), organic species are oxidized at a faster rate than that observed during reactive chlorine oxidation alone.6,31 A more rapid consumption of oxidizable organics also allows for free chlorine to react with NH4+. Thus, the BDD electrode is shown to be more efficient for NH4+ removal than the STI anode, even though the BDD electrode is less active in terms of chlorine production than the STI anode (Fig. S2a†).
The removal of NH4+ during anodic wastewater treatment should enhance the overall rate of chlorine facilitated oxidation of the organic matter. This conjecture is proven by the results of probe compound degradation tests (Fig. S3†). Electrochemical phenol degradation in a NaCl electrolyte is inhibited by NH4+. However, the negative impact of NH4+ is reduced during CEM-separated electrolysis. The use of a CEM-separated cell during the treatment of latrine wastewater using the STI anode is shown in Fig. 2a. From the data shown in Fig. 2, it is clear that CEM-separated electrolysis is faster for COD removal than membrane-less electrolysis. In addition, higher steady-state concentrations of total chlorine are obtained (Fig. 2b). 78% of the initial [NH4+]0 is removed from the anodic wastewater compartment (Fig. 2c), while 51% of the removed NH4+ is recovered in the RS-A chamber via electrodialysis (Fig. 2d). As shown in Fig. 2a–d, an increase in current densities from 5 mA cm−2 to 10 and 15 mA cm−2 results in increased rates of COD reduction and enhanced recovery of NH4+. At 15 mA cm−2, complete removal of NH4+ and COD was achieved after 2 h and 4 h, respectively.
As shown in Fig. 2b, CEM electrolysis produces more total chlorine than membrane-less electrolysis. This is not due to enhanced chlorine production using the STI anode. In contrast, the acidification of the anolyte during CEM electrolysis (vide infra) inhibits the intrinsic chlorine evolution activity of the electrode. As shown in Fig. S2,† the chlorine evolution rate of the STI anode decreases with a reduction of pH. Therefore, the apparent increased chlorine production during CEM electrolysis of latrine wastewater is more likely due to the facile removal of NH4+ from wastewater.
The relative improvement in COD removal using a CEM-separated reactor is greater for the BDD anode than for the STI anode. This is due to the depletion of NH4+, which allows for more chlorine to react directly with oxidizable organics as a removal pathway in addition to the free radical-mediated oxidation pathways obtained during electrolysis using the BDD anode.
During membrane-free electrolysis, the pH of the wastewater remains relatively constant at 8.8 (Fig. 2e), while during CEM electrolysis, the pH of the anolyte decreases while that of the catholyte increases. This is due to the interrupted proton balance between the anodic and cathodic chambers. The sulfonate groups of the Nafion 117 membrane have a higher affinity for other cations than for protons.12,32,33 Moreover, the summation of cation concentrations in the latrine wastewater was 106 times higher than the initial [H+]. These factors allow for the transport of non-proton cations across the CEM during the early stage of electrolysis. This leads to the accumulation of H+ in the anolyte and HO− in the catholyte, respectively. The preferential transport of non-proton cations through the CEM membrane increases the membrane resistance.34–36 As a consequence, a gradual increase of cell voltage is observed during CEM-separated electrolysis (Fig. S4†). The voltage, however, decreases as the pH in the wastewater cell drops to below 2. Under these conditions, H+ transport across the CEM is dominant.
The reduction of pH alters the chlorine chemistry. During membrane-free electrolysis at pH 8.8, OCl− is the dominant reactive chlorine species (Fig. S5†). As the pH decreases during CEM-separated electrolysis, the speciation of free chlorine clearly shifts toward HOCl (pKa = 7.4) and Cl2(aq). Low pH favors chlorine-mediated oxidation due in part to the higher standard-state E0 values for Cl2(aq) (1.36 VSHE) and HOCl (1.49 VSHE) compared with OCl− (0.89 VSHE).8 Thus, at low pH there is a faster organic matter removal rate and improved disinfection potential (vide infra).
Helminth egg inactivation
In under-developed regions of the world, helminth eggs often lead to severe worm infestations.37 Thus, we dosed latrine wastewater with 100 to 200 Ascaris suum eggs per mL for electrolysis. We then observed a 15% reduction in the number of viable Ascaris eggs after 4 h of membrane-free electrolysis at 15 mA cm−2 (Fig. S6a†). Reducing the pH to 1.5 during electrolysis did not improve the reduction of viable helminth eggs. Due to the presence of NH4+, chloramines were the dominant oxidants in a membrane-free reactor. Our results are consistent with previous studies showing that Ascaris eggs are resistant to both low pH and to disinfection using chloramines.26,38However, during CEM-separated electrolysis at 15 mA cm−2, 85% of Ascaris suum eggs were inactivated after 4 h (Fig. S6a†). As discussed above, the CEM electrolysis is able to exclude the quenching effect of NH4+ on Cl2(aq) at low pH. HOCl/OCl− is known to weaken the outer protein layer of the eggshells.26,39 It is reasonable to assume that the more reactive Cl2(aq) may lead to more effective weakening of the Ascaris eggshells and the susceptibility of their inner lipoprotein layers than by HOCl/OCl−. This argument is supported by microscopic images that show a greater uptake of non-cell permeable dye (propidium iodide) by the helminth eggshells after 4 hours of CEM-separated electrolysis (Fig. S6c and d†).
