
Catalytic stereoselective total synthesis of a spiro-oxindole alkaloid and the pentacyclic core of tryptoquivalines†
Tao
Wei
and
Darren J.
Dixon
 *
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: darren.dixon@chem.ox.ac.uk
First published on 15th October 2018
Abstract
An expedient route to the pentacyclic core of the tryptoquivaline alkaloids and the total synthesis of natural product (+)-3′-(4-oxoquinazolin-3-yl)spiro[1H-indole-3,5′-oxolane]-2,2′-dione (1) have been achieved. The route is enabled by a key, highly stereoselective, aldol reaction catalysed by a Ag(I) and cinchona-derived aminophosphine ligand system, forming a highly substituted oxazoline ring, and setting the C1 spirocyclic stereocentre for downstream manipulation.
Among the large family of quinazolinone alkaloids,1–3 tryptoquivalines4–6 represent a unique subset characterised by their [5,5]oxo-spirocyclic core, N,N-aminal moiety and quinazolinone ring systems (Fig. 1). Previous approaches to construct the spirocyclic core have focussed on oxidative cyclisation approaches. For example, Büchi7 (Scheme 1) and Ban8 explored oxidative lactonization for establishing the spirocyclic C1 position, with stereocontrol arising from the existing C7 position. Subsequent introduction of the methyl alanine moiety to form the final N,N-aminal ring structure then afforded tryptoquivaline L and its C7-epimer.‡ The thermodynamically stable epi-1 of natural product 1 was chosen by the Büchi group to complete the synthesis of both tryptoquivaline L and C7-epi-tryptoquivaline L. In an alternative approach, Nakagawa and co-workers9,10 reported an oxidative double cyclization where the generation of the new C1 stereocentre was followed by conversion to tryptoquivaline L, its C7 epimer and ent-tryptoquivaline L within a concise synthetic sequence (Scheme 2).
