
Recent progress in the mechanofluorochromism of distyrylanthracene derivatives with aggregation-induced emission
Juan
Zhao
,
Zhihe
Chi
,
Zhiyong
Yang
*,
Zhu
Mao
,
Yi
Zhang
 *,
Eethamukkala
Ubba
and
Zhenguo
Chi
*
*,
Eethamukkala
Ubba
and
Zhenguo
Chi
*
PCFM Lab, GD HPPC Lab, Guangdong Engineering Technology Research Center for High-performance Organic and Polymer Photoelectric Functional Films, State Key Laboratory of Optoelectronic Material and Technologies, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: chizhg@mail.sysu.edu.cn; ceszy@mail.sysu.edu.cn; yangzhy29@mail.sysu.edu.cn
First published on 21st May 2018
Abstract
Mechanofluorochromic (MFC) luminogens, as a group of evidence-based, practical smart materials, have established immense interest with respect to mechanical stimuli, due to their promising applications in various fields like mechanosensors, safety papers, and optical storage. However, MFC luminogens were rare earlier than 2010. After the discovery that MFC is an essential unique feature of aggregation-induced emission (AIE)-active molecules in 2011, researchers are encouraged to pay more attentions toward MCF luminogens, in which field the investigations are hastened steadily and focused distinctly. As one of the most vital AIE cores, distyrylanthracene (DSA) has been extensively used to assemble MFC motifs. As part of the interest in MFC luminogens, this contemporary overview is targeted on recent advances in the mechanofluorochromism of DSA derivatives with AIE properties and mechanistic study, which encourages more researchers to dedicate themselves to this interesting exploration discipline.
1. Introduction
Mechanofluorochromic (MFC) luminogens are a class of smart fluorescent molecules which can respond to external forces including mechanical stimuli such as pressing, grinding, crushing, or rubbing with alteration of the emission colors or strengths, and have attracted considerable attention given their promising applications in mechano-sensors, security papers, and optical storage.1–3 The emission of MFC luminogens can be altered through changes in the molecular structure or aggregate morphology. Although the first one is a general way to tune the emission of a luminogen, limited examples of MFC luminogens have been reported based on this mechanism due to their incomplete and irreversible chemical reactions in the solid state. Even though each system has its own characteristics, the MFC luminescence of most reported luminogens has been achieved during modulation in their morphology by mechanical stimuli.4 However, prior to this essential discovery that mechanofluorochromism is one of the common and unique properties for most aggregation-induced emission (AIE) luminogens,5 MFC luminogens that are dependent on changes in their physical molecular packing modes were very rare. There are two noticeable reasons:6 firstly, there is a shortage of clear design strategies for their syntheses; secondly, the emission of many luminogens is totally or partially quenched when aggregates are formed due to the aggregation-caused quenching (ACQ) effect.In 2001, Tang et al.7 suggested a few AIE molecules that emit more efficiently in the aggregated state than in the dissolved form, leading to key examples of anti-ACQ luminogens. Since then, many AIE references such as triphenylethylene, tetraphenylethylene, silole, cyano distyrylbenzene and distyrylanthracene have been used for the development of AIE molecules.8–13 AIE luminogens have become one of the hottest research topics due to their potential applications in diverse fields, such as organic light-emitting devices (OLEDs) and chemo-sensors.14 In fact, the development of AIE has surmounted the two aforementioned problems. Recently, a number of AIE luminogens displaying MFC properties have been discovered. For this reason, nowadays the use of AIE moieties provides an important strategy to construct new compounds and also various mechano-responsive AIE luminogens.
During the investigation of AIE, Tang and co-workers found that several AIE luminogens can switch their emission between bright and dark when crystalline and amorphous states are interconverted.15–17 The first reported investigation regarding the mechano-responsive luminescence of an AIE molecule18 attempted to prove the AIE mechanism, which was the restriction of intramolecular rotations, by applying hydrostatic pressure to an amorphous film of silole. It was found that the photoluminescence (PL) emission intensity was enhanced by 9% when the pressure was increased swiftly up to 104 atm, and then decreased slowly when the film was further pressurized. In 2010, Park et al. described the mechanofluorochromism of a cyano-distyrylbenzene derivative of an AIE compound.19 However, at that time, it was not recognized well that there is a relationship between the molecular structure of the AIE molecule and its mechanofluorochromic nature. Indeed, at the same time, Chi and Xu's group (2011)5 demonstrated a number of new mechanofluorochromic compounds with AIE nature and pointed out that mechanofluorochromism should be a common property for most of the AIE luminogens. Since then, new mechano-responsive AIE luminogens have been mushrooming.2–4,20–27
In 2009, distyrylanthracene (DSA) as an AIE unit was first reported by Tian's group,13 and a lot of DSA-based AIE molecules have been developed to date. In 2011, the MFC properties of many DSA derivatives were exemplified by Chi and Xu's group.5,28–30 Afterward, many DSA derivatives were investigated with mechanofluorochromism. This review summarizes the research progress of DSA derivatives along with the relationship between their molecular structures and MFC characteristics. We try our best to provide ideas about designing distyrylanthracene MFC materials.
2. Typical mechanochromic mechanism of DSA derivatives
A possible mechanism based on the planarization of the molecular conformation (Fig. 1) was proposed by Chi and Xu's group5 to explain the mechanochromic phenomenon of a DSA derivative (1). A common structural feature, which is that multiple phenyl peripheries are linked to an olefinic core via rotatable C–C single bonds to form an AIE moiety, has been found in a number of reported AIE compounds. This steric effect between the phenyl rings endows the AIE moieties or the molecules with a twisted conformation, which induces the existence of twist stress. Due to the twisted conformation and the weak C–H⋯π interactions, the molecular packing was relatively loose and numerous defects (cavities) were formed, leading to a low lattice energy. The cavities are the feeblest parts of the crystalline structures. Under the combined effects of these two structural features, low lattice energy and the formation of cavities, the crystals could be easily destroyed when external pressure was applied. After releasing the twist stress, the molecular conformation was planarized and then the molecular conjugation was increased, which resulted in a red-shift of the PL spectrum.5 Based on this hypothesis, Zhang et al. synthesized a number of AIE luminogens, among which mechanofluorochromism is generally observed. This finding of the common or unique nature of AIE molecules could be very imperative and crucial for the development of more mechanoluminochromic molecules.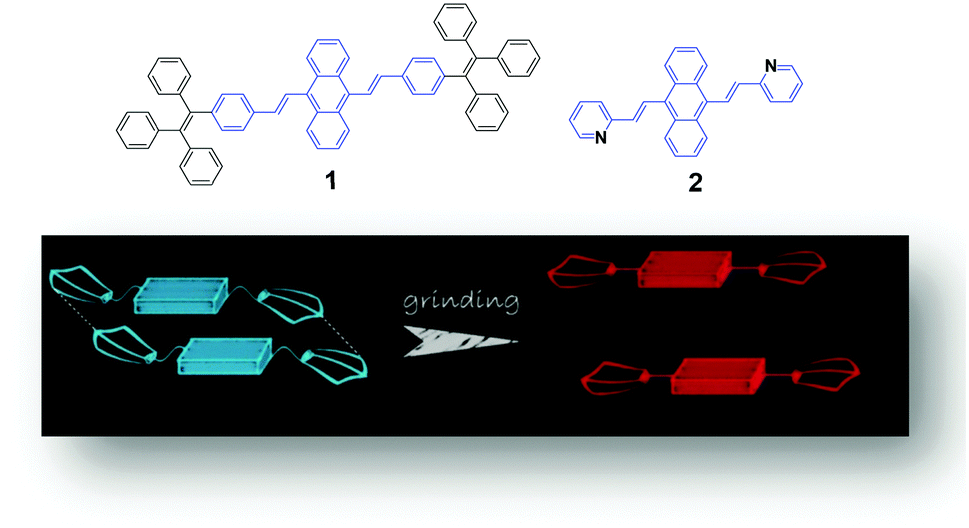 | ||
| Fig. 1 (Top) The molecular structures of compounds 1 and 2. (Bottom) An illustration of the MFC mechanism based on the planarization of the molecular conformation. | ||
Tian et al.31 proposed another mechanism for the mechanochromic luminescence of an AIE molecule depending on the changes in the molecular aggregation state of 9,10-bis((E)-2-(pyrid-2-yl)vinyl)anthracene (2). When an external stimulus like grinding or external pressure is applied on the powder, the compound reveals fabulous luminescence properties, and the emission color turns from green to red, while three crystal polymorphs with different emission colors are obtained and present fluorescence quantum yields (ΦF) of 0.28, 0.37 and 0.48 respectively. By applying pressure, the compound powder can transform between the molecular aggregation states of the three crystals (Fig. 2), showing mechanochromic luminescence. In this process, the molecular aggregation state of the compound powder favourably transforms from J-type aggregation to H-type aggregation by virtue of the external pressure, and further to aggregated dimers stacked in a more tightly bound face-to-face arrangement. At the same time, considering that the intermolecular π–π interaction is strengthened gradually, the PL emission of the powder is shifted from green (no π–π interaction) to orange (weak π–π interaction) and finally to red (strong π–π interaction). Therefore, this work provides another effective way to understand the mechanochromic effect in organic compounds by controlling the molecular aggregation state, which can be changed by external influences.
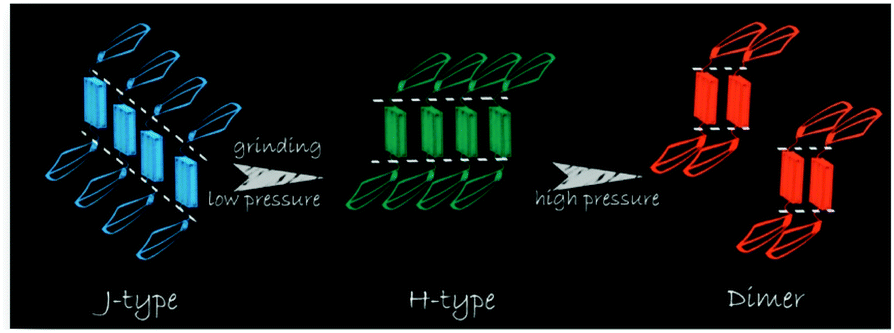 | ||
| Fig. 2 An illustration of the stacking modes and the corresponding emission colors for the various molecular aggregation states of 2. | ||
3. MFC distyrylanthracene derivatives
The spectroscopic properties and morphological structures of luminogen 3 were elucidated upon pressing, grinding, annealing or fuming (Fig. 3A) by Chi and Xu's group in 2011.28 Based on powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC) analysis, the MFC nature of 3 results from the phase transformation. The crystalline structure analysis shows that the luminogen molecules are packed in a head-to-head mode (Fig. 3B), wherein the backbone of the molecule largely deviates from the plane. As a result, the molecule is in a highly twisted conformation accompanied by the presence of steric hindrance of the bulky phenothiazinyl groups, making typical co-facial π–π stacking impossible. The molecules are packed in clusters via weak C–H⋯S, S⋯π and C–H⋯π interactions. The former is helpful to bind the clusters for the formation of lamellar layers, which are connected via the weak and sparse π–π interactions and partial π-overlaps from the phenyl rings of the phenothiazinyl groups. Consequently, under an external force or stimulus, the layer–layer and cluster–cluster interfaces can be readily destroyed through slip-deformation, facilitating the existence of mechanofluorochromism.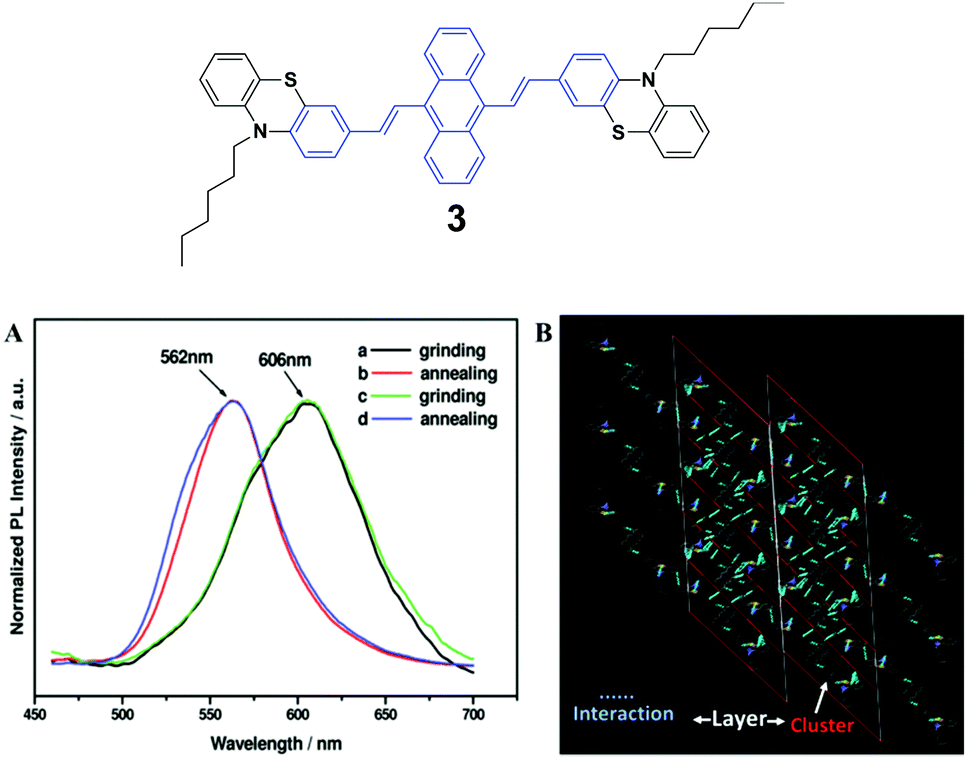 | ||
| Fig. 3 (Top) The molecular structure of compound 3. (Bottom) (A) Normalized PL spectra of 3: (a) grinding, (b) annealing (a) at 100 °C for 1 min, (c) grinding, and (d) annealing (c) at 100 °C for 1 min. (B) Molecular packing of luminogen 3 in the single crystal structure. Adapted with permission from ref. 28. Copyright 2011 American Chemical Society. | ||
Chi and Xu's group32 synthesized DSA derivatives with monocarbazolyl groups (5), tricarbazolyl groups (6) and without any carbazolyl group (4). The AIE compounds 4, 5 and 6 display distinct MFC properties, which are related to the carbazolyl moiety and suggest an important role of the variety of carbazolyl groups in the luminescence changes upon grinding. As for the solid luminogen of 5 with mono-carbazolyl groups, the emission color undergoes a change from strong green (523 nm) to strong yellow (547 nm), and as for 6 with tricarbazolyl groups, the emission color turns from yellowish green (547 nm) to orange (580 nm); therefore luminogens 5 and 6 exhibit MFC properties. However, no MFC is observed in luminogen 4 because of the absence of the carbazolyl group. When the ground samples of 5 and 6 are treated by annealing or fuming, their original colors are nearly recoverable, suggesting a good reversibility of the MFC properties (Fig. 4). The results also indicate that no grinding-induced MFC properties exist in the AIE molecules with strong crystallizability.
