
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and pharmaceutical applications of porous particles
Miaomiao Zhou
ab,
Lan Shena,
Xiao Lin
 *a,
Yanlong Hongb and
Yi Feng
*b
*a,
Yanlong Hongb and
Yi Feng
*b
aCollege of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. E-mail: duotang@163.com
bEngineering Research Center of Modern Preparation Technology of TCM of Ministry of Education, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China. E-mail: shutcmfyi@163.com
First published on 14th August 2017
Abstract
Porous particles (PPs) are a novel kind of carrier with a large surface area, high porosity, stable uniform porous structure, tunable pore sizes with narrow distribution, and well defined surface properties. There are many methods for preparing PPs, mainly including using porogens or templating agents to form pores, self-forming pores by particle matrices without any pore-forming agents, and chemical synthesis. In this review, reports on various PPs are collected, which are applied in various fields of medicine for many purposes, such as improvement of dissolution of poorly soluble drugs, development of novel drug delivery systems, and improvement of the functional properties of drug-loaded particles. In general, these functions of PPs are closely related to their fundamental particle and pore properties, such as morphology, size, surface property, surface area, and porosity. The reasonable application of PPs has markedly facilitated the development of pharmaceutics and the number of published reports has increased progressively from year to year. However, there remains a serious deficiency of summarization and systematic analysis of these reports. Therefore, this review aims to provide a comprehensive and relevant overview on the design and pharmaceutical applications of PPs, which can serve as an informative reference for interested readers.
 Miaomiao Zhou | Miaomiao Zhou received her BS degree from Anhui University of Chinese Medicine in 2016. She is now a master's degree candidate in Shanghai University of Traditional Chinese Medicine. Her current research direction focuses on the applications of porous particles in traditional Chinese medicine. |
 Xiao Lin | Xiao Lin received his Ph.D. degree in Pharmaceutics from Shanghai University of Traditional Chinese Medicine (TCM), China, in 2004. He is currently working as a professor in Shanghai University of TCM. His research interests focus on particle engineering of excipients and TCM for tableting and drug delivery systems for TCM. |
 Yi Feng | Yi Feng is a Professor of the Engineering Research Center of Modern Preparation Technology of TCM of Ministry of Education, Shanghai University of Traditional Chinese Medicine, China. She received her Ph.D. degree from Shanghai University of Traditional Chinese Medicine, China, in 2000. Her research interests focus on physical modification of TCM powders and digitized technology platforms for TCM manufacturing. |
1. Introduction
Nowadays, it is difficult for many newly developed active pharmaceutical ingredients (APIs) to obtain the desired efficacy or be produced in desired dosage forms mainly due to their poor physicochemical, micromeritic, physicomechanical, and/or pharmacokinetic properties. To solve this, scientists have devoted their efforts to investigate a variety of new carriers, dosage forms, and drug delivery systems.1–4 As part of the effort, porous particles (PPs) based on old pharmaceutical materials emerged and have been developed as a novel carrier to improve drug delivery, dissolution, tableting, and so on.5–7PPs are low-density solids with open or closed pore structures that provide a large exposed surface area.8 Originated in the twentieth century, they have been used in various fields, and especially in the pharmaceutical field significant results have been achieved. According to pore diameter, PPs are classified into three categories: macroporous (pore diameter > 50 nm), mesoporous (2–50 nm), and microporous (<2 nm).9 The pore diameter can be tailored by the preparation conditions, which plays a vital role in the properties of PPs.10,11 Compared to common particles, PPs possess several distinct features, such as large surface area, high porosity, uniform and tunable pore structure, and well defined inner and outer surface properties, which render them more suitable for being used as drug carriers.12 Generally, drugs are loaded into the pores and on the surfaces of PPs to improve their dissolution and achieve sustained or targeted release thanks to the distinct structure of PPs.
It is reported that particle engineering involves the operation at three solid state levels, i.e., crystal, particle, and bulk levels.13 PPs mainly belong to the particle level's operation, which involves individual particle properties (e.g. size, shape, surface area, and porosity). Moreover, the particle level can cause changes in the bulk level, which involves properties like tabletability, flowability, etc.13–15 Namely, PPs can be used to improve fundamental properties and functional properties of particles simultaneously. PPs with excellent fundamental properties, such as large surface area and high porosity, can achieve markedly improved functional properties like adsorbability and compactability, which are essential for effective drug loading and property improvement.
Investigations on applications of PPs have been the hot focus of multiple pharmaceutical fields (including drug delivery, particle engineering, treatment of diseases, and separation)16–19 and have provided encouraging opportunities to solve scientific problems once considered intractable. Similarly, the preparations of PPs have also attracted scientists' interests.20–22 The reasonable application of PPs has markedly facilitated the development of pharmaceutics and the number of published reports has increased progressively from year to year, resulting in an increasing understanding of PPs. However, there remains a serious deficiency of summarization and systemic analysis of these reports. Therefore, this review aims to provide a comprehensive and relevant overview on the design and pharmaceutical applications of PPs, which can be served as an informative reference for relevant readers.
2. Design of porous particles
Porous particles (PPs) are usually made from certain parent particles by adding some ingredients followed by removing them or using certain techniques to form pores. In more details, the methods for preparing PPs are summarized and discussed as follows.Firstly, the most common method is the use of porogens or templating agents within particles in the first step, followed by removing them in the second step to produce pores (Fig. 1). In fact, the pore forming mechanism of porogens and templates is different. In terms of porogens, they mainly form pores by producing gas or volatilization during the preparation process. For example, Bae et al.23 manufactured porous poly(lactic-co-glycolic acid) microsphere by utilizing hydrogen peroxide (HP) as porogen. An enzymatic reaction between HP and catalase formed oxygen bubbles which naturally developed many internal pores within microsphere. In the preparation of porous microspheres (PMSs), the physicochemical properties of porogens seem to have a great influence on the size distribution and porous features of microspheres.24 In another study, Al-Khattawi et al.22 fabricated porous mannitol using ammonium bicarbonate (NH4HCO3) as porogen by spray drying. It is reported that NH4HCO3 sublimes above 50 °C and is removed directly during spray drying which leaves voids in particles because of the evolution of H2O↑, NH3↑, and CO2↑.25,26 Camphor was also applied as porogen to produce porous granules, which depended on its sublimation by exposure to vacuum.27 Besides the above, the commonly studied subliming agents also include menthol and thymol.28 As for templating agents, they need to be washed using solvent or removed by heat treatment (including calcination) in the second step where differs from porogens. In some cases, sulfuric acid can also be used for removing a copolymer template.29 There are many kinds of templates, including (i) ethanol-soluble food-grade acids30 and carbohydrate sugars,31 (ii) polystyrene32,33 and other polymers,34,35 and (iii) inorganic salts.36 Moreover, there exists a special templating agent, namely, emulsion, a two-phase system consisting of continuous phase and dispersed phase. Polymerization of the continuous phase of the emulsion followed by removing the dispersed phase using ethanol leads to the formation of pores.37 Generally, porous lactose or mannitol tends to be fabricated using templates. Although using porogens or templates is applicable to the preparation of most PPs, there are still shortcomings. Either porogens or templating agents must be removed in the second processing step and thus require a relatively tedious process. So, it's more difficulty to carry out in large scale and has potential detrimental effects in clinic if they are not completely removed.37 In addition, the removing process must not have any meaningful effects on the other components of PPs.