The effectiveness of Cl2(aq) mediated oxidation during helminth egg inactivation was validated by dosing Cl2(aq) to the NH4+ depleted latrine wastewater at pH 1.5 (Fig. S6a†). It is found that the 5 mM Cl2(aq) dosage leads to 50% egg inactivation. Compared with the homogeneous Cl2(aq) oxidation, CEM electrolysis is more efficient for egg inactivation. This is probably due to a lower pH in the diffusion layer at the anode coupled with a higher Cl2(aq) concentration near the electrode surface than in the bulk solution.30,40
By-product formation
To achieve >90% COD and NH4+ removal, CEM electrolysis with the STI anode requires a shorter reaction time (4 vs. 6 h) and a lower current density (15 vs. 25 mA cm−2) than membrane-free electrolysis (Fig. 2vs. S1†). Moreover, we found that CEM-separated electrolysis produces less ClO3− than membrane-free electrolysis (5.6 vs. 9 mM shown in Fig. S7a†). In addition, CEM separated electrolysis using the BDD anode was observed to produce a considerable amount of ClO4− (0.63 mM), while ClO4− could not be detected in electrolysis tests with the STI anode.The reactor effluent concentrations of indicative halogenated by-products such as trihalomethanes (THMs: chloroform, bromodichloromethane, dichlorobromomethane, and bromoform) and halogenated acetic acids (HAAs: monochloroacetic acid (MCAA), dichloroacetic acid (DCAA), and trichloroacetic acid (TCAA)) were determined (Fig. S8†). In total, 359 μM THMs and HAAs were detected in the effluent of the membrane-free electrolysis reactor, while only 28 μM was found in the effluent of CEM-separated electrolysis reactor.
In general, CEM-separated electrolysis has lower potential for the formation of both inorganic and organic by-products than the result from membrane-free electrolysis. This is most likely due to the fact that less chlorine is utilized during CEM-separated electrolysis to completely remove COD and NH4+. In addition, the low pH in the anodic chamber during CEM-separated electrolysis most likely inhibits the formation of THMs and HAAs due to the hydrolysis of other chlorinated DBPs that were not measured in this study, e.g., haloacetonitriles and haloacetaldehydes.15
Energy consumption and H2 production
Although higher cell voltages are required for CEM-separated electrolysis compared to membrane-free electrolysis, the increased energy consumption using a CEM separator is offset by the faster removal rates for COD and NH4+. As shown in Fig. 3a and b, the specific energy consumption (SEC) levels off during CEM-separated electrolysis for COD and NH4+ removal, respectively. These limiting values were lower than those measured during membrane-free electrolysis at 5 mA cm−2. Moreover, an increase in current density to 15 mA cm−2 did not result in a significant increase in the SEC. The energy consumption of 40–92 kW h kg−1 COD is already among the lowest values reported previously (Table S1†). In previous work, we have demonstrated in the field-testing of onsite wastewater treatment that the energy consumption of the system could be further reduced by biological pretreatment before electrolysis.41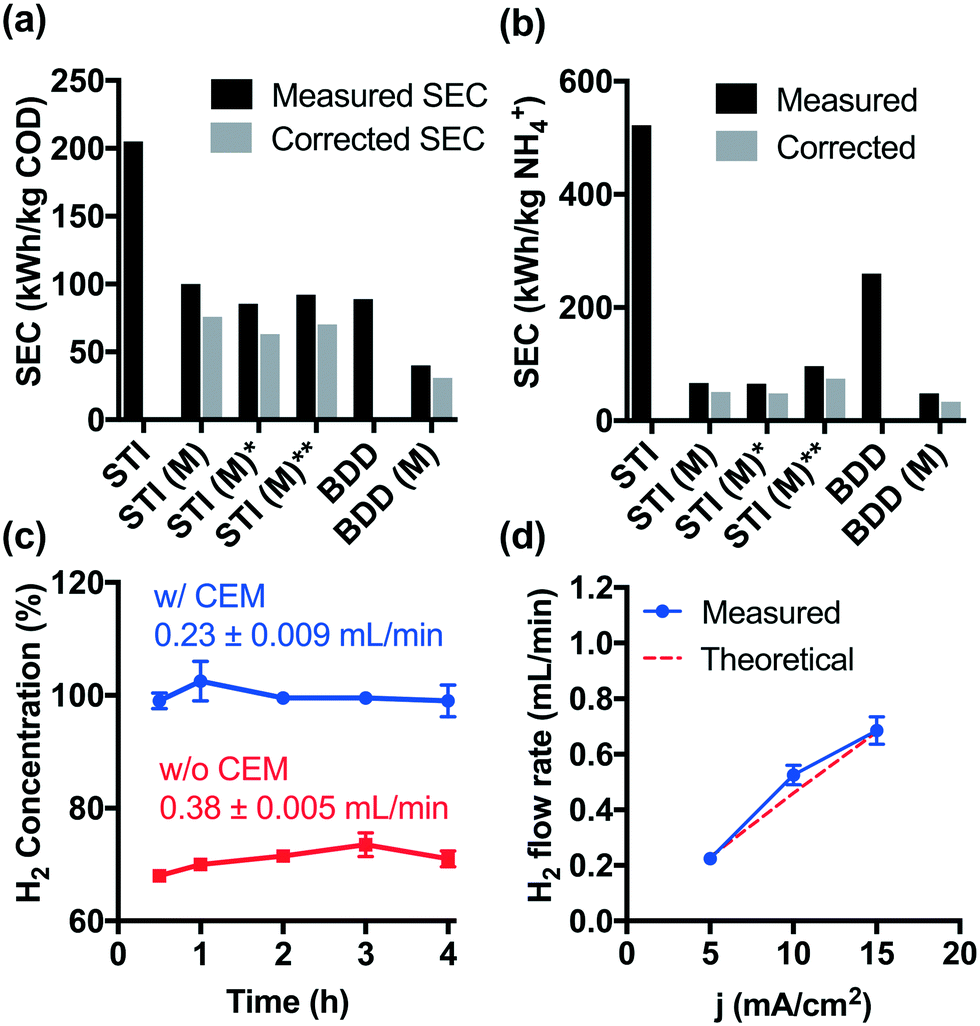 | ||