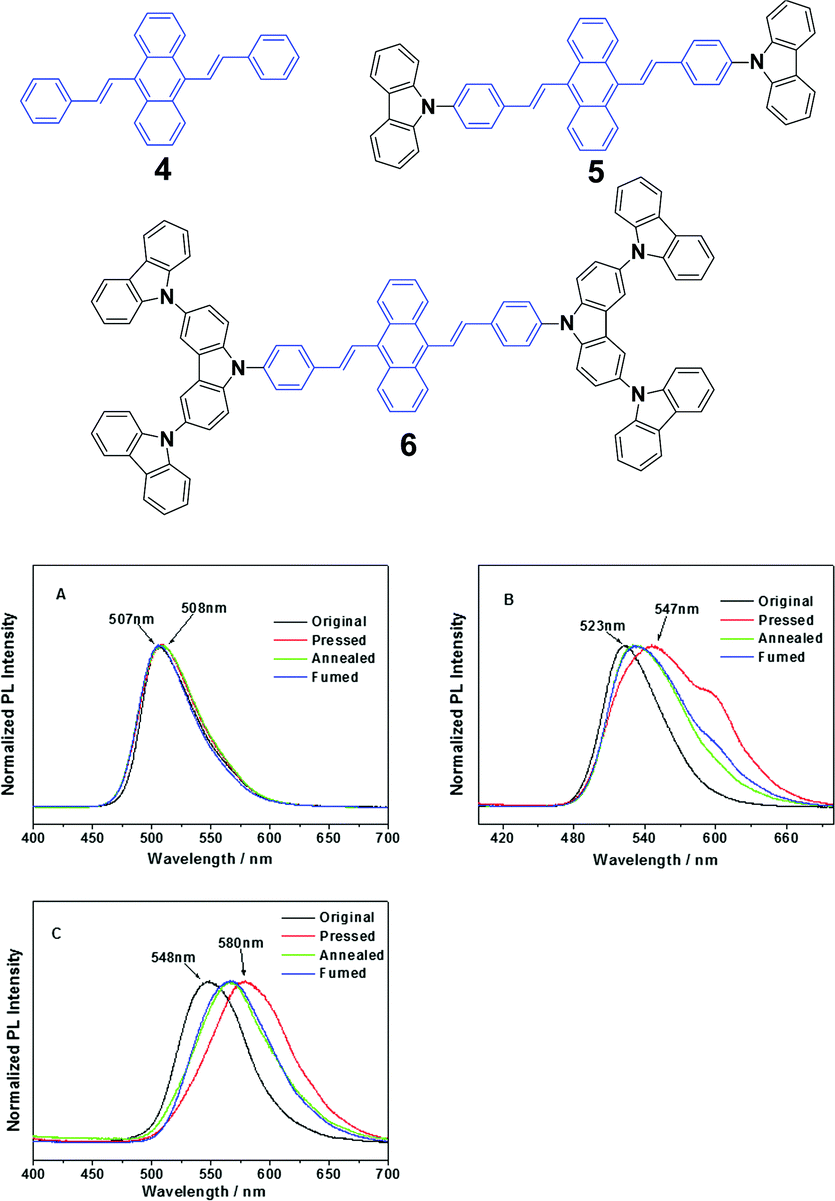 | ||
| Fig. 4 (Top) The molecular structures of compounds 4, 5 and 6. (Bottom) The normalized PL spectra of 4 (A), 5 (B) and 6 (C) samples obtained under different conditions. Original: original sample; ground: ground sample; annealed: after annealing the ground sample at 200 °C for 5 min; and fumed: after fuming the ground sample in CH2Cl2 vapor for 5 min. Reproduced with permission from ref. 32. Copyright 2012 American Chemical Society. | ||
As mentioned above, the strong crystallizability of luminogen 4 makes its crystals very stable; thus it is difficult to trigger the transformation from the crystalline to the amorphous phase, resulting in non-mechanofluorochromism. This implies that the initial state of a luminogen has a significant effect on the observance of mechanofluorochromism. It can be further conjectured that the MFC phenomenon could be hardly observed from a luminogen whose initial state is non-crystalline, considering that there is no transition between crystalline and amorphous phases. This ratiocination has been confirmed by the experimental results on a series of luminogens 7–10, which were designed and synthesized by Chi and Xu's group.29 All of these compounds are AIE-active because of the employed triphenylethylene or distyrylanthracene AIE moiety. After grinding, samples of DSAs 7 and 9 display clear MFC phenomena, and the emission peaks of 7 and 9 red-shift from 534 nm to 572 nm and 566 nm to 580 nm, respectively, whereas the 8 and 10 derivatives are not MFC-active. The PXRD results confirmed that the initial phases of luminogens 7 and 9 were crystalline to a particular extent, while those of luminogens 8 and 10 were amorphous. As the MFC behavior is dependent on the packing change from the crystalline to an amorphous state, compounds 8 and 10 show no MFC properties. In other words, AIE luminogens with strong crystallizability or non-crystallizability could not be good candidates for MFC materials. As a result, the design strategy for MFC molecules remains a crucial issue, i.e., not all luminogens containing AIE moieties exhibit MFC properties.
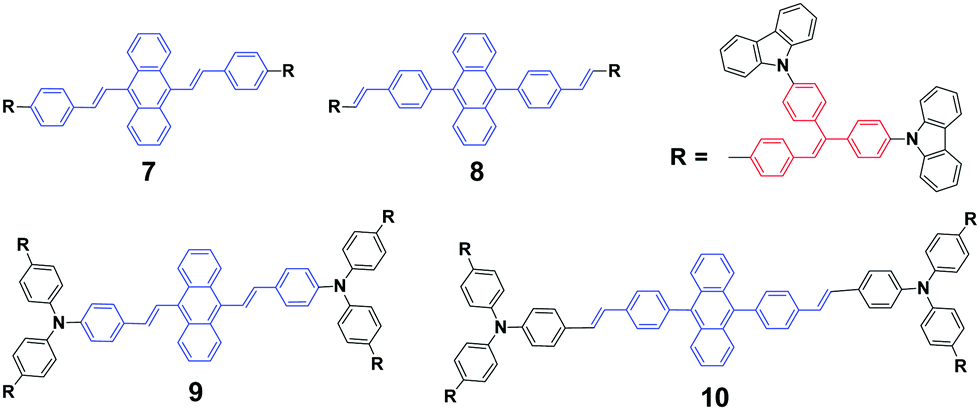
In general, the solubility of materials can be improved through the introduction of an outer edge alkyl chain into the conjugated organic molecule. Recently, some studies have demonstrated that the length of the alkyl chain has effects on the state of the aggregation behavior of conjugated organic small molecules, as well as the photoelectric properties through regulation. The influence of alkyl chains will be considered as an important factor in the controllable preparation of MFC materials.
Chi and Xu's group33 and Yang et al.34 produced a series of multifunctional 9,10-distyrylanthracene derivatives (11, DSAn, n = 1–10, 12, 14, 16, 18) with remarkable AIE properties. Under grinding, the longer alkoxy-containing DSA-OCn (n ≥ 10, Δλem = 46–53 nm) exhibit more obvious spectral variations than those of the shorter alkoxy-containing chains (n ≤ 9, Δλ < 20 nm) (Fig. 5). The single crystal morphology was studied to explore the relationship between the molecular structures of the compounds and their MFC properties; however, the X-ray studies revealed that the molecules crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , which indicates a non-planar conformation in the crystals. On one hand, the DSA10, DSA11, and DSA12 crystals have symmetrical conformation structures, and the shorter PL wavelengths (from 498 to 500 nm) in their original states are associated with larger dihedral angles (>60°) of the molecules. On the other hand, the DSA7 and DSA8 crystals show both symmetric and asymmetric conformation structures, while the DSA9 crystals are only asymmetric. As a result, longer PL wavelengths (from 515 to 531 nm) are produced in DSA7, DSA8 and DSA9 with smaller dihedral angles (<50°). The crystal structure analysis confirms that the above-mentioned difference among the molecular conformation structures of the crystals stems from supramolecular interactions. Among them, C–H⋯π interactions are the most important ones. The backbone of the molecules largely deviates from a plane, and due to the highly twisted conformation and steric hindrance brought about by the bulky aromatic rings in the molecules, the formation of typical co-facial π–π stacking is impossible. There are four, five and four interactions found in the DSA7, DSA8, and DSA9 crystals, respectively; whereas in the DSA10, DSA11, and DSA12 crystals, only one, two and two supramolecular interactions exist, respectively. Thus, the former three crystals show much stronger supramolecular interactions, resulting in not only more planarized and stabilized molecules in the lattice, but also more tightened intermolecular packing. Moreover, the DSA7, DSA8 and DSA9 crystals exhibit higher density d than the DSA10, DSA11, and DSA12 crystals, which indicates that the molecular packings are relatively loosened in the DSA10, DSA11 and DSA12 crystals with more twisted conformations and weaker supramolecular interactions, leading to lower lattice energies. In light of these two structural features, external pressure can easily destruct the crystals by planarization of the molecular conformation or slip deformation. The release of twist stress and the planarization of the molecular conformation tend to increase molecular conjugation, thereby red-shifting the PL spectrum. According to the diffuse reflectance absorption and IR spectral results, the out-of-plane hydrogen bending modes of the phenyl rings disappear after grinding the original DSAn crystals with long alkoxy chains, suggesting that the backbone conformation of the ground samples is less twisted, inducing dichotomous MFC behavior. The results may explain the origin of the more significant mechanofluorochromism of DSAn (n > 10), as compared to the others. Thus, there is a high need for long peripheral aliphatic tails for balancing the intermolecular π–π and aliphatic interactions, in order to achieve remarkable MFC behavior from 9,10-bis(p-alkoxystyryl)anthracene homologs. All the DSAn compounds exhibited thermochromic properties. For example, under 365 nm UV illumination, DSA11 emits green light (514 nm), which quickly changes to dark yellow (566 nm) when it is heated to its transition temperature (Ti), leading to an emission variation of 52 nm; meanwhile, the emission color change is also reversible.
