
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria†
Fabian
Panter
a,
Daniel
Krug
 a,
Sascha
Baumann
a and
Rolf
Müller
*ab
a,
Sascha
Baumann
a and
Rolf
Müller
*ab
aDepartment Microbial Natural Products, Helmholtz-Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research (HZI), Department of Pharmaceutical Biotechnology, Saarland University, Campus E8.1, 66123 Saarbrücken, Germany. E-mail: rolf.mueller@helmholtz-hzi.de
bGerman Centre for Infection Research, partner-site Hannover/Braunschweig, Germany
First published on 3rd May 2018
Abstract
There is astounding discrepancy between the genome-inscribed production capacity and the set of known secondary metabolite classes from many microorganisms as detected under laboratory cultivation conditions. Genome-mining techniques are meant to fill this gap, but in order to favor discovery of structurally novel as well as bioactive compounds it is crucial to amend genomics-based strategies with selective filtering principles. In this study, we followed a self-resistance guided approach aiming at the discovery of inhibitors of topoisomerase, known as valid target in both cancer and antibiotic therapy. A common host self-defense mechanism against such inhibitors in bacteria is mediated by so-called pentapeptide repeat proteins (PRP). Genes encoding the biosynthetic machinery for production of an alleged topoisomerase inhibitor were found on the basis of their collocation adjacent to a predicted PRP in the genome of the myxobacterium Pyxidicoccus fallax An d48, but to date no matching compound has been reported from this bacterium. Activation of this peculiar polyketide synthase type-II gene cluster in the native host as well as its heterologous expression led to the structure elucidation of new natural products that were named pyxidicyclines and provided an insight into their biosynthesis. Subsequent topoisomerase inhibition assays showed strong affinity to – and inhibition of – unwinding topoisomerases such as E. coli topoisomerase IV and human topoisomerase I by pyxidicyclines as well as precise selectivity, since E. coli topoisomerase II (gyrase) was not inhibited at concentrations up to 50 μg ml−1.
Introduction
Natural products of bacterial origin have continuously provided drug leads to combat infectious diseases and cancer.1 However, as certain groups of microorganisms have been screened extensively for natural products, the discovery of novel secondary metabolite scaffolds becomes increasingly challenging. Recent approaches to find valuable novel and biologically active natural products include screening of underexploited bacterial species, genera and families in different culture conditions as well as improving the bioinformatics, genetic and analytical toolset to access the “hidden” secondary metabolome of respective microorganisms.2–4 As the quest for new bioactive compounds is thriving, combinations of these methods are increasingly applied to uncover previously unidentified classes of natural products from a range of microbes, including – amongst others – marine streptomycetes, plant endosymbionts, cyanobacteria and the myxobacteria.3–5 Myxobacteria are ubiquitous Gram-negative, soil dwelling bacteria that exert a unique life-style involving the formation of multicellular fruiting bodies.6 Among myxobacteria the suborder Cystobacterineae – to which the genus Pyxidicoccus investigated in this work belongs – is typically represented by predatory strains displaying a high level of sophisticated multi-cellular coordination and cooperation.6 The complex life-style is reflected by large genome sizes exceeding 10 Mbps as observed from many myxobacteria.7 Moreover, the sheer number of biosynthetic gene clusters present in myxobacterial genomes according to bioinformatics analysis suggests myxobacteria constitute a highly prolific source of natural products. Nonetheless, the number of characterised compound families is still strikingly lower than expected from the genome-encoded capacity.8 Even for myxobacterial model strains such as Myxococcus xanthus DK1622 or Stigmatella aurantiaca DW4/3-1 only a fraction of the more than 15 secondary metabolite biosynthesis pathways found in their genomes have been assigned a natural product to date, indicating that the corresponding natural products are absent or below detection limits under standard laboratory conditions.8,9 Whereas several reasons might account for this observable discrepancy, it has emerged as a common picture for secondary metabolite-producing microorganisms. A range of genome-mining approaches has consequently been devised in order to “harvest” the untapped genome-encoded promise for novel natural products.10–12One straight-forward concept to bring to light the natural products supposedly originating from biosynthetic pathways that are repressed under standard laboratory conditions employs gene cluster activation via insertion of a heterologous promotor upstream of biosynthesis genes (Fig. 1).
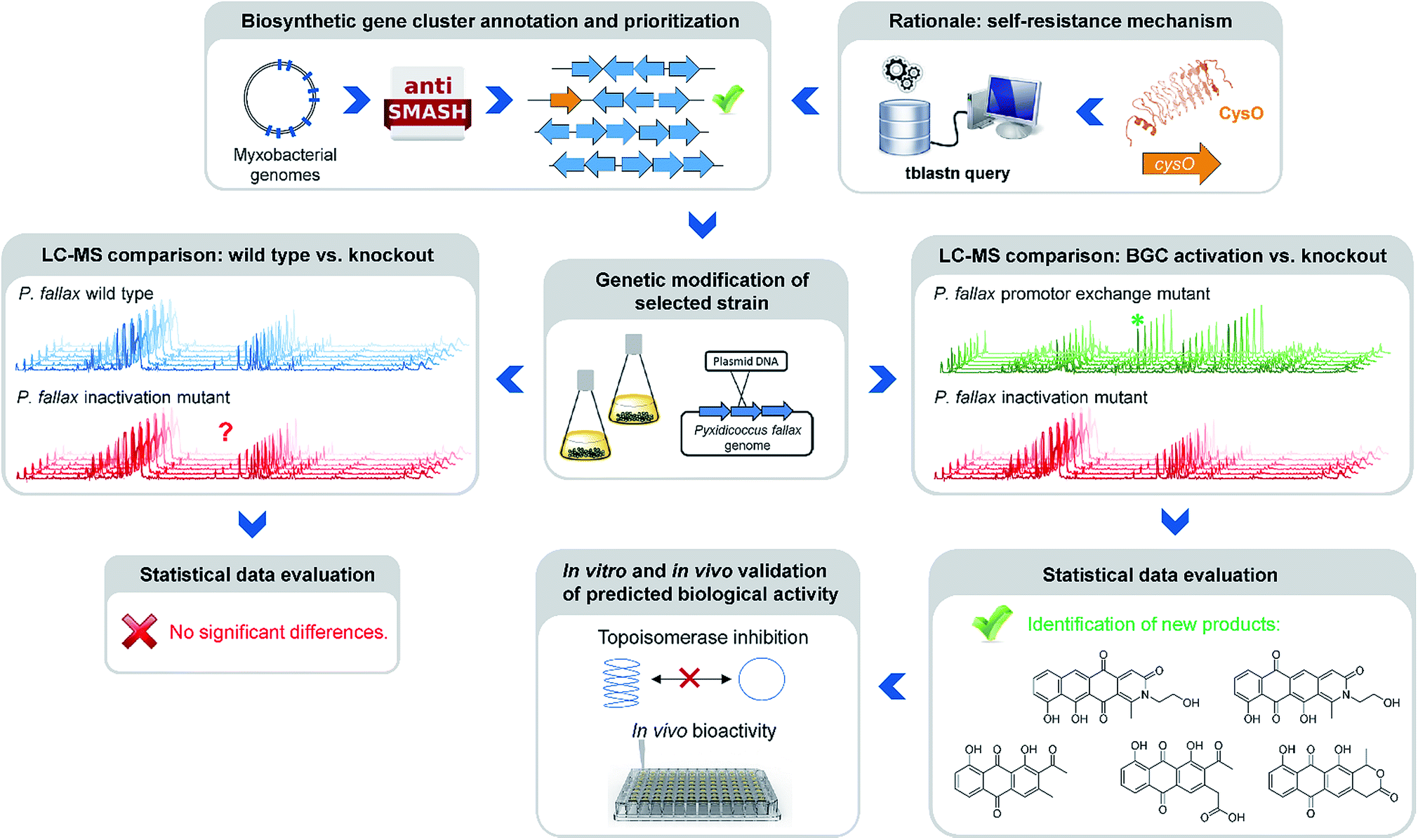 | ||
| Fig. 1 Workflow scheme illustrating the genomics-based gene cluster prioritization and activation approach towards novel bioactive natural products from ‘silent’ gene clusters. | ||
This approach however is typically laborious and time-consuming. Success is critically depending on pre-established cultivation conditions, an understanding of the regulation of the respective pathway and genetic manipulation protocols specific to the target organism. A strong rationale is therefore required to prioritise the plethora of eligible biosynthetic gene clusters for well-directed gene cluster activation. A conceivable prioritization approach is based on the finding that potential genetic self-resistance determinants are often co-located to candidate biosynthetic gene clusters.13 Considering topoisomerase inhibitors, the occurrence of a gene encoding a pentapeptide repeat protein (PRP) might serve as an indicator for the nearby BGC to potentially produce a congener of this compound class. Adopting this strategy in the current study was inspired by a specific example recently reported from myxobacteria, i.e. the cystobactamid gene cluster where a pentapeptide repeat protein is responsible for self-resistance against the gyrase inhibitor cystobactamid, even allowing for simplified mode of action determination of this compound class.14
Along these lines, we report here the identification and full structural characterization of a new bioactive secondary metabolite family connected to a biosynthetic gene cluster not previously attributed to any natural product from the myxobacterium Pyxidicoccus fallax An d48. In agreement with the rationale used for their discovery, the new polyketide-type compounds named pyxidicyclines A (1) and B (2) display potent inhibition of bacterial and human topoisomerases. In addition, the heterologous expression of the underlying pcy gene cluster permits insights into the intriguing biosynthesis of a new myxobacterial secondary metabolite class featuring an unprecedented tetracene quinone scaffold.