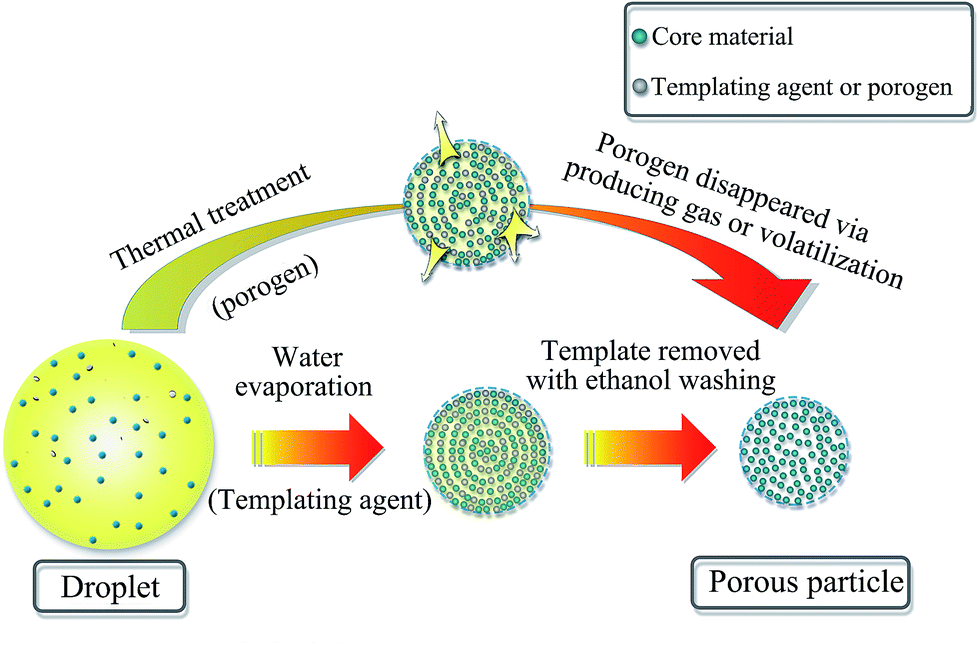 | ||
| Fig. 1 The preparation of porous particles by using porogen or templating agent. | ||
In comparison with the above facts, the second method does not use porogens or templates anymore (Fig. 2). Wei et al.21 utilized amphiphilic mPEG-based polymers with different hydrophobic segments as microspheres matrix to prepare PMSs with narrow size distribution based on a double emulsion-premix membrane emulsification technique. This process can self-form the pores without any porogens or templates. The mechanisms of the pores-forming process possibly go as follows: (i) water molecules were absorbed by hydrophilic PEG segments and then evaporated, forming the pores on the microsphere surface, (ii) local explosion of microspheres occurred due to fast evaporation of solvent used as oil phase in emulsion, which played a critical role in determining the porous structure of microspheres.38 Results showed that the property of the hydrophobic segment had an important impact on structure characteristics of pores, which can be used to prepare different PMSs. This method involves in amphiphilic polymers which can be employed as permanent geometric porous matrix, so it is applicable to the preparation of PMSs. Nevertheless, theoretical and experimental studies on this method are still deficient, and thus, it is less used so far.
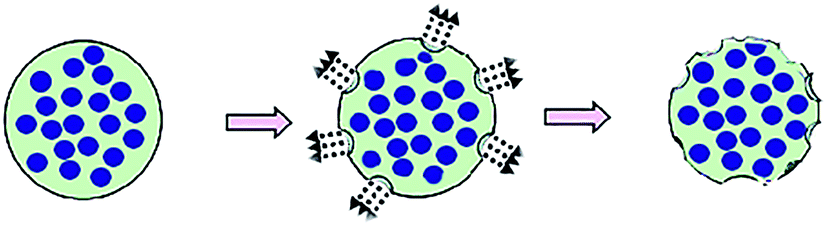 | ||
| Fig. 2 The preparation of porous particles by using amphiphilic polymers as particle matrix. | ||
The third method is to prepare PPs by synthesis, which mainly involves in Si-related PPs. For instance, the basic method to produce porous silicon is electrochemical dissolution of Si in HF based solutions obtained by monitoring either the anodic current (galvanostatic) or voltage (potentiostatic).39 Generally, constant current can control the porosity and thickness better. Mesoporous silica nanoparticles (MS-NPs) are usually synthesized by reacting a self-assembling silica TEOS (tetraethyl ortho-silicate) or sodium silicate and a surfactant (usually quaternary ammonium salt) micelle.20 And then the surfactant, which acts as a structure directing agent, is removed by calcination or extraction, causing a porous silicate network. The combination between the negatively charged silicate and the positively charged surfactant micelles contributes to an ordered structure of MS-NPs. Furthermore, porous calcium silicate hydrate (CSH) was fabricated based on reactivity-enhanced SiO2 (i.e. silica/polyethylene glycol (PEG2000) composites) as the source silica material at a low hydrothermal temperature of 110 °C.40 The pore formation mechanism of CSH was believed to be ascribed to that cavitation, which resulted from sonication, enabled PEG2000 (via intercalation on silica) to break apart Si–O–Si bonds and, thus, enhanced SiO2 reactivity at a low hydrothermal temperature.
Besides the above three methods, there are also some specific ways to manufacture certain PPs. For example, porous calcium carbonate can be created by re-precipitation of inorganic mixed salts (e.g., calcium phosphate and calcium carbonate) under controlled conditions.41 Enzymatic reaction between raw starches and starch hydrolyzing enzymes (e.g., amylase or amyloglucosidase) at sub-gelatinization temperatures offers an attractive alternative for producing porous starch.42 In addition, PPs can be even manufactured only via adopting specific preparation conditions such as spray freeze drying or vacuum freeze drying.43,44
In general, each approach has its applicability, advantages, and disadvantages, so researchers should choose the most appropriate method to prepare the corresponding PPs according to their own purposes.
3. Types of porous particles (PPs) (Table 1)
3.1. Porous silicon (PSiN)
PSiN is a extensively studied biomaterial fabricated by the electrochemical dissolution of Si in HF-based solutions.39 Intriguingly, its essential properties, such as pore size, surface area, and particle size, can be regulated in the preparation process, which make it a very multifunctional material used in medical filed.45 Different from most PPs, PSiN can enhance the antimicrobial effect of drugs. In addition, it can also be used for oral drug delivery, especially for cancer therapy.PSiN shows excellent in vivo stability, nonimmunogenic profiles, and low cytotoxicity, which makes it ideal for oral drug delivery.46 The loading and release behaviors of five model drugs (antipyrine, ibuprofen, griseofulvin, ranitidine and furosemide) incorporated into PSiN microparticles were studied.47 In the process of loading, besides factors affecting the stability of the particles, surface properties would affect compound affinity towards the particles.47 In addition, the loading solution and the chemical nature of the drug seemed to be vital to the loading process. On the other hand, the drug release rate of drug-loaded PSiN microparticles depended on the drug's own dissolution behavior. Namely, the PSiN microparticles caused a delayed release when the plain drug had a high dissolution rate. In contrast, the release rate of poorly dissolving drugs loaded into the PSiN microparticles was clearly improved.47
Modified PSiN nanoparticles play an important role in cancer therapy. They can be used for controlled and targeted anti-cancer drug delivery, which implies that carriers can be triggered to release the drug once it has reached its target. For example, PSiN nanoparticles grafted with a temperature responsive (TR) polymer could be triggered externally by infrared (IR) or radiofrequency (RF) radiation.48 Inhibition of tumor growth and a prolongation of the survival time of animals were obtained after using this carrier system in combination with radiation. Both IR and RF radiations well triggered the release of drug. The conformation of the TR polymer implanted on the surface of PSiN nanoparticles changed in response to the heating induced by radiation, which allowed the loaded drug to escape from the PSiN pores and, thus, achieved controlled and targeted drug release. High efficacy of such nanoparticles against malignant cells were observed both in vitro and in vivo.48 In another study, Xia et al.49 fabricated photothermal nanocarriers, i.e., polyaniline/PSiN hybrid nanocomposites (P/PSiN-NPs), for combined chemo-photothermal therapy of cancer. These biodegradable and non-toxic PSiN-based nanocomposites could be completely cleared in mouse body after intravenous injection. Doxorubicin hydrochloride (DOX), a model anticancer drug, was loaded into P/PSiN-NPs efficiently. Most importantly, the combination of chemotherapy and photothermal therapy based on DOX-loaded P/PSiN-NPs showed a synergistic inhibition effect on both the growth of tumors in vivo and the proliferation of cancer cells in vitro.49 As a whole, these PSiN-based composite carriers are promising for cancer treatments in clinic.
