
Palladium-catalyzed reductive electrocarboxylation of allyl esters with carbon dioxide†
Xue-Tao
Xu,  b
Tian-Sheng
Mei
*a
b
Tian-Sheng
Mei
*a
b
Tian-Sheng
Mei
*a
Abstract
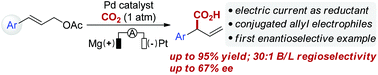
Palladium-catalyzed regioselective electrocarboxylation of homostyrenyl acetates with CO2 has been successfully developed, providing α-aryl carboxylic acids with good selectivity and yield. In addition, the catalytic asymmetric carboxylation of cinnamyl acetate has been demonstrated, despite moderate enantioselectivity.
- This article is part of the themed collections: Celebrating 70 Years of Shanghai Institute of Organic Chemistry and Celebrating the 90th birthday of Professor Lu Xiyan
 Please wait while we load your content...
Please wait while we load your content...

