
A solid-supported arylboronic acid catalyst for direct amidation†
Yihao
Du
a,
Thomas
Barber
a,
Sol Ee
Lim
a,
Henry S.
Rzepa
 b,
Ian R.
Baxendale
*a and
Andrew
Whiting
*a
b,
Ian R.
Baxendale
*a and
Andrew
Whiting
*a
aCentre for Sustainable Chemical Processes, Department of Chemistry, Durham University, Science Laboratories, South Road, Durham, DH1 3QZ, UK. E-mail: andy.whiting@durham.ac.uk
bDepartment of Chemistry, Imperial College, South Kensington Campus, London, SW7 2AZ, UK
First published on 13th February 2019
Abstract
An efficient heterogeneous amidation catalyst has been prepared by co-polymerisation of styrene, DVB with 4-styreneboronic acid, which shows wide substrate applicability and higher reactivity than the equivalent homogeneous phenylboronic acid, suggesting potential cooperative catalytic effects. The catalyst can be easily recovered and reused; suitable for use in packed bed flow reactors.
The amide bond is by far the most widely occurring structural motif incorporated into pharmaceutical compounds with amide formation reactions constituting around 16% of all reactions used by medicinal chemists.1 Most amidation methods developed for amide bond formation between carboxylic acids and amines, particularly those for peptide synthesis involve the use of carboxylic acid activating agents,2,3 however, such approaches generate stoichiometric amounts of waste, which has led to the development of more environmentally friendly processes and methods.4,5 The challenge for new, sustainable amidation is to achieve high conversion and catalyst reactivity, coupled with recyclability. Such developments in the direct amidation of carboxylic acids and amines have been realised in recent years, enabling major improvements in atom economy since water is the only by-product, with particular success being derived from metal-catalysed direct amidations.6–9
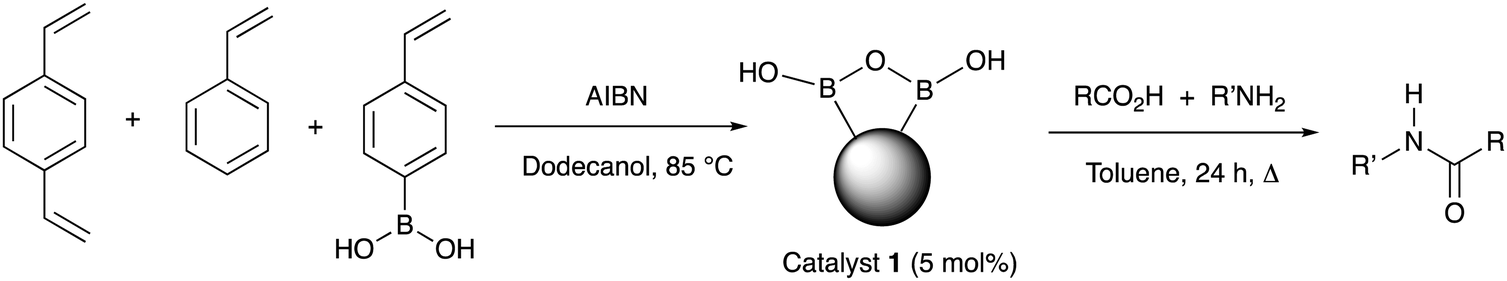
From as early as 1970, a role for boron-based catalysts in direct amidation has been recognised, however, years have seen a considerable number of new developments involving borate and boronate systems, showing increasing generality and utility.10 Our interests have involved developing new generations of boron-based catalysts,11 together with mechanistic understanding of the likely role of these systems in direct amidation.12 Boronic acid are particularly attractive for developing new amidation catalysts because they are reasonably stable, readily accessed and show good applicability and substrate scope when used in inert organic solvents and coupled with azeotropic water removal or molecular sieve drying.10 However, there are still major challenges to be overcome before boron-based catalysts can become the system of choice for routine amide formation, including: (1) developing lower temperature reactions; (2) efficient water removal; (3) low catalyst loading; and (4) simple catalyst recovery and re-use. An approach targeted at addressing all these issues is to employ a heterogeneous catalyst and indeed, the Ishihara group have reported such systems, including a polystyrene-bound borono-pyridinium salt and supported ortho-fluorophenyl boronic acid, systems which have shown good efficiency, yet both systems are non-trivial to prepare.13 Based on our recent mechanistic proposals and understanding about the way in which boronic acids form reactive dimeric B–X–B species in situ, we surmised that the inherent and facile drive for boronic acids to form B–O–B bridged and boroxine systems could be used as a templating effect to cluster boronate functions in close proximity within a polymer matrix, making the resulting polymer pre-organised towards amidation catalysis. In addition, the inherently hydrophobic nature of the bulk polymer might render the resulting supported catalyst system capable of not requiring additional water removal from the reaction. Herein, we report the realisation of this hypothesis through a simple and inexpensive polystyrene-based phenylboronic acid catalyst that is readily prepared, easily recycled and can even be employed in a flow reactor.