Peptides have great potential in the treatment of severe illnesses. However, an effective delivery system, such as thermally hydrocarbonized mesoporous silicon microparticles (THC-PSiNs), is necessary to prolong their action after administration. D-lys-GHRP6 (ghrelin antagonist, GhA), used as a model peptide, was loaded into THC-PSiNs.50 Food intake and blood pressure are two distinct parameters used to examine the pharmacological activity of GhA. Results showed that GhA-loaded THC-PSiNs inhibited food intake of mice and rats for a longer period and increased their blood pressure more slowly than a control GhA solution, thus exhibiting a stronger pharmacological activity. Cytokines are a part of the immune system whose appearance relates to infections and inflammatory conditions. Since plain THC-PSiNs do not increase cytokine activity, they are believed to be a suitable carrier for drug delivery.50 Besides, a sustained and tailorable release system for peptide YY3-36 using PSiN was also achieved.51
As mentioned above, PSiN has a unique characteristic, i.e., it can enhance the antimicrobial effect of drugs. Vale et al.52 reported that the loading of ethionamide (ETH), a drug used to treat Mycobacterium tuberculosis, into PSiN enhanced the permeability and solubility of ETH, as well as its metabolism process. Then, carboxylic acid functionalized thermally hydrocarbonized porous silicon nanoparticles conjugated with ETH were synthesized, which exhibited much better antimicrobial effect than free ETH.52 It is supposed that the essence of the synergy between the nanoparticles and ETH is owing to the weakening of the bacterial cell wall, which enhances conjugate penetration.
3.2. Porous silica (PSi)
PSi has large specific surface area and high porosity due to its porous structure. It was reported that PSi could be synthesized using emulsions as template.37 Sylysia 350, a common type of PSi, with pore size at 21.0 nm and pore volume at 1.6 mL g−1, has many silanol groups on its surface and can be applied as a pharmaceutical excipient for oral administration because of its good biocompatibility, stability, and nontoxicity.7,53 In addition, it has an excellent capacity to improve the powder properties and the dissolution of drugs. Therefore, PSi is widely used as a carrier to enhance various properties of drugs in pharmaceutical field.Solid dispersion (SD) is commonly used for improving the dissolution rate of poorly water-soluble drugs. Recently, there are many literatures reported that PSi combined with the SD technology (PSi-SD) achieved preferable solubilization of drugs.7,53–55 For example, Yan et al.7 reported that tanshinone IIA (tanIIA) in PSi-SD with different ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to 1:8) showed significantly improved dissolution rates (∼4-fold at maximum) compared to pure tanIIA and corresponding physical mixtures. Besides, the AUC0–24 h of tanIIA from PSi-SD increased by 1.97 times compared with pure tanIIA, demonstrating that its oral bioavailability was improved by SD with PSi. Takeuchi et al.53 prepared the SDs of indomethacin with non-porous Si or PSi. The latter showed faster drug dissolution rate. When the latter achieved complete drug dissolution, the former only released 80% of the drug. Similar results were also reported in other literatures.54,55 The mechanisms of improved drug dissolution are generally analyzed as follows. The amorphous state of a drug is conducive to the increase of its dissolution. Generally, amorphous drugs in SDs are unstable and tend to crystallisation.56 However, this process could be prevented by forming hydrogen bonds between the carbonyl groups of the drug and the silanol groups existed on the surface of PSi. Meanwhile, the formation of hydrogen bonds could improve the wettability of the drug and further increase drug dissolution rates.7,57 On the other hand, the enhancement of dissolution seems to be negatively affected by drug loading.58
:2 to 1:8) showed significantly improved dissolution rates (∼4-fold at maximum) compared to pure tanIIA and corresponding physical mixtures. Besides, the AUC0–24 h of tanIIA from PSi-SD increased by 1.97 times compared with pure tanIIA, demonstrating that its oral bioavailability was improved by SD with PSi. Takeuchi et al.53 prepared the SDs of indomethacin with non-porous Si or PSi. The latter showed faster drug dissolution rate. When the latter achieved complete drug dissolution, the former only released 80% of the drug. Similar results were also reported in other literatures.54,55 The mechanisms of improved drug dissolution are generally analyzed as follows. The amorphous state of a drug is conducive to the increase of its dissolution. Generally, amorphous drugs in SDs are unstable and tend to crystallisation.56 However, this process could be prevented by forming hydrogen bonds between the carbonyl groups of the drug and the silanol groups existed on the surface of PSi. Meanwhile, the formation of hydrogen bonds could improve the wettability of the drug and further increase drug dissolution rates.7,57 On the other hand, the enhancement of dissolution seems to be negatively affected by drug loading.58
PSi can improve the tableting properties of powders. It can combine with an active pharmaceutical ingredient to form composite particles (CPs) with improved tabletability. For example, CPs were formed by embedding erythritol in PSi pores with different ratios (erythritol/PSi: 1:1, 2:1, and 3:1) and then were applied for the preparation of orally disintegrating tablets via direct tableting.19 The CPs showed excellent compactability because of the strong binding property between PSi and erythritol. In comparison, the parent erythritol powder had poor compactability and caused problems during tableting. Compacts could not be produced using the physical mixtures between erythritol and PSi due to capping, while the CPs could be compacted successfully and the tablets formed showed excellent tensile strength (∼2.7–4.5 MPa).19 This phenomenon should be attributed to both an increased number of contact sites between CPs and an enhanced interparticulate bonding force. Similarly, in another report, CPs of indomethacin and PSi also showed enhanced tabletability compared to the corresponding physical mixture.59 What's more, PSi can also be exploited for improving flowability of dry powders during the tableting process, which makes dosages more accurate.54
Sustained and controlled drug release can be obtained by using PSi as carrier. Chlorhexidine diacetate was incorporated into the pores of PSi.60 This drug loaded-PSi sustained a slow release of chlorhexidine over more than 1 week. The release of chlorhexidine was controlled by configurational diffusion in the PSi pores and could be tuned by adjusting the particle size and pore diameter of PSi. In addition, Jang et al.1 investigated the release behaviors of methylene blue (MB) after it was introduced into PSi. The release of MB from PSi was maintained for more than 16 days, which was more sustained (approximately five times) than that from MB loaded-nonporous silica.1 With the pore volume of PSi increasing, the release rate of MB also increased. Because of a large number of pores in PSi, drugs tend to be adsorbed not on the surface but in the interior of PSi and, thus, obtain a controlled and sustained release, which can prolong its half-life and avoid multiple dosing.
Intriguingly, PSi can be even used as an oral vaccine immunological adjuvant to enhance mucosal and systemic immune responses. PSi incorporated with a model antigen, bovine serum albumin (BSA), has the capacity to produce a sustained-release vaccine delivery system.5 Results showed that the PSi/BSA formulation by oral immunization produced a stimulated humoral and mucosal response comparable to that induced by parenteral administration with BSA emulsified in Freund's Complete Adjuvant. The immunity greatly relied on the architecture of PSi, which was related to the uptake and release profile of the antigen as well as the uptake by M cells (a portal of the mucosal immune system).5 There are several advantages for the application of PSi as an oral vaccine immune adjuvant. Firstly, because of the high surface area and large pore volume of PSi, it can load a large amount of antigens. Secondly, PSi can provide slow release of antigens and thus create a “depot” effect. Thirdly, its stable and rigid framework can provide suitable protection against the effects of gastric acid and proteases in the alimentary tract. Fourthly, when antigens are loaded into the silica pores using phosphate buffered solution instead of organic solvent, they generally maintain their full activity.5 These aspects prove that PSi is an excellent candidate for oral vaccine delivery.
Generally, PSi refers to fully porous silica whose pores extend from the surface to the core of particles. But there are still superficially porous silica (SPSi) which consists of a non-porous silica core surrounded by a thin shell of porous silica (typically of 1–2 μm thickness), with overall particle size being ∼30 μm.61,62 Such particles are usually used as a stationary phase for chromatography to separate biomolecules. Those with pore diameter of ∼100 Å can be used for separating small molecules and those with 160–200 Å pore diameter are available for separating peptides and small proteins.18 The thin porous shell provides excellent mass transfer (kinetics) for large molecules, leading to superior separation efficiency compared to conventional PSi.18,63 So, SPSi has become popular as a stationary phase for chromatography over the last decade.