, which indicates a non-planar conformation in the crystals. On one hand, the DSA10, DSA11, and DSA12 crystals have symmetrical conformation structures, and the shorter PL wavelengths (from 498 to 500 nm) in their original states are associated with larger dihedral angles (>60°) of the molecules. On the other hand, the DSA7 and DSA8 crystals show both symmetric and asymmetric conformation structures, while the DSA9 crystals are only asymmetric. As a result, longer PL wavelengths (from 515 to 531 nm) are produced in DSA7, DSA8 and DSA9 with smaller dihedral angles (<50°). The crystal structure analysis confirms that the above-mentioned difference among the molecular conformation structures of the crystals stems from supramolecular interactions. Among them, C–H⋯π interactions are the most important ones. The backbone of the molecules largely deviates from a plane, and due to the highly twisted conformation and steric hindrance brought about by the bulky aromatic rings in the molecules, the formation of typical co-facial π–π stacking is impossible. There are four, five and four interactions found in the DSA7, DSA8, and DSA9 crystals, respectively; whereas in the DSA10, DSA11, and DSA12 crystals, only one, two and two supramolecular interactions exist, respectively. Thus, the former three crystals show much stronger supramolecular interactions, resulting in not only more planarized and stabilized molecules in the lattice, but also more tightened intermolecular packing. Moreover, the DSA7, DSA8 and DSA9 crystals exhibit higher density d than the DSA10, DSA11, and DSA12 crystals, which indicates that the molecular packings are relatively loosened in the DSA10, DSA11 and DSA12 crystals with more twisted conformations and weaker supramolecular interactions, leading to lower lattice energies. In light of these two structural features, external pressure can easily destruct the crystals by planarization of the molecular conformation or slip deformation. The release of twist stress and the planarization of the molecular conformation tend to increase molecular conjugation, thereby red-shifting the PL spectrum. According to the diffuse reflectance absorption and IR spectral results, the out-of-plane hydrogen bending modes of the phenyl rings disappear after grinding the original DSAn crystals with long alkoxy chains, suggesting that the backbone conformation of the ground samples is less twisted, inducing dichotomous MFC behavior. The results may explain the origin of the more significant mechanofluorochromism of DSAn (n > 10), as compared to the others. Thus, there is a high need for long peripheral aliphatic tails for balancing the intermolecular π–π and aliphatic interactions, in order to achieve remarkable MFC behavior from 9,10-bis(p-alkoxystyryl)anthracene homologs. All the DSAn compounds exhibited thermochromic properties. For example, under 365 nm UV illumination, DSA11 emits green light (514 nm), which quickly changes to dark yellow (566 nm) when it is heated to its transition temperature (Ti), leading to an emission variation of 52 nm; meanwhile, the emission color change is also reversible.
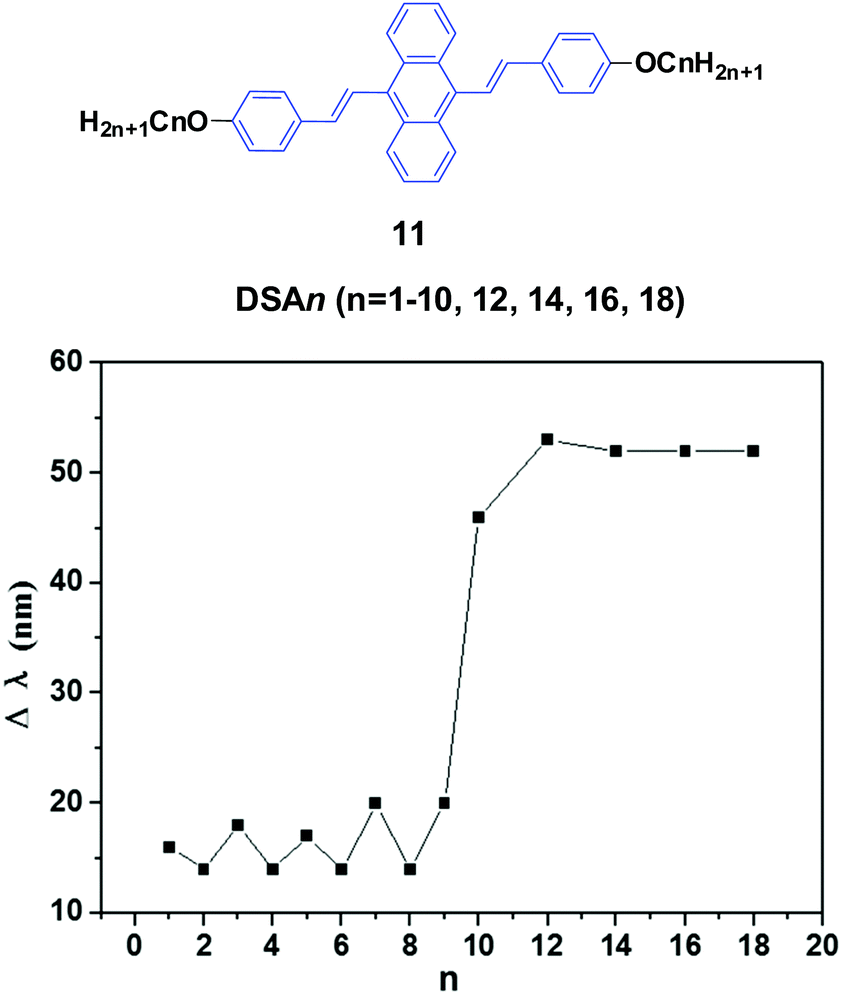 | ||
| Fig. 5 (Top) The molecular structure of compound 11. (Bottom) Wavelength change of the luminogen 11 series versus n after grinding. Data from ref. 34. | ||
Yang et al.35 also designed and synthesized a series of new alkyl length-dependent mechanochromic 9,10-bis[(9,9-dialkylfluorene-2-yl)vinyl]anthracenes (12, FLA-Cn) with propyl, pentyl and dodecyl side chains. FLA-Cn exhibit AIE properties, wherein the weak emission in solutions (ΦF = 3.8–4.9%) is intensified in THF/water (1/9) mixtures (ΦF = 21–29%). After pressing, the emission colors of the FLA-C12, FLA-C5 and FLA-C3 solids changed from green to yellow, from yellowish-green to yellow and from yellow to yellowish-orange, respectively, while these emission changes could be recovered when the pressed samples were annealed below the isotropic melt transition temperature or exposed to solvent vapor (fuming above dichloromethane). With further repressing the fumed or annealed samples, the same emission change as that shown in the first pressing was observed, indicating that the MFC behavior was highly reversible. Upon pressing, the spectra of the FLA-C12, FLA-C5 and FLA-C3 solids shifted by 40, 26 and 18 nm, respectively (Fig. 6), indicating that the color change was more remarkable in the FLA-Cn with longer alkyl chains. The MFC properties of the FLA-Cn solids had a high dependence on the chain length, which was like that of series 11; thus it was possible to tune the MFC behavior of FLA-Cn through adjustment of the alkyl chains. In addition, the original green emission of FLA-C12 could be spontaneously and gradually restored from its just ground solid that was yellow emissive, upon increasing the standing time at room temperature. However, in the cases of the ground FLA-C3 (orange) and FLA-C5 (yellow) solids, the emission colors showed no change for over 24 h at room temperature. It is considered that it is an effective approach to control the stabilization or self-recovery of the MFC behavior of the 9,10-diarylvinyl-anthracene luminogens by tuning the alkyl length.
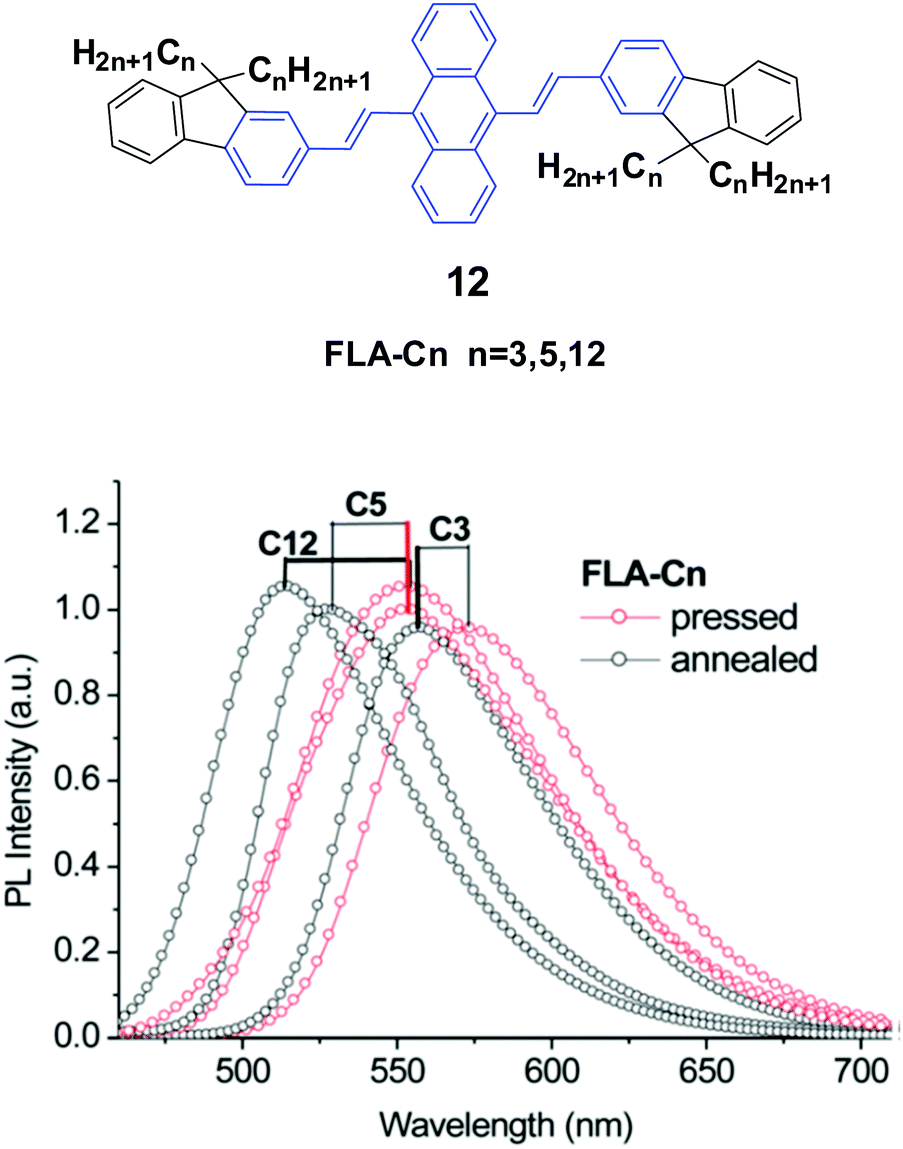 | ||
| Fig. 6 (Top) The molecular structure of compound 12. (Bottom) Emission spectra of FLA-Cn solids mixed with KBr upon pressing and annealing. Reproduced with permission from ref. 35. Copyright 2013 The Royal Society of Chemistry. | ||
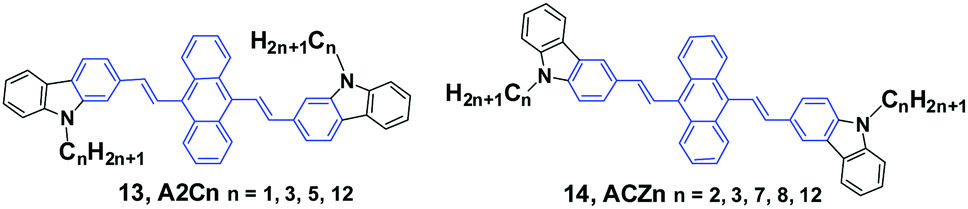
Yang et al.36 reported the mechanofluorochromism of a series of 9,10-bis(N-alkylcarbazol-2-yl-vinyl-2)anthracenes (13, A2Cn) by changing the lengths of the N-alkyl chains (methyl, propyl, pentyl, and dodecyl). The fluorescence quantum efficiencies of A2Cn in THF range from 4.8 to 6.7%, while they show a low AIE effect considering the moderate enhancement of PL intensity in THF/water mixtures. Upon external press stimuli, all the A2Cn samples demonstrate the same fluorescence change from green to yellow, which can return back to the initial green color through thermal-annealing (at 120 °C) or solvent-fuming (exposed to CH2Cl2 vapor at room temperature) treatment. The color changes can be reversibly switched between green and yellow by repeatedly pressing and annealing/solvent-fuming. All the A2Cn samples show remarkable pressing-induced spectral shifts (Δλ = λpressed − λannealed) of 36–45 nm (Table 1). In comparison with A2Cn, as the alkyl length is increased, the emission wavelength peaks of the pristine (as-prepared) solids (λpristine) are slightly blue-shifted (from 548 to 530 nm), which is consistent with the results observed for FLA-Cn described above. In spite of this, the emission peaks of the A2Cn samples in the pressed (λpressed) and annealed states (λannealed) are not significantly sensitive to the alkyl lengths, endowing the A2Cn samples with slightly increased Δλ upon increasing the alkyl lengths. Therefore, the MFC behavior of A2Cn shows a weak dependence on the alkyl length, which is obviously different from those presented in the DSAn and FLA-Cn systems in which the Δλ values are strongly dependent on the alkyl length and double changed.