| Fig. 3 Specific energy consumption with regard to the removal of a) COD and b) NH4+. SEC values for the experiments with STI (M)*, STI (M)** and BDD (M) were calculated based on the decay of COD and NH4+ after 2 h of electrolysis. The remainder of the SEC is based on the removal efficiency after 4 h. c) Concentration of evolved H2 during wastewater electrolysis. d) H2 flow rate as a function of applied current density. The red line is the predicted H2 flow rate produced at 100% current efficiency. | ||
Hydrogen produced at the cathode can be utilized directly in a fuel cell provided that the gaseous effluent is free of molecular oxygen. However, membrane-free electrolysis produces H2 at volumetric concentrations ranging from 68 to 73% (Fig. 3c). The other gaseous components include CO2 produced during organic compound oxidation, N2 formed by breakpoint chlorination, and O2 as a product of water oxidation.22,23,42 In contrast, during CEM-separated electrolysis, high purity H2 (99–102%) was produced in the cathodic chamber (Fig. 3c). The H2 flow rate is found to be proportional to the current density with an apparent current efficiency close to 100% (Fig. 3d). As a proof-of-concept, Fig. S9† shows that the H2 produced in the cathodic chamber can be directly utilized as a hydrogen fuel to produce usable electric power. The hydrogen energy value can be calculated as follows:
| Estimated H2 Energy = 78 W h mol−1 × QH2 × t | (1) |
Phosphate recovery, desalination, and pH adjustment by AEM electrolysis
The acidic effluents from the anodic chamber of a CEM-separated electrolysis reactor are not suitable for non-potable reuse or discharge to surface waters. In order to address this problem, an additional AEM-separated electrolysis reactor was used to adjust the pH of the effluent of the CEM-separated anodic chamber. The acidic effluent was transferred to the cathodic chamber of an AEM-separated electrolysis reactor. The AEM-separated reactor was operated at a constant voltage of +6 V. Protons were reduced to hydrogen in the cathodic chamber, while anions were transferred via electrodialysis. This combination resulted in neutralization of the acidic effluent to a final pH of 8 after 1.5 h of treatment (Fig. 4a).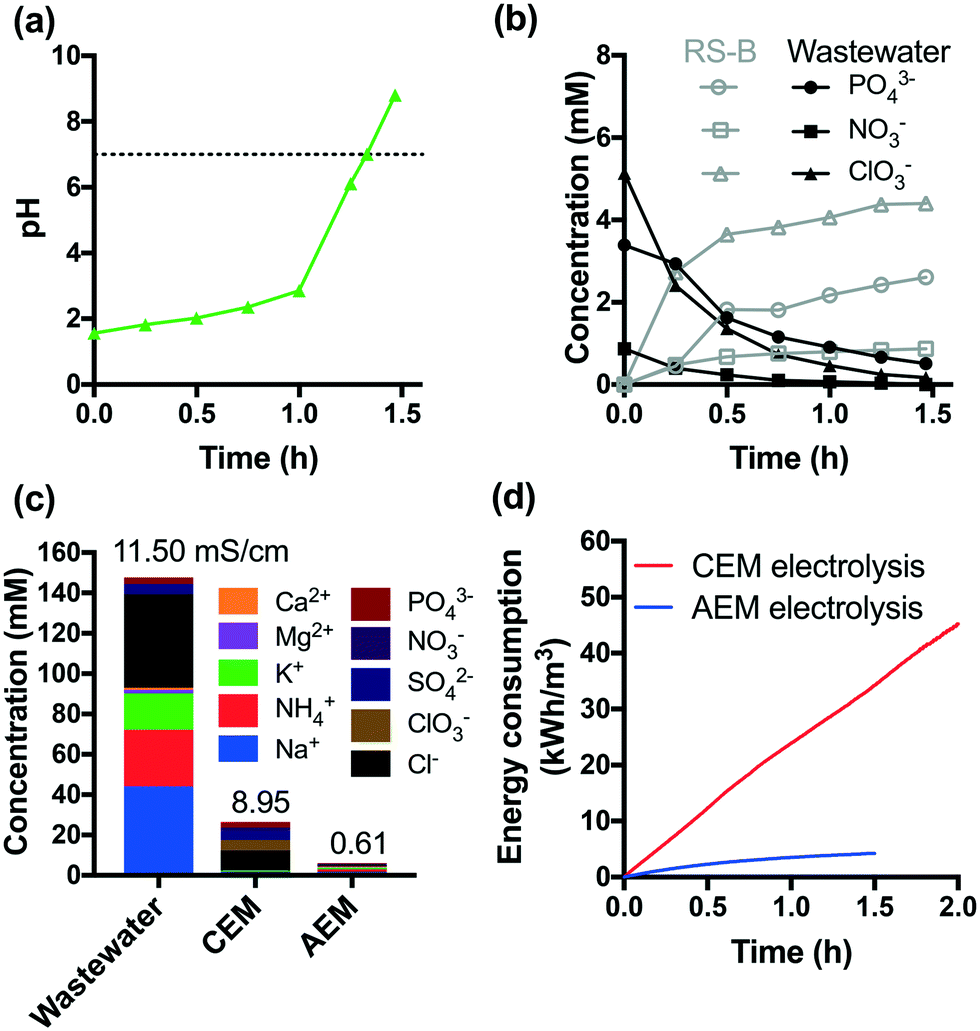 | ||
