
Palladium-catalyzed reductive electrocarboxylation of allyl esters with carbon dioxide†
Ke-Jin
Jiao
a,
Zhao-Ming
Li
a,
Xue-Tao
Xu
 b,
Li-Pu
Zhang
b,
Yi-Qian
Li
b,
Kun
Zhang
b and
Tian-Sheng
Mei
*a
b,
Li-Pu
Zhang
b,
Yi-Qian
Li
b,
Kun
Zhang
b and
Tian-Sheng
Mei
*a
aState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, China. E-mail: mei7900@sioc.ac.cn
bSchool of Chemical & Environmental Engineering, Wuyi University, Jiangmen, 529020, China
First published on 22nd June 2018
Abstract
Palladium-catalyzed regioselective electrocarboxylation of homostyrenyl acetates with CO2 has been successfully developed, providing α-aryl carboxylic acids with good selectivity and yield. In addition, the catalytic asymmetric carboxylation of cinnamyl acetate has been demonstrated, despite moderate enantioselectivity.
Introduction
Catalytic carbon–carbon (C–C) bond forming reactions that utilize carbon dioxide (CO2) to directly access carboxylic acids have received extensive attention in the past decades due to the attractiveness of CO2 as a C1 synthon.1,2 In particular, transition metal-catalyzed allylation of carbon dioxide has emerged as a promising tool to construct β,γ-unsaturated carboxylic acids.3 Most allylic carboxylation reactions that incorporate CO2 require highly reactive substrates such as allylstannanes or allylboron reagents, which is due to both the thermodynamic stability and kinetic inertia of CO2.4 Recently, Martin and co-workers elegantly demonstrated Ni-catalyzed carboxylation of allyl acetates with CO2 utilizing Mn as a reducing agent (Scheme 1a).5 Similarly, Sato and co-workers have successfully developed Pd-catalyzed carboxylation of allylic alcohols using Et2Zn as a reducing agent.6 More recently, the Martin group7 and our group8 independently reported Ni-catalyzed carboxylation of allyl alcohols using Mn powder. Unfortunately, the use of super-stoichiometric amounts of reducing agents like Mn or Et2Zn is a drawback of these reactions. Furthermore, these developed protocols are not suitable for homostyrenyl electrophiles (Scheme 1b). Enantio- and regioselective carboxylations of homostyrenyl electrophiles are needed as an efficient way to deliver α-aryl carboxylic acids, which are found in numerous medicines (Scheme 1c). Toward that end, the development of catalytic carboxylation reactions using more environmentally benign reducing agents is highly desirable.9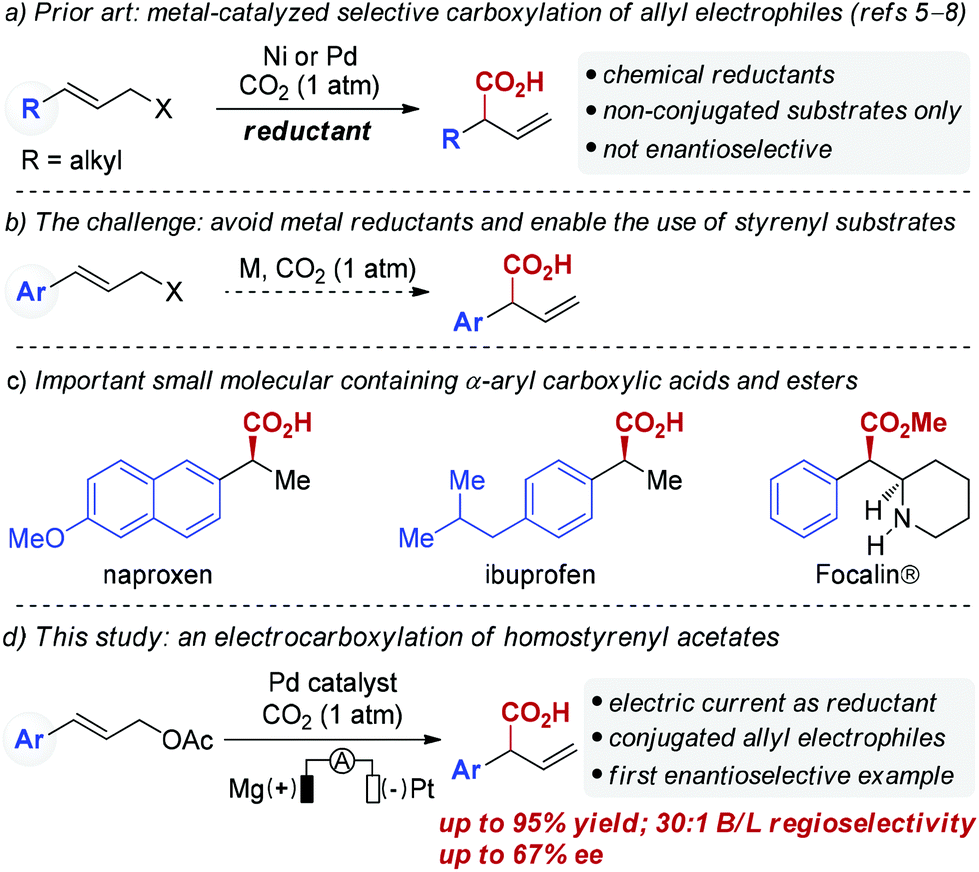 | ||
| Scheme 1 Catalytic carboxylation of allyl electrophiles. | ||