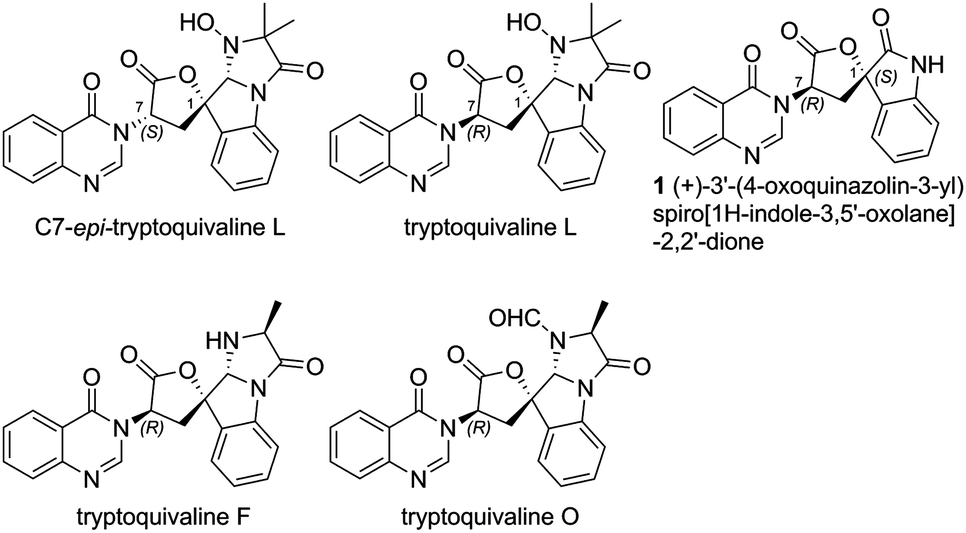 | ||
| Fig. 1 Selected quinazolinone alkaloids. | ||
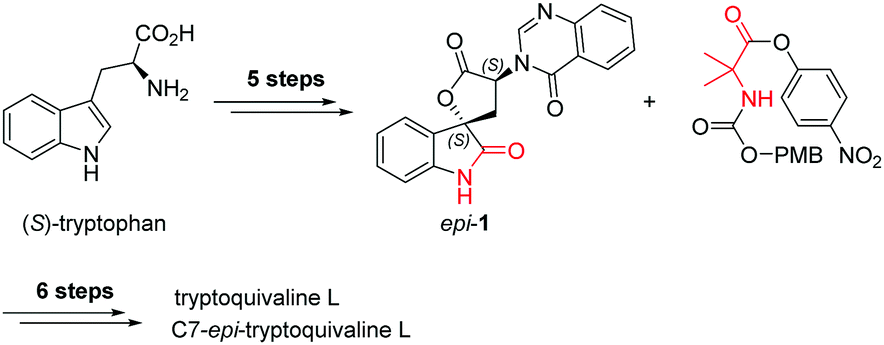 | ||
| Scheme 1 Previous synthetic route by Büchi et al. | ||
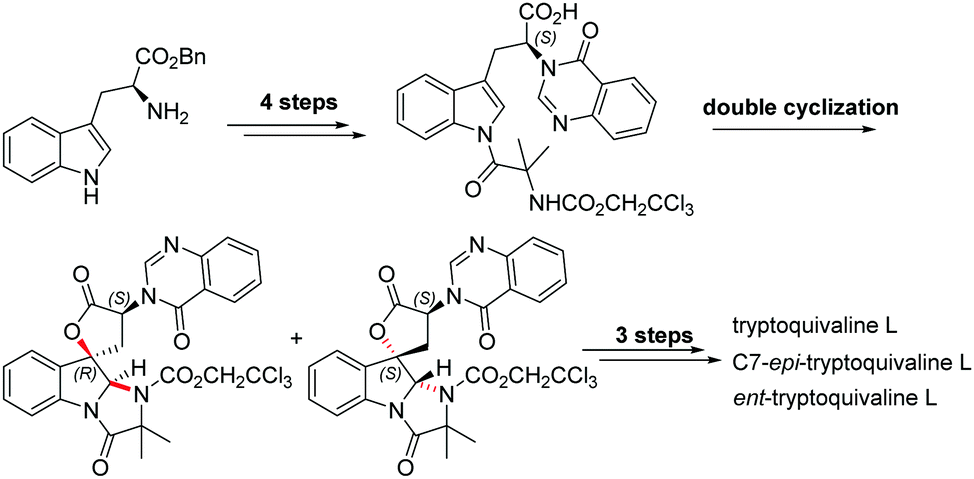 | ||
| Scheme 2 Previous synthetic route by Nakagawa et al. | ||
Against these precedents our aim was to develop a new total synthesis of 1 based on a catalytic stereoselective approach to generate the C1 stereocentre which would be independent of any substrate bias, thus potentially enabling access to other family members. Our general strategy to natural product 1, (+)-3′-(4-oxoquinazolin-3-yl)spiro[1H-indole-3,5′-oxolane]-2,2′-dione, is presented in Scheme 3. Pivotal to our approach was the stereoselective addition reaction of a masked one-carbon nucleophilic unit into a suitably functionalised quinazolinone-containing aryl ketone derivative where the resulting tertiary alcohol was poised to form the C-ring in a lactonisation process. In turn, the spiro oxindole D-ring could be accessed through a suitable intramolecular C–N coupling reaction. We recognized that the broad-scope silver catalysed enantio- and diastereoselective isocyanoacetate aldol methodology recently developed in our group,11,12Scheme 4, could be applied to deliver the necessary one carbon unit through oxidative manipulation en route to the target.