| Fig. 4 a) Neutralization of acid effluent of CEM electrolysis (i.e. treated wastewater). b) Recovery of anions by AEM-separated electrolysis. c) Desalination achieved using the two-step membrane-separated electrolysis. d) Comparison of energy consumption of CEM and AEM electrolysis. | ||
As shown in Fig. 4b, 85% of the initial total PO43− is recovered in the anodic RS-B chamber (vide supraFig. 1). In addition, NO3− and ClO3− were also separated from the treated wastewater. Fig. 4c shows the removal and selective extraction of ionic components along with the change in conductivity after CEM- and AEM-separated electrolysis. The combination of CEM and AEM electrolysis together removed both cations and anions from the wastewater resulting in a 95% reduction in the measured conductivity. The effluent conductivity is 0.65 mS cm−1, which is within the range of river water conductivity in the United States (0.05 to 1.5 mS cm−1).44 We also observed that cathodic reduction eliminates residual total and free chlorine (Fig. S10†). Moreover, the stainless-steel cathode catalyzes the dehalogenation of HAAs, which is consistent with previous studies.31,45 The total amount of HAAs decreases by 43% (Fig. S8†) and the dominant species shifts from trichloroacetic acid to monochloroacetic acid. Chloroform is completely removed during AEM-separated electrolysis. Since chloroform is relatively resistant to dechlorination, the major removal mechanism appears to be volatilization.
Fig. 4d compares the energy consumption of CEM and AEM electrolysis in the scale of kW h m−3. The removal of 90% of COD and NH4+ requires 2 h of CEM electrolysis, which leads to an energy consumption of 45 kW h m−3. In contrast, AEM electrolysis consumes much less energy (4 kW h m−3).
Durability of the membrane electrolysis system
The durability of CEM electrolysis was validated in a one-month continuous operation. According to Fig. 2, CEM-separated electrolysis with an HRT of 3 h at 15 mA cm−2 current density removes >90% of COD and NH4+ from 25 mL latrine wastewater. Thus, given the system size, the treatment capacity over a one-week period is 1.4 L. For durability testing, 1.4 L of wastewater and 100 mL of recovery solution were circulated in the anodic and cathodic chamber, respectively, using peristaltic pumps at a rate of 25 mL min−1. Off-gas produced from the cathodic chamber was passed through a 100 mL 0.5 M H2SO4 acid trap to collect the volatilized NH3. Tests were repeated for four weeks without changing the Nafion membrane or the electrodes.As shown in Fig. 5a, the pH of wastewater gradually decreases down to 1–1.5 and the cell voltage peaks at 8 V then decreases with an increase in electrolysis time. Four repetitive runs show similar pH and voltage profiles. Over 90% of NH4+ and COD are removed in each run (Fig. 5b).