Electrochemistry has been demonstrated to be an efficient and environmentally friendly approach for organic chemistry that employs electrons as redox reagents.10–12 As part of our ongoing interest in using electric currents as redox reagents in palladium catalyzed reactions,13 we questioned whether catalytic carboxylations of allyl acetates could be achieved using electric current as a reducing agent.14 Herein, we describe the first Pd-catalyzed regioselective carboxylation of homostryrenyl acetates utilizing electric current as the reductant, thus providing an environmentally benign method for accessing α-aryl carboxylic acids. In addition, we demonstrate the first catalytic asymmetric carboxylation of cinnamyl acetate, which provides a heretofore unavailable opportunity to construct chiral phenyl acetic acids from homostyrenyl acetates (Scheme 1d).
Results and discussion
Our initial studies focused on Pd-catalyzed carboxylation of (E)-3-(4-bromophenyl)allyl acetate 1a with CO2 (Table 1). We chose 1a as the model substrate because it contains relatively reactive C–Br and C–O bonds, which may have different reactivities under electrochemical conditions, and the retained bromide may be subsequently elaborated into useful products by employing various metal-catalyzed cross-coupling reactions.15 To our delight, at 30 °C, using Pd(OAc)2 as catalyst and 1,2-bis(diphenylphosphino)benzene (DPPPh) as ligand, with Et4NOTs as electrolyte, EtOH as an additive, and DMF as solvent, the reaction of 1a with CO2 gave 81% isolated yield of branched product 2a with 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioselectivity under constant current electrolysis conditions at 8.0 mA (J = 8.0 mA cm−2) at 3 F mol−1 (entry 1). Lower regioselectivity was observed when other phosphine ligands were used, including PPh3, and bidendate DPPE, DPPP, DPPB (entries 2–5). Without EtOH, the regioselectivity and yield were significantly reduced (entry 6). Decreasing or increasing the amount of EtOH to 0.5 or 5.0 equivalents resulted in a slight decrease in yield (entries 7 and 8). Cathodic reduction of EtOH could take place to stabilize the reaction system, although the real role of EtOH is not clear at this stage. Furthermore, both Pd(OAc)2 and phosphine ligand are required for achieving regioselective carboxylation (entries 9 and 10). Finally, a control experiment gave the results that no reaction was observed in the absence of electric current (entry 11). It is worth noting that chemical reductant such as Mn or Zn is not suitable for this carboxylation reaction (entry 12). This implies that electrochemistry could provide unique reactivity by enhancing the nucleophilicity of resulting palladium species (vide infra).