| Compound | λ pristine | λ pressed | λ annealed | λ repressed | λ fumed | Δλa |
|---|---|---|---|---|---|---|
| a Δλ = λpressed − λannealed. Reproduced with permission from ref. 36 and 37. Copyright 2013 Elsevier and 2013 The Royal Society of Chemistry. | ||||||
| A2C1 | 548 | 572 | 536 | 572 | 541 | 36 |
| A2C3 | 543 | 579 | 537 | 577 | 540 | 42 |
| A2C5 | 535 | 578 | 533 | 575 | 533 | 45 |
| A2C12 | 530 | 571 | 527 | 572 | 530 | 44 |
| ACZ2 | 511 | 600 | 512 | 598 | 513 | 88 |
| ACZ3 | 514 | 594 | 518 | 591 | 515 | 76 |
| ACZ7 | 534 | 583 | 535 | 582 | 532 | 48 |
| ACZ8 | 536 | 576 | 530 | 573 | 532 | 46 |
| ACZ12 | 543 | 569 | 541 | 572 | 541 | 28 |
As the linking isomers of A2Cn, ACZn (14) were also synthesized by Yang et al.37 and displayed unusual chain length-dependent solid-state fluorescence properties under various external stimuli. ACZn (14) are AIE-active, and their ΦF values are 0.7–0.9% in solution but highly increased to 19–31% in the aqueous nano-aggregate state. In contrast to the results found in A2Cn, a shorter N-alkyl chain was favourable for more remarkable MFC behavior (Table 1). It was believed that upon grinding not only the close stacking of molecules in pristine crystals and aqueous suspensions, but also the sufficient amorphization was obstructed by the long alkyl chains, which made the fluorescence properties have an unusual dependence on the chain length and brought about MFC behaviors. This work presents that the unique solid-state optical properties of the 9,10-divinylanthracene derivatives could be tuned through subtle manipulation of the end groups in the molecules.
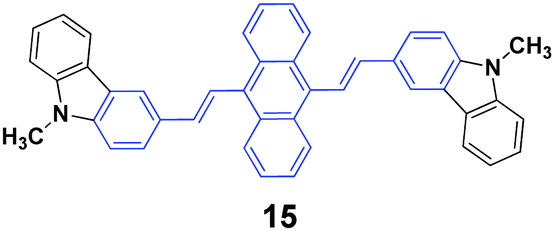
High contrasts in fluorescence color and large spectral shifts under simple mechanical forces are desirable. Yang et al.38 investigated the mechanofluorochromic behavior of 9,10-bis(N-methylcarbazol-3-yl-vinyl-2)anthracene (15), which exhibited strong AIE and crystallization-enhanced emission (CEE) effects. The initial 15 solid with a ΦF of 47% gave off green emission (512 nm), which turned to red emission (602 nm) under a simple mechanical force, and thus a large spectral shift (90 nm) confirmed high contrast mechanofluorochromism. The red emission could return back to the original green emission upon fuming the pressed sample with dichloromethane at room temperature. The emission color could be switched between green and red by repeating repressing and re-fuming treatments, affording high reversibility of the conspicuous color change. It is noted that the pressed solid that was bright red emissive exhibited a commendable fluorescence efficiency of 0.31, which was measured on a fluorescence integrating sphere. Wide-angle X-ray diffraction and differential scanning calorimetry results confirmed the presence of some crystal defects and some amorphous states in the pressed samples.
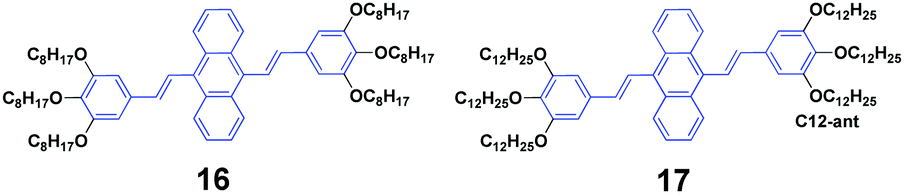
Siva et al.39 synthesized symmetrical linear conjugated molecules of 9,10-bis-(trisalkoxystyryl)anthracenes with different lengths of alkoxy chains, which are octyloxy (16) and dodecyloxy (17) that are linked at the para- and meta-positions of the phenyl rings, respectively. When dissolved in the same solution, the PL spectrum of 16 (ΦF = 71%) slightly red-shifts compared to that of 19 (ΦF = 56%), which is possibly caused by the steric hindrance between the neighbouring groups of the alkyl chain. Besides AIE properties, both 16 and 17 exhibit mechanofluorochromism and thermochromism features. The length of the alkoxy substitution has a significant effect on the fluorescence properties and piezofluorochromic behaviors of the two compounds in solution and aqueous dispersion. After pressing, 16 and 17 show spectral shifts of 1 and 30 nm, respectively, but the origin of this dichotomous alkyl chain length-dependent MFC behavior is still not clear. When the pressed samples were treated by annealing, the fluorescence of pressed 16 turned from green to light brown, while that of 17 changed from yellow to bright brown, affording a more obvious color change.
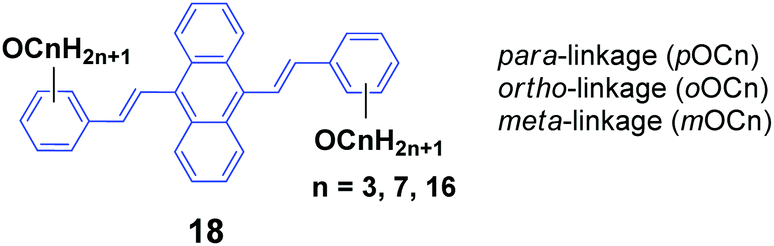
As seen, the MFC behavior of the above AIE molecules is highly alkyl length-dependent. Therefore, it is quite possible that the linking positions of the alkoxy chains at the peripheral aryl rings have a great influence on the molecular backbone conformation and intermolecular stacking structure of the AIE molecules, which can change the optical properties. Thus, Yang et al.40 also designed and synthesized a series of 9,10-bis(alkoxystyryl)-anthracenes (18, OCn) with different linking positions (ortho, meta and para) and lengths of alkoxy chains (propoxy, heptyloxy and hexadecyloxy) (denoted oOCn, mOCn and pOCn; among them, pOC7 is also reported by Chi and Xu's group, which is denoted as DSA7 above). The AIE properties of 18 can be evidenced from the enhanced ΦF in THF/water mixtures with high water ratios: the ΦF values of oOCn in THF solutions (3.2–3.9%) are increased to 21.4–25.1% in 1/9 THF/water solutions, while those of mOCn (10.8–11.4%) are increased to 41.8–46.1%, and those of pOCn (0.72–0.81) are increased to 47.2–66.6% under the same conditions. The fluorescence spectroscopic data of the oOCn, mOCn and pOCn samples upon a cycle of pressing, annealing, repressing and solvent-fuming are summarized in Table 2. Under external stimuli, all the OCn solids presented a fluorescence color change along with a spectral shift, which is obviously dependent on both the alkoxy-position and length. For the pOCn series, a longer alkoxy chain endows the compound with larger Δλ; however, for the oOCn and mOCn isomers, a shorter alkoxy chain tends to induce significant MFC behavior in OCn (oOC3 and mOC3). According to the X-ray single crystal analysis of pOC3, mOC3 and oOC3, there are two classes of C–H⋯π hydrogen bonds formed between two OC3 molecules. One is the hydrogen in the benzene moiety along the long axis of one molecule as the H-donor with the corresponding phenyl ring of the anthrylene moiety of the adjacent molecule as the H-acceptor (interaction type I); the other is the OCH moiety of one molecule as the H-donor with the phenyl ring of the adjacent molecule as the H-acceptor (interaction type II). Type I and type II interactions appear in pOC3 and oOC3, respectively, while both type I and II interactions exist in mOC3. Considering the easy-mechanofluorochromism and large Δλ found in oOC3 and mOC3, it seems that the OCH⋯π interaction is weaker and is more likely to be destroyed, which probably results from the inherent flexibility of the alkoxy chains. It is observed that one molecule slides to its neighbor along the long axis of the molecule with angles of 36.81°, 34.71° and 66.11° and vertical distances of 3.37, 3.51 and 3.51 Å for pOC3, mOC3, and oOC3, respectively. This indicates a J-aggregation mode adopted for the pOC3 and mOC3 molecules and an H-aggregation mode for the oOC3 molecule in the crystals. On the other hand, pOC3, mOC3, and oOC3 have similar crystal densities (1.219, 1.218 and 1.213 mg m−3, respectively), but they have different selected dihedral angles (phenyl ring vs. anthryl ring), which are in the order of oOC3 (87.0°) > mOC3 (83.6°) > pOC3 (77.6°). The more twisted backbones make the molecular conjugation degree decrease and then enable more blue-shifted emission of the fluorophores. Thus, it was concluded that the molecular conformations and packing structures can be significantly affected by tuning the linking-positions of the alkoxy chains at the phenyl rings of the OCn derivatives, and under external stimuli, the crystals are endowed with stronger inner stress, easily-destructible structures and large conformation change due to the strongly twisted conjugated backbone and weaker intermolecular interaction.
| Compound | λ pressed | λ annealed | λ repressed | λ fumed | Δλa |
|---|---|---|---|---|---|
| a Δλ = λpressed − λannealed. Reproduced with permission from ref. 40. Copyright 2013 The Royal Society of Chemistry. | |||||
| oOC3 | 550 | 495 | 546 | 495 | 55 |
| oOC7 | 529 | 503 | 529 | 503 | 26 |
| oOC16 | 530 | 497 | 527 | 495 | 33 |
| mOC3 | 538 | 493 | 535 | 492 | 45 |
| mOC7 | 514 | 499 | 511 | 498 | 22 |
| mOC16 | 523 | 484 | 523 | 484 | 39 |
| pOC3 | 525 | 511 | 522 | 511 | 14 |
| pOC7 | 525 | 508 | 525 | 509 | 17 |
| pOC16 | 549 | 499 | 549 | 501 | 50 |
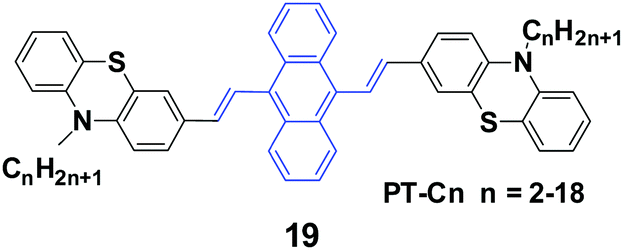
Yang et al.41 reported 9,10-bis(N-alkylphenothiazin-3-yl-vinyl-2)anthracenes (19, PT-Cn) with different carbon numbers (n) of the alkyl chains (n = 2, 3, 5, 6, 7, 9, 12, 18) to further understand the effect of alkyl lengths on the solid-state fluorescence and piezochromic luminescence of alkyl-containing 9,10-bis(arylvinyl)anthracenes. The results show that the spectra of pressed PT-Cn are blue-shifted after annealing, while the annealed PT-Cn containing longer alkyl chains exhibit more remarkable mechanofluorochromism (Table 3). Based on the PXRD and DSC results, the transformation between crystalline and amorphous states is responsible for the MFC and restoration behaviors under various external stimuli. This work demonstrates once again that it is possible to tune the solid-state optical properties of some organic fluorophores by simply altering the molecular chemical structure and changing the aggregate morphology under external stimuli. Wei et al. (2014)42 also reported the MFC behaviors of three 9,10-bis[(N-alkylphenothiazin-3-yl)vinyl]anthracene derivatives with propyl, hexyl, and dodecyl side chains.