Results and discussion
Genome mining for topoisomerase inhibitors
The starting point of this study is defined by our aim to find novel topoisomerase inhibitors connected to matching BGCs hidden in the genomes of myxobacteria and guided by signature genes likely to encode the required self-resistance mechanism. We reasoned that CysO from the cystobactamide pathway in Cystobacter ferrugineus and AlbG from the albicidin pathway in Xanthomonas albilineans should be suitable templates to identify putative pentapeptide repeat proteins conferring self-resistance to topoisomerase poisons.14,15 Searching for genes encoding topoisomerase-targeting pentapeptide repeat protein (TTPRP) sequences similar to AlbG and CysO yielded 31 candidate sequences among myxobacterial genomes in our in-house database.15 Eight of those hit sequences were located close to uncharacterised biosynthetic gene clusters as predicted by antiSMASH (Fig. 3).10,11 Based on primary sequence similarity, these genes were regarded as candidates which could plausibly be responsible for topoisomerase inhibitor self-resistance against the products afforded by said BGCs. In view of desired structural novelty of products to be discovered, our attention was drawn to a candidate encoded near a type II polyketide synthase (PKS) gene cluster present in the myxobacterial strains P. fallax An d30 and An d48. However, no compound resembling a type II PKS product has been found and no topoisomerase inhibitor was reported in these strains. While type II PKS compounds are well known from Gram-positive actinobacteria, there are only few examples of type II PKS products from Gram-negative bacteria as exemplified by anthraquinones from Phodorhabdus luminescens or aurachin from Stigmatella aurantiaca.16 Therefore, the envisaged BGC was considered likely to be responsible for the biosynthesis of hitherto unknown secondary metabolites with uncommon structures. Besides the biosynthetic genes encoding polyketide synthase machinery, the pcy cluster features an obvious operon comprising ABC transporter genes and the 639 bp open reading frame (ORF) encoding the proposed PRP (pcyQ-T, Fig. 3). Thus, this gene ensemble hints at a combined export- and topoisomerase protection mechanism allowing immunity of the bacterium to the hypothetical product. The proposed PRP in this cluster named PcyT was compared via MUSCLE alignment to CysO, AlbG as well as E. coli McbG, M. tuberculosis MfpA and Qnr proteins known to mediate resistance against the fluoroquinolone gyrase and topoisomerase IV inhibitor drug class (Fig. 2).17,18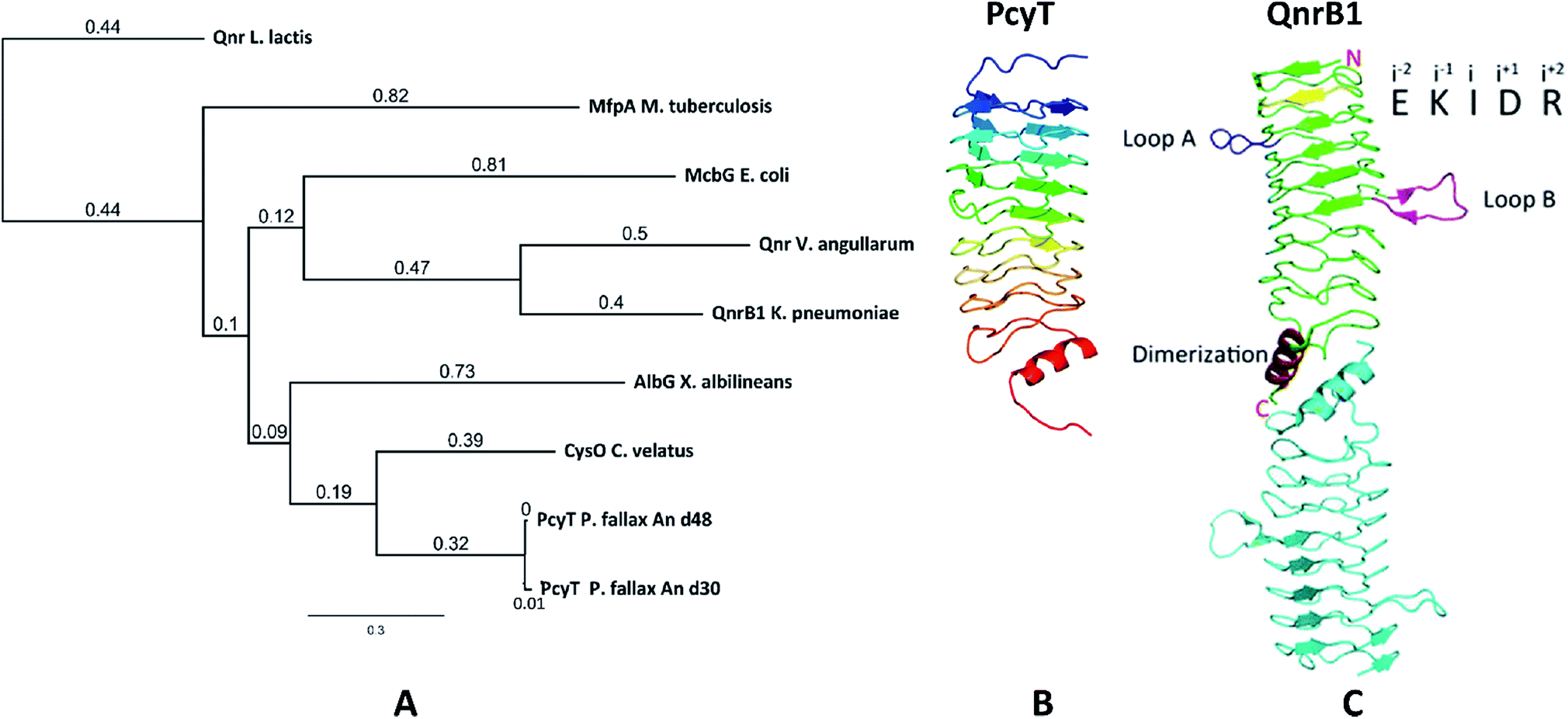 | ||
| Fig. 2 (A) Neighbour joining tree constructed from a MUSCLE alignment including TTPRP primary sequences known to confer topoisomerase inhibitor resistance, (B) comparison of the Phyre-2 3D structure homology model of PcyT from Pyxidicoccus fallax to (C) the protein crystal structure of the topoisomerase targeting pentapeptide repeat protein (TTPRP) QnrB1 from K. pneumoniae. | ||
The alignment tree clearly supports functional similarities among these proteins, although amino acid sequence similarity between these short (200–300 AA) PRP is generally not higher than 50%.19 Their 3D structures are characterised by a consensus motif of five [S,T,A,V][D,N][L,F][S,T,R][G] repeats forming a right handed quadrilateral helix, indicating this 3D fold to be more important than the primary sequence.19 Therefore, the 3D structure of PcyT was further analysed using the Phyre-2 homology modelling tool (Section S2.1 and 2.2†).20 The modelled structure of PcyT suggests the presence of the right handed quadrilateral helix structure thought to account for DNA mimicry plus a dimerization domain consisting of the alpha helical part and the terminal loop (depicted in red in Fig. 2), both of which are required for mediating topoisomerase poison resistance.15,19 The striking difference between the quinolone resistance protein QnrB1 (PDBe: 2xtx) and PcyT is the predicted absence of loop structures Loop A and Loop B suggesting PcyT falls into the category of “loopless” TTPRPs like CysO and AlbG.19
Thus, since in silico analysis suggested the possibility for production of a topoisomerase inhibitor by P. fallax we set out to identify the small molecules associated with said type II PKS BGC to ultimately assess their bioactivity. P. fallax An d48 has been the subject of natural product screening in the past and turned out to be a viable bioactive secondary metabolite producer.21 However, no known small molecule from this strain resembles a structure likely to stem from type II polyketide synthase machinery. Furthermore, we could not detect any inhibition of E. coli DNA gyrase and E. coli DNA topoisomerase IV in assays using wild type crude extract (data not shown), pointing towards absence of a matching bioactive compound under standard laboratory conditions.
We thus took a closer look at the prioritised gene cluster (designated pcy) which comprises 20 ORFs encoded within three apparent operons (Fig. 3). Operon 1 contains the minimal PKS genes pcyA-I, encoding a ketosynthase unit composed of the ketosynthase α (KSα, pcyE), the chain length factor (CLF or KSβ, pcyF) and the acyl carrier protein (ACP, pcyG) which are fundamental for the function of any type II PKS assembly line (Fig. 3, Table 1, and KS assignments in Section S2.3 and S2.4†).22 Operon 2 encodes proposed tailoring enzymes (pcyJ-P) while operon 3 encodes ABC transporter-related genes as well as the putative resistance protein (pcyQ-T).