:1 regioselectivity under constant current electrolysis conditions at 8.0 mA (J = 8.0 mA cm−2) at 3 F mol−1 (entry 1). Lower regioselectivity was observed when other phosphine ligands were used, including PPh3, and bidendate DPPE, DPPP, DPPB (entries 2–5). Without EtOH, the regioselectivity and yield were significantly reduced (entry 6). Decreasing or increasing the amount of EtOH to 0.5 or 5.0 equivalents resulted in a slight decrease in yield (entries 7 and 8). Cathodic reduction of EtOH could take place to stabilize the reaction system, although the real role of EtOH is not clear at this stage. Furthermore, both Pd(OAc)2 and phosphine ligand are required for achieving regioselective carboxylation (entries 9 and 10). Finally, a control experiment gave the results that no reaction was observed in the absence of electric current (entry 11). It is worth noting that chemical reductant such as Mn or Zn is not suitable for this carboxylation reaction (entry 12). This implies that electrochemistry could provide unique reactivity by enhancing the nucleophilicity of resulting palladium species (vide infra).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Deviation from above conditions | F mol−1 | Conv.b (%) | Yieldb (%) | 2a/2aac |
| a Reaction conditions: 1a (0.3 mmol), Pd(OAc)2 (5.0 mol%), DPPPh (5.3 mol%), Et4NOTs (0.15 g, 0.07 M) and EtOH (1.0 equiv.), DMF (6 mL) in an undivided cell with a platinum electrode and a magnesium rod as sacrificial anode. b The ratio of B/L products was determined by 1H-NMR. c The yields were determined by 1H-NMR with CH2Br2 as the internal standard. d Isolated yield of 2a. | |||||
| 1 | None | 3 | 100 | 85 (81)d | 20:1 |
| 2 | PPh3 (12 mol%) in lieu of DPPPh | 3 | 79 | 36 | 2:1 |
| 3 | DPPE in lieu of DPPPh | 3 | 85 | 70 | 15:1 |
| 4 | DPPE in lieu of DPPPh | 3 | 81 | 65 | 18:1 |
| 5 | DPPE in lieu of DPPPh | 3 | 68 | 51 | 6:1 |
| 6 | No EtOH | 3 | 38 | 21 | 2:1 |
| 7 | EtOH (0.5 equiv.) | 3 | 94 | 64 | 20:1 |
| 8 | EtOH (5.0 equiv.) | 3 | 81 | 59 | 11:1 |