The heterogeneous polymeric catalyst 1 was prepared by heating a mixture of 4-styreneboronic acid, styrene, DVB (1,4-divinylbenzene) and AIBN in 1-dodecanol at 85 °C (Table 1).14 The resulting white polymeric catalyst was washed, dried and ground into a powder, and further treated through solvent extraction to remove residual monomers and 1-dodecanol (see ESI†). The polymer was analysed by solid state 11B NMR and showed a very broad peak centered at δ 17 ppm (range 30–5 ppm) suggesting a range of boron species (see ESI†).15 Application of catalyst 1 in direct amidation reactions (5% catalyst loading, calculated according to the estimated theoretical maximum boron content of the catalyst, see ESI†) under azeotropic refluxing conditions in toluene with a range of different carboxylic acids and benzylamine (Table 1) showed effective catalysis. Further reactions were also conducted with additional amines and comparative reactions were performed against phenylboronic acid, since this has previously been demonstrated to not be a particularly effective catalyst.11d The results are reported in Table 1.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Carboxylic acid | Amine | Yield/% | Entry | Carboxylic acid | Amine | Yield/%a |
| a General conditions: 2.86 mmol (1 equiv.) carboxylic acid, (1 equiv.) amine, 75.8 mg (5 mol%) catalyst 1, 20 mL toluene, Dean–Stark reflux, 24 h. Purified by low pressure Kugelrohr distillation. b Benzylamine (2 equiv.) was used due to both amidation and conjugate addition occurring to give a mixture of both PhCHCHCONHBn and PhCH(NHBn)CH2CONHBn. c Homogeneous catalysis using 5 mol% phenylboronic acid (see ESI). d Reflux without azeotropic water removal. NR = No reaction. | |||||||