3.3. Mesoporous silica nanoparticles (MS-NPs)
MS-NPs are a kind of porous nanoparticles which have tunable pore size in the mesopore range (2–50 nm).64 They are synthesized by reacting a self-assembling silica tetraethyl ortho-silicate or sodium silicate and a surfactant (usually quaternary ammonium salts) micelle.20 Generally, MS-NPs can be classified as: M41S type MS-NPs family (the first MSN reported in the early 1990s), organically modified silica (ORMOSIL) nanoparticles, and hollow type MS-NPs.17,65 They are mostly applied for drug delivery and, most importantly, for cancer therapy.MS-NPs as drug delivery carriers offer several distinct advantages: (i) MS-NPs with large internal surface area and pore volume can be used as drug delivery carriers for a large range of drugs; (ii) MS-NPs exhibit biodegradation and low cytotoxicity; (iii) a tunable porous structure can help tailor drug loading and release kinetics; (iv) MS-NPs with a highly hydrophobic and rigid matrix structure can be uniformly dispersed in water and avoid variations due to heat, pH, mechanical stress, and hydrolysis-induced breakdown; (v) an easily modified surface enables MS-NPs to offer targeted and controlled drug delivery.17,66–68 Qu et al.69 investigated the release profiles of ibuprofen loaded into MS-NPs. The results showed that the drug-loading amount and drug release profiles were directly correlated to the specific surface area, pore volume, pore geometry, and morphologies of MS-NPs. With increasing the specific surface area of MS-NPs, the drug-loading amount also increased and a smaller spherical particle size led to a faster drug release rate.69 It was also found that loaded ibuprofen was in an amorphous state and was present both on the surface and within the core of MS-NPs, which should also contribute to the enhanced dissolution.64
MS-NPs have been increasingly popular as drug carriers owing to the promising in vivo results related to cancer therapy. MS-NPs now play an important role in cancer diagnosis and therapy because of their tunable pore size, high drug loading capacity, biodegradation, and nontoxicity. For example, in early cancer detection and diagnosis, MS-NPs can be used as imaging contrast agents, which displays encouraging results; MS-based chips with inert properties and specific pore size can act as a filter in protein mass spectrometry for recognizing cancer biomarkers; and the combination of silica nanoparticles with fluorescent materials can be used for optical imaging.17 These MSN-based approaches exhibit better efficacies than those commonly used with several limitations. As for cancer therapy, MS-NPs have been commonly used as anti-cancer drug delivery carriers, including passive delivery systems, active delivery systems, and controlled-release drug delivery systems.17 In general, MS-NPs can not only improve efficacy but also reduce side effects of anticancer drugs. They can also deliver gene and achieve molecular targeted therapy. Particularly, MS-NPs can be used to achieve significant anti-cancer efficacy by co-delivering several drugs, meanwhile, avoiding cancer multidrug resistance, which is a major cause of therapy failure in cancer patients.70
MS-NPs also have the ability to enhance the dissolution of drugs. For example, aceclofenac was loaded into MS-NPs.20 The dissolution efficiency of the drug from MS-NPs was ∼2 times higher than that of the plain drug or the marketed tablets. Correspondingly, the drug-loaded MS-NPs achieved a significant improvement of 189% in oral bioavailability (AUC0–8 h) than the plain drug in rats.20 So, the efficacy of aceclofenac was remarkably improved due to the enhanced bioavailability.
MS-NPs can also be used as a potential vaccine adjuvant. Oliveira et al.71 evaluated the function of MS-NPs in the prevention of Schistosoma mansoni. Compared to conventional adjuvants, MS-NPs triggered stronger immune response, which is believed to be related to some distinct characteristics of MS-NPs, such as spherical morphology, high surface area, high volume of large pores, stability, and biocompatibility.71
3.4. Porous calcium silicate (PCS)
PCS, which has both a highly porous structure of assembled petal-like flakes and a large individual pore volume, can be synthesized at a low hydrothermal temperature, using reactivity-enhanced SiO2 as the raw material.40,72 Florite RE (FLR) is a type of PCS that possesses many intraparticle and interparticle pores (sizes being approximately 12 and 0.15 μm, respectively) on its surface.73 FLR has been widely used in pharmaceutical field to improve dissolution, mechanical properties, and delivery of drugs.Firstly, FLR has the ability of adsorption and hence is able to improve the dissolution of drugs. Meloxicam, a poorly water soluble drug, was adsorbed on FLR in 2 proportions (meloxicam/FLR, 1:1 and 1:3). The results showed that the time required for 80% drug dissolution of the 2 proportions displayed 29.8- and 344.9-fold reduction, respectively, compared with the plain drug, clearly demonstrating significantly improved drug dissolution caused by FLR.8 Improvement in dissolution rate may be owing to (i) large surface area of FLR for adsorption, which reduced the chances of agglomeration of drug particles; (ii) a reduction in crystal size of drug; and (iii) the possible improvement in the wettability of drug because of FLR's dispersibility.8,74 The result of increased drug dissolution caused by PCS was also found in other studies.75 Furthermore, FLR could even improve the solubility of drugs. Kinoshita et al.72 studied the dissolution characteristics of 3-bis(4-methoxyphenyl) methylene-2-indolinone (TAS-301) after it was melt-adsorbed on FLR. It was found that not only the dissolution of melt-adsorbed products was improved, but also the solubility was approximately 20-fold greater than that of pure drug.72 Correspondingly, the oral bioavailability of TAS-301 melt-adsorbed on FLR was also markedly enhanced, which could be demonstrated by the evidence of increased AUC0–12 h (1.8–4.6-fold) and Cmax (2.3–5.1-fold) compared with pure drug in any feeding conditions.
PCS can also be applied for drug delivery to achieve sustained drug release. Gentamicin was loaded on calcium silicate (GENT-CS) and mesoporous calcium silicate (GENT-MCS), respectively.16 The release curves of both were investigated. On the first day, the burst release of both groups was visible. However, the drug release rate decreased significantly for GENT-CS. In contrast, the drug released from GENT-MCS on the fifth day was about 2-fold than that on the first day, indicating that the sustained-release profile could be obtained by GENT-MCS.16 The result is believed to attribute to the presence of mesopores that seem to provide a higher surface area for GENT adsorption.8
In addition, MCS itself has an antibacterial effect. The GENT-MCS group also showed a better antibacterial activity against a S. aureus strain than the GENT-CS group when the culture period of bacteria was 12 hours or longer.16 The reason for the antibacterial effect of MCS may be the appearance of an alkaline microenvironment and an increased pH value due to the release of calcium ions of MCS, which has certain inhibition effect on bacterial growth.16 Therefore, MCS can be applied as carries for antibacterial agents to achieve synergistic and preferable antibacterial effects.
Though PCS exhibits many benefits in pharmaceutical field, shortcomings still exist. The most prominent drawback of PCS is its low density, which limits its bulk volume and can make it easily become airborne during manufacturing. Additionally, low-density PCS may blend nonuniformly with other materials due to marked differences in density.75 So, novel techniques should be adopted to improve the bulk density of PCS.
3.5. Porous mannitol (PM)
PM is commonly produced through the spray drying of mannitol solutions containing templating agents or porogens.22,76 Mannitol has been widely used as a pharmaceutical excipient because of its prominent advantages, such as good taste and mouth feel, high compactibility, low hygroscopicity, chemical inertness towards both API and other excipients, etc.77 For the same reason, the application of PM also becomes increasingly popular in pharmacy.PM can improve the dissolution behavior of poorly water-soluble drugs and make the drug content uniform by nano-confinement of drugs inside it. For example, acetaminophen was nano-confined into PM. The results showed an improved dissolution rate and blending uniformity of acetaminophen. 80% of the drug loaded in PM was released within the first 5 min.78 Similar results are also found in the experiment on nano-confinement of nifedipine and indomethacin into PM.6 Solubilization mechanisms of PM are similar to other PPs. The improved blending uniformity of drug is attributed to nano-confinement of drug inside PM, which not only results in the reduction of drug particle size but also guarantees the highly homogenous dispersion of drug particles.