| 9 | no Pd(OAc)2 | 3 | 32 | 19 | 2:1 |
| 10 | no DPPPh | 3 | 72 | 38 | 3:1 |
| 11 | no electric current | — | 0 | NP | — |
| 12 | Mn or Zn in lieu of electric current | — | 0 | NP | — |
|
|||||
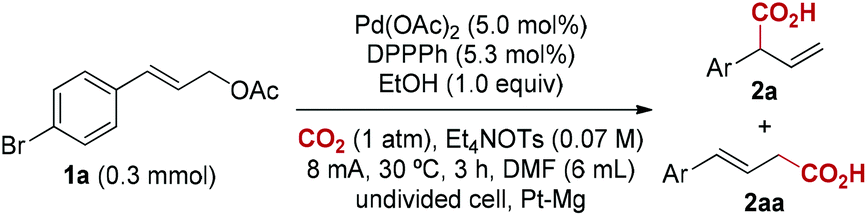
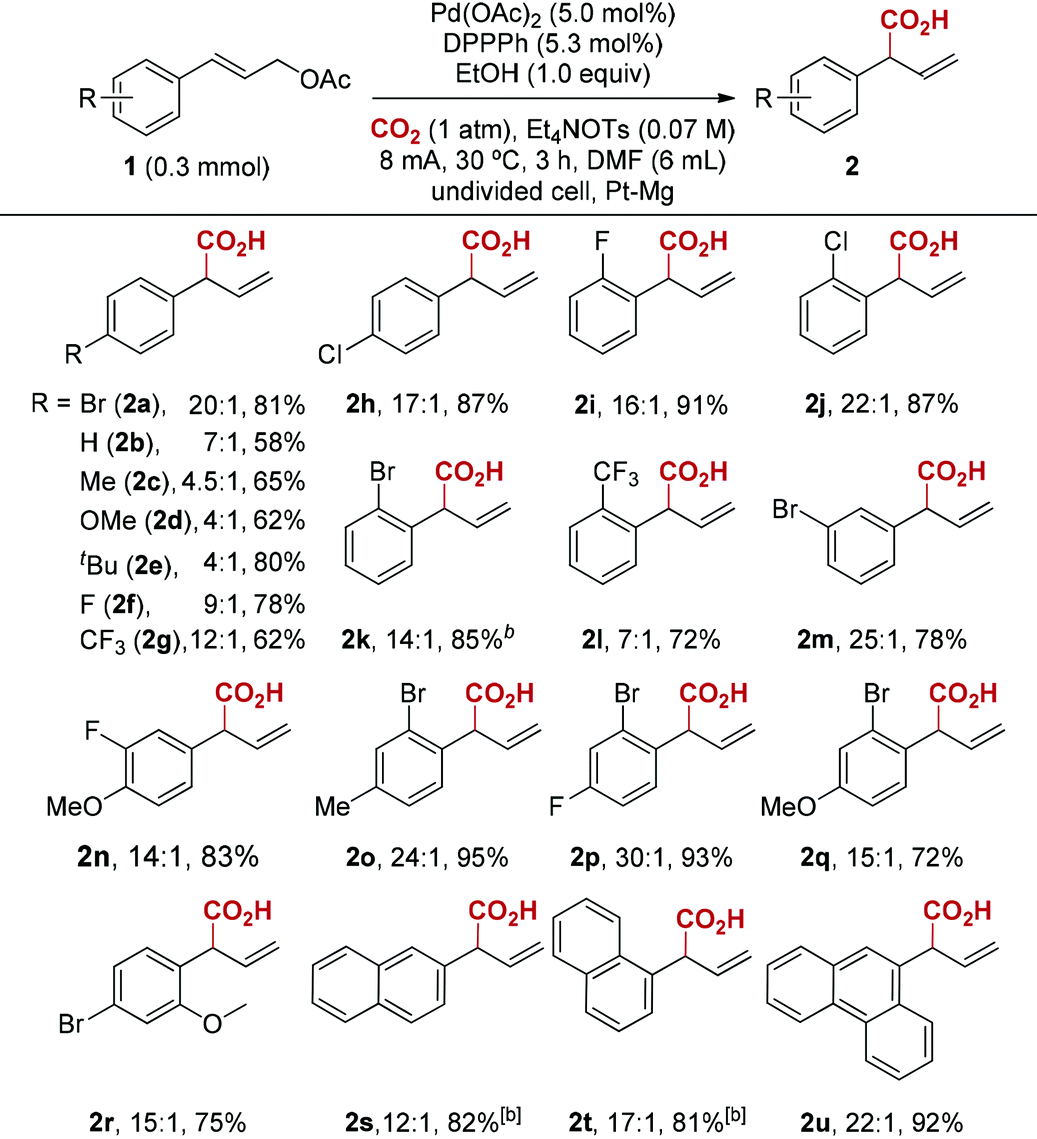
With the optimized reaction conditions in hand, we next investigated the scope of this reductive electrocarboxylation reaction (Table 2). In addition to Br, our conditions were compatible with numerous other functional groups, including F, Cl, Me, OMe, t-Bu, and CF3 (2a–2r). Substrates containing electron-withdrawing groups (2a, 2f–2h) deliver better regioselectivity than electronrich ones (2b–2e). Interestingly, this electronic factor is different from Pd-catalyzed allylic alkylation, in which electron-withdrawing groups on aryl rings reduce formation of the branched product.16 It is worth emphasizing that polysubstituted arenes also deliver high regioselectivity (2n–2r). Finally, substrates bearing polycyclic aromatics such as naphthalene and anthracene were well tolerated in the reaction (2s–2u). Unfortunately, alkyl-substituted allylic esters or 3,3-disubstituted allylic esters are not effective at current reaction conditions.
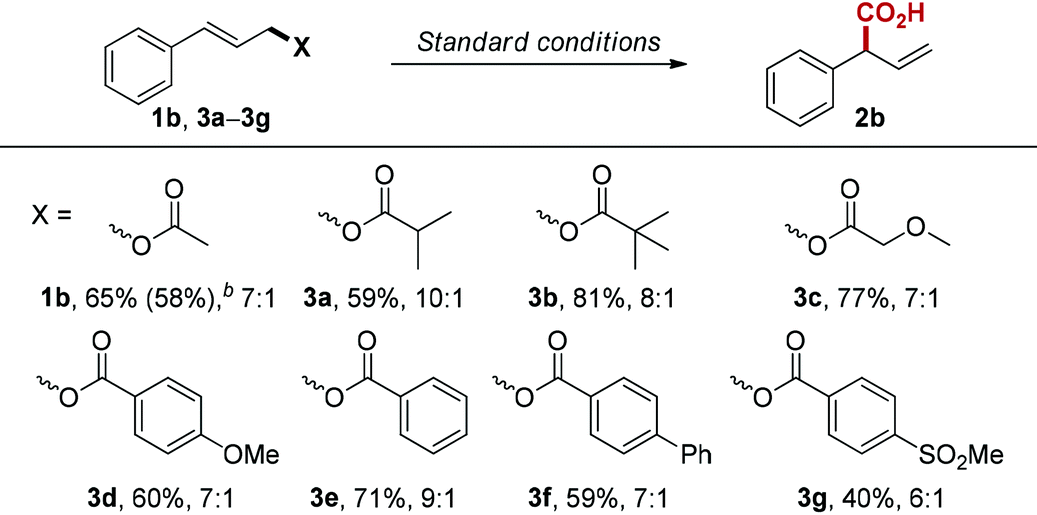
Next, we examined the influence of the leaving group on the reaction, since many allylic compounds with appropriate leaving groups can form π-allylpalladium intermediate via oxidative addition to Pd(0) under mild conditions.17 Unsurprisingly, the different leaving groups influence reaction performance under the optimized conditions (Table 3). Among the esters we tested, cinnamyl isobutyrate (3a) gave the best regioselectivity (10:1). However, cinnamyl pivalate (3b) provided the best yield (81%) with slightly lower regioselectivity (8:1). We also investigated the electronic influence of substituted benzoate leaving groups (3d–3g), but we did not observe any correlation between Hammett values and regioselectivity.