| 1 | PhCO2H | BnNH2 | 90 (25c 22d) | 13 |
|
BnNH2 | 40 |
| 2 |
|
BnNH2 | 5 | 14 |
|
BnNH2 | 70 (42c) |
| 3 |
|
BnNH2 | 25 (16d) | 15 |
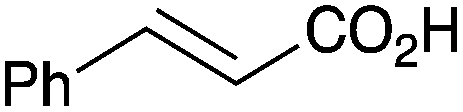
|
BnNH2 | 83b |
| 4 |
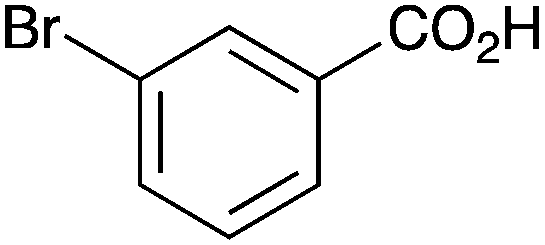
|
BnNH2 | 30 | 16 |
|
BnNH2 | NR |
| 5 |
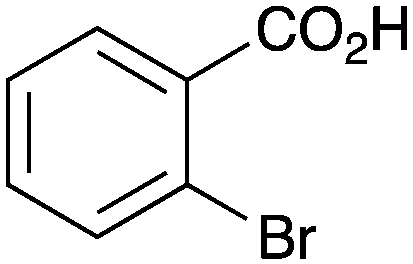
|
BnNH2 | 27 | 17 |
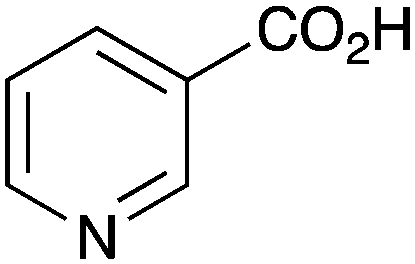
|
BnNH2 | NR |
| 6 |
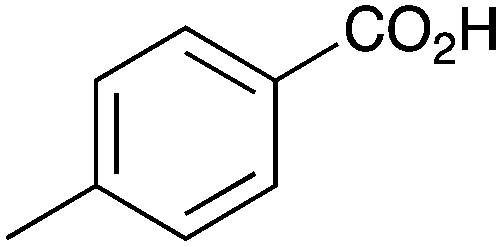
|
BnNH2 | 32 | 18 |
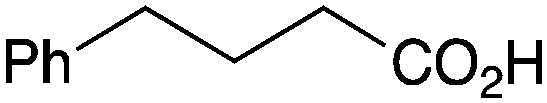
|
BnNH2 | 82 (64c) |
| 7 |
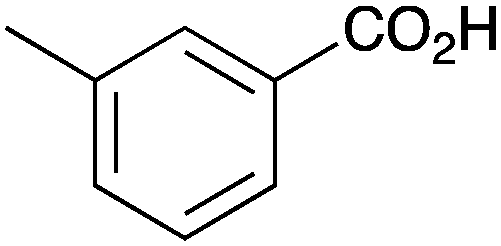
|
BnNH2 | 46 | 19 |
|
BnNH2 | 76 |
| 8 |
|
BnNH2 | 20 | 20 |
|
BnNH2 | 42 |
| 9 |
|
BnNH2 | NR | 21 |
|
PhNH2 | 75 |
| 10 |
|
BnNH2 | 29 | 22 | PhCO2H | PhNH2 | 15 |
| 11 |
|
BnNH2 | 92 (75c) | 23 |
|
Morpholine | 79 |
| 12 |
|
BnNH2 | 67 | 24 |
|
NR | |
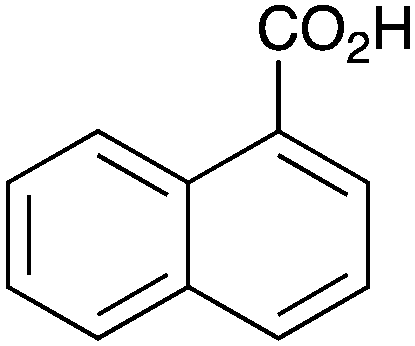
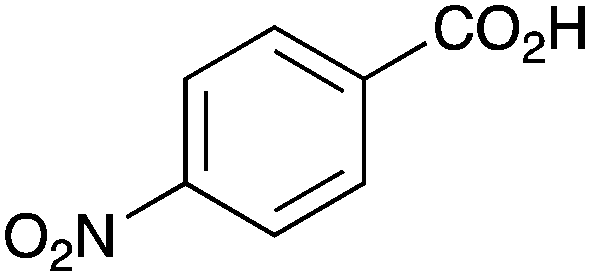
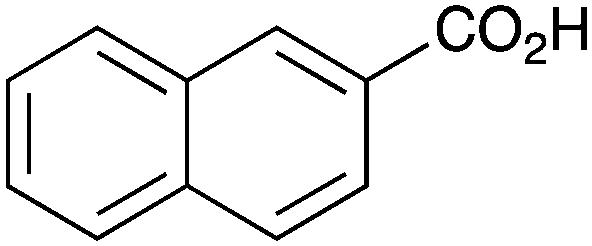
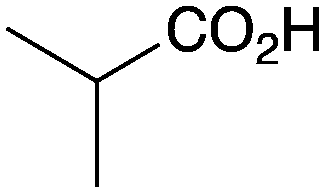
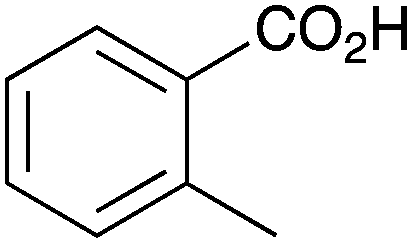
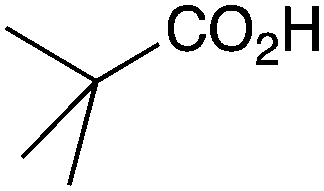
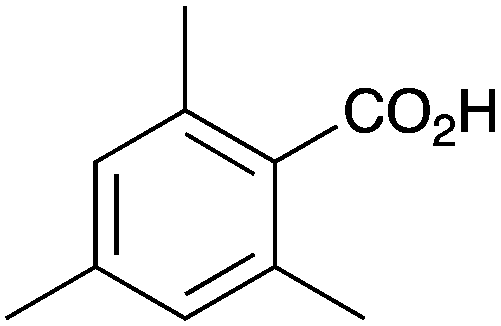
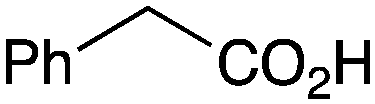
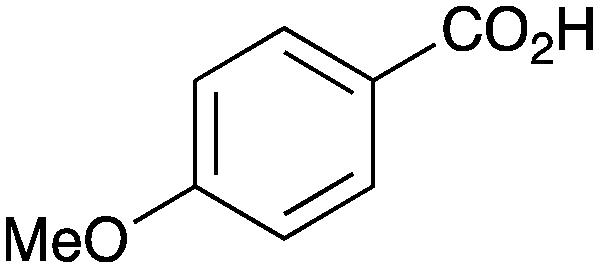
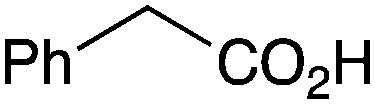
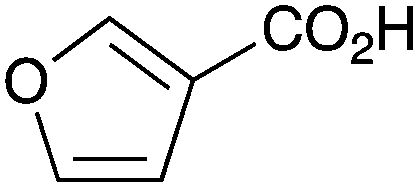