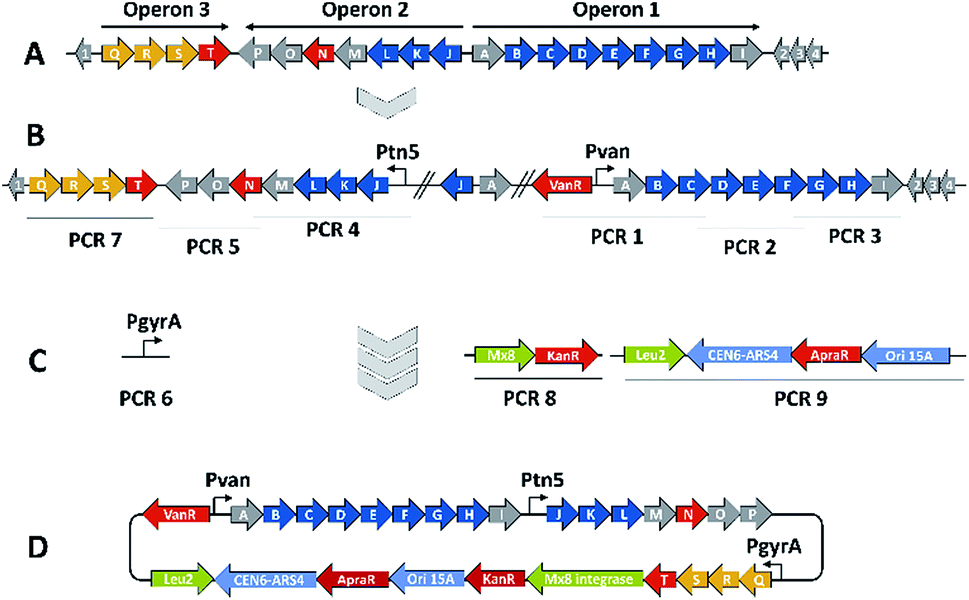 | ||
| Fig. 3 BGC activation and heterologous expression strategy for the pcy biosynthetic gene cluster. (A) The native gene cluster in An d48 and its operon structure, (B) the gene cluster activated by two promotor exchanges, (C) the TAR cloning strategy including PCR pieces necessary to complete the pClY vector and (D) the assembled heterologous expression plasmid. PKS type-II biosynthesis genes depicted in blue, regulation and resistance genes in red, exporter genes in orange, auxiliary genes for the cloning strategy in light green and ORFs of unknown function in grey colour. | ||
| Gene | Putative function | Closest annotated homologue – organism of origin |
|---|---|---|
| pcyA | Precursor release from the acyl carrier protein | Acyl-CoA thioester hydrolase – multispecies |
| pcyB | Polyketide cyclisation (B-ring cyclisation) | Polyketide cyclase – Streptomyces nogalater |
| pcyC | Polyketide cyclisation (A-ring cyclisation) | Polyketide cyclase – Streptomyces atratus |
| pcyD | C9′ type ketoreductase | Ketoacyl reductase – Sorangium cellulosum |
| pcyE | Ketoacyl synthase KSα subunit | 3-Oxoacyl-ACP synthase II – Dendrosporobacter quericolus |
| pcyF | Ketoacyl synthase KSβ subunit (chain length factor) | 3-Oxoacyl-ACP synthase II – Pelosinus sp. |
| pcyG | Acyl carrier protein | Acyl carrier protein – Terriglobus saanensis |
| pcyH | 4′-Phosphopanthenyl transferase | Holo-ACP synthase – Sorangium cellulosum |
| pcyJ | Serine incorporation | 4-Coumarate-CoA ligase family protein – Modestobater sp. |
| pcyK | Heterocycle formation | Muconolactone delta-isomerase – Acidovorax valerianellae |
| pcyL | Heterocycle formation | Polyketide cyclase – Streptomyces sp. |
| pcyQ | Export | ABC transporter, ATP binding protein – Myxococcus xanthus |
| pcyR | Export | ABC transporter permease – Myxococcus xanthus |
| pcyS | Export | ABC transporter substrate binding protein – Myxococcus xanthus |
| pcyT | Putative self-resistance mechanism | Pentapeptide repeat containing protein – Myxococcus fulvus |
In order to connect a chemotype to the candidate gene cluster we initially followed a strategy combining single crossover inactivation with subsequent LC-MS profiling of ‘inactivation’ mutants vs. the wild type strain. Strikingly, disruption of either pcyE or pcyC located within operon 1 did not lead to any detectable change in the P. fallax An d48 metabolome, as corroborated by in-depth statistical evaluation of the LC-MS chromatograms of replicate mutant extracts compared to wild type extracts (Fig. 4 and Section S3.1 to 3.4†).
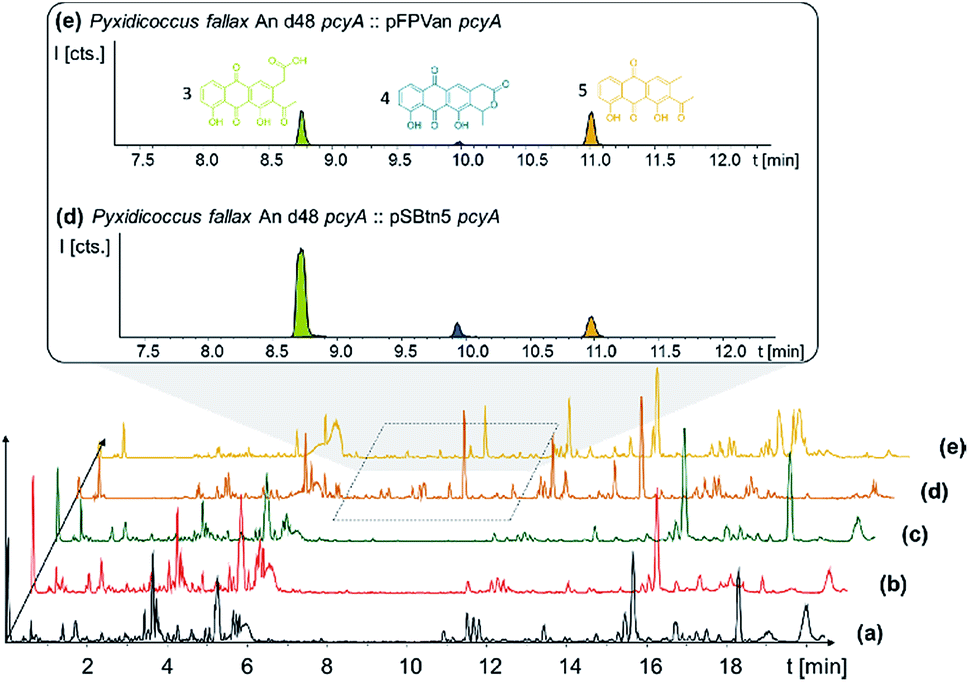 | ||
| Fig. 4 BPC comparison of (a) Pyxidicoccus fallax An d48 wild type extract, the extracts of the (b) pcyC and (c) pcyE gene inactivation mutants, and anthraquinones produced in P. fallax An d48 (d) vanillate-induced and (e) tn5 activation mutants. The three anthraquinone metabolites 340 (3), 296 (5) and 325 (4) are displayed as LC-MS EICs at 341.06 [M + H]+ (light green), 325.07 [M + H]+ (dark blue) and 297.075 [M + H]+ (orange). The corresponding structure formula is shown in the same colour. Differences visible in traces (a) to (e) at retention times later than 12 minutes are unrelated to the investigated biosynthetic route. | ||
Reviving a ‘silent’ type II PKS gene cluster
Expecting the small molecule product of the pcy cluster to be absent or below the detection limit, gene cluster activation by insertion of a heterologous promotor was performed.23 Since operon 1 contains the minimal PKS genes, promotor exchange via single crossover homologous recombination upstream of this operon should boost transcription of these genes and thus facilitate subsequent detection of the matching PKS-type metabolites. Similar pathway-refactoring strategies have been successfully employed for the activation of ‘silent’ gene clusters in Streptomyces and Burkholderia.24,25 Furthermore, promotor insertions have been reported to improve secondary metabolite production by myxobacteria, although the approach did not afford a previously unknown myxobacterial natural product class to date.2,26
P. fallax An d48 mutants controlling the expression of the minimal PKS operon by a heterologous tn5 promotor and alternatively via the vanillate promotor/repressor system were created through single crossover homologous recombination using the plasmids pSBtn5 pcyA and pFPvan pcyA, respectively (Section S1.10†). The vanillate promotor repressor system was chosen as it has been shown to work in myxobacteria.27 LC-MS analysis of the corresponding crude extracts revealed three newly emerging substances exhibiting strong UV absorption, indicating the presence of a chromophore as it would be expected from a type II PKS derived compound (Fig. 4 and S13†). Their intensity is reduced to basal level shown by both LC-UV and LC-MS measurements if the An d48 pcyA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pFPVan pcyA mutant is grown without induction of the vanillate promotor, further indicating a direct link of found compounds to the target type II PKS machinery. Comparison of LC-UV and LC-MS peak areas suggests the tn5 promotor leads to higher expression levels than the induced vanillate promotor/repressor system (Table S21†). Purification of the new compounds appearing as a result of promotor integration into An d48 using a combination of chromatographic methods and NMR-based structure elucidation consequently revealed a series of anthraquinones (3–5) (Fig. 4; Section S3.5.1 and 3.8†), resembling literature-known secondary metabolites described from actinomycetes.28,29 UV/Vis spectra and NMR signals agree well with the previously published data (Section S3.5.1, 3.8 and 3.9†). These anthraquinones show moderate in vivo activity against Gram-positive pathogens and efflux deficient E. coli (Table 2).