The enantioselective carboxylation of allylic acetates is a major challenge.18 To our delight, chiral bidentate triarylphosphine ligands were found to be efficient for this catalytic asymmetric carboxylation, and moderate enantioselectivity (67% ee) was achieved (Scheme 2). Interestingly, chiral bidentate phosphine ligands are known to be inactive in Pd-catalyzed allylation of aldehydes.19
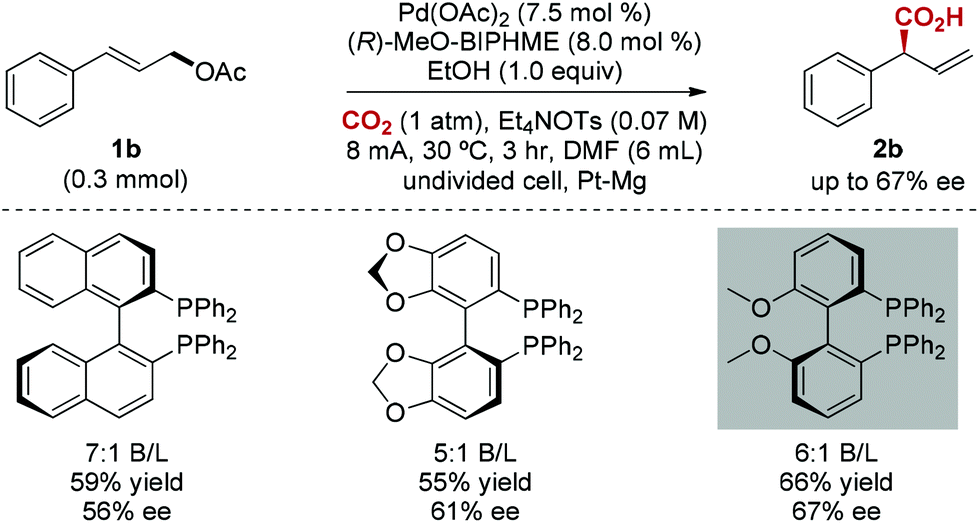 | ||
| Scheme 2 Enantioselective carboxylation. | ||
To gain insight into the reaction mechanism, a cyclic voltammogram (CV) was conducted. The CV of [(R)-BINAP]Pd(OAc)2 in DMF reveals one irreversible reduction wave at −0.60 V vs. Ag/AgI, corresponding to the reduction of Pd(II) to Pd(0) (b, Fig. 1). The addition of homostyrenyl acetate 1b causes an increase in the peak current, which indicates oxidative addition of Pd(0) to allyl acetate 1b (c, Fig. 1). Furthermore, we also observed the second reduction wave at −0.90 V, which is tentatively assigned to the reduction of the resulting allylPd(II) species (c, Fig. 1). The further increase of peak currents (both around −0.65 V and −1.05 V) after the addition of CO2 suggests that the carboxylation takes place (d, Fig. 1).