As shown higher pKa carboxylic acids were more reactive towards direct amide formation as discussed elsewhere,16 and importantly, the new heterogeneous catalyst 1 followed the same trend. The aromatic substrates benzoic, furoic, and 2-naphthoic acids showed excellent reactivity (Table 1, entries 1, 12 and 14). However, a reduction in conversion was observed for those aryl carboxylic acids with a range of substituents including bromine, methoxy and methyl (entries 3–8, 10, Table 1). While some of the effects could be steric in nature 1-naphthoic acid versus 2-naphthoic acid for example (Table 1, entries 13 and 14) and mesityl carboxylic acid (Table 1, entry 9), there are clearly other, more subtle effects at work. Furthermore, the more electron deficient aryl carboxylic acids were much less reactive, as seen with nitrobenzoic acid and 1 nicotinic acid, both of which were unreactive (Table 1, entries 2 and 17). Nonetheless, the general utility of catalyst 1 was further demonstrated through amidation of cinnamic acid (Table 1, entry 15) and several other alkyl carboxylic acids (Table 1, entries 11, and 18–20).
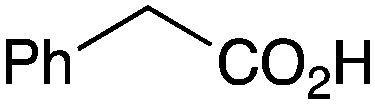
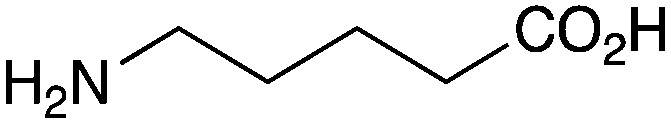
The less reactive amines such as aniline and morpholine, also showed good reactivity with phenylacetic acid (Table 1, entries 21 and 23), although the combination of benzoic acid and aniline (Table 1, entry 22) gave low conversion, as might be expected for a lower pKa carboxylic acid reacting with a less nucleophilic amine. Unfortunately, the potential lactamisation reaction of 5-aminovaleric acid failed (Table 1, entry 24).