:pFPVan pcyA mutant is grown without induction of the vanillate promotor, further indicating a direct link of found compounds to the target type II PKS machinery. Comparison of LC-UV and LC-MS peak areas suggests the tn5 promotor leads to higher expression levels than the induced vanillate promotor/repressor system (Table S21†). Purification of the new compounds appearing as a result of promotor integration into An d48 using a combination of chromatographic methods and NMR-based structure elucidation consequently revealed a series of anthraquinones (3–5) (Fig. 4; Section S3.5.1 and 3.8†), resembling literature-known secondary metabolites described from actinomycetes.28,29 UV/Vis spectra and NMR signals agree well with the previously published data (Section S3.5.1, 3.8 and 3.9†). These anthraquinones show moderate in vivo activity against Gram-positive pathogens and efflux deficient E. coli (Table 2).
| Test Organism | 1 | 2 | 3 | 5 | 4 |
|---|---|---|---|---|---|
| S. aureus newman | 16 μg ml−1 | 32 μg ml−1 | 2 μg ml−1 | 16 μg ml−1 | n.d. |
| C. albicans DSM 1665 | 64 μg ml−1 | 64 μg ml−1 | n.d. | n.d. | n.d. |
| M. luteus DSM 1790 | 16 μg ml−1 | 32 μg ml−1 | n.d. | n.d. | n.d. |
| B. subtilis DSM 10 | 16 μg ml−1 | 32 μg ml−1 | 64 μg ml−1 | 64 μg ml−1 | 16 μg ml−1 |
| M. smegmatis mc2-155 | 32 μg ml−1 | 64 μg ml−1 | n.d. | n.d. | n.d. |
| E. coli ΔTolC | 4 μg ml−1 | 2 μg ml−1 | 8 μg ml−1 | 64 μg ml−1 | 16 μg ml−1 |
| P. aeruginosa PA 14 | >64 μg ml−1 | >64 μg ml−1 | n.d. | n.d. | n.d. |
| C. violaceum DSM 30191 | >64 μg ml−1 | >64 μg ml−1 | n.d. | n.d. | n.d. |
| E. coli DSM 1116 | >64 μg ml−1 | >64 μg ml−1 | n.d. | n.d. | n.d. |
| M. hiemalis DSM 2656 | 16 μg ml−1 | 16 μg ml−1 | n.d. | n.d. | n.d. |
| P. anomala DSM 6766 | >64 μg ml−1 | >64 μg ml−1 | n.d. | n.d. | n.d. |
| HCT-116 (human colon carcinoma) | 0.06 μg ml−1 | 0.03 μg ml−1 | n.d. | n.d. | n.d. |
From a biosynthetic perspective 3 was described as a polyketide synthesis derailment product occurring when the frenolicin minimal PKS was complemented with the actinorhodin ketoreductase (KR) and the cyclases CYC1 and CYC2 from the griseorhodin and oxytetracycline pathways, respectively, and homologues of these genes also exist within the activated minimal PKS operon of the pcy gene cluster.29 We thus reasoned that the products observed upon promotor insertion upstream of the pcyA-I operon could stem from incomplete processing of polyketide biosynthesis intermediates as a consequence of non-expressed tailoring enzymes located within the second operon pcyJ-P. Therefore, a second promotor was integrated via single crossover homologous recombination upstream of pcyJ, whereby this operon was activated in addition to the pcyA-I operon (Fig. 3). The resulting ‘dual promotor’ mutant strain P. fallax An d48 pcyA:pFPVan pcyA, pcyJ:pFPtettn5 pcyJ (named An d48 pcy in short) was cultivated in small scale following successful confirmation of correct promotor integration via PCR. Notably, we found that this strain lysed upon vanillate induction when XAD-16 adsorber resin was absent from the medium, while cultivation and induction was possible with added XAD-16. Thus, the second promotor exchange apparently led to the production of biologically active entities other than the previously detected anthraquinones, and these yet unidentified products seemed to exhibit significant autotoxicity. Concomitantly, it was not possible to introduce simultaneously two constitutive tn5 promotors upstream of both operons pcyA-I and pcyJ-P despite several attempts (data not shown), further indicating autotoxicity effects associated with the observed metabolites.
Structure elucidation of the pyxidicycline class of natural products
Induction of the newly engineered double-promotor mutant strain with vanillate and subsequent LC-MS analysis of XAD-16 extracts led to the detection of two novel peaks with an UV absorption at 500 nm (Fig. S16†). Isolation and structure elucidation of both candidate compounds (Section S3.5.2, 3.6, 3.7 and 3.9†) ultimately revealed them as two novel type II PKS derived nitrogen containing tetracene quinones named pyxidicycline A (1) and B (2) (Fig. 5). The two anthraquinones 3 and 5 referred to as polyketide derailment products are still present even though their yields are significantly reduced (Fig. 5, Table S21†), whereas 4 could not be detected in the extract of An d48 pcy.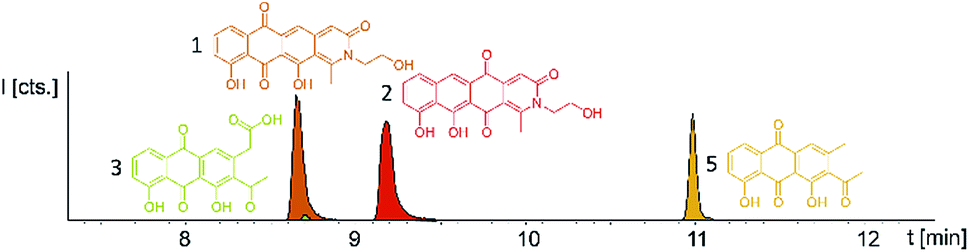 | ||
| Fig. 5 Overlay of three EICs of the two novel mature type two PKS compounds (1 and 2) in Pyxidicoccus fallax An d48 pcy extract accompanied by two anthraquinones (3 and 5). 3 and 5 are displayed as LC-MS EICs at 341.06 [M + H]+ (light green) and 297.075 [M + H]+ (orange); the two pyxidicyclines are displayed as LC-MS EICs at 366.10 [M + H]+ (1 in orange; 2 in red). | ||
The pyxidicyclines are difficult to dissolve in organic solvents except for DMSO and DMF, which unfortunately both degrade the compound over time. However, they are well soluble and stable in pure formic acid. Dissolution in formic acid leads to reversible formation of formate esters as shown by an increase of the compound's masses by 28 daltons as seen in the LC-MS chromatogram. Fortunately, both isomers formed dark red crystals from formic acid solution and the corresponding crystal structures obtained by single-crystal Xray diffraction experiments afforded the structures depicted in Fig. 6 (Section S3.6 and 3.7†). The structures of both pyxidicycline formate esters were subsequently confirmed by NMR analysis in formic acid-d2 (Section S3.11†). These compounds represent a novel type II PKS scaffold, as nitrogen-containing tetracene quinone-like four ring structures have – to the best of our knowledge – not been reported to date. Among the rare examples of type II PKS products containing nitrogen in their backbone structure (albeit not as part of a tetracene system) are the jadomycins and nitrogen-containing congeners of fredericamycins.32,33
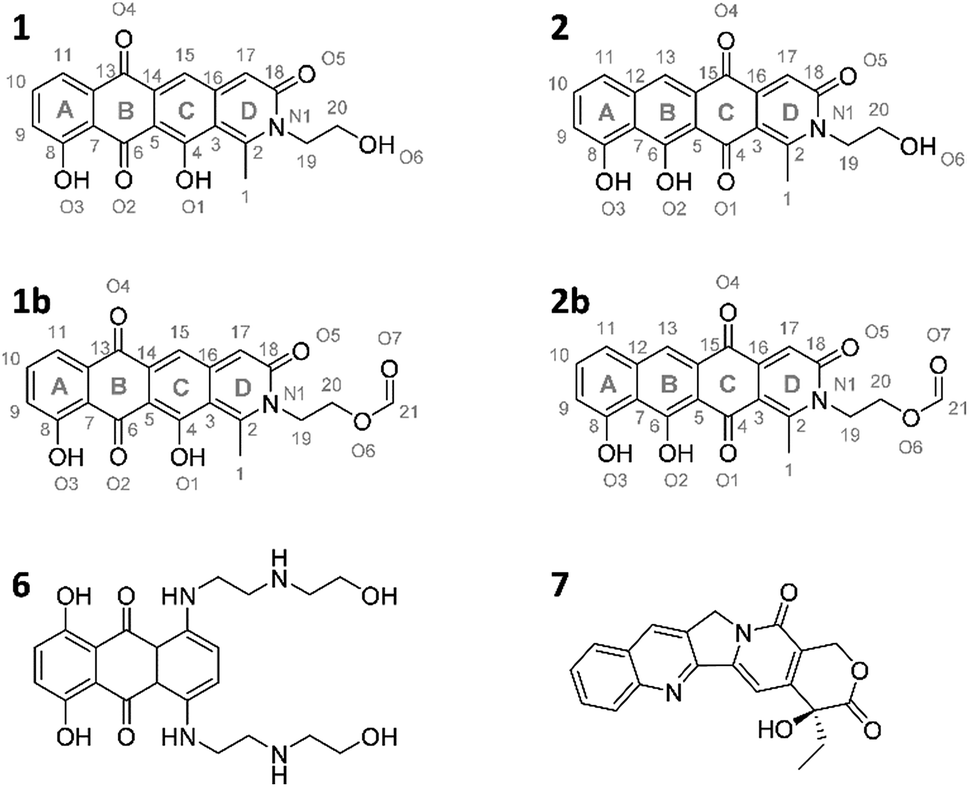 | ||
| Fig. 6 Structures of the two pyxidicycline derivatives A (1) and B (2) as well as the corresponding formate esters (1b and 2b) as determined by structure elucidation using NMR and X-ray crystallography. 6 and 7, structures of mitoxanthrone and camptothecin.30,31 | ||
The pyxidicyclines were subjected to antimicrobial and cytotoxic activity testing against a panel of bacterial pathogens and cancer cells. The anthraquinone precursors were tested against a reduced set since low isolation yields did not allow full bioactivity profiling. 1 and 2 show moderate antibacterial activity but strong cytotoxicity in the nano molar range (Table 2). The complete lack of activity against wild type Gram-negative pathogens is likely due to compound efflux as the efflux deficient E. coli ΔTolC strain shows the lowest MIC values among bacteria. While the yeast strains C. albicans and P. anomala show very high MIC values, the pyxidicyclines inhibit the growth of filamentous fungi displayed by M. hiemalis at intermediate concentrations (Table 2).