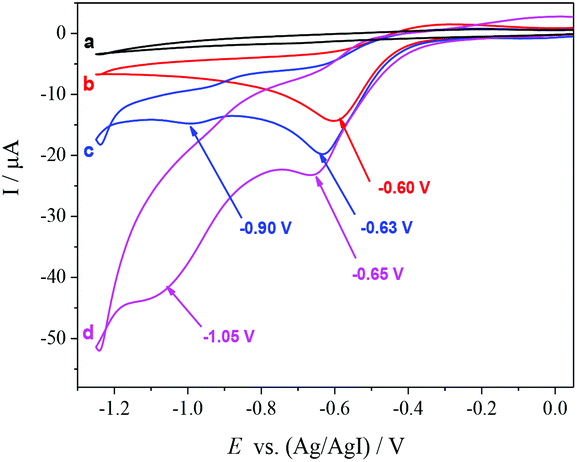 | ||
| Fig. 1 Cyclic voltammograms recorded on a Pt electrode at 100 mV s−1 in: (a) DMF containing 0.07 M Bu4NPF6; (b) solution (a) after addition of 2 mM [(R)-BINAP]Pd(OAc)2; (c) solution (b) after addition of 20 mM 1b; (d) solution (c) saturated with CO2. | ||
Based on Wendt's and Hazari's elegant studies on η1-allylPd species reacting with CO2 and our observation, a plausible mechanism for this Pd-catalyzed carboxylation reaction via electrochemical reduction is proposed in Scheme 3.4 Initially, oxidative addition of Pd(0) to the allyl electrophile generates a cationic π-allylpalladium(II) complex (A) with leaving group (OAc) as the counteranion,20 which is in equilibrium to the terminal η1-allylpalladium(II) species (B and C).4 The formation of species C, which leads to linear carboxylation product, is less favored due to the steric hindrance between catalyst and aryl group. Next, complex B is directly reduced at the cathode to form an anionic η1-allylpalladium(0) species (D) by overall two-electron transfer, which is consistent with the studies on carboxylation of aryl halides by Amatore, Jutand and co-workers.21 Finally, the nucleophilic complex D reacts with CO2 at the γ-position to deliver product E in the presence of Mg2+ derived from anodic oxidation, thus regenerating Pd(0). Since carboxylation did not occur in the absence of electric current, the terminal η1-allylpalladium(II) species (B) attack CO2 directly to deliver the palladium(II) carboxylate is not likely in our reaction system.22
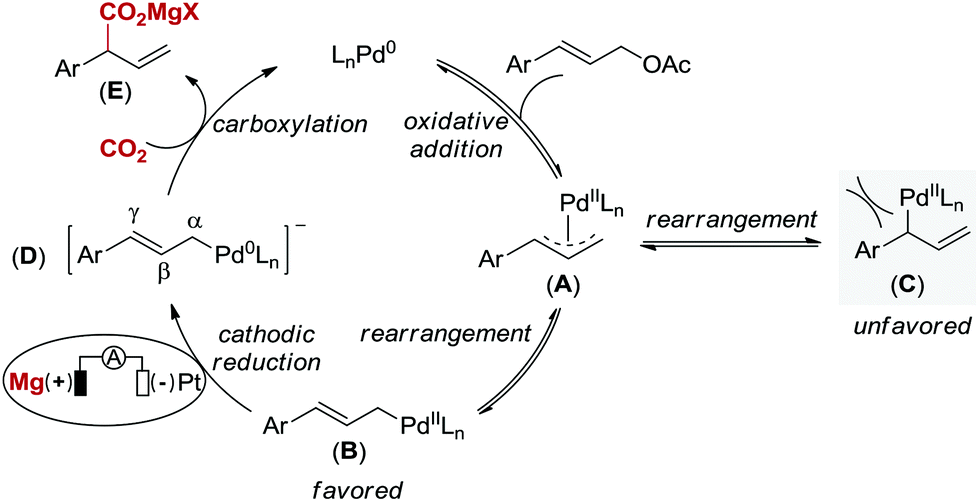 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In summary, we have demonstrated the first example of Pd(0)-catalyzed regioselective carboxylation reaction of homostyrenyl acetates under electrochemical reduction. Compared to established catalytic methods, we employed raw electrons to facilitate this transformation. In addition, an enantioselective carboxylation was also realized via Asymmetric Organometallic Electrochemistry (AOE). These findings provide a new avenue for transition metal-catalyzed electrochemical reactions using chiral ligands. Investigations aimed at understanding the reaction mechanism and further increasing the scope of these reactions are currently underway in our laboratory.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant XDB20000000),“1000-Youth Talents Plan”, NSF of China (Grant 21572245, 21772222, 21772220), and S&TCSM of Shanghai (Grant 17JC1401200, 18JC1415600).References
- (a) F. Juliá-Hernández, M. Gaydou, E. Serrano, M. Van Gemmeren and R. Martin, Top. Curr. Chem., 2016, 374, 45 CrossRef PubMed; (b) M. Börjesson, T. Moragas, D. Gallego and R. Martin, ACS Catal., 2016, 6, 6739 CrossRef PubMed; (c) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (d) C. S. Yeung and V. M. Dong, Top. Catal., 2014, 57, 1342 CrossRef; (e) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC; (f) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (g) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (h) Y.-G. Chen, X.-T. Xu, K. Zhang, Y.-Q. Li, L.-P. Zhang, P. Fang and T.-S. Mei, Synthesis, 2018, 50, 35 CrossRef; (i) Z. Zhang, T. Ju, J.-H. Ye and D.-G. Yu, Synlett, 2017, 28, 741 CrossRef.