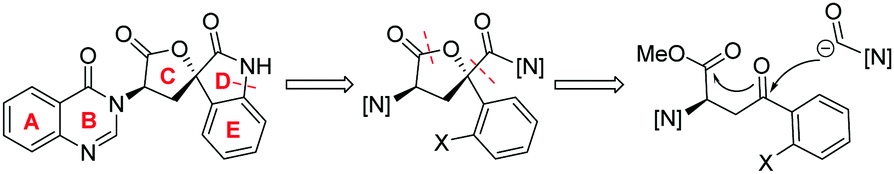 | ||
| Scheme 3 This work synthetic plan. | ||
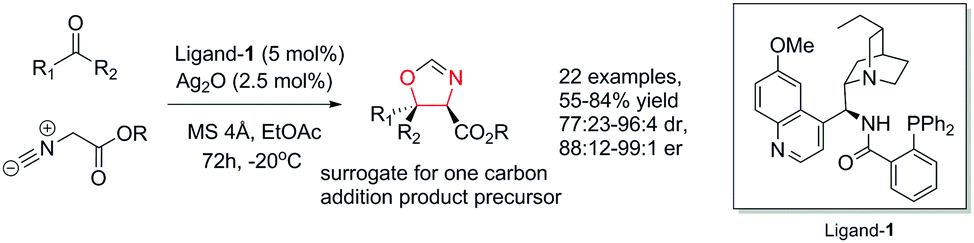 | ||
| Scheme 4 Underpinning enantioselective isocyanoacetate ketone aldol methodology. | ||
Our route to the key functionalised aryl ketone substrate 5 (poised for the stereoselective isocyanoacetate ketone aldol reaction) is shown in Scheme 5. Starting from commercially available D-tryptophan methyl ester hydrochloride, amine acylation with 2-nitrobenzoyl chloride gave the amide 2 in excellent yield. The indole ring was then subjected to an oxidative cleavage13,14 with sodium periodate to afford a mixture of free aniline 3 and its formamide derivative, which upon methanolysis with methanolic HCl, gave aniline 3 in 84% yield. Diazotisation with tert-butyl nitrite and subsequent treatment with KI afforded iodoarene 4 in 63% yield. The nitro group of 4 was reduced using iron powder in the presence of ammonium chloride, a method that was sufficiently mild to leave the previously installed iodide intact. Subsequent treatment with trimethyl orthoformate and p-toluenesulfonic acid lead to the efficient formation of the quinazolinone ring15 of desired ketone 5.
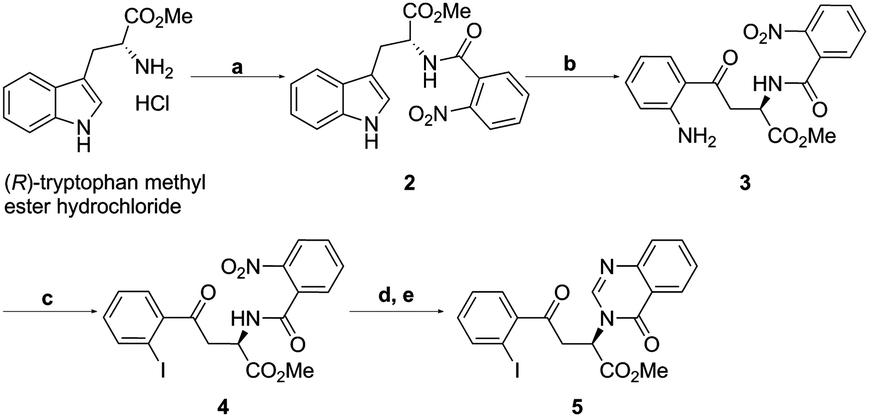 | ||
| Scheme 5 (a) 2-Nitrobenzoyl chloride, NaHCO3, CH2Cl2/H2O, 99%; (b) NalO4, MeOH/H2O; then SOCl2, MeOH, 84%; (c) HBF4, tBuONO, EtOH, then Kl, acetone, 63%; (d) Fe, NH4Cl, EtOH/H2O, 95%; (e) pTSA, CH(OMe)3, CH3OH, 82%. | ||
Before carrying out the key stereoselective isocyanoacetate aldol reaction on 5, the viability of the planned methodology was first investigated on a readily prepared model substrate 6,§ as shown in Scheme 6. In the study, both quinine and quinidine-derived aminophosphine ligands,16 ligand-1 and ligand-2 respectively, were tested to ascertain reactivity and stereocontrol. It was found that ligand-1 resulted in a mismatched outcome as shown by a 12.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, whereas ligand-2 afforded a matched result with a 20:1 dr.¶ This study uncovered that the natural diastereoselectivity of the substrate was weak and could be readily overridden with ligand control, thus giving us confidence to perform the reaction on real substrate 5 with accurate prediction of stereochemical outcome.