Investigating the pyxidicyclines' molecular target
Our motivation for activating the pcy BGC was based on the finding of pentapeptide repeat protein PcyT, which suggested the compounds afforded by this gene cluster could act as topoisomerase inhibitors. Structural similarity of pyxidicyclines to clinically used mitoxanthrone (6, Fig. 6) – a topoisomerase II and topoisomerase IV inhibitor used in breast cancer treatment with an IC50 of 12 μM and 2 μM against E. coli topoisomerase II (gyrase) and topoisomerase IV, respectively31 – increased our suspicion that pyxidicyclines might indeed target topoisomerases. Hence their topoisomerase inhibition activity towards two bacterial topoisomerases was tested, namely E. coli topoisomerase II (gyrase) and E. coli topoisomerase IV (Section S4†). Contrary to the assumption based on the predicted structure of PcyT, the pyxidicyclines showed no inhibition of E. coli gyrase up to 60 μg ml−1. We thus conclude that gyrase is not the molecular target as the in vitro MIC values of these substances are significantly lower (Table 2). On the other hand, topoisomerase IV was inhibited with IC50 values of 6.25–3.2 μg ml−1 for 1 and 3.2–1.6 μg ml−1 for 2 (Section S4†). These IC50 values match the observed bactericidal activity against E. coli ΔTolC, the prokaryote in our testing panel that is most sensitive to pyxidicyclines. Therefore, topoisomerase IV inhibition is likely their main mode of action with respect to bactericidal effects. We reason that Gram-negative pathogens are likely pyxidicycline-resistant due to compound efflux, as topoisomerase IV inhibition should otherwise also kill wild type E. coli strains. As the pyxidicyclines exhibited pronounced cytotoxicity in our bioassays, inhibition of human topoisomerase I was evaluated. This enzyme is the human counterpart for unwinding topoisomerases such as topoisomerase IV. 1 and 2 exert potent activity on this protein, which is an established target in anticancer research.34The IC50 values of 1 and 2 against human topoisomerase I were 1.6–0.4 μg ml−1 for 1 and 0.2–0.05 μg ml−1 for 2, respectively. The MIC of pyxidicyclines against HCT-116 cells was determined to 0.06 μg ml−1 and 0.03 μg ml−1 for 1 and 2, respectively, which is significantly lower than the on-target IC50 values determined in the cell free assay. This discrepancy has already been reported for the topoisomerase I inhibitor camptothecin (7, Fig. 6),30 which shows some structural similarity to the pyxidicyclines and this comparison also underlines topoisomerase I as the pyxidicycline's likely molecular target in mammalian cells.
Taken together, the pyxidicyclines are a novel class of type II PKS products with promising in vitro bioactivity based on the unprecedented nitrogen-containing tetracene quinone scaffold. Since the preferential inhibition of topoisomerase I by pxidicyclines might inspire further development of the compounds towards an anti-cancer drug, biotechnological production of the molecule is desirable. In order to pave the way for optimizing pyxidicycline production we thus decided to pursue the in-depth investigation of pyxidicycline biosynthesis. This was however met with challenges, as available genetic tools for the producer strain An d48 are limited to single-crossover recombination. We therefore sought to establish a heterologous expression system to facilitate investigation of this particular type II PKS biosynthetic pathway.
Heterologous expression of the pcy gene cluster
The myxobacterium P. fallax An d48 is related to model strains M. xanthus DK1622 and S. aurantiaca DW4/3-1 through membership of the same suborder Cystobacterinae and these two strains were thus chosen as host for heterologous expression. Since the promotor-activated pcy cluster in the P. fallax An d48 pcy mutant strain was already constructed, genomic DNA of this strain was used for PCR-based transformation assisted recombination (TAR) assembly of the gene cluster as described by Bylik et al. (Fig. 3).35 Through this assembly method, the whole cluster is reassembled during the cloning step and additional modifications can be engineered into the final construct by co-transformation of PCR products (Fig. 3). The method also allows reorganising the native operon structure of the biosynthetic gene cluster. Thus, the minimal PKS operon (operon 1, pcyA-I) responsible for the creation of the polyketide chain was placed upstream of the tailoring enzyme operon (operon 2, pcyJ-P) likely responsible for the incorporation of an nitrogen-containing moiety into the molecule backbone (Section S1.12,†Table 1) and subsequent dehydration to complete the fourth ring of the tetracyclic ring system. The putative resistance and export operon (operon 3, pcyQ-T) was relocated downstream of the tailoring enzyme operon. As already achieved in the An d48 double mutant, the minimal PKS system was kept under the control of the vanillate promotor/repressor system to control compound production through induction, thereby avoiding self-toxicity effects. The tailoring enzymes were subjected to control by the tn5 promotor as in the An d48 pcy mutant to ensure sufficient supply of tailoring enzymes for formation of mature pyxidicyclines. To boost the host's self-resistance towards the pcy cluster derived compounds, the putative resistance operon consisting of the ABC-transporter subunits and the pentapeptide repeat protein PcyT was placed under control of the native P. fallax An d48 gyrase subunit A promotor (PGyrA) to achieve strong and stable expression of the putative export and self-resistance system. That way, all pcy cluster related genes are transcribed in the same direction and the vanillate repressor's transcription and translation cannot interfere with the transcription of operon 2 or 3, and vice versa (Fig. 3). The heterologous promotors for operon 1 and 2 were PCR amplified from the double mutant gDNA together with their gene starts, while the gyrase promotor was taken out of P. fallax An d48 by an additional PCR (Section S1.11†). Finally, in addition to the vector described by Bilyk et al.35 the pClY vector backbone used in this study does not only carry two replication origins ori15A for E. coli and Cen-Ars6 for replication in S. cerevisiae, but also a yeast auxotrophy marker plus kanamycin and apramycin resistance genes. The vector additionally contains the Mx8 integrase system for later integration of the cloned construct into S. aurantiaca DW4/3-1 and M. xanthus DK1622 (Fig. 3).36 Therefore the vector assembled in yeast can be used for M. xanthus transformation following retransformation in E. coli. The assembled vector was subsequently inserted into M. xanthus DK1622 and S. aurantiaca DW4/3-1 via Mx8 integrase mediated chromosome integration (Section S1.9.3 and 1.9.4†). The M. xanthus DK1622 and S. aurantiaca DW4/3-1 attB:attP pClY pyxidicycline cluster (M. xanthus DK1622 pcy and S. aurantiaca DW4/3-1 pcy in short) integration clones were cultivated, induced with vanillate, extracted and measured by LC-MS according to the standard protocol (Section S3.1 to 3.4†). Compared to the non-induced strain we noted the appearance of two novel UV and MS signals that correspond to pyxidicycline A and B by retention time, high resolution mass and UV pattern indicating successful heterologous production of the pyxidicyclines.
Comparison of the relative LC-UV and LC-MS peak areas (Fig. 7) reveals another peculiarity of this biosynthesis. The ratio of 1 to 2 shifts strongly depending on the host strain: 1 is by far the major product in the P. fallax An d48 pcy mutant while this ratio shifts to 2 in M. xanthus DK1622 pcy. Finally, S. aurantiaca DW4/3-1 pcy produces almost exclusively 2 (Fig. 5 and 7, Table S21†) suggesting the corresponding oxidation to the quinone scaffold to be loosely controlled. This observation may be seen as a hint that this oxidation reaction is a spontaneous process as has been described for many anthraquinone type secondary metabolites such as steffimycin,37 and that the regioselectivity of this reaction depends on the strain-specific cytoplasmic redox power.