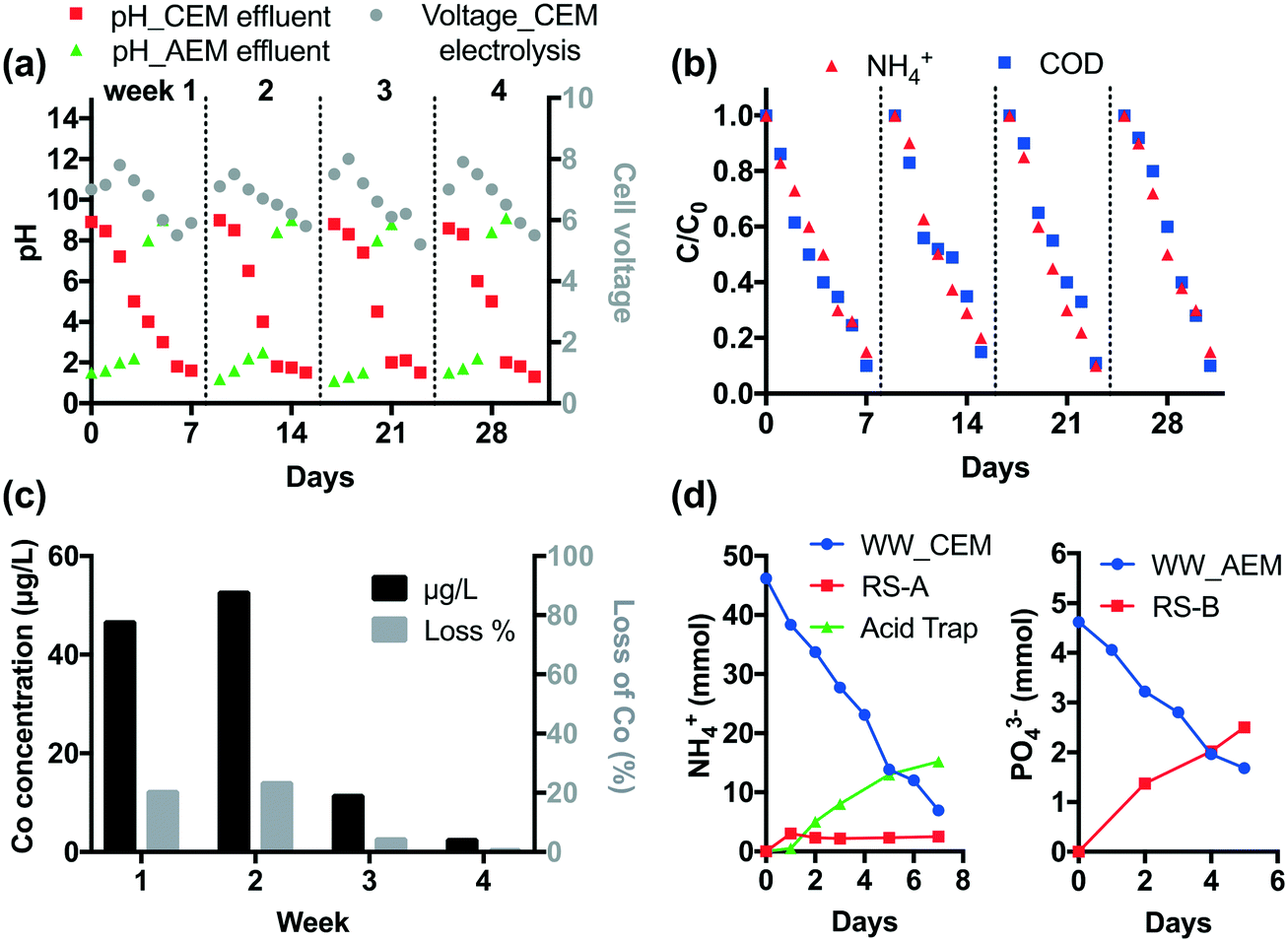 | ||
| Fig. 5 a) Profiles of pH and cell voltage during one-month CEM and AEM electrolysis. b) The removal of NH4+ and COD during one-month CEM electrolysis. c) Leaching of cobalt from the STI anode during CEM electrolysis. d) Removal and recovery of NH4+ and PO43− in CEM and AEM electrolysis processes in one-week operation shown as an example. | ||
The STI anode employs a Co dopant in the TiO2 layer in order to enhance the CER. The Co concentration in wastewater was monitored by ICP-MS (Agilent 8800) weekly. As shown in Fig. 5c, 45% of Co leaches from the STI anode in the first two weeks. After that the leaching is insignificant (<1.3%). This is probably because the residual Co dopant is immobilized in the TiO2 matrix due to the strong metal–metal oxide interactions.7 The electrode performance is not affected by Co leaching given the steady COD and NH4+ removal performance.
In CEM electrolysis, part of the NH4+ in the wastewater is recovered in the cathodic chamber. The pH of the catholyte (RS-A) rapidly rises to 13 in the first day. With the increase of electrolysis time, volatilization of NH3 from alkalized RS-A becomes significant as the [NH4+] increases in the acid trap (Fig. 5d). In total, 39.3 mmol NH4+ is removed from the wastewater, of which 17.7 mmol can be recovered in RS-A and the acid trap.