| Compound | λ pressed | λ annealed | λ repressed | λ fumed | Δλa |
|---|---|---|---|---|---|
| a Δλ = λpressed − λannealed. Reproduced with permission from ref. 41. Copyright 2014 Elsevier. | |||||
| PT-Cn2 | 620 | 577 | 621 | 572 | 43 |
| PT-Cn3 | 608 | 568 | 614 | 553 | 40 |
| PT-Cn5 | 607 | 563 | 608 | 560 | 44 |
| PT-Cn6 | 605 | 554 | 605 | 541 | 51 |
| PT-Cn7 | 600 | 546 | 604 | 537 | 54 |
| PT-Cn9 | 599 | 547 | 600 | 546 | 52 |
| PT-Cn12 | 595 | 537 | 601 | 534 | 58 |
| PT-Cn18 | 588 | 517 | 593 | 514 | 71 |
Yang et al.43 reported the synthesis and optical properties of 9,10-bis(N-alkylindole-3-yl-vinyl-2)anthracenes (20, IACn) with different alkyl lengths. These homologs exhibit strong and negative alkyl length-dependent spectral shifts of 27–65 nm; that is to say, the compounds containing shorter N-alkyl chains present more significant spectral shifts induced by grinding or pressing (Fig. 7). As evidenced from PXRD results, after mechanical grinding, the pristine ordered structures are destroyed, crystal defects are formed, and some content of amorphous states emerges, which are the reasons for the MFC mechanism. It is found that the ground states could rapidly return back to the pristine states upon annealing or solvent-fuming, which is probably due to the existence of residual crystal seeds in the ground states. Given the fact that no cold-crystallization phenomenon occurred upon heating the ground samples, the amorphization should not play an essential role in determining the MFC behavior of IACn. It is noted that the IACn with shorter N-alkyl chains demonstrate more remarkable MFC behavior, and this phenomenon is the same as that observed in ACZn. It is only concluded that the different types of π-conjugated backbones and aromatic substituents might involve a different balance between the π–π and aliphatic interactions through the variation of electron density in the central ring and the length of the peripheral aliphatic tails, which makes the design of MFC materials and the understanding of the MFC phenomenon at the molecular level more complicated but represents a rich new approach to side chain engineering.43
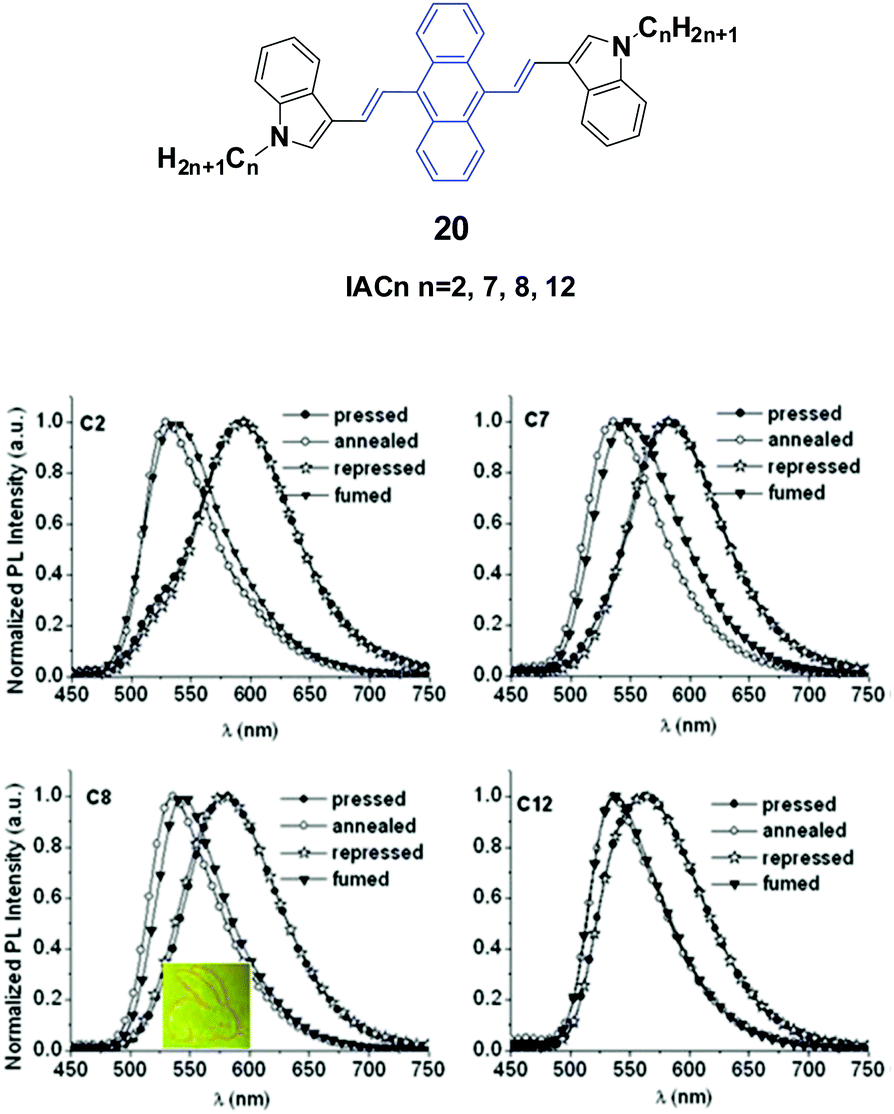 | ||
| Fig. 7 (Top) The molecular structure of compound 20. (Bottom) Normalized emission spectra of IACn samples upon external stimuli (excited at 470 nm). Inset: A piece of filter paper soaked with IAC8 on which a rabbit is drawn using a metal spatula and illuminated under UV light. Reproduced with permission from ref. 43. Copyright 2015 The Royal Society of Chemistry. | ||
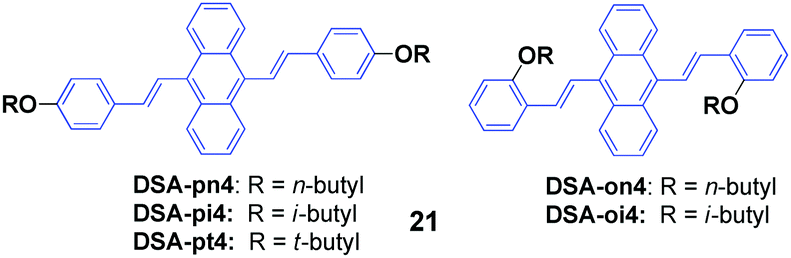
Isomers of 9,10-bis(butoxystyryl)anthracene (21, DSA4), including n-butyl, i-butyl, and t-butyl at the ortho or para positions, were designed and synthesized by Chen et al.44 All these DSA4 derivatives are AIE-active, but respond differently to pressing, leading to spectral shifts of 3–53 nm (Table 4), which indicates that the MFC behaviors are obviously dependent on the alkyl chains. Compared to the para-substituted ones, the ortho-substituted 9,10-bis(butoxystyryl)anthracenes display more prominent MFC properties. The molecular packing structures of the crystals change somewhat to amorphous features, as evidenced from the PXRD and DSC results. Single crystal analysis of the X-ray spectra reveals that their conformations maintained torsion angles between the anthracene core and peripheral aryl rings, which was consistent with the speculation that the PL emission peak could be shortened by enlarging the dihedral angle (DHA) of the molecule. This is because of a more twisted pattern that is formed in the molecules with a larger DHA, which hinders the internal molecular conjugation, leading to blue-shifted spectra. Moreover, all the crystals exhibit different packing structures. On one hand, DSA-pn4 adopted a stacking mode with J-type aggregation along the long axis of the molecule, and thus π–π interaction is hardly formed. The vertical distance between two close neighboring molecules is relatively short (3.476 Å), and thus tight intermolecular packing might account for its poor mechanofluorochromism (Δλ = 8 nm). On the other hand, the DSA-pt4 and DSA-oi4 crystals probably adopted a stacking mode with H-type aggregation; the vertical distances between two adjacent molecules are 3.637 Å and 3.546 Å, respectively. Given that DSA-pt4 and DSA-oi4 are stacked in loose molecular aggregations, their crystals are very sensitive to external pressure, which produces significant mechanofluorochromism.
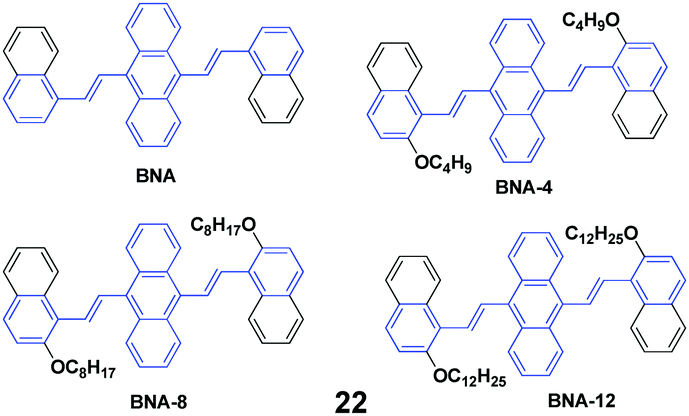
Four AIE-active derivatives of 9,10-bis[2-(2-alkoxynaphthalen-1-yl)vinyl]anthracene (22, BNAs) were synthesized and investigated by Chen et al.45 Upon pressing, the emission colors of these compounds underwent a red-shift from yellow to orange, which could revert to the original yellow by virtue of an annealing process, indicating the reversibility of the mechanofluorochromic properties. Notably, the length of the alkoxy group has a crucial effect on the MFC behaviors. BNA displayed the largest mechanofluorochromic spectral shift (26 nm), while the bathochromic shift decreased with increasing length of the alkoxyl chain (20 nm, 18 nm and 11 nm for BNA-4, BNA-8, and BNA-12, respectively). Therefore, changing the length of the alkoxyl chain provides an alternative way to tune the mechanofluorochromic behaviors, which is due to the steric effect caused by the alkoxyl chain. Meanwhile, the steric effect can also inhibit the planarization and intermolecular π–π overlap of BNAs. Therefore, under a mechanical stimulus, BNA can more easily transform from crystalline into an amorphous state than the micro-crystalline state of BNA-12. Thus, the mechanofluorochromic behaviors are related to the transformation from crystalline to an amorphous state, which is also evidenced by the PXRD profiles.
Lu et al.46 designed a series of 10,10′-bis(2-(N-alkylphenothiazine-3-yl)vinyl)-9,90-bianthracene compounds (23, PVBAn, n = 2, 8, 12 and 16) with different lengths of the N-alkyl chains and systematically studied the relationship between the chain length and the solid-state fluorescence properties. These compounds are strongly emissive in both solution and the solid state, and the emission spectral peaks are highly sensitive to the solvent polarity, implying intramolecular charge transfer (ICT) transitions. The ΦF values of the compounds also demonstrate a high dependence on the solvent polarity, while more polar solvents induced lower ΦF values. When the solvent is changed from n-hexane to dimethylformamide by increasing the solvent polarity, as for PVBA2, PVBA8, PVBA12 and PVBA16, their ΦF values are decreased from 36% to 1%, from 40% to 2%, from 59% to 3% and from 54% to 2%, respectively. Although the PVBAn solids exhibit similar mechanochromic behaviors, the grinding-induced spectral shifts of PVBAn show a high dependence on the alkyl length (Fig. 8). Compared to that of PVBA2, PVBAn with longer alkyl chains display higher fluorescence contrasts before and after grinding. Moreover, the fluorescence of ground solid PVBA16 can return back to the initial emission at room temperature due to its low cold-crystallization temperature, whereas high temperatures are required for the other compounds to recover the original emission. According to the PXRD and DSC analysis, the transformation between crystalline and amorphous states is responsible for the MFC behavior upon various external stimuli.