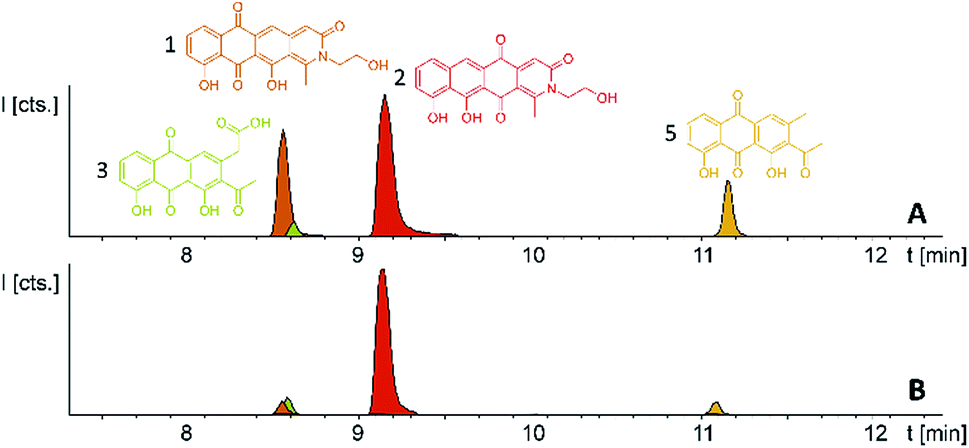 | ||
| Fig. 7 (A) Heterologous expression of the pyxidicycline gene cluster in M. xanthus DK1622 pcy and (B) S. aurantiaca DW4/3-1 pcy. Compounds 3 and 5 are displayed as LC-MS EICs at 341.06 [M + H]+ (light green) and 297.075 [M + H]+ (orange); the two pyxidicyclines are displayed as LC-MS EICs at 366.10 [M + H]+ (1 in orange; 2 in red). | ||
Characterization of pyxidicycline biosynthesis
Following successful heterologous expression of the pyxidicyclines we set out to further investigate their biosynthesis. Formation of the nitrogen-containing D ring is probably mediated by the tailoring enzyme operon comprising pcyJ, pcyK and pcyL. Considering the structure of pyxidicyclines and in light of biosynthetic logic, the nitrogen atom involved in fourth ring closure could stem from ethanolamine, serine or glycine incorporation. However, it was not obvious whether this moiety originally derives directly from incorporated ethanolamine, from serine that is decarboxylated after incorporation or from glycine that is subsequently reduced. LC-MS analysis of extracts following addition of stable isotope labelled precursors to a M. xanthus DK1622 pcy mutant culture showed that feeding of 3,3,2-serine-d3 leads to a +2 daltons mass shift explained by incorporation of serine into the molecule. In contrast, feeding of 2,2-glycine-d2 or 13C2 ethanolamine did not lead to any labelling of pyxidicyclines. The nitrogen atom present in the D ring of the tetracene quinone-like backbone as well as the attached side chain are therefore originating from serine, which is decarboxylated following incorporation. In order to localise the position of the two incorporated deuterium atoms the labelled molecule was subjected to LC-MS2 analysis revealing the position of the incorporated deuterium atoms in the molecule (Section S3.10†). This finding is additional proof that the ring nitrogen N1 and the adjacent 2-hydroxyethyl moiety consisting of C19, C20 and O6 derive from serine.To investigate the effect of the three genes pcyJ, pcyK and pcyL on pyxidicycline biosynthesis they were deleted by λ red prophage mediated recombination (RedE/T) (Section 1.12†). The resulting plasmids called pClY pyxidicycline cluster ΔpcyJ, ΔpcyK and ΔpcyL were subsequently transformed into M. xanthus DK1622 to study the consequences of deleting these tailoring enzymes for pyxidicycline biosynthesis. The corresponding M. xanthus DK1622 Mx8 integration mutants are called M. xanthus DK1622 pcy ΔpcyJ, ΔpcyK and ΔpcyL. As shown in Fig. 8 presence of PcyJ is indispensable for formation of the final product. Therefore, the An d48 mutants where only the minimal PKS is activated as well as the M. xanthus DK1622 pcy ΔpcyJ mutant show no pyxidicycline formation. Interestingly, the activation mutants of the minimal PKS in An d48 show production of 4 that cannot be observed in any of the heterologous expression systems. However, the connection of this metabolite to the biosynthesis of 3 is explained by reduction of the ketone moiety by An d48 enzyme, which does not have an isoenzyme in DK1622 or DW4/3-1, and subsequent intramolecular lactone formation (Fig. 9). Furthermore, deletion of pycK and pcyL clearly impairs the production of the pyxidicyclines whereas not only the peaks of 1 and 2 become significantly smaller, but at the same time their biosynthetic precursors 3 and 5 accumulate significantly (Fig. 8, Table S21†). Therefore, although the gene pcyJ is sufficient to obtain the final product from the reduced analogue of 3 (Fig. 9), efficient recruitment of serine into the pyxidicyclines and subsequent decarboxylation is only possible if the biosynthetic gene cluster possesses functional copies of pcyK and pcyL. Since the role of pcyO in the biosynthesis was unclear, the gene was placed under the control of a strong constitutive T7A1 promotor to boost its expression. The pcyO gene shows similarities to transglycosylases that might be involved in glycosyl transfer to the pyxidicycline core. However, no novel products were obtained from this overexpression, but the mutant afforded slightly better yields of 1 and 2 when compared to the gene cluster on the initial pClY pyxidicycline cluster vector. A complete summary of the relative pyxidicycline and anthraquinone precursor productivities in the different strains can be found in Table S21.†
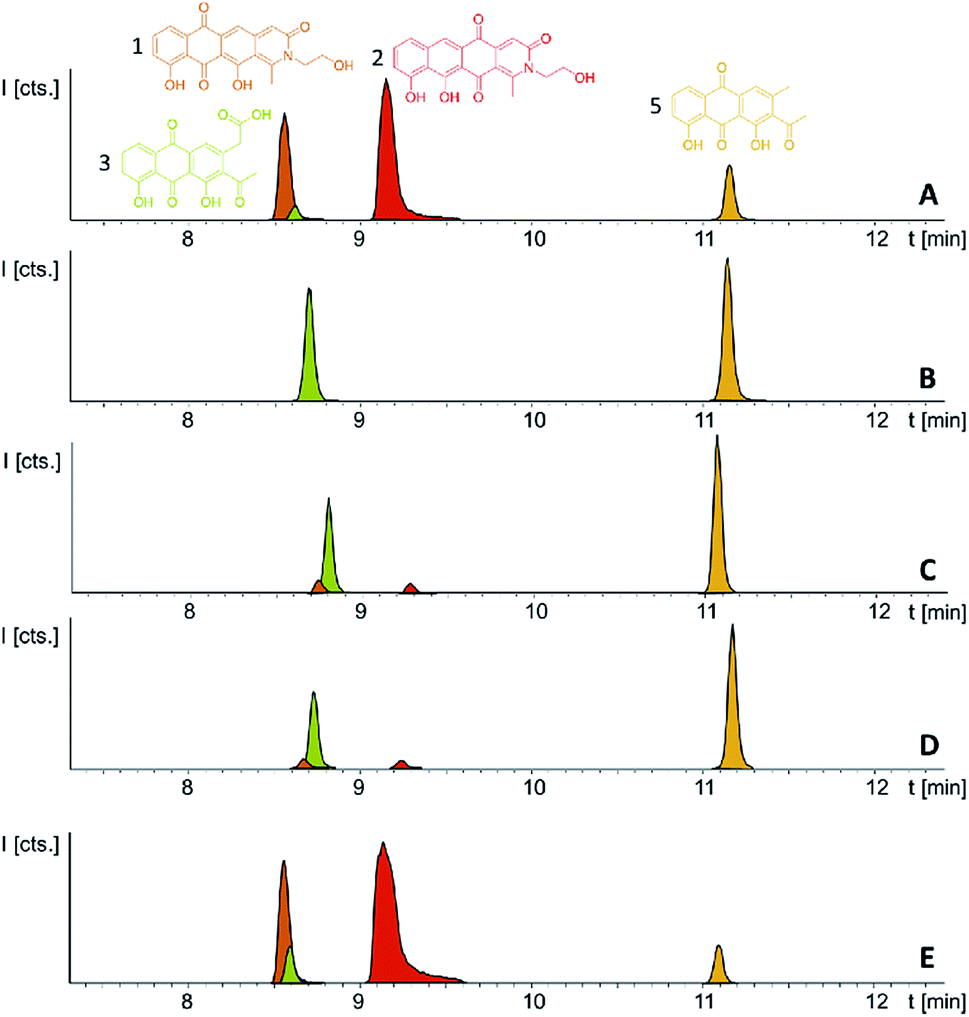 | ||
| Fig. 8 (A) LC-MS chromatograms of M. xanthus DK1622 pcy; (B) pcy ΔpcyJ; (C) pcy ΔpcyK; (D) pcy ΔpcyL and (E) pcy T7A1 pcyO, producing the pyxidicyclines and pyxidicycline precursors. Compounds 3 and 5 are displayed as LC-MS EICs at 341.06 [M + H]+ (light green) and 297.075 [M + H]+ (orange); the two pyxidicyclines are displayed as LC-MS EICs at 366.10 [M + H]+ (1 in orange; 2 in red). | ||
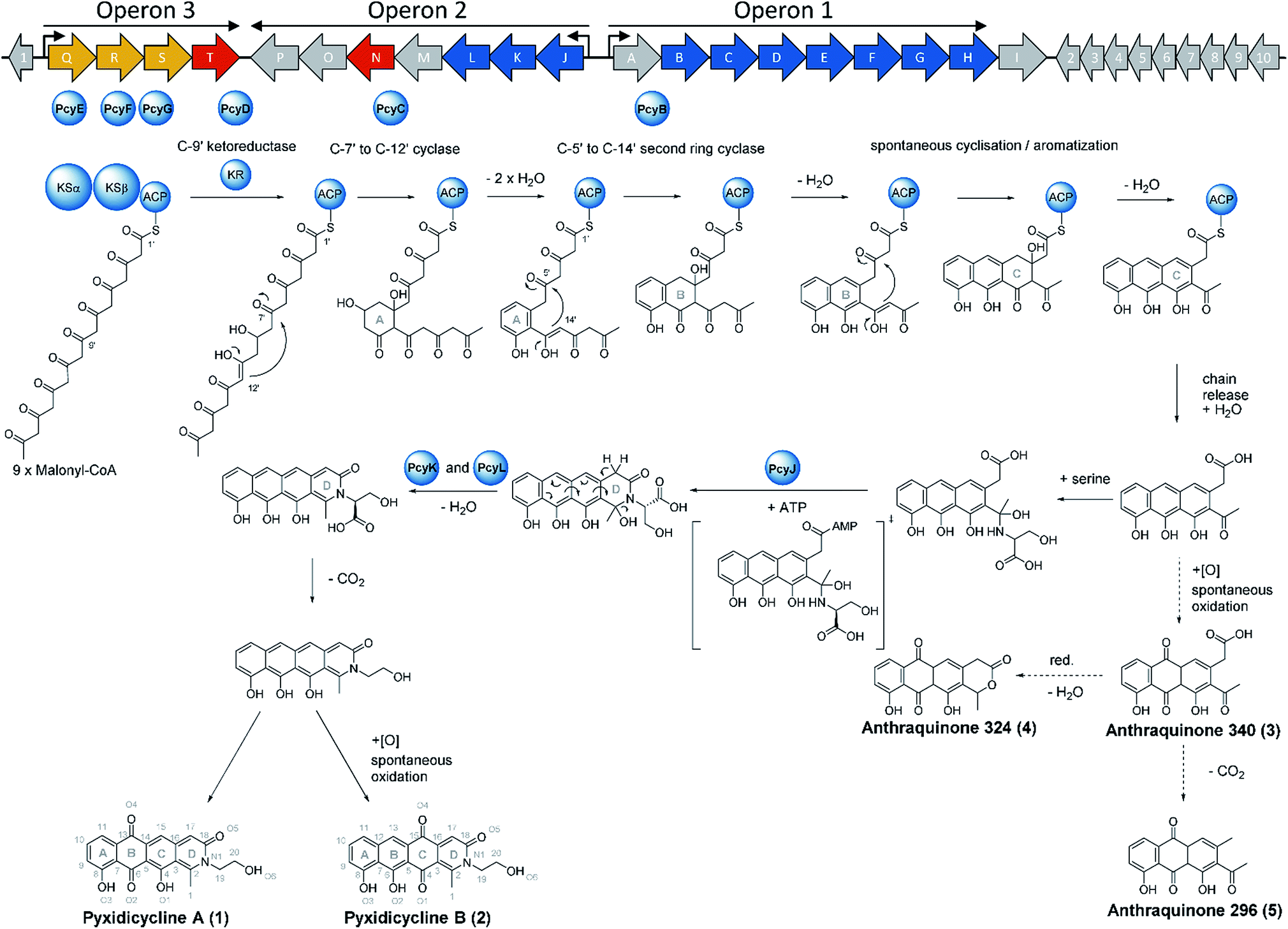 | ||
| Fig. 9 Biosynthetic gene cluster organization in An d48 wild type and biosynthesis proposal for the pyxidicyclines (1 and 2) and the detected anthraquinone shunt products (3–5), according to experiments made in this study. PKS type II biosynthesis genes are depicted as blue arrows, regulatory and resistance elements are depicted as red arrows, exporter genes are depicted as yellow arrows and orfs of unknown function are depicted as grey arrows. Proteins participating in polyketide biosynthesis are depicted as blue spheres. | ||
To complete the biosynthetic model for pyxidicyclines the assignment of the different cyclase proteins to their respective cyclisation reaction was done starting from in silico considerations. The anthraquinone backbone of the pyxidicyclines is probably assembled by two cyclases responsible for first ring and second ring cyclisation, while third ring formation is thought to occur spontaneously. Zhang et al. could show this for structure 3, called SEK 26 in their study.29 To determine which cyclase enzyme is responsible for which cyclisation/aromatisation reaction of the pyxidicyclines we used a combination of gene deletion studies on the pClY pyxidicycline cluster plasmid and in silico analysis. From the deletion experiments, it is evident that aromatase/cyclase like enzymes PcyJ and PcyL cannot affect this biosynthetic step as they have been deleted in the heterologous expression host – showing only effects on the total pyxidicycline production but not on the strain's potential to produce the precursor anthraquinones (Fig. 8). Furthermore, upon activation of the minimal PKS operon in the An d48 mutant strains via tn5 or vanillate promotor the precursor anthraquinones were still produced but not the full-length pyxidicyclines. Therefore, we conclude that genes pcyJ to pcyP are not involved in anthraquinone precursor formation. The two remaining candidate cyclase/aromatase genes are pcyB and pcyC. The pyxidicyclines undergo C-7′ to C-12′ “S-type” first ring cyclisation like steffimycin and tetracycline.38 The corresponding cyclase genes were compared to the first and second ring cyclases of the steffimycin and oxytetracycline pathway by MUSCLE alignment18 (Fig. S12†), since first and second ring cyclases typically differ by primary sequence.38 As the cyclase