In terms of heterogeneous versus homogeneous phase catalyst, the solid catalyst 1 showed considerably improved reactivity over phenylboronic acid. Hence, although catalyst 1 is electronically similar to phenylboronic acid, its reactivity was shown to be considerably higher (Table 1, entries 1, 3, 14 and 18). In addition, the recovery of catalyst 1 (vide infra) added to the general utility of the solid supported system.
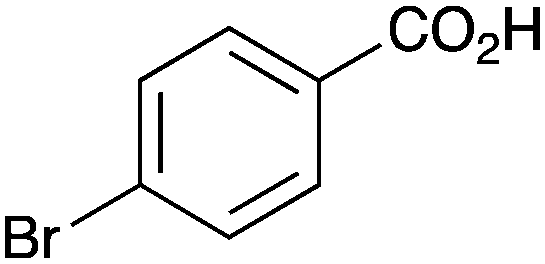
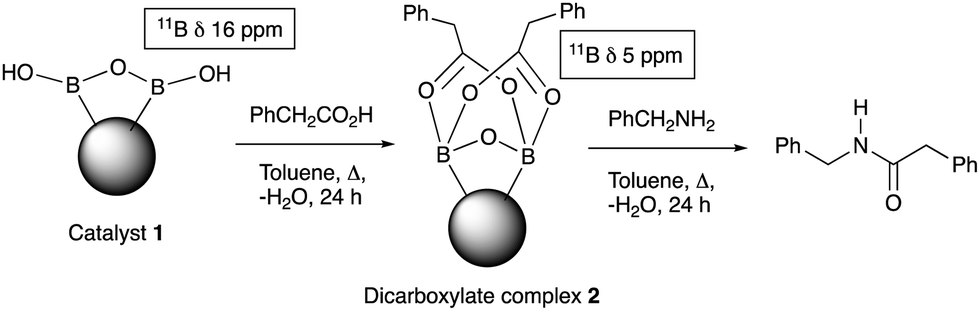
Further examination of the results Table 1, it is clear that the reaction timescales varied considerably, depending upon the substrate, which was further investigated. Therefore, two amidation reactions involving phenylacetic and 4-bromobenzoic acids (these reactions have quite different reaction timescales. See entries 3 and 11, Table 1) were repeated in parallel, under identical reaction conditions. Individual reactions representing different reaction times were terminated and their conversion determined (see the ESI†). The faster reaction of phenylacetic acid with benzylamine is shown in Fig. 1 and shows a high conversion level within the first 3 h, eventually yielding 80% amide formation. In contrast, the less reactive combination of 4-bromobenzoic acid and benzylamine only reached ∼70% conversion even over a prolonged timescale, i.e. 70 h (Fig. 1). These contrasting results demonstrate the strong correlation between carboxylic acid pKa and reactivity even using the solid phase catalyst 1 which mirrors the previously reported results of Loupy et al.16 for homogeneous catalysis. However, these results also show that both reactions, albeit on different timescales, stalled at a maximum of 70–80% conversion. Based on recent mechanistic studies,12 boronic acid-mediated catalysis is likely dependent upon the formation of B–X–B-type intermediates, that react with carboxylic acids to form doubly bridged carboxylate complexes which are attacked by the amine nucleophiles. However, the formation of such complexes necessarily means that excess carboxylate is tied up through boron coordination and hence, not all the carboxylic acid can subsequently react, resulting in incomplete conversion to the amide when equimolar quantities of the two substrates are used. Further demonstration that catalyst 1 comprises of proximal boroxine-like species that can indeed form carboxylate complexes resulting in catalytic reactivity was obtained through solid state NMR experiments using a stoichiometric amidation reaction. Hence, catalyst 1 was taken step-wise through the amidation reaction of phenylacetic acid with benzylamine, as shown in Scheme 1.