:1 dr, whereas ligand-2 afforded a matched result with a 20:1 dr.¶ This study uncovered that the natural diastereoselectivity of the substrate was weak and could be readily overridden with ligand control, thus giving us confidence to perform the reaction on real substrate 5 with accurate prediction of stereochemical outcome.
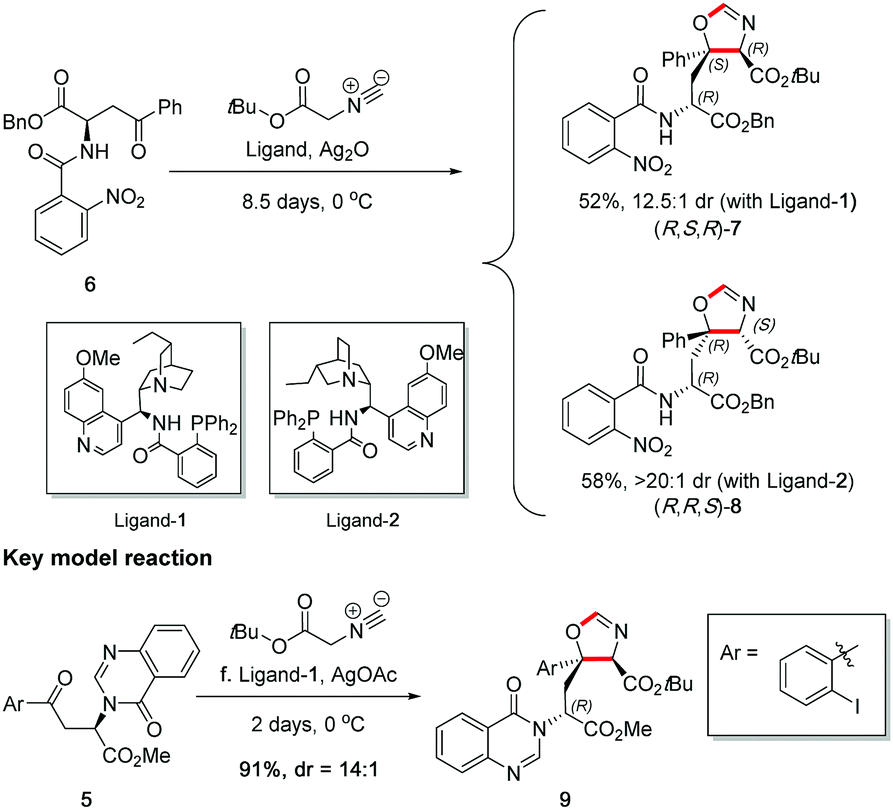 | ||
| Scheme 6 Key catalyst-controlled isocyanoacetate stereoselective Aldol reaction. | ||
The key stereoselective reaction was then performed between the chiral aryl ketone 5 and tert-butyl isocyanoacetate, catalysed by silver(I) acetate and quinine-derived aminophosphine ligand-1 (Scheme 6) which pleasingly afforded oxazoline 9 bearing two contiguous stereocentres in high yield (91%) and high diastereoselectivity (dr = 14:1) on gram scale. The application of this robust and general methodology on the highly functionalized ketone substrate demonstrates its excellent performance, adaptability and functional group tolerance.