PM can improve mechanical properties of tablets. The tabletability of drug-loaded PM was much better than that of the physical mixture of non-porous mannitol and indomethacin.6 With increasing compaction pressure from 100 MPa to 300 MPa, the crushing strength of tablets with the physical mixture increased only from 20 N to 45 N, while that with the drug-loaded PM increased from 55 N to 90 N.6 This result is believed to be related to the much higher porosity and larger surface area of drug-loaded PM than those of the physical mixture. Besides, Al-khattawi et al.22 prepared orally disintegrating tablets using spray dried PM. Results showed that compared with pure mannitol, PM could make tablet hardness increased by about 150%. On the other hand, PM could make tablet disintegration time dropped by 77% from 135 s (pure mannitol) to 31.6 s.22 For PM, the higher hardness of orally disintegrating tablets was mainly attributed to changed particle densification mechanism that enhanced bonding between particles, and the faster disintegration time was ascribed to axial elastic recovery and highly porosity.22
PM can also be used as a carrier for pulmonary drug delivery. For example, hydrophobic cyclosporine A (CsA)-loaded PM nanoparticles exhibited good aerosol performance. Moreover, delivering them to the lungs not only achieved an improved drug dissolution profile but also reduced the side effects associated with the systemic exposure of CsA.79
3.6. Porous lactose (PL)
PL is commonly produced using the templating approach through spray drying.30,80,81 Lactose is usually applied in oral dosage formulations because of its unique properties, such as water solubility, availability, high physical and chemical stability in crystalline form, and cost effectiveness.82 Besides these properties, PL displays some additional advantages in pharmaceutical field.Like PM, PL can improve the dissolution rate of poorly water-soluble drugs and make drug dosage uniform. For example, 85% of acetaminophen that loaded in PL could be released within the only first 5 min.82 Moreover, the tabletability of acetaminophen-loaded PL was also much better than that of the physical mixture of non-porous lactose and acetaminophen, which was demonstrated by the 15 times and 4 times increased crushing strength of tablets produced at 100 and 300 MPa, respectively.82
3.7. Porous starch (PS)
PS is a biodegradable porous biomaterial derived from natural starch. PS possesses abundant micro-sized pores from the surface of starch granules extending to the center, resulting in a large specific surface area. The diameter of pores is about 1 μm, and the total volume of pores is about half that of starch. PS exhibits distinctive advantages such as tunable pore size, good biocompatibility, stable pore structure, and high pore volume.83–85 Currently, a freezing-solvent exchange technique and enzyme treatment have been mostly used to prepare porous starch.11,86–89 Generally, the pore structure of PS can be manipulated by altering the solvent ratio or the enzyme type and its level under different preparation conditions.10,11 PS is wildly used to enhance drug dissolution, adsorb drugs, and improve drug stability in the field of medicine.First, PS can be used as a novel carrier for dissolution enhancement of poorly soluble drugs. Carbamazepine (CBZ), which is practically insoluble in water, was loaded into PS.90 The drug release from CBZ-loaded PS showed to be complete within only 20 min. The significantly improved dissolution is believed to be due to (a) high surface area of drug; (b) geometric confinement of CBZ within the pores of PS and, thus, the reduction of the CBZ particle size; and (c) improved wettability and decreased crystallinity of drug, both of which originally make CBZ insoluble.90 Moreover, the improved dissolution rate of CBZ from CBZ-loaded PS made CBZ readily available for absorption and, thus, led to an enhanced in vivo anticonvulsant activity as compared to neat CBZ. Similarly, the dissolution of another poorly water soluble drug, lovastatin, was also improved when a biodegradable porous starch foam (BPSF) was used as a carrier.84 In vitro drug release studies showed that the drug-loaded BPSF system exhibited ∼3 times faster release rate than the current commercial product. Besides, the oral bioavailability value of the developed system was about 1.77- and 1.48-fold higher than those of the pure lovastatin and the commercial capsules, respectively.84
Second, PS has excellent adsorption property because of its honeycomb structure and large specific surface area. Zhang et al.83 observed the micrographs of native corn starch and corn PS. They found that native corn starch had the relatively smooth surface while the surface of corn PS became rough.83 Moreover, large internal cavities and many pores exist in the starch granules of corn PS, resulting in a large specific surface area. Thus, PS can be wildly used as adsorbents in medicine in terms of its excellent adsorption property compared to native starch.
Third, PS can be used as shell material to improve stability and water-solubility of compounds. Lutein microcapsules were well prepared by using a combined shell material consisting of gelatin and porous starch.91 Results showed that when lutein was embedded in this shell material, its solubility increased and became directly dissolved in water. Further, the stability of lutein loaded in microcapsules against heat, light, pH, and oxygen was also remarkably improved and its retention rates were enhanced by ∼15–50% than that of free lutein.91 This is believed to be due to the proper properties of PS in terms of film-formation, honeycomb structure, water-solubility, high adsorbability, and a tendency to form a network upon drying. These properties improve the adsorbability and adhesive property of the shell material, making all drug well dispersed and encapsulated.91
3.8. Porous chitosan (PC)
PC, fabricated by a microemulsion and thermally induced phase separation technique, has some special applications in medicinal field.92 PC is possibly the most hopeful hemostats because of its greatly effective blood clotting potential, inexpensiveness, biodegradability, easy storage and long shelf-life, antibacterial activity, stimulatory effect on tissue regeneration, and the abundance of naturally-occurred chitin.92 For example, the hemostatic function of PC with regard to hemostatic time, formation of blood clot, and blood loss had been shown to be improved both in vitro and in vivo compared with the conventional chitosan particles. PC made the hemostatic time and blood loss decreased by 1.5- and 9.8-fold, respectively, in the rat liver laceration model.92 The blood clotting mechanism of PC is attributed to the electrostatic interaction between negatively charged red blood cells (RBC) and positively charged chitosan, as well as adhesion/aggregation of platelets to form a soft RBC-platelet plug; the clotting factors released from activated platelets can induce coagulation cascade to produce fibrils, reinforcing the RBC-platelet plug to form a robust clot.92,933.9. Others
Besides the above, there are some other PPs, such as porous calcium carbonate, functionalized calcium carbonate, and porous ceramics, which are also applied in various pharmaceutical aspects.41,94–96 (Table 1) In addition, porous microspheres (PMSs) have attracted great interest for medical application. There are many methods for preparation of PMSs, including non-solvent induced phase separation, double emulsion-premix membrane emulsification technique combined with a solvent evaporation method without any porogens, solvent evaporation method using porogen, and so on.21,97,98 Various preparation methods make PMSs versatile as carriers for small molecules, macromolecules, and drug delivery systems. For example, PMSs containing salmon calcitonin can obtain sustained release and overcome some release problems.21 PMSs can control the delivery of human serum albumin, efficiently lowering the burst release and achieving a sustained protein release in vitro.97 Besides, when aspirin was loaded into PMSs fabricated by chitosan modified halloysite nanotubes, the loading capacity (42.4 wt%) of such PMSs was around 20 times higher than that of the pristine halloysite (2.1 wt%) and controlled drug release was also obtained.99| Types | Key properties | Preparations | Functions and applications |
|---|---|---|---|
| a Drug-loaded porous particles were compared with the corresponding physical mixture.b Drug-loaded porous particles were compared with the corresponding non-porous particles.c Drug-loaded porous particles were compared with the corresponding commercial product.d Unlabelled data indicated that they were compared with the pure drug. Data in the italic form indicated that they were estimated from the figure in the published paper. Sylysia 350, a type of porous silica; Florite RE, a type of porous calcium silicate; TAS-301, 3-bis(4-methoxyphenyl)methylene-2-indolinone; CC 330, ground calcium carbonate; AUC, area under the concentration–time curve; Cmax, maximum plasma concentration. | |||
| Porous silicon | The porosity and pore size can be tuned | The electrochemical dissolution of Si into HF based solutions | Dissolution: time for 80% ibuprofen release, ↓ 3-fold;47 release amount of furosemide, ↑ 14-fold (at 1 h)47 |
| Sustained release: release amount of ranitidine, ↓ ∼1.28-fold (at 30 min)47; release time of peptide (YY3-36), ↑ 3-fold51 | |||
| Improved pharmacological activity of ghrelin antagonist: ↓ food intake for a longer period, ↑ blood pressure more slowly50 | |||
| Cancer therapy: ↑ efficacy, ↓ side effects48,49 | |||
| Antimicrobial effect of drugs: ↑52 | |||
| Porous silica | Sylysia 350: pore size, 21.0 nm; pore volume, 1.6 mL g−1 | Using emulsions as template | Dissolution: release amount of tanshinone IIAa, ↑ ∼4-fold at maximum (at 180 min), AUC0–24 h, ↑ 1.97-fold;7 release amount of indomethacinb, ↑ ∼25% (at 10 min)59; release amount of carvedilol, ↑ ∼5.7-fold (at 2 h)55 |