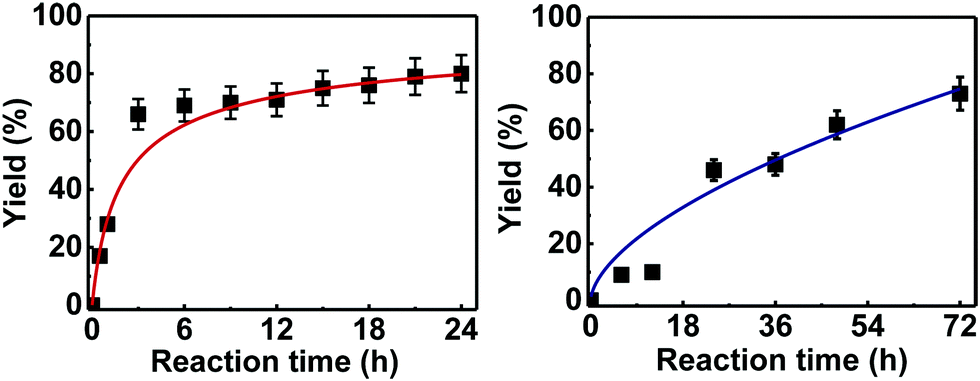 | ||
| Fig. 1 Kinetic studies on the heterogeneous catalytic direct amide formation between benzylamine and phenylacetic acid (left) or 4-bromobenzoic acid (right). | ||
 | ||
| Scheme 1 Activation process of polymer catalyst 1. | ||
Firstly, catalyst 1 was heated at reflux with the carboxylic acid (1 equiv.), followed by 24 h reaction with benzylamine to yield 92% of phenylacetamide. Most of the amide was generated within 30 min of the addition of the benzylamine (see ESI†). Solid-state 11B NMR and in situ IR (ReactIR, see ESI†) was carried out at each stage of the reaction. The catalyst showed a broad δ 17 ppm 11B NMR shift after preparation and solvent extraction, which may be due to a B–O–B-like water-associated complex (see ESI†). After reaction with a carboxylic acid a species of type 212 was indicted by a δ 5 ppm shift relating to the reactive double acyl-bridged intermediate. The amount of the reactive species 2 decreased after addition of benzylamine, synchronised with the amide formation, and indicative of partial consumption of the B–O–B coordinated bridged species (see ESI†). The in situ IR helped unveil the amidation process further, wherein the same solid phase reaction, addition of carboxylic acid to the reaction mixture resulted in the appearance of a peak at 1719 cm−1 (typical of phenylacetic acid), which also reduced as the reaction proceeded after benzylamine addition, and deriving a new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak at 1686 cm−1 for the amide within 30 minutes.
O peak at 1686 cm−1 for the amide within 30 minutes.
Catalyst recovery and re-use was demonstrated in a series of amidations, followed by filtration and recovery. The recovered catalyst showed high, reproducible reactivity through several cycles (Fig. 2). The first 3 reactions gave essentially identical yields and although there was some drop-off in yield over later reactions (4th and 5th reactions, Fig. 2), this was minimal. Indeed, accounting for minor mass loss at each flitration of the catalyst explains the slight decline in yield. These results show that the catalyst was robust, suitable for repeated use, and importantly, for potential application in a packed bed reactor as part of a flow system. Hence, a vapourtec R2+/R4 flow reactor (see ESI†), consisting of a binary pumping unit driving liquid flow through a reactor was utilised to examine the feasibility of flow-based amidations. To accomplish this, a heated column unit was loaded with catalyst 1 and the system pressure regulated (in-line 75-psi back-pressure regulator, BPR). A secondary digital pressure monitor was added for precise pressure monitoring (sited before column). Finally, a liquid handling robot allowing collection and fractionation of the fluidic output. After safety checking and reliability tests, the system was checked for precipitation and blocking due to ammonium salt formation upon mixing the carboxylic acid and amines which could limit this approach. Prior substrate screening informed us that phenylbutyric acid and benzylamine was a soluble pairing (57.2 mmol L−1 solution). Fraction collection was set for 100 min intervals to allow NMR analysis to determine conversion and estimation of catalytic efficiency. The output pattern indicated rapid initial formation of the amide product (as in batch) but delayed in the output due to a chromatographic effect of the column (fractions 2–6, see ESI†). In a typical run, this unoptimised reactor system was run for 20 h reaching steady state at ∼10 h, giving an output comprising 17% conversion (31 min residence time). In total, this reactor produced 1.80 g of amide over 116 h (15.5 mg h−1), with activity right to the end of this operational period.