The manipulation of isocyanoacetate aldol product 9 into spirocyclic products is shown in Scheme 7. The oxazoline was readily hydrolysed to amino formamide 10 with 1 N hydrogen chloride solution11 in quantitative yield. Then, an acetic acid mediated lactonisation gave lactone 11 in 70% yield. Formamide deprotection with HCl in methanol took place smoothly, and then the amino ester intermediate was oxidized with IBX17 to give imine 12 in 77% yield. Copper mediated Buchwald type C–N bond formation, to ester indolenine 13 was achieved in 52% yield as shown in Scheme 5. With 13 in hand and inspired by Bode18 and others,19–21 we conceived the decarboxylative rearrangement of oxaziridine intermediate 19 to directly give natural product 1. However, various conditions failed to realise this transformation.
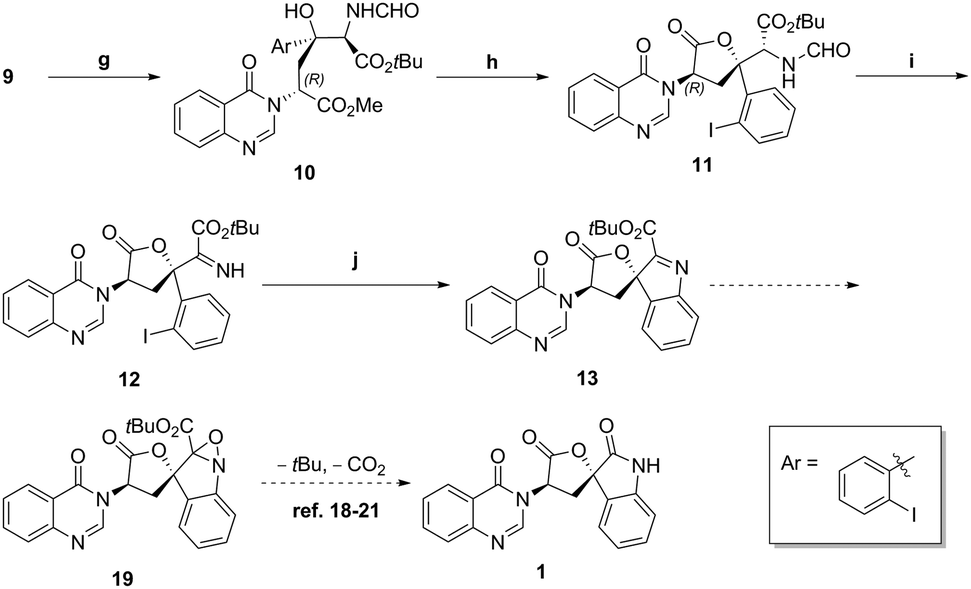 | ||
| Scheme 7 (g) 1 N HCI, THF, quant; (h) AcOH, toluene, 70%; (i) SOCl2, MeOH, then IBX, CH2Cl2/DMSO, 77%; (j) Cul, Mg(OAc)2·4H2O, DMSO, 52%. | ||
Accordingly, an alternative sequence to 1 was followed as shown in Scheme 8. Imine 12 was first hydrolysed to α-ketoester 14 with acetic acid in good yield, then subsequently treated with TFA to afford α-keto acid 15 in quantitative yield. The generated α-keto acid moiety was oxidatively cleaved with PIDA22 to give carboxylic acid 16 in good yield (pyridinium dichromate23 was far less efficient). Acid 16 was then converted to amide 17 in 70% yield, using a HATU mediated coupling with 4-methoxybenzylamine. The final D-ring was formed by a copper(I) mediated Buchwald type C–N bond coupling condition, giving spiro oxindole 18 in 70% yield. Due to the ease of epimerization at the C7 position, only weak bases were tolerated, magnesium acetate tetrahydrate being found to be optimal. Natural product 1 was finally obtained by deprotection of the PMB group with triflic acid24,25 in good yield. The spectroscopic data and specific rotation of the synthetic material were in good agreement with that of the isolated material4 confirming the total synthesis of 1 in 15 steps from D-tryptophan methyl ester.