Membrane fouling was observed after one month of operation (Fig. S11†). The fouling does not increase the internal resistance given that the maximum cell voltage of four repetitive runs does not increase. This is probably because the foulant is loosely attached to the membrane. The foulant can be readily dissolved with 1 M HCl (Fig. S11†). The ionic analysis of acid digestion indicates that membrane fouling is caused by the precipitation of Ca2+ and Mg2+. Scaling was observed on the cathode as well. Microscopic observation indicates that only a part of the cathode surface is covered by Ca2+ and Mg2+ precipitates (Fig. S12†), suggesting that scaling has little impact on the cathodic reactions.
We further investigated the durability during AEM electrolysis. After each one-week CEM electrolysis period, the treated wastewater was transferred to the cathodic chamber of the AEM cell operated at 6 V. The wastewater (1.4 L) and recovery solution (RS-B, 100 mL) were circulated in the cathodic and anodic chamber, respectively, for five days. Tests were repeated four times without changing the AEM membrane and electrodes.
It is found that Ca2+ and Mg2+ were completely removed by CEM electrolysis, therefore membrane fouling and electrode scaling were not observed during the downstream AEM electrolysis. The AEM electrolysis adjusts the pH of the wastewater effluent to 8–9 (Fig. 5a). At the same time, anions are removed from the treated wastewater. As illustrated in Fig. 5d, 2.5 mmol PO43− is recovered in the RS-B chamber after one week of electrolysis.
In general, we demonstrated that the combined membrane electrolysis processes are durable for one month of continuous operation. Membrane cleaning by acid washing is not required, but should be considered as a precautionary measure for a longer period of operation.
Feasible approaches to produce fertilizer products are discussed below. It is known that the majority of NH4+ is captured by the H2SO4 acid trap. This approach directly produces an aqueous N-fertilizer solution of (NH4)2SO4. As for the production of a solid P-fertilizer, the concentrated PO43− in RS-B can be recovered as hydroxyapatite (Ca5(PO4)3OH) by the electrochemical process described previously.46 The residual RS-A and -B can be recharged into wastewater to increase the electrolyte conductivity.
Conclusions
This study found that introducing ionic exchange membranes to an electrolysis cell brings multiple benefits to latrine wastewater treatment. The presence of a CEM-separator in electrolysis reduces by 51% and 87% the energy consumption for COD and NH4+ removal, respectively. Compared with membrane-free electrolysis, CEM-separated electrolysis achieves enhanced helminth egg inactivation and has lower potential for the formation of unwanted by-products. Using a combination of CEM-separated and AEM-separated electrolysis allows recovery of NH4+ and PO43− for potential use as fertilizers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Bill and Melinda Gates Foundation (BMGF RTTC Grants OPP1111246 and OPP1149755).Notes and references
- J. Radjenovic and D. L. Sedlak, Challenges and Opportunities for Electrochemical Processes as Next-Generation Technologies for the Treatment of Contaminated Water, Environ. Sci. Technol., 2015, 49, 11292–11302 CrossRef CAS PubMed.
- Y. Yang, J. Shin, J. T. Jasper and M. R. Hoffmann, Multilayer Heterojunction Anodes for Saline Wastewater Treatment: Design Strategies and Reactive Species Generation Mechanisms, Environ. Sci. Technol., 2016, 50, 8780–8787 CrossRef CAS PubMed.
- K. Cho, Y. Qu, D. Kwon, H. Zhang, C. m. A. Cid, A. Aryanfar and M. R. Hoffmann, Effects of Anodic Potential and Chloride Ion on Overall Reactivity in Electrochemical Reactors Designed for Solar-Powered Wastewater Treatment, Environ. Sci. Technol., 2014, 48, 2377–2384 CrossRef CAS PubMed.
- J. T. Jasper, O. S. Shafaat and M. R. Hoffmann, Electrochemical Transformation of Trace Organic Contaminants in Latrine Wastewater, Environ. Sci. Technol., 2016, 50, 10198–10208 CrossRef CAS PubMed.
- X. Huang, Y. Qu, C. A. Cid, C. Finke, M. R. Hoffmann, K. Lim and S. C. Jiang, Electrochemical Disinfection of Toilet Wastewater Using Wastewater Electrolysis Cell, Water Res., 2016, 92, 164–172 CrossRef CAS PubMed.
- Y. Yang and M. R. Hoffmann, Synthesis and Stabilization of Blue-Black TiO2 Nanotube Arrays for Electrochemical Oxidant Generation and Wastewater Treatment, Environ. Sci. Technol., 2016, 50, 11888–11894 CrossRef CAS PubMed.
- Y. Yang, L. C. Kao, Y. Liu, K. Sun, H. Yu, J. Guo, S. Y. H. Liou and M. R. Hoffmann, Cobalt-Doped Black TiO2 Nanotube Array as a Stable Anode for Oxygen Evolution and Electrochemical Wastewater Treatment, ACS Catal., 2018, 8, 4278–4287 CrossRef CAS PubMed.