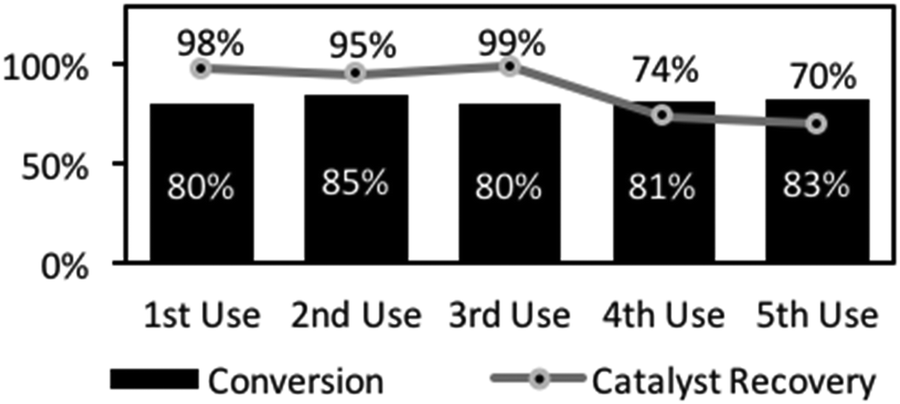 | ||
| Fig. 2 Catalyst recovery test of catalyst 1 in the reaction of phenylacetic acid with benzylamine. | ||
In summary, a novel polystyrene-supported boronic acid catalyst, prepared by co-polymerisation, shows the propensity of boronic acids to form anhydrides, resulting in clustering of the boronate sites within the catalyst, and hence, enabling efficient catalytic amidation reactivity. Evidence for B–O–B systems forming di-acyl bridged structures mirrors that of the homogeneous systems previously reported.12 Application of the heterogeneous catalyst under standard amidation conditions (24 h, reflux) gave up to 92% of the amide products. More than 20 substrates showed direct applicability including aryl and aliphatic carboxylic acids, and alkyl and aryl amines. The catalyst could also be recovered by filtration and reused multiple times without loss of reactivity. This stability also enabled the use of the catalyst in a flow reaction using a non-precipitating substrate combination. The flow reaction proceeded smoothly, maintaining catalytic activity for over 4.5 days of continuous operation.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr., R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS.
- A. Correa and R. Martin, J. Am. Chem. Soc., 2014, 136, 7253–7256 CrossRef CAS PubMed.
- Y. Kawagoe, K. Moriyama and H. Togo, Tetrahedron, 2013, 69, 3971–3977 CrossRef CAS.
- S. Ghosh, A. Bhaumik, J. Mondal, A. Mallik, S. Sengupta (Bandyopadhyay) and C. Mukhopadhyay, Green Chem., 2012, 14, 3220–3229 RSC.
- (a) A. Pelter, T. Levitt and P. Nelson, Tetrahedron, 1970, 26, 1539–1544 CrossRef CAS; (b) K. Ishihara, S. Ohara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS PubMed; (c) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386 CrossRef CAS PubMed; (d) S. Fatemi, N. Gernigon and D. G. Hall, Green Chem., 2015, 17, 4016–4028 RSC; (e) M. T. Sabatini, L. T. Boulton and T. D. Sheppard, Sci. Adv., 2017, 3, e1701028 CrossRef PubMed; (f) H. Noda, M. Furutachi, Y. Asada, M. Shibasaki and N. Kumagai, Nat. Chem., 2017, 9, 571–577 CrossRef CAS PubMed; (g) H. Charville, D. Jackson and A. Whiting, Chem. Commun., 2010, 46, 1813–1823 RSC; (h) K. Ishihara, Synthesis and Application of Organoboron Compounds, 2015, 243–270 CAS ; and references therein.