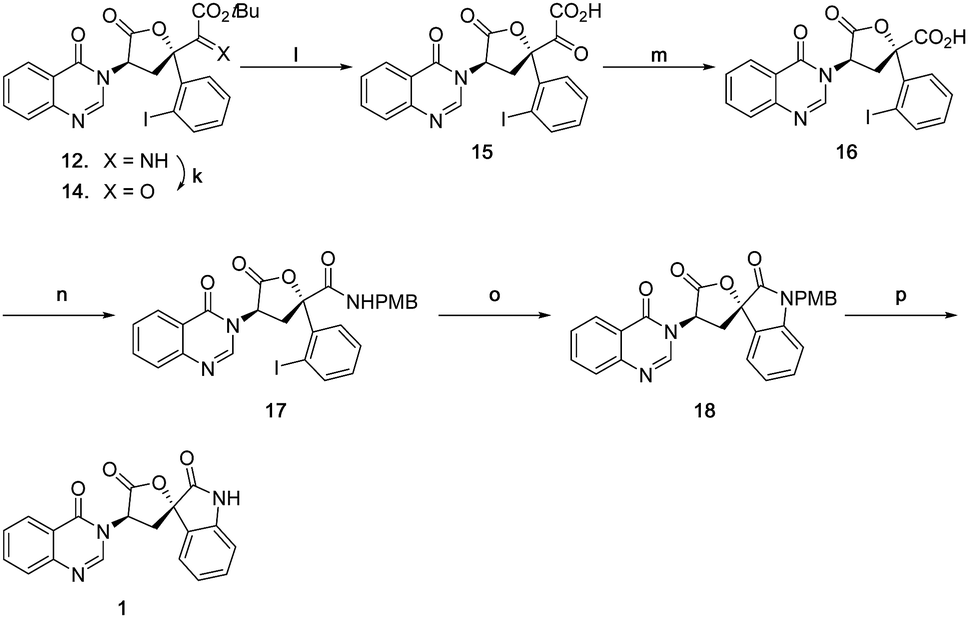 | ||
| Scheme 8 (k) AcOH, THF/H2O, 73%; (l) TFA/CH2Cl2, quant; (m) Phl(OAc)2, AcOH/H2O, 80%; (n) PMBNH2, DIPEA, HATU, CH2Cl2/DMF, 70%; (o) Cul, Mg(OAc)2·4H2O. DMSO, 70%; (p) TfOH, CH2Cl2, 64%. | ||
In summary, the catalytic stereoselective total synthesis of natural product 1, 3′-(4-oxoquinazolin-3-yl)spiro[1H-indole-3,5′-oxolane]-2,2′-dione, was achieved from D-tryptophan methyl ester hydrochloride in a linear route of 15 steps with an overall yield of 6.4%. The key step featured a gram scale catalyst controlled stereoselective aldol reaction to construct the highly substituted oxazoline ring bearing two contiguous stereocentres in 91% yield with a dr 14:1, poised for downstream manipulation. Our approach to 1 lays the foundation for further synthetic approaches to quinazolinone alkaloids and the details will be reported in due course.
TW wishes to thank the China Scholarship Council for PhD funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Numata, C. Takahashi, T. Matsushita, T. Miyamoto, K. Kawai, Y. Usami, E. Matsumura, M. Inoue, H. Ohishi and T. Shingu, Tetrahedron Lett., 1992, 33, 1621–1624 CrossRef CAS.
- C. Takahashi, T. Matsushita, M. Doi, K. Minoura, T. Shingu, Y. Kumeda and A. Numata, J. Chem. Soc., Perkin Trans. 1, 1995, 2345 RSC.
- G. Büchi, K. C. Luk, B. Kobbe and J. M. Townsend, J. Org. Chem., 1977, 42, 244–246 CrossRef.
- S. Buttachon, A. Chandrapatya, L. Manoch, A. Silva, L. Gales, C. Bruyère, R. Kiss and A. Kijjoa, Tetrahedron, 2012, 68, 3253–3262 CrossRef CAS.