| Superficially porous silica (particle diameter ∼ 30 μm) with a thin shell of porous silica (1–2 μm) | Tabletability: tablet tensile strength of erythritola, ↑ from 0 to ∼2.7–4.5 MPa;19 tablet tensile strength of indomethacina, ↑ ∼1.5-fold;59 flowability of dry powders, ↑54 | ||
| Sustained release: release time of chlorhexidine diacetate, more than 1 week;60 release time of methylene blueb, ↑ ∼5-fold1 | |||
| Mucosal and systemic immune responses: ↑5 | |||
| Using for separating biomolecules18 | |||
| Mesoporous silica nanoparticles | Pore size, 2–50 nm | Reacting a self-assembling silica tetraethyl ortho-silicate or sodium silicate and a surfactant micelle | Dissolution: release amount of aceclofenac, ↑ ∼2 times (at 2 h), AUC0–8 h, ↑ 189% (ref. 20) |
| Achievement of controlled and sustained ibuprofen release69 | |||
| Targeted TAT peptide release100 | |||
| Cancer therapy: ↑ efficacy, ↓ side effects17 | |||
| Immune responses, ↑71 | |||
| Porous calcium silicate | Porous structure, assembled petal-like flakes | Using reactivity-enhanced SiO2 as the silica materials at a low hydrothermal temperature | Dissolution: time for 80% meloxicam release, ↓ 29.8-fold (meloxicam/FLR, 1:1), ↓ 344.9-fold (meloxicam/FLR, 1:3);8 solubility of TAS-301, ↑ ∼20-fold, AUC0–12 h, ↑ 1.8–4.6-fold, Cmax, ↑ 2.3–5.1-fold72 |
| Florite RE: pore sizes, particularly 12 μm and 0.15 μm | Sustained release: release time of gentamicin, more than 5 days16 | ||
| Antibacterial effect of drugs, ↑16 | |||
| Porous mannitol | The porosity and pore size can be tuned | Spray drying of mannitol solutions containing templating agents or porogen | Dissolution: 80% acetaminophen was released within the first 5 min;78 80% nifedipine and indomethacin were released within the first 15 min (ref. 6) |
| Tabletability: tablet crushing strength of indomethacina, ↑ 175% (compaction force: 100 MPa) and ↑ 100% (compaction force: 300 MPa);6 tablet hardness of PM, ↑ 46.6–148.3%, tablet disintegration time of PM, ↓ 50–77% (compared to parent mannitol)22 | |||
| Pulmonary delivery: release amount of hydrophobic cyclosporine A, ↑ ∼9-fold (at 20 min)79 | |||
| Porous lactose | The porosity and pore size can be tuned | Spray drying of lactose solutions containing templating agents | Dissolution: 85% acetaminophen was released within 7 min (ref. 82) |
| Tabletability: tablet crushing strength of acetaminophen, ↑ ∼15-fold (compaction force: 100 MPa) and ∼4-fold (compaction force: 400 MPa)82 | |||
| Porous starch | Honeycomb structure; pore size, ∼1 μm; total volume, ∼50% of starch | A freezing-solvent exchange technique or enzyme treatment | Dissolution: carbamazepine, from insoluble in water to dissolved completely within 20 min;90 time for 80% lovastatin releasec, ↓ ∼3-fold, AUC0–8 h, ↑ 1.77-fold (compared to the pure drug) and 1.48-foldc,84 |
| Stability of drug: retention rates, ↑ 15–50% (compared to lutein)91 | |||
| Porous chitosan | — | Microemulsion combined with thermally induced phase separation technique | Hemostatic effectb: hemostatic time, ↓ 1.5-fold; blood loss, ↓ 9.8-fold92 |
| Porous microspheres | — | Non-solvent induced phase separation, or double emulsion-premix membrane emulsification technique combined with a solvent evaporation method without any porogens, or solvent evaporation method using porogen | Achievement of sustained/controlled release of proteins, peptides, etc.21,97,99 |
| Porous ceramics | Comprised of a three-dimensional array of hollow polygons | Unavailable | Achievement of sustained drug release96 |
| Functionalized calcium carbonate | Size, 5–15 mm; a specific lamellar surface area, 40–80 m2 g−1; porosity, ∼70% | Re-precipitation of calcium phosphate incorporating calcium carbonate under controlled conditions | Tabletability: tablet tensile strength, ↑ ∼10-fold (compaction force: ∼400 MPa) (compared to CC 330)41 |
4. Pharmaceutical applications of porous particles
Owing to a broad range of useful properties, such as regular and tunable pores, large surface area, high porosity, and excellent adsorbability, PPs have been applied in pharmaceutics for various purposes including development of novel drug delivery systems, enhancement of dissolution of poorly soluble drugs, improvement of the micromeritic characteristics of particles, and so on.5,8,16,19,20 The corresponding application of each PP had been collected in Table 1. Next, a brief summarization was presented from the perspective of each application of PPs.Firstly, PPs have been used for drug delivery, which is the most major application of PPs. Almost all of the PPs can be used as carriers to deliver drugs for certain purpose(s). The carriers for drug delivery are required to possess some specific pharmaceutical properties. For example, they should be able to combine drugs effectively; they should have certain transmembrane transport capacity that helps deliver the drug to the body and target organs; they should be able to release drugs at a controlled and sustained rate after getting to target sites; they must be inert for drugs and bodies, and should be non-toxic, biodegradable, etc. In general, many PPs possess almost all of these properties. Because of their large surface area, high porosity, and tunable pores, they often have prominent adsorption capacity. Thus, they can load drug effectively and control or target drug release as expected. PPs-based drug delivery systems mainly include gastrointestinal drug delivery systems, intravenous drug delivery systems, and pulmonary delivery systems, which are capable to deliver small molecule drugs, macromolecule drugs (mainly proteins and polypeptides), and other substances.46,49,79,97 Most importantly, they, such as MS-NPs and PSiN,17,48 can deliver the anti-cancer drug effectively for enhancing the curative efficacy and reducing the side effects. In addition, some PPs even deliver gene and improve the antibacterial activity when delivering the antibacterial drug.16,17,52 Overall, PPs play an important role in drug delivery, which necessitates further studies in future.
Secondly, PPs can significantly improve the dissolution of many drugs due to their structural characteristics. For example, when a drug is loaded into the pores of PPs, it tends to keep in an amorphous state.7 Nano-confinement can also happen when the pore size of PPs is nano-scaled.90 Moreover, large surface area, high porosity, and suitable pore size of PPs can facilitate water infiltration into the inner of PPs and thus increase the contact area between dissolution media and drug.84
Thirdly, the applications of PPs also involve in tableting. For PPs, both interparticle and intraparticle contact points can be significantly increased during compaction, which increases the bonding force between and within particles and thus make the particles bond together firmly.19 High porosity and large surface area also provide strong adhesion sites for components of a tablet formulation, resulting in less segregation within the powder mixtures. Therefore, PPs are often used for direct powder compaction thanks to their markedly improved micromeritic and physicomechanical characteristics. What's more, PPs can facilitate the rapid disintegration of tablets owing to rapid permeation of water through capillary action into their porous structure.22,101 This together with their excellent compactibility makes them especially suitable for the preparation of orally disintegrating tablets.
Besides, several PPs possess some special functions. For instance, SPSi can be used as a stationary phase for chromatography to separate biomolecule;18,63 MS-NPs and PSi are used as immunological adjuvants due to their function of enhancing immunological reaction;5,71 and PC has the hemostatic function and can be used as a hemostat.92 These unique applications are also mainly attributed to the structural characteristics of these PPs. In a word, the importance of PPs in pharmaceutics has become increasingly apparent and further studies are worth being carried out to boost and extend the practical applications of such particles.
5. Conclusion
PPs have been applied as novel carriers in pharmaceutical field by more and more researchers. There are many methods for preparing PPs, mainly including using porogens or templating agents to form pores, self-forming pores by particle matrices without any pore-forming agents, and chemical synthesis. In this review, various reported PPs are collected, which are applied in various fields of medicine for many purposes, mainly including improvement of dissolution of poorly soluble drugs, development of novel drug delivery systems (e.g., sustained/controlled drug delivery systems and targeted drug delivery systems), and improvement of functional properties of drug-loaded particles (e.g., tabletability, disintegration time, and flowability). In addition, individual PPs also have some special applications, such as molecule separation, vaccine adjuvant, hemostat, etc.In general, these functions of PPs are closely related to their fundamental particle and pore properties, such as morphology, size, surface property, surface area, and porosity. As carriers to deliver drugs, PPs with large internal surface area can not only obtain high drug loadings, but also control the sustained or targeted drug release by adjusting pore structure and surface property. Moreover, the porous structure of PPs can maintain the amorphous state of drugs, improve drug' wettability, avoid aggregation of drug particles, and, in some cases, even achieve nano-confinement of drugs, all of which help to enhance the dissolution of drugs and, thus, improve their oral bioavailability. As for tableting, it is amazing that PPs can improve the tensile strength of tablets and decrease the disintegration time of tablets simultaneously, which is often unachievable by ordinary particles. For ordinary particles, the improvement in compactibility is often accompanied by the decrease in disintegration ability; however, the porous nature of PPs not only significantly increases bonding points during compaction, but also facilitates permeation of water by capillary action during disintegration. As a whole, unique fundamental properties of PPs make them versatile, superior, and promising in many pharmaceutical aspects.