- C. A. Martínez-Huitle, M. A. Rodrigo, I. Sirés and O. Scialdone, Single and Coupled Electrochemical Processes and Reactors for the Abatement of Organic Water Pollutants: A critical review, Chem. Rev., 2015, 115, 13362–13407 CrossRef PubMed.
- B. P. Chaplin, Critical review of electrochemical advanced oxidation processes for water treatment applications, Environ. Sci.: Processes Impacts, 2014, 16, 1182–1203 RSC.
- M. Paidar, V. Fateev and K. Bouzek, Membrane electrolysis—History, current status and perspective, Electrochim. Acta, 2016, 209, 737–756 CrossRef CAS.
- A. Y. Bagastyo, D. J. Batstone, I. Kristiana, B. I. Escher, C. Joll and J. Radjenovic, Electrochemical Treatment of Reverse Osmosis Concentrate on Boron-Doped Electrodes in Undivided and Divided Cell Configurations, J. Hazard. Mater., 2014, 279, 111–116 CrossRef CAS PubMed.
- R. A. Rozendal, H. V. Hamelers and C. J. Buisman, Effects of membrane cation transport on pH and microbial fuel cell performance, Environ. Sci. Technol., 2006, 40, 5206–5211 CrossRef CAS PubMed.
- F. Harnisch, U. Schröder and F. Scholz, The suitability of monopolar and bipolar ion exchange membranes as separators for biological fuel cells, Environ. Sci. Technol., 2008, 42, 1740–1746 CrossRef CAS PubMed.
- P. Ledezma, J. Jermakka, J. Keller and S. Freguia, Recovering Nitrogen as a Solid without Chemical Dosing: Bio-Electroconcentration for Recovery of Nutrients from Urine, Environ. Sci. Technol. Lett., 2017, 4, 119–124 CrossRef CAS.
- A. Y. Bagastyo, D. J. Batstone, I. Kristiana, W. Gernjak, C. Joll and J. Radjenovic, Electrochemical Oxidation of Reverse Osmosis Concentrate on Boron-Doped Diamond Anodes at Circumneutral and Acidic pH, Water Res., 2012, 46, 6104–6112 CrossRef CAS PubMed.
- J. Xu, G.-P. Sheng, H.-W. Luo, W.-W. Li, L.-F. Wang and H.-Q. Yu, Fouling of proton exchange membrane (PEM) deteriorates the performance of microbial fuel cell, Water Res., 2012, 46, 1817–1824 CrossRef CAS PubMed.
- A. K. Luther, J. Desloover, D. E. Fennell and K. Rabaey, Electrochemically driven extraction and recovery of ammonia from human urine, Water Res., 2015, 87, 367–377 CrossRef CAS PubMed.
- W. A. Tarpeh, J. M. Barazesh, T. Y. Cath and K. L. Nelson, Electrochemical stripping to recover nitrogen from source-separated urine, Environ. Sci. Technol., 2018, 52, 1453–1460 CrossRef CAS PubMed.
- J. Desloover, J. De Vrieze, M. Van de Vijver, J. Mortelmans, R. Rozendal and K. Rabaey, Electrochemical nutrient recovery enables ammonia toxicity control and biogas desulfurization in anaerobic digestion, Environ. Sci. Technol., 2015, 49, 948–955 CrossRef CAS PubMed.
- X. Chen, Y. Gao, D. Hou, H. Ma, L. Lu, D. Sun, X. Zhang, P. Liang, X. Huang and Z. J. Ren, Microbial Electrochemical Current Accelerates Urea Hydrolysis for Nutrient Recovery from Source-separated Urine, Environ. Sci. Technol. Lett., 2017, 4(7), 305–310 CrossRef CAS.
- Y. Zhang, E. Desmidt, A. Van Looveren, L. Pinoy, B. Meesschaert and B. Van der Bruggen, Phosphate separation and recovery from wastewater by novel electrodialysis, Environ. Sci. Technol., 2013, 47, 5888–5895 CrossRef CAS PubMed.
- H. Park, C. D. Vecitis and M. R. Hoffmann, Electrochemical Water Splitting Coupled with Organic Compound Oxidation: The role of Active Chlorine Species, J. Phys. Chem. B, 2009, 113, 7935–7945 CAS.
- H. Park, C. D. Vecitis and M. R. Hoffmann, Solar-powered electrochemical oxidation of organic compounds coupled with the cathodic production of molecular hydrogen, J. Phys. Chem. A, 2008, 112, 7616–7626 CrossRef CAS PubMed.
- J. Jiang, M. Chang and P. Pan, Simultaneous hydrogen production and electrochemical oxidation of organics using boron-doped diamond electrodes, Environ. Sci. Technol., 2008, 42, 3059–3063 CrossRef CAS PubMed.
- H. Park, C. D. Vecitis, W. Choi, O. Weres and M. R. Hoffmann, Solar-powered production of molecular hydrogen from water, J. Phys. Chem. B, 2008, 112, 885–889 CrossRef CAS PubMed.