- M. Yamazaki, H. Fujimoto and E. Okuyama, Chem. Pharm. Bull., 1978, 26, 111–117 CrossRef CAS.
- J. Clardy, J. P. Springer, G. Béchi, K. Matsuo and R. Wightman, J. Am. Chem. Soc., 1975, 97, 663–665 CrossRef CAS PubMed.
- G. Büchi, P. R. DeShong, S. Katsumura and Y. Sugimura, J. Am. Chem. Soc., 1979, 101, 5084–5086 CrossRef.
- T. Ohnuma, Y. Kimura and Y. Ban, Tetrahedron Lett., 1981, 22, 4969–4972 CrossRef CAS.
- M. Nakagawa, M. Ito, Y. Hasegawa, S. Akashi and T. Hino, Tetrahedron Lett., 1984, 25, 3865–3868 CrossRef CAS.
- M. Nakagawa, M. Taniguchi, M. Sodeoka, M. Ito, K. Yamaguchi and T. Hino, J. Am. Chem. Soc., 1983, 105, 3709–3710 CrossRef CAS.
- R. de la Campa, I. Ortin and D. J. Dixon, Angew. Chem., Int. Ed., 2015, 54, 4895–4898 CrossRef CAS PubMed.
- R. de la Campa, A. D. Gammack Yamagata, I. Ortín, A. Franchino, A. L. Thompson, B. Odell and D. J. Dixon, Chem. Commun., 2016, 52, 10632–10635 RSC.
- M. Takemoto, Y. Iwakiri, Y. Suzuki and K. Tanaka, Tetrahedron Lett., 2004, 45, 8061–8064 CrossRef CAS.
- T. He, X. Tao, J. Yang, D. Guo, H. Xia, J. Jia and M. Jiang, Chem. Commun., 2011, 47, 2907 RSC.
- D. Zhao, T. Wang and J.-X. Li, Chem. Commun., 2014, 50, 6471–6474 RSC.
- F. Sladojevich, A. Trabocchi, A. Guarna and D. J. Dixon, J. Am. Chem. Soc., 2011, 133, 1710–1713 CrossRef CAS PubMed.
- C. Zheng, I. Dubovyk, K. E. Lazarski and R. J. Thomson, J. Am. Chem. Soc., 2014, 136, 17750–17756 CrossRef CAS PubMed.
- I. Pusterla and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 513–516 CrossRef CAS PubMed.
- M. Bucciarelli, A. Forni, I. Moretti, G. Torre, A. Prosyanik and G. Remir, J. Chem. Soc., Chem. Commun., 1985, 998–999 RSC.
- J. S. Splitter and M. Calvin, J. Org. Chem., 1958, 23, 651–652 CrossRef CAS.
- J. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
- B. Podolesov, J. Org. Chem., 1984, 49, 2644–2646 CrossRef CAS.
- L. Y. Shi, J. Q. Wu, D. Y. Zhang, Y. C. Wu, W. Y. Hua and X. M. Wu, Synthesis, 2011, 3807–3814 CAS.
- A. El Bouakher, S. Massip, C. Jarry, Y. Troin, I. Abrunhosa-Thomas and G. Guillaumet, Eur. J. Org. Chem., 2015, 556–559 CrossRef CAS.
- H. Takada, N. Kumagai and M. Shibasaki, Org. Lett., 2015, 17, 4762–4765 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07479h |
| ‡ Originally reported tryptoquivaline G was structurally revised to tryptoquivaline L (ref. 4) and accordingly in this paper the originally reported tryptoquivaline L was referred to as C7-epi-tryptoquivaline L for clarity. |
| § For the preparation of 6, see the ESI.† |
| ¶ Stereochemical configuration was predicted by analogy to previous work and confirmed by NOE on a lactam derivative of (R,S,R)-7; see S3 in the ESI.† |
| This journal is © The Royal Society of Chemistry 2018 |