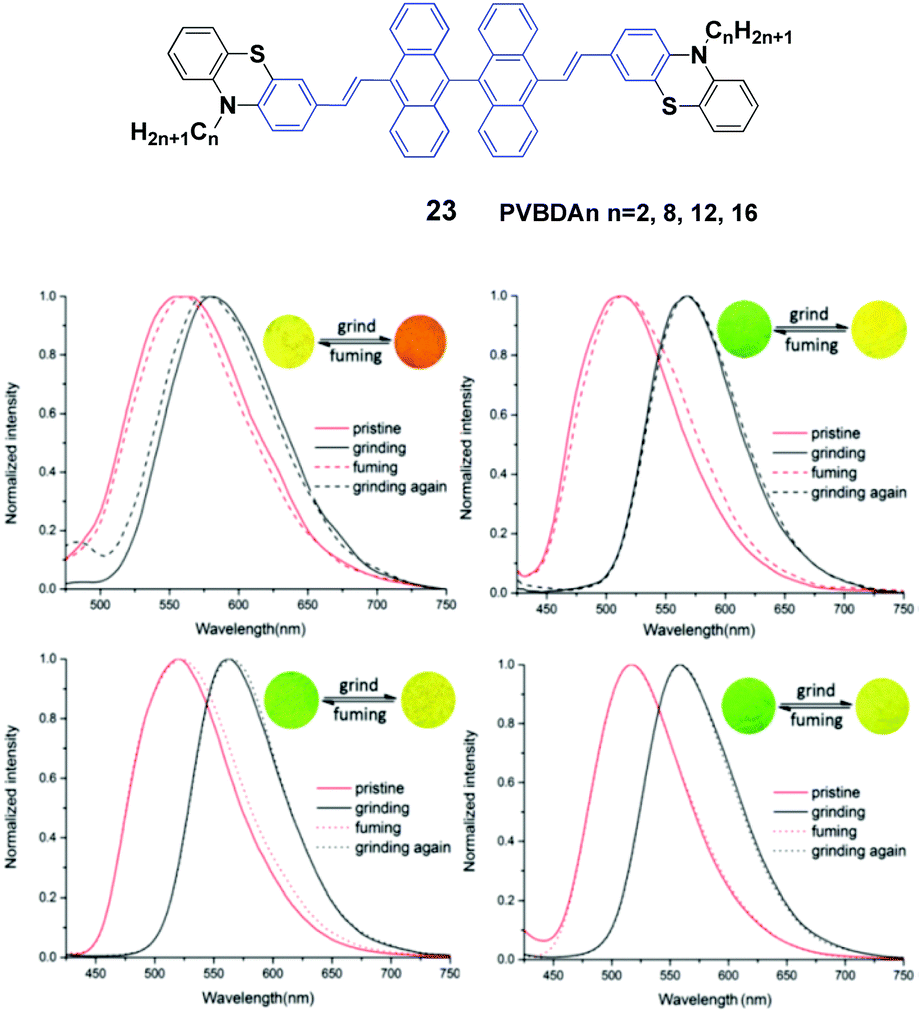 | ||
| Fig. 8 (Top) The molecular structure of compound 23. (Bottom) Normalized fluorescence spectra of (a) PVBA2, (b) PVBA8, (c) PVBA12 and (d) PVBA16 in the solid state under external stimuli. Inset: Schematic diagrams of photos under 365 nm light. Reproduced with permission from ref. 46. Copyright 2015 The Royal Society of Chemistry. | ||
Ouyang et al.47 synthesized two aggregation-induced enhanced emission (AIEE)-active compounds, BQVA (24) and BNVA (25). Upon increasing the solvent polarity, the emission color of the 24 solution red-shifts from green (527 nm) to orange (565 nm), suggesting the solvatochromism of the heteroatom-assisted 24. Notably, 24 exhibits reversible chromism properties, including mechano- and thermo-chromism. The as-prepared 24 powders emit green fluorescence (λem = 525 nm), which changes to orange emission (λem = 573 nm) after grinding, while the original color can be restored by annealing the ground sample at high temperature. Based on these reversible chromism properties, a simple and convenient erasable board has been designed. Different from 24, the non-heteroatom-assisted 25 exhibits no clear chromism properties (Fig. 9). Based on the PXRD, DSC, crystal structure and theoretical calculation results, the multi-chromism of 24 is closely related to the incorporated heteroatoms. Firstly, due to the heteroatoms in 24, an acceptor (A)–π–A structure is formed and produces solvatochromism. Secondly, due to the nitrogen atoms, intermolecular C–H⋯N bonds are formed and stabilize the molecular sheets in the 24 packing structures. These molecular sheets will slip upon external stimuli such as pressure or temperature, resulting in reversible multi-chromism. This work provides a new strategy to design and develop more multi-chromism materials. Additionally, 24 also exhibits excellent self-assembly effects in different solvents. Amino-functionalized nanoparticles (24@AFNPs) can be obtained by doping homogeneous nanospheres, which are formed in mixtures of THF and water, with silica nanoparticles and treating with 3-aminopropyltriethoxysilane, and thus 24@AFNPs can stain protein markers in polyacrylamide gel electrophoresis.
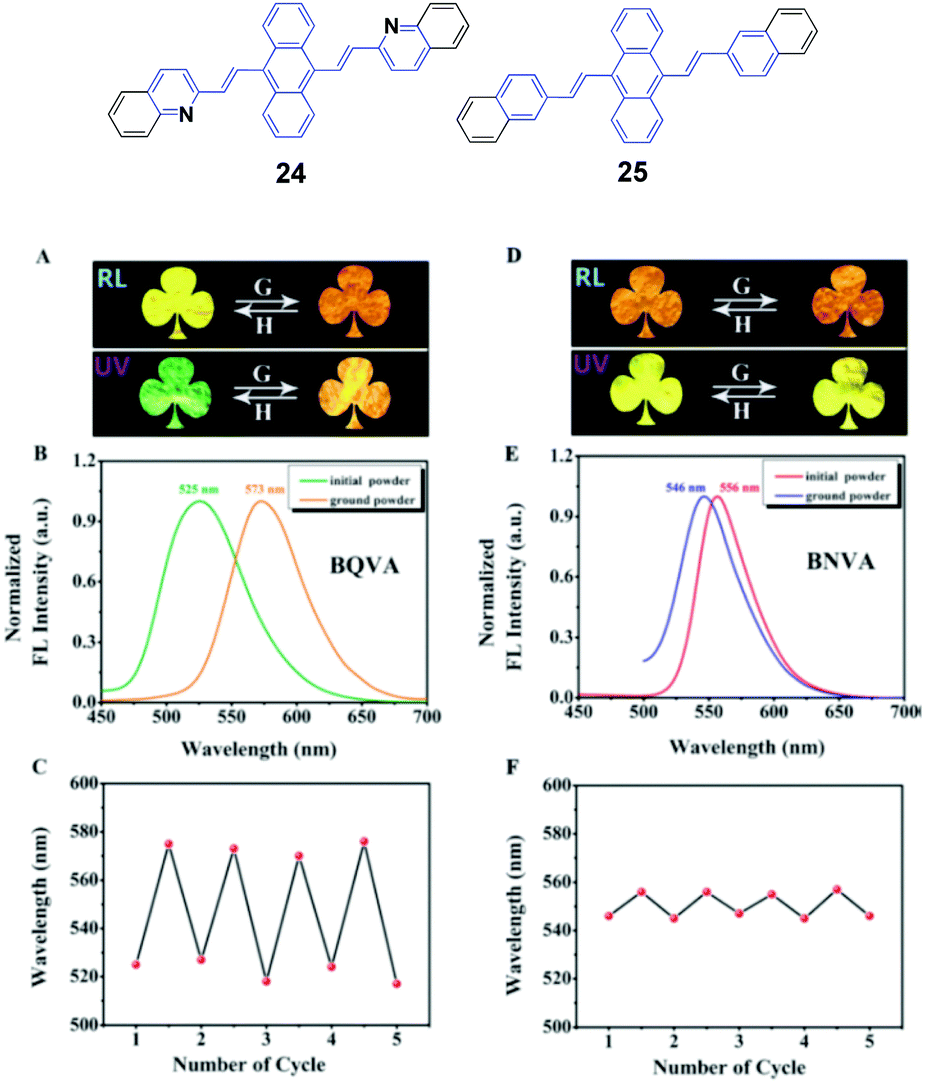 | ||
| Fig. 9 (Top) The molecular structures of compounds 24 and 25. (Bottom) (A) Photographs of the initial and ground powders of BQVA taken in room light (RL) and under UV light (UV). (B) Fluorescence (FL) spectra of the initial and ground powders of BQVA, excited at 365 nm. (C) Reversible switching of the emission wavelengths of BQVA after repeated grinding and heating cycles. (D) Photographs of the initial and ground powders of BNVA taken under RL and UV. (E) FL spectra of the initial and ground powders of BNVA, excited at λ = 365 nm. (F) Reversible switching of the emission wavelengths of BNVA after repeated grinding and heating cycles. The heating temperature was 130 °C. Reproduced with permission from ref. 47. Copyright 2015 Wiley-VCH. | ||
Yang et al.48 synthesized three N-phenylcarbazole-capped 9,10-divinylanthracene isomers through a change in the linking positions of N-phenylcarbazole, 9,10-bis(N-phenylcarbazol-2-/3-yl-vinyl-2)anthracenes 26 (CZ2) and 27 (CZ3), and 9,10-bis(4-(carbazol-9-yl)styryl)anthracene 28 (CZ9) with solid fluorescence quantum yields of 21%, 26% and 34.3%, respectively, which are all CEE-active and isomeric effects on the fluorescence and electroluminescence (EL) properties were examined. It is evidenced that the linking position of N-phenylcarbazole plays an important role in the fluorescence emission properties of the luminogens in both the crystalline and amorphous states. The crystalline state of CZ9 exhibits the strongest fluorescence efficiency, while that of the amorphous state of CZ9 is the weakest. After pressing, the emission peaks of the CZ2, CZ3, and CZ9 samples are 600, 593, and 565 nm, and when the pressed samples are annealed, the emission peaks change to 562, 552, and 528 nm, respectively, which are similar to those of the as-prepared solids. With further repressing, the emission colors show the same changes as that observed in the first pressing. Therefore, the CZ2, CZ3, and CZ9 luminogens exhibit reversible MFC properties upon an external stimulus. As confirmed by PXRD experiments, the MCF properties are associated with the grinding-induced change in aggregate structures. The diffractograms of the ground samples reveal notable amorphous features, considering the presence of broad and featureless reflections along with a series of overlapped peaks. The OLED devices are implemented by adopting the isomers as bulk emitting layers through vapor deposition. The CZ9-based device shows a turn-on voltage of 7.8 V, and a maximal luminance and luminous efficiency of 550 cd cm−2 and 0.10 cd A−1, respectively. In sharp contrast, the best EL performance is achieved in the CZ3-based device, which corresponds to 3.2 V, 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770 cd cm−2 and 3.1 cd A−1, respectively. The evident isomeric effect indicates that the subtle manipulation of peripheral groups is a feasible and efficient way to adjust the optical and optoelectronic properties of CEE luminogens.
770 cd cm−2 and 3.1 cd A−1, respectively. The evident isomeric effect indicates that the subtle manipulation of peripheral groups is a feasible and efficient way to adjust the optical and optoelectronic properties of CEE luminogens.
To investigate the effects of the molecular symmetry and isomerization on the light-emitting properties, Yang et al.49 described three isomeric 10-(arylvinyl)anthracenes with N-phenylcarbazole as the monoaryl moiety whose 2-, 3-, or carbazole-9-yl-phenyl positions are linked, leading to 29, 30, and 31, which show strong fluorescence in THF solutions with ΦF values of 78%, 41% and 85%, respectively. The optical and EL properties are investigated and compared with those of analogous 9,10-bis(arylvinyl)anthracenes. Contrary to the dual N-phenylcarbazole-capped analogs, the three mono N-phenylcarbazole-capped isomers are neither AIE-active nor MFC-active despite the fact that they feature a twisted π-backbone and grinding-induced amorphization. To study the origin of the stabilized fluorescence colors under the influence of a changed aggregate morphology, amorphous states of 29, 30, and 31 films are obtained through vacuum deposition. Fig. 10 shows that the 29, 30, and 31 give out green emission with a peak at 495, 500, and 490 nm, respectively, which is similar to those observed in the crystalline states. Thus, the fluorescence emission is relatively stabilized even though amorphization occurs after grinding, leading to the absence of mechanofluorochromism. The fluorescence decay profiles of 29, 30, and 31 solids show that the luminescence lifetime for each luminophore hardly changes before and after grinding at room temperature. Therefore, the fluorescence properties and stimuli-responsive behaviors of the materials are closely dependent on the substitution patterns, and the grinding-induced phase transition cannot guarantee emission change.