- (a) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (b) M. Gaydou, T. Moragas, F. Juliá-Hernández and R. Martin, J. Am. Chem. Soc., 2017, 139, 12161 CrossRef PubMed.
- (a) B. Miao and S. Ma, Org. Chem. Front., 2015, 2, 65 RSC; (b) B. Miao and S. Ma, Chem. Commun., 2014, 50, 3285 RSC.
- (a) H. A. Duong, P. B. Huleatt, Q.-W. Tan and E. L. Shuying, Org. Lett., 2013, 15, 4034 CrossRef PubMed; (b) J. Wu and N. Hazari, Chem. Commun., 2011, 47, 1069 RSC; (c) D. P. Hruszkewycz, J. Wu, N. Hazari and C. D. Incarvito, J. Am. Chem. Soc., 2011, 133, 3280 CrossRef PubMed; (d) R. Johansson and O. F. Wendt, Dalton Trans., 2007, 4, 488 RSC; (e) M. Shi and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 5057 CrossRef.
- T. Moragas, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 17702 CrossRef PubMed.
- T. Mita, Y. Higuchi and Y. Sato, Chem. – Eur. J., 2015, 21, 16391 CrossRef PubMed.
- M. vanGemmeren, M. Börjesson, A. Tortajada, S.-Z. Sun, K. Okura and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 6558 CrossRef PubMed.
- Y.-G. Chen, B. Shuai, C. Ma, X.-J. Zhang, P. Fang and T.-S. Mei, Org. Lett., 2017, 19, 2969 CrossRef PubMed.
- For carboxylation via photocatalysis, see: (a) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915 CrossRef PubMed; (b) K. Shimomaki, K. Murata, R. Martin and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 9467 CrossRef PubMed; (c) J.-H. Ye, M. Miao, H. Huang, S.-S. Yan, Z.-B. Yin, W.-J. Zhou and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15416 CrossRef PubMed; (d) K. Murata, N. Numasawa, K. Shimomaki, J. Takaya and N. Iwasawa, Chem. Commun., 2017, 53, 3098 RSC; (e) H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2016, 9, 453 CrossRef PubMed; (f) N. Ishida, Y. Masuda, S. Uemoto and M. Murakami, Chem. – Eur. J., 2016, 22, 6524 CrossRef PubMed; (g) Y. Masuda, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2015, 137, 14063 CrossRef PubMed; (h) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644 CrossRef; (i) F. Tan and G. Yin, Chin. J. Chem., 2018, 36, 545 CrossRef; (j) Q.-Y. Meng, S. Wang, G. S. Huff and B. Konig, J. Am. Chem. Soc., 2018, 140, 3198 CrossRef PubMed; (k) Z.-B. Yin, J.-H. Ye, W.-J. Zhou, Y.-H. Zhang, L. Ding, Y.-Y. Gui, S.-S. Yan, J. Li and D.-G. Yu, Org. Lett., 2018, 20, 190 CrossRef PubMed; (l) L. Sun, J.-H. Ye, W.-J. Zhou, X. Zeng and D.-G. Yu, Org. Lett., 2018, 20, 3049 CrossRef PubMed; (m) Y.-Y. Gui, W.-J. Zhou, J.-H. Ye and D.-G. Yu, ChemSusChem, 2017, 10, 1337 CrossRef PubMed.
- For selected reviews on electroorganic synthesis, see: (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef PubMed; (b) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef PubMed; (c) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef PubMed; (d) S. R. Waldvogel and M. Selt, Angew. Chem., Int. Ed., 2016, 55, 12578 CrossRef PubMed; (e) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC; (f) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265 CrossRef PubMed; (g) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605 RSC; (h) K. D. Moller, Tetrahedron, 2000, 56, 9527 CrossRef.