PcyC resembles the oxytetracycline and steffimycin first ring cyclases it is safe to assume that PcyC is responsible for first (A) ring cyclisation (Section S2.6†). On the contrary, PcyL is not part of the minimal PKS operon, which produces the anthraquinone core and has already been demonstrated to be involved in the formation and aromatisation of the nitrogen-containing D heterocycle by gene inactivation experiments. It is therefore not involved in A- and B-ring cyclisation steps. The second ring cyclases StfY and OxyN share high similarity with PcyB and all three proteins form a group with high phylogenetic distance to other proteins in the alignment tree (Section S2.6†). PcyB is therefore likely to be the second (B) ring cyclase in pyxidicycline biosynthesis. The enzyme PcyJ is singled out in this alignment; this is unsurprising as our gene inactivation experiments determined the protein to be responsible for serine integration and heterocyclization, an unprecedented step in type II polyketide biosynthesis. This enzyme was therefore expected to contain a rather uncommon cyclase/aromatase fold that does not fit very well to the enzymes known from streptomycetes (Section S2.5 and 2.6†).
The biosynthetic route to pyxidicyclines
Based on the results of activation studies, stable isotope labelling, in silico analysis as well as the knockout experiments we are able to devise a comprehensive biosynthetic model for the formation of pyxidicyclines (Fig. 9). The biosynthesis route shown with dotted arrows on the lower right hand side of the picture additionally illustrates formation of the anthraquinone shunt products mainly occurring if pcyJ is inactivated or repressed. Similar to known type II polyketide synthase pathways like the oxytetracycline pathway,39 the biosynthesis of pyxidicyclines starts with polycondensation of nine malonyl-CoA units by the KSα PcyE with its chain length factor PcyF, whereas intermediates are tethered to the ACP PcyG. The next acting enzyme is the C-9′ type ketoreductase (KR) PcyD reducing the nascent polyketide chain at C-9′ counting from the thioester bond (C-10 in the final molecule). This step is necessary to pre-orientate the polyketide chain for the first ring cyclisation/aromatization catalysed by the C-7′ to C-12′ ‘S type’ cyclase PcyC that forms the pyxidicyclines' A ring.38 The second ring cyclase/aromatase PcyB is subsequently responsible for ring closure and aromatization of the B ring by C–C bond formation between C5′ and C14′ on the nascent polyketide chain. As it has been shown for tetracycline biosynthesis, third ring (C) formation occurs spontaneously with these scaffolds.39 This is also likely to be the case for the pyxidicyclines as the minimal PKS operon does not contain additional cyclases and it was shown experimentally that the minimal PKS operon is sufficient to produce anthraquinones 340, 296 and 324, all of which contain the C ring. Following C ring cyclisation the product is released and the AMP-dependent synthetase and ligase protein and aromatase/cyclase di-domain protein, PcyJ, attaches a serine moiety and closes the D ring with the help of PcyK and PcyL (Section S2.5†). Serine recruitment into pyxidicyclines is notably different from amino acid incorporation during jadomycin biosynthesis, as it is not a spontaneous process but relies on catalysis by PcyJ.33 Fredericamycin A formation on the other hand invloves catalysis by the asparagine synthetase homologue FdmV, leading to the nitrogen-containing six membered ring structure. FdmV does not transfer an amino acid but synthesizes a cyclic amide by transfer of an ammonium equivalent.32Our feeding experiments unambiguously showed that only 3,3,2 serine-d3 feeding leads to labelling of the pyxidicyclines, indicating that serine is incorporated and decarboxylated on the scaffold, since 13C2 monoethanolamine did not lead to any labelling (Section S3.10†). Subsequent aromatization of the D ring can be spontaneous as indicated by the gene inactivation experiment of the muconolactone δ-isomerase type enzyme PcyK and the aromatase-like enzyme PcyL (Table 1). On the other hand, those experiments also revealed that the yield of pyxidicyclines compared to the corresponding anthraquinone shunt products drops dramatically upon inactivation of PcyK or PcyL (Table S21†). However, the exact timing for decarboxylation remains unresolved, as decarboxylation is too efficient to detect carboxylated derivatives by LC-MS. It is therefore unclear whether decarboxylation occurs before or after D ring aromatization. From the enzyme characteristics, we assume that PcyK – a muconolactone δ-isomerase homolog – is more likely to help with decarboxylation while the cyclase/aromatase homolog PcyL is more likely to assist D ring aromatization. Other enzymes cannot be involved in these transformation steps, as proven by successful production in the heterologous hosts S. aurantiaca DW 4/3-1 and M. xanthus DK1622, both considered unlikely to furnish suitable isoenzymes encoded in their genomes.
Conclusion
This study presents an efficient approach for prioritizing particular BGCs out of over 93 myxobacterial genomes containing 10 to 30 BGCs each with focus on desired bioactivity and chemical novelty of the products to be discovered. Using the known host-defense mechanism of pentapeptide repeat proteins as the rationale behind our genome-mining strategy, a new small-molecule product was uncovered featuring an intriguing nitrogen-containing tetracene quinone scaffold constructed by a type II polyketide synthase pathway, a biosynthetic route rarely found in Gram-negative bacteria. Comprehensive in vitro bioactivity profiling ultimately allowed us to conclude the pyxidicyclines' molecular target. Consequently, the pyxidicyclines could become interesting leads mainly for anticancer research as they share the target and the order magnitude of activity with the camptothecins, an approved anti-cancer drug for an established anti-cancer drug target.30,34As much as this work highlights the promise of contemporary genome-mining techniques applied to myxobacteria as a still underexploited group of microbes, it hints at the same time at the limitations currently faced by genomics-based methods for natural products discovery. While it was reasonably likely that the new compounds would exhibit biological activity in the direction of topoisomerases, it was impossible to forecast the precise range of bioactivities as well as the type of topoisomerase that would be targeted. Future developments in bioinformatics analysis are undoubtly needed for well-directed genome-mining, but this notion just as much underlines the importance to advance knowledge of biosynthetic pathway enzymatics and to develop the required genetic tools further, in order to increase our chances for translating genome-encoded potential for novel natural products into real candidate molecules, ideally for human therapy.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Viktoria Schmitt for performing bioassays and Volker Huch for acquiring and interpreting X-ray diffraction data.References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef PubMed.