- J. Barrett, Studies on the induction of permeability in Ascaris lumbricoides eggs, Parasitology, 1976, 73, 109–121 CrossRef CAS PubMed.
- S. A. Brownell and K. L. Nelson, Inactivation of single-celled Ascaris suum eggs by low-pressure UV radiation, Appl. Environ. Microbiol., 2006, 72, 2178–2184 CrossRef CAS PubMed.
- J. M. Kalbermatten, D. S. Julius and C. G. Gunnerson, Appropriate technology for water supply and sanitation, World Bank Washington, DC, 1980 Search PubMed.
- C. T. Jafvert and R. L. Valentine, Reaction scheme for the chlorination of ammoniacal water, Environ. Sci. Technol., 1992, 26, 577–586 CrossRef CAS.
- Y. Gendel and O. Lahav, Revealing the mechanism of indirect ammonia electrooxidation, Electrochim. Acta, 2012, 63, 209–219 CrossRef CAS.
- J. T. Jasper, Y. Yang and M. R. Hoffmann, Toxic Byproduct Formation during Electrochemical Treatment of Latrine Wastewater, Environ. Sci. Technol., 2017, 51, 7111–7119 CrossRef CAS PubMed.
- T. Okada, S. Møller-Holst, O. Gorseth and S. Kjelstrup, Transport and equilibrium properties of Nafion® membranes with H+ and Na+ ions, J. Electroanal. Chem., 1998, 442, 137–145 CrossRef CAS.
- T. Okada, N. Nakamura, M. Yuasa and I. Sekine, Ion and water transport characteristics in membranes for polymer electrolyte fuel cells containing H+ and Ca2+ cations, J. Electrochem. Soc., 1997, 144, 2744–2750 CrossRef CAS.
- K. Hongsirikarn, J. G. Goodwin, S. Greenway and S. Creager, Effect of cations (Na+, Ca2+, Fe3+) on the conductivity of a Nafion membrane, J. Power Sources, 2010, 195, 7213–7220 CrossRef CAS.
- T. Okada, G. Xie, O. Gorseth, S. Kjelstrup, N. Nakamura and T. Arimura, Ion and water transport characteristics of Nafion membranes as electrolytes, Electrochim. Acta, 1998, 43, 3741–3747 CrossRef CAS.
- R. Halseid, P. J. Vie and R. Tunold, Influence of ammonium on conductivity and water content of Nafion 117 membranes, J. Electrochem. Soc., 2004, 151, A381–A388 CrossRef CAS.
- P. J. Hotez, P. J. Brindley, J. M. Bethony, C. H. King, E. J. Pearce and J. Jacobson, Helminth infections: the great neglected tropical diseases, J. Clin. Invest., 2008, 118, 1311–1321 CrossRef CAS PubMed.
- N. D. Levine, Chemical control of soil stages of animal-parasitic nematodes, Trans. Am. Microsc. Soc., 1969, 135–141 CrossRef CAS.
- K.-S. Oh, G.-T. Kim, K.-S. Ahn and S.-S. Shin, Effects of disinfectants on larval development of Ascaris suum eggs, Korean J. Parasitol., 2016, 54, 103 CrossRef CAS PubMed.
- B. P. Chaplin, G. Schrader and J. Farrell, Electrochemical Destruction of N-Nitrosodimethylamine in Reverse Osmosis Concentrates using Boron-doped Diamond Film Electrodes, Environ. Sci. Technol., 2010, 44, 4264–4269 CrossRef CAS PubMed.
- C. A. Cid, Y. Qu and M. Hoffmann, Design and preliminary implementation of onsite electrochemical wastewater treatment and recycling toilets for the developing world, Environ. Sci.: Water Res. Technol., 2018, 4, 1439–1450 RSC.
- K. Cho and M. R. Hoffmann, Urea Degradation by Electrochemically Generated Reactive Chlorine Species: Products and Reaction Pathways, Environ. Sci. Technol., 2014, 48, 11504–11511 CrossRef CAS PubMed.
- M. R. Hoffmann, A. Aryanfar, K. Cho, C. A. Cid, D. Kwon and Y. Qu, Self-contained, pv-powered domestic toilet and wastewater treatment system, U.S. Patent Application 14/048,163, filed July 31, 2014 Search PubMed.
- USEPA, https://archive.epa.gov/water/archive/web/html/vms59.html, (accessed 0322, 2018).
- M. Esclapez, M. Díez-García, V. Sáez, P. Bonete and J. González-García, Electrochemical degradation of trichloroacetic acid in aqueous media: influence of the electrode material, Environ. Technol., 2013, 34, 383–393 CrossRef CAS PubMed.
- C. A. Cid, J. T. Jasper and M. R. Hoffmann, Phosphate Recovery from Human Waste via the Formation of Hydroxyapatite during Electrochemical Wastewater Treatment, ACS Sustainable Chem. Eng., 2018, 6, 3135–3142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ew00698a |
| This journal is © The Royal Society of Chemistry 2019 |