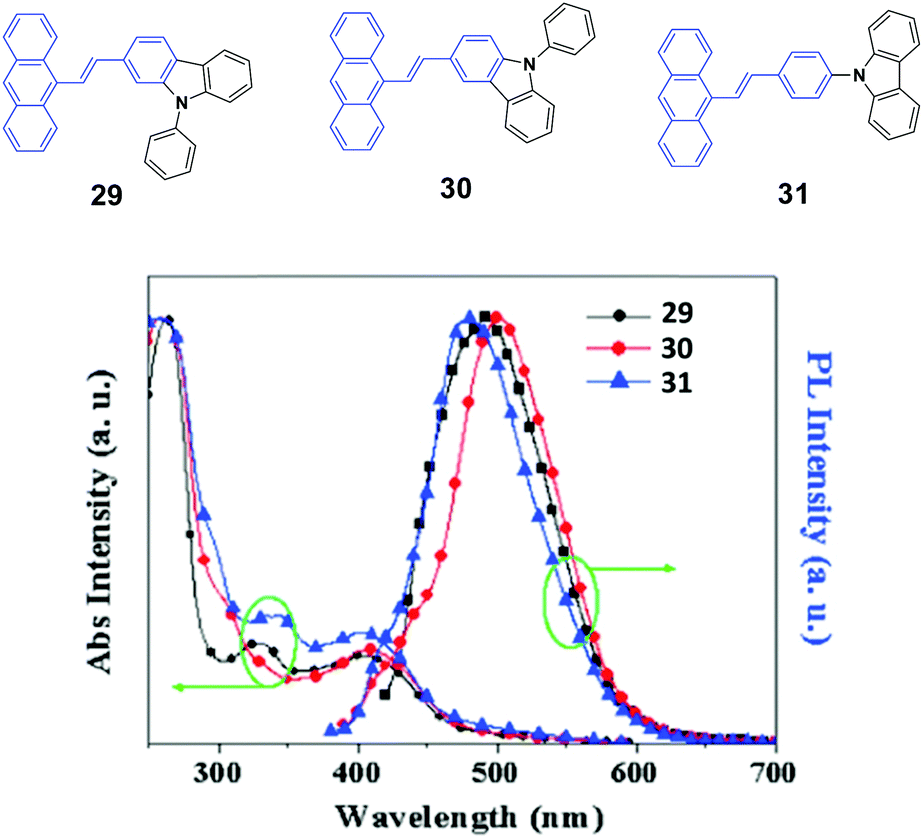 | ||
| Fig. 10 (Top) The molecular structures of compounds 29, 30 and 31. (Bottom) The normalized absorption and PL emission spectra of vacuum deposited 29, 30, and 31 films. Reproduced with permission from ref. 49. Copyright 2016 Elsevier. | ||
Chen et al. (2017)50 reported a series of 9-styrylanthracene-based luminophores with different geometries (unsymmetrical 31 and 32, symmetrical 33). The AIE-active 32 and 33 exhibit high solid-state fluorescence quantum efficiencies but contrary emission shifts under external force. When the pristine powder of 33 was ground with a spatula, the emission color underwent a bathochromic shift from yellow (548 nm) to orange (591 nm), and the mechanofluorochromic process is reversible by fuming the ground sample with dichloromethane for several minutes. Similarly, the emission color of 31 could be reversibly switched between 483 nm and 498 through grinding–fuming treatments. Unlike the mechanofluorochromic behavior with bathochromic-shifts generally observed from most reported AIE-active small molecules, a hypsochromic shift from 546 nm to 530 nm occurred after grinding 32. For traditional AIE small organic molecules with mechanofluorochromic properties, the reasons for the bathochromically shifted emission are the grinding-induced destruction of the intermolecular interactions in the crystals through slip deformation and enhanced molecular conjugations. In the case of 32, according to single crystal analysis, the authors speculated that the hypsochromically shifted emission was related to the grinding-induced decrease in excimer emission. In addition, the pristine states of 32, 31, and 33 exhibited ΦF values of 63%, 25% and 37%, respectively, which were 53%, 21% and 26% for their ground states respectively. For unsymmetrical molecules of 32 and 31, the fluorescence lifetime (τ) values of both were reduced after grinding. However, as for symmetrical luminophore 33, the τ value was slightly prolonged after grinding, which indicated the existence of efficient excitonic and excimeric coupling between the ground 33 molecules. Based on the PXRD and DSC results, the mechanofluorochromism of these molecules was probably associated with the partial transformation between the highly ordered form and the amorphous state.
Xu and Tian's group have reported a series of divinylanthracene derivatives with fascinating properties.51 Due to the twisted conformation and multiple supramolecular interactions, these molecules present high solid-state fluorescence quantum efficiencies and special molecular aggregation states. By modifying the molecular structures, interesting phenomena including AIE properties, self-assembly behavior, and amplified spontaneous emission are observed. In particular, when pyridyl groups are further incorporated into the molecular structure, more supramolecular interactions such as N–H hydrogen bonds are induced and also different crystal polymorphs are formed. Such polymorphs are helpful for investigating the structural changes in molecular aggregates under mechanical force and understanding the intrinsic process of the piezochromic fluorescence. Moreover, the basicity of the pyridyl group also provides an ideal model to reveal the effect of a protonation stimulus on tuning the fluorescence properties. Xu and Tian's group52 also implemented a combined study based on a novel derivative of 9,10-bis((E)-2-(pyridin-4-yl)vinyl)anthracene (34, BP4VA), which is the isomer of 2 and has multistimuli responsive fluorescence properties. 34 possesses two polymorphs with high fluorescence efficiencies, and the different polymorphs can be used to explain its piezochromic properties by means of high pressure-PL experiments. By grinding the initial 34 powder with a pestle and mortar, the fluorescence emission color turns from green (523 nm) to yellow (555 nm), while the original green is fully restored when the ground sample is heated at 170 °C for about 20 min. Thus switchable emission change between green and yellow can endure repeated cycles. PXRD of the initial powder displays sharp and intense reflections that are consistent with the simulated PXRD pattern from the crystal data of C1, where the molecular arrangement is J-type aggregation along the molecular long axis and correspondingly π–π interactions between the central anthracene planes are absent (Fig. 11a and b). The PXRD profiles of the ground samples are the same as those of the unground samples; however, the weakened intensities of the peaks and broadened shape suggest grinding-induced change in the aggregation state. This indicates that the crystal structure of 34 can be readily influenced by an external stimulus. To further understand the mechanochromic behavior, the luminescence of 34 was investigated under the effect of applied pressure. As shown in Fig. 11c, upon increasing the applied pressure from 0 to 6 GPa, the luminescence color of the 34 powder gradually red-shifted from green (523 nm) to red (650 nm), along with the appearance of multiple fluorescence peaks. Meanwhile, the crystal sample also exhibits an obvious transition of the fluorescence emission, implying that the packing structure is varied under high pressure. Therefore, the external pressure has an important influence on the luminescence colors of the 34 powder and crystal samples, resulting from the changed molecular aggregation states under high pressure. Additionally, fluorescence changes are investigated based on controlling protonation–deprotonation, bringing about switched emission from green to red under different acidic/basic atmospheres.
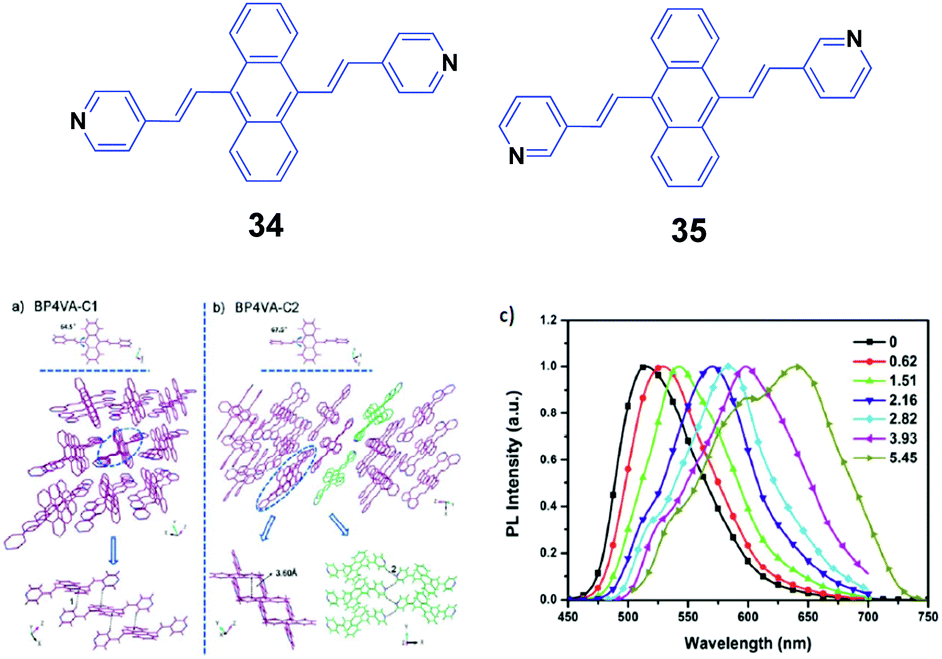 | ||
| Fig. 11 (Top) The molecular structures of compounds 34 and 35. (Bottom) The aggregation state of 34 in the two single crystals: (a) C1 and (b) C2. (c) Normalized solid-state PL spectra of the 34 powder after the application of pressure (GPa). Reproduced with permission from ref. 52. Copyright 2013 The Royal Society of Chemistry. | ||
Tian et al.53 investigated the self-assembly behavior, molecular stacking structure and photophysical properties of two polymorphs of a supramolecular co-crystal (C1 and C2), consisting of an AIEE-active luminogen 34 as the luminescent host molecule and 1,3,5-trifluoro-2,4,6-triiodobenzene as the guest molecule. The ΦF of the 34 crystal is 50.3%, which is only 36.5% in THF solution. As for the block-like crystal C1 which is packed in segregated stacking, there are strong π–π interactions between the host and guest molecules, and it gives off weak green emission with a low efficiency of 2%. However, for the needle-like crystal C2 which is packed in segregated stacking, there are no obviously strong intermolecular interactions, and it emits bright yellow emission with a high efficiency of 34%. Furthermore, C1 exhibits a unique mechanochromic behavior with the spontaneous recovery of the grinding-induced fluorescence change. After grinding, the emission peak red-shifts from the initial 510 nm to the final 546 nm, which can return back to 515 nm after 24 hours. Notably, the ground C1 exhibits more intense emission than the C1 crystal, and the sufficient difference between the emission intensities of the pristine and the ground crystal means that it can be easily distinguished by the naked eye. Thus, manipulating the supramolecular interactions of the co-crystal can not only control the stacking mode but also tune the optical properties. Given the merits of diversity and versatility for organic molecular crystals, it is expected that highly efficient supramolecular luminescent systems and new functions for organic luminescent materials can be developed by combining the AIE with supramolecular self-assembly, which is a new flexible and controllable design strategy.
A similar study was done by Zhao's group.54 They successfully developed two mechanochromic assemblies from BP4VA (34) or BP3VA (35) and 1,2,4,5-tetrafluoro-3,6-diiodobenzene via halogen bonding and π–π stacking interactions. The two molecules have slightly different backbones, and enable two different assembly structures with linear wires and networks. The two assemblies exhibit significant piezochromic behavior because they can respond to different external pressures along with a change in emission color, while such color changes are reversible through thermal annealing treatment, indicating their potential applications in practical devices. Due to the advantages of unique features such as high sensitivity and easy handling, the present supramolecular assemblies are helpful to design and prepare touch memory performance systems through noncovalent bonding strategies.
In 2014, Yang et al. synthesized a series of anthracene-centered cruciforms 3655 with a tetra-donor, 3756 with a di-donor and 3857 with a di-donor and di-acceptor. All the luminogens are AIEE-active, and light- and heat-stable. 36 shows weak fluorescence in THF solution with a ΦF of 2.9%, which reaches up to 17.6% when a high content of water (90%) is added. 36 shows large and enhanced two-photon absorption cross sections (σ), fluorescence sensing selectivity to transition metal ions such as Zn2+ and Cu2+, and appreciable mechanofluorochromism. Upon pressing, the emission peak of the 36 solid red-shifts from the original 606 nm to the final 615 nm, which reverts back to ∼582 nm when the pressed sample is treated by solvent-fuming or heat-annealing. With repeated cycles of pressing/fuming or pressing/annealing, the fluorescence color shows the same change as that observed in the first cycle. As for 37, its fluorescence efficiency in 1/9 THF/water (ΦF = 18.1%) is 12 times as high as that in THF solution (ΦF = 1.53%). Both 37 and 38 solids are endowed with MFC behaviors upon pressing and annealing, and the emission color can be switched reversibly between orange-red (602 nm) and red (632 nm), and between red (617 nm) and deep red (654 nm), respectively. Additionally, 38 exhibits a strong solvatochromic effect, as well as solvent-dependent two-photon absorption cross sections, whose maximal σ values reach 670, 1840, and 2030 GM in dichloromethane, tetrahydrofuran, and toluene solutions, respectively. According to PXRD and DSC measurements, the phase transformation between crystalline and amorphous states is responsible for the MFC behaviors of the series of anthracene-centered cruciforms under an external stimulus.