So far, most reported researches on PPs were still in the experimental stage. Therefore, more attention should be paid to the scaleup of production and application of PPs in future. It is also short of extensive and in-depth investigation on the relationship between the pore parameters (including size and size distribution, morphology, surface property, and porosity) and functional properties of PPs. Understanding of this relationship will help the design of PPs for certain application, stabilization of functionality of PPs, and control of application quality of PPs by means of the quality by design principles. In addition, the APIs that have been applied to PPs are almost chemical drugs at present. In fact, Chinese drugs and herbal extracts also have such an urgent need to utilize PPs as carriers. In general, there will be two ways for the application of PPs in traditional Chinese medicine (TCM). One is to load TCM into excipient-based PPs, just like the reports in this review. This way should be performed easily; however, its applicability will be very limited in terms of both the high dose and the multicomponent nature of most TCM. The other way, which is much more applicable for TCM, is to directly develop porous TCM particles. Such TCM-based PPs could be used for direct tableting, making tablets with high TCM doses possible. They could also be used to improve the disintegration of TCM tablets and the dissolution of TCM, two common problems suffered by many TCM preparations currently. With significant progresses in all of these aspects, the future of PPs is bound to be brighter.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Program for New Century Excellent Talents in University (NCET-13-0906), the funds from Shanghai Municipal Commission of Health and Family Planning (ZY3-CCCX-3-5001) and Science and Technology Commission of Shanghai Municipality (15DZ2292000), and the Science and Technology Development Fund of Pudong New Area (PKF-2013-003).References
- H. D. Jang, D. S. Kil, H. Chang, K. Cho, S. K. Kim, K. J. Oh and J. H. Choi, J. Nanosci. Nanotechnol., 2011, 11, 4169–4173 CrossRef CAS PubMed.
- M. Irani, G. M. M. Sadeghi and I. Haririan, Int. J. Biol. Macromol., 2017, 97, 744–751 CrossRef CAS PubMed.
- I. I. Krasnyuk, A. V. Beliatskaya, I. I. Krasnyuk, O. I. Stepanova, L. A. Korol, A. M. Valeeva, V. V. Grikh, L. V. Ovsyannikova and T. M. Kosheleva, Bionanoscience, 2017, 7, 340–344 CrossRef.
- L. Fan, H. Ge, S. Zou, Y. Xiao, H. Wen, Y. Li, H. Feng and M. Nie, Int. J. Biol. Macromol., 2016, 93, 582–590 CrossRef CAS PubMed.
- T. Wang, H. Jiang, Q. Zhao, S. Wang, M. Zou and G. Cheng, Int. J. Pharm., 2012, 436, 351–358 CrossRef CAS PubMed.
- M. Saffari, A. Ebrahimi and T. Langrish, Eur. J. Pharm. Sci., 2016, 83, 52–61 CrossRef CAS PubMed.
- H. M. Yan, E. Sun, L. Cui, X. B. Jia and X. Jin, J. Pharm. Pharmacol., 2016, 67, 1207–1214 CrossRef PubMed.
- S. Sharma, P. Sher, S. Badve and A. P. Pawar, AAPS PharmSciTech, 2005, 6, E618–E625 CrossRef PubMed.
- X. Du and J. He, Langmuir, 2010, 26, 10057–10062 CrossRef CAS PubMed.
- Y. Benavent-Gil and C. M. Rosell, Carbohydr. Polym., 2017, 157, 533–540 CrossRef CAS PubMed.
- P. R. Chang, J. Yu and X. Ma, Carbohydr. Polym., 2011, 83, 1016–1019 CrossRef CAS.
- P. Sher, G. Ingavle, S. Ponrathnam and A. P. Pawar, Int. J. Pharm., 2007, 331, 72–83 CrossRef CAS PubMed.
- A. G. Mirani, S. P. Patankar, V. S. Borole, A. S. Pawar and V. J. Kadam, Curr. Drug Delivery, 2011, 8, 426–435 CrossRef CAS.
- P. York, Drug Dev. Ind. Pharm., 1992, 18, 677–721 CrossRef CAS.
- Z. Li, X. Lin, L. Shen, Y. L. Hong and Y. Feng, Int. J. Pharm., 2017, 519, 272–286 CrossRef CAS PubMed.
- C. Y. Huang, T. H. Huang, C. T. Kao, Y. H. Wu, W. C. Chen and M. Y. Shie, J. Endod., 2017, 43, 69–76 CrossRef PubMed.
- Y. Feng, N. Panwar, D. J. H. Tng, S. C. Tjin, K. Wang and K. T. Yong, Coord. Chem. Rev., 2016, 319, 86–109 CrossRef CAS.
- B. M. Wagner, S. A. Schuster, B. E. Boyes and J. J. Kirkland, J. Chromatogr. A, 2012, 1264, 22–30 CrossRef CAS PubMed.
- S. Tanimura, K. Tahara and H. Takeuchi, Powder Technol., 2015, 286, 444–450 CrossRef CAS.
- D. Kumar, S. V. S. Chirravuri and N. R. Shastri, Int. J. Pharm., 2014, 461, 459–468 CrossRef CAS PubMed.
- Y. Wei, Y. Wang, H. Zhang, W. Zhou and G. Ma, J. Colloid Interface Sci., 2016, 478, 46–53 CrossRef CAS PubMed.
- A. Al-Khattawi, J. Koner, P. Rue, D. Kirby, Y. Perrie, A. Rajabisiahboomi and A. R. Mohammed, Eur. J. Pharm. Biopharm., 2015, 94, 1–10 CrossRef CAS PubMed.
- S. E. Bae, J. S. Son, K. Park and D. K. Han, J. Controlled Release, 2009, 133, 37–43 CrossRef CAS PubMed.
- D. X. Hao, F. L. Gong, W. Wei, G. H. Hu, G. H. Ma and Z. G. Su, J. Colloid Interface Sci., 2008, 323, 52–59 CrossRef CAS PubMed.
- C. Gervelas, A. L. Serandour, S. Geiger, G. Grillon, P. Fritsch, C. Taulelle, G. B. Le, H. Benech, J. R. Deverre and E. Fattal, J. Controlled Release, 2007, 118, 78–86 CrossRef CAS PubMed.
- D. M. El-Sherif and M. A. Wheatley, J. Biomed. Mater. Res., Part A, 2003, 66, 347–355 CrossRef PubMed.
- M. Gohel, M. Patel, A. Amin, R. Agrawal, R. Dave and N. Bariya, AAPS PharmSciTech, 2004, 5, 10–15 CrossRef PubMed.
- A. Al-Khattawi and A. R. Mohammed, Expert Opin. Drug Delivery, 2013, 10, 651–663 CrossRef CAS PubMed.
- S. Fiorilli, F. Tallia, L. Pontiroli, C. Vitale-Brovarone and B. Onida, Mater. Lett., 2013, 108, 118–121 CrossRef CAS.
- M. Saffari, A. Ebrahimi and T. Langrish, J. Food Eng., 2015, 164, 1–9 CrossRef CAS.
- A. Ebrahimi, M. Saffari and T. Langrish, Powder Technol., 2015, 283, 171–177 CrossRef CAS.
- S. Y. Lee, L. Gradon, S. Janeczko, F. Iskandar and K. Okuyama, ACS Nano, 2010, 4, 4717–4724 CrossRef CAS PubMed.
- A. B. D. Nandiyanto, O. Arutanti, T. Ogi, F. Iskandar, T. O. Kim and K. Okuyama, Chem. Eng. Sci., 2013, 101, 523–532 CrossRef CAS.