- (a) S. Liu, Y. Yang and A. Whiting, Eur. J. Org. Chem., 2013, 5692–5700 CrossRef CAS; (b) B. M. Monks and A. Whiting, Sustainable Catal., 2013, 89–110 CAS; (c) K. Arnold, B. Davies and A. Whiting, Angew. Chem., 2008, 120, 2713 CrossRef; (d) K. Arnold, A. S. Batsanov and A. Whiting, Green Chem., 2008, 10, 124–134 RSC; (e) K. Arnold, B. Davies and A. Whiting, Adv. Synth. Catal., 2006, 348, 813–820 CrossRef CAS; (f) I. Georgiou, G. Ilyashenko and A. Whiting, Acc. Chem. Res., 2009, 42, 756–768 CrossRef CAS.
- S. Arkhipenko, M. T. Sabatini, A. S. Batsanov, V. Karaluka, T. D. Sheppard, H. S. Rzepa and A. Whiting, Chem. Sci., 2018, 9, 1058–1072 RSC.
- (a) F. Jäkle, Synthesis and Application of Organoboron Compounds, 2015, 297–325 Search PubMed; (b) T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2005, 7, 5043–5046 CrossRef CAS PubMed.
- Examples of bespoke polymers as immobilized monolithic reagents see: (a) F. Svec and J. M. J. Frechet, Anal. Chem., 1992, 62, 820–822 CrossRef; (b) F. Svec and J. M. J. Frechet, Chem. Mater., 1995, 7, 707–715 CrossRef CAS; (c) P. Hodge, Curr. Opin. Chem. Biol., 2003, 7, 362–373 CrossRef CAS PubMed; (d) G. Jas and A. Kirschning, Chem. – Eur. J., 2003, 9, 5708–5723 CrossRef CAS PubMed; (e) A. Kirschning, W. Solodenko and K. Mennecke, Chem. – Eur. J., 2006, 12, 5972–5990 CrossRef CAS PubMed; (f) K. F. Bolton, A. J. Canty, J. A. Deverell, R. M. Guijt, E. F. Hilder, T. Rodemann and J. A. Smith, Tetrahedron Lett., 2006, 47, 9321–9324 CrossRef CAS; (g) N. Nikbin, M. Ladlow and S. V. Ley, Org. Process Res. Dev., 2007, 11, 458–462 CrossRef CAS; (h) M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin and C. D. Smith, Org. Biomol. Chem., 2008, 6, 1587–1593 RSC; (i) C. J. Smith, N. Nikbin, C. D. Smith, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2011, 9, 1927–1937 RSC; (j) K. A. Roper, H. Lange, A. Polyzos, M. B. Berry, I. R. Baxendale and S. V. Ley, Beilstein J. Org. Chem., 2011, 7, 1648–1655 CrossRef CAS PubMed; (k) R. J. Ingham, E. Riva, N. Nikbin, I. R. Baxendale and S. V. Ley, Org. Lett., 2012, 14, 3920–3923 CrossRef CAS PubMed; (l) M. V. Rojo, L. Guetzoyan and I. R. Baxendale, Org. Biomol. Chem., 2015, 13, 1768–1777 RSC.
- H. S. Rzepa, S. Arkhipenko, E. Wan, M. T. Sabatini, V. Karaluka, A. Whiting and T. D. Sheppard, J. Org. Chem., 2018, 83, 8020–8025 CrossRef CAS PubMed.
- L. Perreux, A. Loupy and F. Volatron, Tetrahedron, 2002, 58, 2155–2162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc09913h |
| This journal is © The Royal Society of Chemistry 2019 |