Yang et al.58 also synthesized a series of 2,6-bis(p-dialkylaminostyryl)-9,10-distyrylanthracene (39, FCn) cruciforms consisting of N-alkyl chains with different lengths, and aggregation-enhanced fluorescence and MFC behaviors were investigated. FCn exhibit higher ΦF values in aqueous dispersions than those in THF solutions. For instance, the ΦF of FC4 increases from 12% in THF solution to 25% in a THF/water (1/9) mixture. The FCn exhibit MFC properties, as confirmed by the variable spectral shifts Δλ (23–54 nm) that are induced by grinding and pressing, while the FCn with longer alkyl chains are likely to trigger larger spectral shifts (Table 5). Based on PXRD and DSC results, the reversible MFC behavior is associated with the transformation between the crystalline and amorphous states. It is found that increasing the N-alkyl length can effectively decrease the cold-crystallization temperature of the ground states; thus the MFC states are endowed with a tunable heat-recovering temperature, which accounts for the spontaneous recovery of the original FC10 and FC12 solids from their ground states at room temperature.
| Compound | λ pristine | λ pressed | λ annealed | λ repressed | λ fumed | Δλa |
|---|---|---|---|---|---|---|
| a Δλ = λpressed − λannealed. Reproduced with permission from ref. 58. Copyright 2014 The Royal Society of Chemistry. | ||||||
| FC1 | 591 | 616 | 593 | 615 | 593 | 23 |
| FC4 | 597 | 613 | 588 | 613 | 588 | 25 |
| FC7 | 563 | 595 | 565 | 593 | 563 | 30 |
| FC8 | 541 | 598 | 544 | 597 | 547 | 54 |
| FC10 | 538 | 597 | 552 | 597 | 554 | 45 |
| FC12 | 551 | 596 | 547 | 597 | 555 | 49 |
In 2017, Chi's group59 successfully synthesized an AIEE-active polymer, 40, wherein the backbone 9,10-distyrylanthracene and side tetraphenylethene units were adopted as two monomers. This polymer is AIEE-active, and the solid powder has a high ΦF of 90%, which is only 12% in THF solution. 40 exhibits mechanochromic properties, showing an emission peak change from 541 nm to 602 nm upon grinding the original sample. It is revealed that the crystalline structure of the polymer is destroyed upon external stimuli, and then the molecular conformation becomes planarized and molecular conjugation gets increased in the polymer chain segments, resulting in red-shifted emission. Considering that it is difficult to fully recover the destroyed crystalline structure, the initial fluorescence color cannot be restored completely by thermal annealing or solvent fuming of the ground polymer.
4. Summary and outlook
Research on MFC materials has been expanding rapidly due to their propitious applications in diverse fields. In this review, we discuss recent research progress on MFC materials based on AIE molecules derived from distyrylanthracene. The structure–property relationships and mechanisms of the MFC materials have been described. Although distyrylanthracene itself showed no MFC, its derivatives with substitutions or modifications on its phenyl groups exhibited fascinating MFC properties. A high contrast emission color change with a large spectral shift of 90 nm has been achieved in an N-alkyl carbazoyl group substituted distyrylanthracene derivative. However, their MCF properties could be affected by a variety of structure modifications, including the replaced groups for the phenyl units, the new substituted groups and their positions on the phenyl units, and the attached alkyl chains in these groups. Generally, large and crowded groups endow the compounds with more twisted conformations, leading to more obvious MCF properties with larger spectral shifts. Longer alkyl chains could also enhance their MCF features, except for some carbazoyl (or indolyl) substituted compounds. When heteroatom-containing phenyl groups, such as pyridyl groups, were used instead of normal phenyl groups in these dipyridinylanthracene compounds, their MCF characteristics could be enhanced as well. It is the additional C–H⋯N bonds that stabilize the molecular sheets and assist them to form some special stackings, which plays a critical role in their MCF properties. Therefore, their MCF behaviors will become more various when these stackings are destroyed by external mechanical stimuli. Besides, the ability of crystallization also affects the MCF feature of these distyrylanthracene derivatives. Although great success has been achieved, MFC materials are still in their infancy; thus, their MFC mechanism is not very clear and their applications need to be further explored.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the NSFC (51733010, 61605253, 21672267 and 51603232), the Science and Technology Planning Project of Guangdong (2015B090913003 and 2015B090915003) and the Fundamental Research Funds for the Central Universities.References
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429–3436 CrossRef PubMed.
- X. Q. Zhang, Z. G. Chi, H. Y. Li, B. J. Xu, X. F. Li, W. Zhou, S. W. Liu, Y. Zhang and J. R. Xu, Chem. – Asian J., 2011, 6, 808–811 CrossRef PubMed.
- J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119–122 CrossRef.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef PubMed.
- J. Chen, B. Xu, X. Ouyang, B. Z. Tang and Y. Cao, J. Phys. Chem. A, 2004, 108, 7522–7526 CrossRef.
- F. Wang, M.-Y. Han, K. Y. Mya, Y. Wang and Y.-H. Lai, J. Am. Chem. Soc., 2005, 127, 10350–10355 CrossRef PubMed.
- B. Xu, Z. Chi, H. Li, X. Zhang, X. Li, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. C, 2011, 115, 17574–17581 Search PubMed.
- B. J. Xu, Z. G. Chi, X. F. Li, H. Y. Li, W. Zhou, X. Q. Zhang, C. C. Wang, Y. Zhang, S. R. Liu and J. R. Xu, J. Fluoresc., 2011, 21, 433–441 CrossRef PubMed.
- J. He, B. Xu, F. Chen, H. Xia, K. Li, L. Ye and W. Tian, J. Phys. Chem. C, 2009, 113, 9892–9899 Search PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef PubMed.
- Y. Dong, J. Y. Lam, Z. Li, A. Qin, H. Tong, Y. Dong, X. Feng and B. Tang, J. Inorg. Organomet. Polym., 2005, 15, 287–291 CrossRef.
- Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40–42 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Sun, J. Liu, Z. Li, J. Sun, H. H. Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Chem. Commun., 2007, 3255–3257 RSC.
- X. Fan, J. Sun, F. Wang, Z. Chu, P. Wang, Y. Dong, R. Hu, B. Z. Tang and D. Zou, Chem. Commun., 2008, 2989–2991 RSC.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef PubMed.
- L. Ma, X. Feng, S. Wang and B. Wang, Mater. Chem. Front., 2017, 1, 2474–2486 RSC.
- P. C. Xue, J. P. Ding, P. P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688–6706 RSC.
- S. Mukherjee and P. Thilagar, J. Mater. Chem. C, 2016, 4, 2647–2662 RSC.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef.
- V. Sathish, A. Ramdass, P. Thanasekaran, K. L. Lu and S. Rajagopal, J. Photochem. Photobiol., C, 2015, 23, 25–44 CrossRef.
- Z. Y. Ma, Z. J. Wang, M. J. Teng, Z. J. Xu and X. R. Jia, Chem. Phys. Chem., 2015, 16, 1811–1828 CrossRef PubMed.
- J. Mei, Y. N. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef PubMed.
- X. Q. Zhang, Z. G. Chi, Y. Zhang, S. W. Liu and J. R. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC.
- X. Q. Zhang, Z. G. Chi, J. Y. Zhang, H. Y. Li, B. J. Xu, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef PubMed.
- H. Y. Li, X. Q. Zhang, Z. G. Chi, B. J. Xu, W. Zhou, S. W. Liu, Y. Zhang and J. R. Xu, Org. Lett., 2011, 13, 556–559 CrossRef PubMed.
- H. Y. Li, Z. G. Chi, B. J. Xu, X. Q. Zhang, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, J. Mater. Chem., 2011, 21, 3760–3767 RSC.
- Y. J. Dong, B. Xu, J. B. Zhang, X. Tan, L. J. Wang, J. L. Chen, H. G. Lv, S. P. Wen, B. Li, L. Ye, B. Zou and W. J. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef PubMed.
- X. Q. Zhang, Z. G. Chi, X. Zhou, S. W. Liu, Y. Zhang and J. R. Xu, J. Phys. Chem. C, 2012, 116, 23629–23638 Search PubMed.
- X. Q. Zhang, Z. G. Chi, B. J. Xu, L. Jiang, X. Zhou, Y. Zhang, S. W. Liu and J. R. Xu, Chem. Commun., 2012, 48, 10895–10897 RSC.
- W. Liu, Y. L. Wang, L. Y. Bu, J. F. Li, M. X. Sun, D. T. Zhang, M. Zheng, C. Yang, S. F. Xue and W. J. Yang, J. Lumin., 2013, 143, 50–55 CrossRef.
- L. Y. Bu, M. X. Sun, D. T. Zhang, W. Liu, Y. L. Wang, M. Zheng, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2013, 1, 2028–2035 RSC.
- L. Y. Bu, Y. P. Li, J. F. Wang, M. X. Sun, M. Zheng, W. Liu, S. F. Xue and W. J. Yang, Dyes Pigm., 2013, 99, 833–838 CrossRef.
- Y. L. Wang, W. Liu, L. Y. Bu, J. F. Li, M. Zheng, D. T. Zhang, M. X. Sun, Y. Tao, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2013, 1, 856–862 RSC.
- W. Liu, S. Ying, Q. K. Sun, X. Qiu, H. C. Zhang, S. F. Xue and W. J. Yang, Dyes Pigm., 2016, 125, 8–14 CrossRef.
- K. Duraimurugan, J. Sivamani, M. Sathiyaraj, V. Thiagarajan and A. Siva, J. Fluoresc., 2016, 26, 1211–1218 CrossRef PubMed.
- W. Liu, Y. L. Wang, M. X. Sun, D. T. Zhang, M. Zheng and W. J. Yang, Chem. Commun., 2013, 49, 6042–6044 RSC.
- M. Zheng, M. X. Sun, Y. P. Li, J. F. Wang, L. Y. Bu, S. F. Xue and W. J. Yang, Dyes Pigm., 2014, 102, 29–34 CrossRef.
- X. Q. Zhang, Z. Y. Ma, Y. Yang, X. Y. Zhang, Z. G. Chi, S. W. Liu, J. R. Xu, X. R. Jia and Y. Wei, Tetrahedron, 2014, 70, 924–929 CrossRef.
- Q. K. Sun, W. Liu, S. A. Ying, L. L. Wang, S. F. Xue and W. J. Yang, RSC Adv., 2015, 5, 73046–73050 RSC.
- Y. Xiong, X. L. Yan, Y. W. Ma, Y. Li, G. H. Yin and L. G. Chen, Chem. Commun., 2015, 51, 3403–3406 RSC.
- X. Y. Teng, X. C. Wu, Y. Q. Cao, Y. H. Jin, Y. Li, X. L. Yan, B. W. Wang and L. G. Chen, Chin. Chem. Lett., 2017, 28, 1485–1491 CrossRef.
- P. C. Xue, B. Q. Yao, X. H. Liu, J. B. Sun, P. Gong, Z. Q. Zhang, C. Qian, Y. Zhang and R. Lu, J. Mater. Chem. C, 2015, 3, 1018–1025 RSC.
- C. X. Niu, Y. You, L. Zhao, D. C. He, N. Na and J. Ouyang, Chem. – Eur. J., 2015, 21, 13983–13990 CrossRef PubMed.
- S. F. Xue, W. Liu, X. Qiu, Y. Y. Gao and W. J. Yang, J. Phys. Chem. C, 2014, 118, 18668–18675 Search PubMed.
- Q. L. Hu, J. F. Wang, L. Yin, M. S. Chen, S. F. Xue and W. J. Yang, J. Lumin., 2017, 183, 410–417 CrossRef.
- X. Zhang, Y. X. Wang, J. Zhao, P. F. Duan, Y. L. Chen and L. Chen, Chem. – Asian J., 2017, 12, 830–834 CrossRef PubMed.
- Y. Dong, B. Xu, J. Zhang, H. Lu, S. Wen, F. Chen, J. He, B. Li, L. Ye and W. Tian, CrystEngComm, 2012, 14, 6593–6598 RSC.
- Y. J. Dong, J. B. Zhang, X. Tan, L. J. Wang, J. L. Chen, B. Li, L. Ye, B. Xu, B. Zou and W. J. Tian, J. Mater. Chem. C, 2013, 1, 7554–7559 RSC.
- Y. J. Liu, S. Q. Ma, B. Xu and W. J. Tian, Faraday Discuss., 2017, 196, 219–229 RSC.
- L. Bai, P. Bose, Q. Gao, Y. Li, R. Ganguly and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 436–441 CrossRef PubMed.
- W. Liu, J. F. Wang, Y. Y. Gao, Q. K. Sun, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2014, 2, 9028–9034 RSC.
- M. X. Sun, D. T. Zhang, Y. P. Li, J. F. Wang, Y. Y. Gao and W. J. Yang, J. Lumin., 2014, 148, 55–59 CrossRef.
- D. T. Zhang, Y. Y. Gao, J. Dong, Q. K. Sun, W. Liu, S. F. Xue and W. J. Yang, Dyes Pigm., 2015, 113, 307–311 CrossRef.
- M. Zheng, D. T. Zhang, M. X. Sun, Y. P. Li, T. L. Liu, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2014, 2, 1913–1920 RSC.
- J. R. Chen, J. Zhao, B. J. Xu, Z. Y. Yang, S. W. Liu, J. R. Xu, Y. Zhang, Y. C. Wu, P. Y. Lv and Z. G. Chi, Chin. J. Polym. Sci., 2017, 35, 282–292 CrossRef.
| This journal is © the Partner Organisations 2018 |