- N. S. Cortina, D. Krug, A. Plaza, O. Revermann and R. Müller, Angew. Chem., Int. Ed. Engl., 2012, 51, 811 CrossRef PubMed.
- D. Krug and R. Müller, Nat. Prod. Rep., 2014, 31, 768 RSC.
- S. Bertrand, O. Schumpp, N. Bohni, A. Bujard, A. Azzollini, M. Monod, K. Gindro and J.-L. Wolfender, J. Chromatogr. A, 2013, 1292, 219 CrossRef PubMed.
- (a) A. W. Schultz, D. C. Oh, J. R. Carney, R. T. Williamson, D. W. Udwary, P. R. Jensen, S. J. Gould, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2008, 130, 4507 CrossRef PubMed; (b) F. Jüttner, A. K. Todorova, N. Walch and W. von Philipsborn, Phytochemistry, 2001, 57, 613 CrossRef; (c) J. Herrmann, A. A. Fayad and R. Müller, Nat. Prod. Rep., 2017, 34, 135 RSC.
- J. Munoz-Dorado, F. J. Marcos-Torres, E. Garcia-Bravo, A. Moraleda-Munoz and J. Perez, Front. Microbiol., 2016, 7, 781 Search PubMed.
- (a) N. Zaburannyi, B. Bunk, J. Maier, J. Overmann and R. Müller, Appl. Environ. Microbiol., 2016, 82, 1945 CrossRef PubMed; (b) S. Schneiker, O. Perlova, O. Kaiser, K. Gerth, A. Alici, M. O. Altmeyer, D. Bartels, T. Bekel, S. Beyer, E. Bode, H. B. Bode, C. J. Bolten, J. V. Choudhuri, S. Doss, Y. A. Elnakady, B. Frank, L. Gaigalat, A. Goesmann, C. Groeger, F. Gross, L. Jelsbak, L. Jelsbak, J. Kalinowski, C. Kegler, T. Knauber, S. Konietzny, M. Kopp, L. Krause, D. Krug, B. Linke, T. Mahmud, R. Martinez-Arias, A. C. McHardy, M. Merai, F. Meyer, S. Mormann, J. Muñoz-Dorado, J. Perez, S. Pradella, S. Rachid, G. Raddatz, F. Rosenau, C. Rückert, F. Sasse, M. Scharfe, S. C. Schuster, G. Suen, A. Treuner-Lange, G. J. Velicer, F.-J. Vorhölter, K. J. Weissman, R. D. Welch, S. C. Wenzel, D. E. Whitworth, S. Wilhelm, C. Wittmann, H. Blöcker, A. Pühler and R. Müller, Nat. Biotechnol., 2007, 25, 1281 CrossRef PubMed.
- S. C. Wenzel and R. Müller, Mol. BioSyst., 2009, 5, 567 RSC.
- S. C. Wenzel and R. Müller, Nat. Prod. Rep., 2009, 26, 1385 RSC.
- K. Blin, T. Wolf, M. G. Chevrette, X. Lu, C. J. Schwalen, S. A. Kautsar, H. G. Suarez Duran, E. L. C. de Los Santos, H. U. Kim, M. Nave, J. S. Dickschat, D. A. Mitchell, E. Shelest, R. Breitling, E. Takano, S. Y. Lee, T. Weber and M. H. Medema, Nucleic Acids Res., 2017, 45, W36–W41 CrossRef PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Müller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef PubMed.
- (a) M. Alanjary, B. Kronmiller, M. Adamek, K. Blin, T. Weber, D. Huson, B. Philmus and N. Ziemert, Nucleic Acids Res., 2017, 45, W42–W48 CrossRef PubMed; (b) M. A. Skinnider, N. J. Merwin, C. W. Johnston and N. A. Magarvey, Nucleic Acids Res., 2017, 45, W49–W54 CrossRef PubMed.
- (a) X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, s. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841 CrossRef PubMed; (b) H.-H. Yeh, M. Ahuja, Y.-M. Chiang, C. E. Oakley, S. Moore, O. Yoon, H. Hajovsky, J.-W. Bok, N. P. Keller, C. C. C. Wang and B. R. Oakley, ACS Chem. Biol., 2016, 11, 2275 CrossRef PubMed.
- S. Baumann, J. Herrmann, R. Raju, H. Steinmetz, K. I. Mohr, S. Hüttel, K. Harmrolfs, M. Stadler and R. Müller, Angew. Chem., Int. Ed. Engl., 2014, 53, 14605 CrossRef PubMed.
- M. W. Vetting, S. S. Hegde, Y. Zhang and J. S. Blanchard, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2011, 67, 296 CrossRef PubMed.
- (a) A. O. Brachmann, S. A. Joyce, H. Jenke-Kodama, G. Schwär, D. J. Clarke and H. B. Bode, ChemBioChem, 2007, 8, 1721 CrossRef PubMed; (b) D. Pistorius, Y. Li, A. Sandmann and R. Müller, Mol. BioSyst., 2011, 7, 3308 RSC.
- (a) G. A. Jacoby, K. E. Walsh, D. M. Mills, V. J. Walker, H. Oh, A. Robicsek and D. C. Hooper, Antimicrob. Agents Chemother., 2006, 50, 1178 CrossRef PubMed; (b) M. W. Vetting, S. S. Hegde, M. Wang, G. A. Jacoby, D. C. Hooper and J. S. Blanchard, J. Biol. Chem., 2011, 286, 25265 CrossRef PubMed.
- R. C. Edgar, BMC Bioinf., 2004, 5, 113 CrossRef PubMed.
- S. Shah and J. G. Heddle, Appl. Microbiol. Biotechnol., 2014, 98, 9545 CrossRef PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845 CrossRef PubMed.
- (a) W. Trowitzsch, G. Reifenstahl, V. Wray and G. Höfle, J. Antibiot., 1980, 33, 1480 CrossRef PubMed; (b) B. Kunze, W. Kohl, G. Höfle and H. Reichenbach, J. Antibiot., 1985, 38, 1649 CrossRef PubMed; (c) Y. Chai, D. Pistorius, A. Ullrich, K. J. Weissman, U. Kazmaier and R. Müller, Chem. Biol., 2010, 17, 296 CrossRef PubMed.
- A. Das and C. Khosla, Acc. Chem. Res., 2009, 42, 631–639 CrossRef PubMed.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509 CrossRef PubMed.
- T. S. Freestone, K.-S. Ju, B. Wang and H. Zhao, ACS Synth. Biol., 2017, 6, 217 CrossRef PubMed.
- (a) J. B. Biggins, X. Liu, Z. Feng and S. F. Brady, J. Am. Chem. Soc., 2011, 133, 1638 CrossRef PubMed; (b) J. Franke, K. Ishida and C. Hertweck, Angew. Chem., Int. Ed. Engl., 2012, 51, 11611 CrossRef PubMed.
- C. Olano, I. García, A. González, M. Rodriguez, D. Rozas, J. Rubio, M. Sánchez-Hidalgo, A. F. Braña, C. Méndez and J. A. Salas, Microb. Biotechnol., 2014, 7, 242 CrossRef PubMed.
- A. A. Iniesta, F. García-Heras, J. Abellón-Ruiz, A. Gallego-García and M. Elías-Arnanz, J. Bacteriol., 2012, 194, 5875 CrossRef PubMed.
- (a) V. Prikrylova, M. Podojil, P. Sedmera, J. Vokoun, Z. Vanek and C. H. Hassal, J. Antibiot., 1978, 31, 855 CrossRef PubMed; (b) J. R. D. McCormick and E. R. Jensen, Adv. Ceram. Mater., 1968, 90, 7126 Search PubMed.
- W. Zhang, Y. Li and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20683 CrossRef PubMed.
- M. J. Luzzio, J. M. Besterman, D. L. Emerson, M. G. Evans, K. Lackey, P. L. Leitner, G. McIntyre, B. Morton, P. L. Myers, M. Peel, J. M. Sisco, D. D. Sternbach, W.-Q. Tong, A. Truesdale, D. E. Uehling, A. Vuong and J. Yates, J. Med. Chem., 1995, 38, 395 CrossRef PubMed.
- J. A. Taylor, L. A. Mitchenall, M. Rejzek, R. A. Field and A. Maxwell, PLoS One, 2013, 8, e58010 Search PubMed.
- E. Wendt-Pienkowski, Y. Huang, J. Zhang, B. Li, H. Jiang, H. Kwon, C. R. Hutchinson and B. Shen, J. Am. Chem. Soc., 2005, 127, 16442 CrossRef PubMed.
- N. Tibrewal, P. Pahari, G. Wang, M. K. Kharel, C. Morris, T. Downey, Y. Hou, T. S. Bugni and J. Rohr, J. Am. Chem. Soc., 2012, 134, 18181 CrossRef PubMed.
- Y. Pommier, ACS Chem. Biol., 2013, 8, 82 CrossRef PubMed.
- O. Bilyk, O. N. Sekurova, S. B. Zotchev and A. Luzhetskyy, PLoS One, 2016, 11, e0158682 Search PubMed.
- V. Magrini, C. Creighton and P. Youderian, J. Bacteriol., 1999, 181, 4050 Search PubMed.
- S. Gullón, C. Olano, M. S. Abdelfattah, A. F. Braña, J. Rohr, C. Méndez and J. A. Salas, Appl. Environ. Microbiol., 2006, 72, 4172 CrossRef PubMed.
- H. Zhou, Y. Li and Y. Tang, Nat. Prod. Rep., 2010, 27, 839 RSC.
- L. B. Pickens and Y. Tang, J. Biol. Chem., 2010, 285, 27509 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1831467 and 1831468. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01325j |
| This journal is © The Royal Society of Chemistry 2018 |