- For selected recent examples on electroorganic synthesis, see: (a) Y. Kawamata, M. Yan, Z. Liu, D.-H. Bao, J. Chen, J. T. Starr and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 7448 CrossRef PubMed; (b) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77 CrossRef PubMed; (c) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575 CrossRef PubMed; (d) P. Xiong, H.-H. Xu and H.-C. Xu, J. Am. Chem. Soc., 2017, 139, 2956 CrossRef PubMed; (e) A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406 CrossRef PubMed; (f) A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 11801 CrossRef PubMed; (g) S. Lips, A. Wiebe, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 10872 CrossRef PubMed; (h) R. Hayashi, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400 CrossRef PubMed; (i) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452 CrossRef PubMed; (j) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009 CrossRef PubMed; (k) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195 CrossRef PubMed.
- (a) C. Li, Y. Kawamata, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 13088 CrossRef PubMed; (b) R. J. Perkins, D. J. Pedro and E. C. Hansen, Org. Lett., 2017, 19, 3755 CrossRef PubMed; (c) Y.-L. Lai and J.-M. Huang, Org. Lett., 2017, 19, 2022 CrossRef PubMed; (d) E. Duñach, D. Franco and S. Olivero, Eur. J. Org. Chem., 2003, 1605 CrossRef.
- (a) Q.-L. Yang, Y.-Q. Li, C. Ma, P. Fang, X.-J. Zhang and T.-S. Mei, J. Am. Chem. Soc., 2017, 139, 3293 CrossRef PubMed; (b) Y.-Q. Li, Q.-L. Yang, P. Fang, T.-S. Mei and D. Zhang, Org. Lett., 2017, 19, 2905 CrossRef PubMed; (c) C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189 RSC; (d) K.-J. Jiao, C.-Q. Zhao, P. Fang and T.-S. Mei, Tetrahedron Lett., 2017, 58, 797 CrossRef; (e) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338 CrossRef.
- (a) M. J. Medeiros, C. Pintaric, S. Olivero and E. Duñach, Electrochim. Acta, 2011, 56, 4384 CrossRef; (b) D. Franco, D. Panyella, M. Rocamora, M. Gomez, J. C. Clinet, G. Muller and E. Duñach, Tetrahedron Lett., 1999, 40, 5685 CrossRef; (c) J. Torii, H. Tanaka, T. Hamatani, K. Morisaki, A. Jutand, F. Pfluger and J.-F. Fauvarque, Chem. Lett., 1986, 169 CrossRef.
- (a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef PubMed; (b) A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020 CrossRef.
- For Pd-catalyzed allylic alkylation, see: (a) S.-L. You, X.-Z. Zhu, Y.-M. Luo, X.-L. Hou and L.-X. Dai, J. Am. Chem. Soc., 2001, 123, 7471 CrossRef PubMed; (b) R. Pretot and A. Pfaltz, Angew. Chem., Int. Ed., 1998, 37, 323 CrossRef.
- (a) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef PubMed; (b) G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336 CrossRef PubMed.
- (a) S. Kawashima, K. Aikawa and K. Mikami, Eur. J. Org. Chem., 2016, 3166 CrossRef; (b) B.-L. Chen, H.-W. Zhu, Y. Xiao, Q.-L. Sun, H. Wang and J.-X. Lu, Electrochem. Commun., 2014, 42, 55 CrossRef; (c) M. Takimoto, Y. Nakamura, K. Kimura and M. Mori, J. Am. Chem. Soc., 2004, 126, 5956 CrossRef PubMed.
- (a) S.-F. Zhu, X.-C. Qiao, Y.-Z. Zhang, L.-X. Wang and Q.-L. Zhou, Chem. Sci., 2011, 2, 1135 RSC; (b) G. P. Howell, A. J. Minnaard and B. L. Feringa, Org. Biomol. Chem., 2006, 4, 1278 RSC; (c) G. Zanoni, S. Gladiali, A. Marchetti, P. Piccinini, I. Tredici and G. Vidari, Angew. Chem., Int. Ed., 2004, 43, 846 CrossRef PubMed.
- C. Amatore, A. Jutand, G. Meyer and L. Mottier, Chem. – Eur. J., 1999, 5, 466 CrossRef.
- (a) C. Amatore, A. Jutand, F. Khalil and M. F. Nielsen, J. Am. Chem. Soc., 1992, 114, 7076 CrossRef; (b) A. Jutand, Chem. Rev., 2008, 108, 2300 CrossRef PubMed.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2008, 130, 15254 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, product characterization, copies of NMR spectra. See DOI: 10.1039/c8qo00507a |
| This journal is © the Partner Organisations 2018 |