- G. Majano, A. Restuccia, M. Santiago and J. Pérezramírez, CrystEngComm, 2012, 14, 5985–5991 RSC.
- H. R. Jang, H. J. Oh, J. H. Kim and K. Y. Jung, Microporous Mesoporous Mater., 2013, 165, 219–227 CrossRef CAS.
- I. Emmanuelawati, J. Yang, J. Zhang, H. Zhang, L. Zhou and C. Yu, Nanoscale, 2013, 5, 6173–6180 RSC.
- H. Gustafsson and K. Holmberg, Adv. Colloid Interface Sci., 2017 DOI:10.1016/j.cis.2017.03.002.
- X. Shi, L. Sun and Z. Gan, Acta Polym. Sin., 2011, 31, 866–873 CrossRef.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V. P. Lehto, J. Pharm. Sci., 2008, 97, 632–653 CrossRef CAS PubMed.
- W. Guan, F. Ji, Z. Fang, D. Fang, Y. Cheng, P. Yan and Q. Chen, Ceram. Int., 2014, 40, 4415–4420 CrossRef CAS.
- T. Stirnimann, S. Atria, J. Schoelkopf, P. A. C. Gane, R. Alles, J. Huwyler and M. Puchkov, Int. J. Pharm., 2014, 466, 266–275 CrossRef CAS PubMed.
- A. Dura, W. Błaszczak and C. M. Rosell, Carbohydr. Polym., 2014, 101, 837–845 CrossRef CAS PubMed.
- A. R. Parsian, A. Vatanara, M. R. Rahmati, K. Gilani and K. M. Khosravi, Powder Technol., 2014, 260, 36–41 CrossRef CAS.
- W. Yin and M. Z. Yates, Langmuir, 2008, 24, 701–708 CrossRef CAS PubMed.
- M. Kaasalainen, E. Mäkilä, J. Riikonen, M. Kovalainen, K. Järvinen, K. H. Herzig, V. P. Lehto and J. Salonen, Int. J. Pharm., 2012, 431, 230–236 CrossRef CAS PubMed.
- L. M. Bimbo, M. Sarparanta, H. A. Santos, A. J. Airaksinen, E. Mäkilä, T. Laaksonen, L. Peltonen, V. P. Lehto, J. Hirvonen and J. Salonen, ACS Nano, 2010, 4, 3023–3032 CrossRef CAS PubMed.
- J. Salonen, L. Laitinen, A. M. Kaukonen, J. Tuura, M. Björkqvist, T. Heikkilä, K. Vähä-Heikkilä, J. Hirvonen and V. P. Lehto, J. Controlled Release, 2005, 108, 362–374 CrossRef CAS PubMed.
- K. Tamarov, W. Xu, L. Osminkina, S. Zinovyev, P. Soininen, A. Kudryavtsev, M. Gongalsky, A. Gaydarova, A. Närvänen and V. Timoshenko, J. Controlled Release, 2016, 241, 220–228 CrossRef CAS PubMed.
- B. Xia, B. Wang, J. Shi, Y. Zhang, Q. Zhang, Z. Chen and J. Li, Acta Biomater., 2017, 51, 197–208 CrossRef CAS PubMed.
- M. Kilpeläinen, J. Riikonen, M. A. Vlasova, A. Huotari, V. P. Lehto, J. Salonen, K. H. Herzig and K. Järvinen, J. Controlled Release, 2009, 137, 166–170 CrossRef PubMed.
- M. Kovalainen, J. Mönkäre, E. Mäkilä, J. Salonen, V. P. Lehto, K. H. Herzig and K. Järvinen, Pharm. Res., 2012, 29, 837–846 CrossRef CAS PubMed.
- N. Vale, A. Correia, S. Silva, P. Figueiredo, E. Mäkilä, J. Salonen, J. Hirvonen, J. Pedrosa, H. A. Santos and A. Fraga, Bioorg. Med. Chem. Lett., 2016, 27, 403–405 CrossRef PubMed.
- H. Takeuchi, S. Nagira, H. Yamamoto and Y. Kawashima, Int. J. Pharm., 2005, 293, 155–164 CrossRef CAS PubMed.
- B. Kovačič, F. Vrečer and O. Planinšek, Chem. Pharm. Bull., 2011, 59, 427–433 CrossRef.
- O. Planinšek, B. Kovačič and F. Vrečer, Int. J. Pharm., 2011, 406, 41–48 CrossRef PubMed.
- H. Al-Obaidi, P. Ke, S. Brocchini and G. Buckton, Int. J. Pharm., 2011, 419, 20–27 CrossRef CAS PubMed.
- D. Pahovnik, S. Reven, J. Grdadolnik, R. Borštnar, J. Mavri and E. Žagar, J. Pharm. Sci., 2011, 100, 4700–4709 CrossRef CAS PubMed.
- Q. Wei, C. M. Keck and R. H. Müller, Int. J. Pharm., 2017, 518, 253–263 CrossRef CAS PubMed.
- H. Takeuchi, S. Nagira, S. Tanimura, H. Yamamoto and Y. Kawashima, Chem. Pharm. Bull., 2005, 53, 487–491 CrossRef CAS PubMed.
- E. Verraedt, M. Pendela, E. Adams, J. Hoogmartens and J. A. Martens, J. Controlled Release, 2010, 142, 47–52 CrossRef CAS PubMed.
- D. V. Mccalley, J. Chromatogr. A, 2008, 1193, 85–91 CrossRef CAS PubMed.
- G. G. Perrenoud, W. P. Farrell, C. M. Aurigemma, N. C. Aurigemma, S. Fekete and D. Guillarme, J. Chromatogr. A, 2014, 1360, 275–287 CrossRef PubMed.
- M. Yi, Z. Sui, L. Zhen, L. Zhang and Y. Zhang, J. Pharm. Biomed. Anal., 2015, 114, 247–253 CrossRef PubMed.
- A. Hillerström, J. V. Stam and M. Andersson, Green Chem., 2009, 11, 662–667 RSC.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- Y. Wang, Q. Zhao, N. Han, L. Bai, J. Li, J. Liu, E. Che, L. Hu, Q. Zhang and T. Jiang, Nanomedicine, 2015, 11, 313–327 CrossRef CAS PubMed.
- S. Hashemikia, Drug Delivery, 2014, 21, 164–172 CrossRef PubMed.
- F. Tang, L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- F. Qu, G. Zhu, H. Lin, W. Zhang, J. Sun, S. Li and S. Qiu, J. Solid State Chem., 2006, 179, 2027–2035 CrossRef CAS.
- V. Mamaeva, C. Sahlgren and M. Lindén, Adv. Drug Delivery Rev., 2013, 65, 689–702 CrossRef CAS PubMed.
- D. C. D. P. Oliveira, A. L. B. D. Barros, R. M. Belardi, A. M. D. Goes and D. C. F. Soares, J. Drug Delivery Sci. Technol., 2016, 35, 234–240 CrossRef CAS.
- M. Kinoshita, K. Baba, A. Nagayasu, K. Yamabe, T. Shimooka, Y. I. Takeichi, M. Azuma, H. Houchi and K. Minakuchi, J. Pharm. Sci., 2002, 91, 362–370 CrossRef CAS PubMed.
- H. Yuasa, Y. Takashima and Y. Kanaya, Chem. Pharm. Bull., 1996, 44, 1361–1366 CrossRef CAS.
- S. Charumanee, S. Okonoki and J. Sirithunyalug, CMU J., 2004, 3, 77–84 Search PubMed.
- Y. Fujimoto, N. Hirai, T. Takatani-Nakase and K. Takahashi, J. Drug Delivery Sci. Technol., 2016, 33, 51–57 CrossRef CAS.
- M. Saffari, A. Ebrahimi and T. Langrish, Food Res. Int., 2015, 67, 44–51 CrossRef CAS.
- H. L. Ohrem, E. Schornick, A. Kalivoda and R. Ognibene, Pharm. Dev. Technol., 2014, 19, 257–262 CrossRef CAS PubMed.
- M. Saffari, A. Ebrahimi and T. Langrish, Microporous Mesoporous Mater., 2016, 227, 95–103 CrossRef CAS.
- S. S. Leung, J. Wong, H. V. Guerra, K. Samnick, R. K. Prud'Homme and H. K. Chan, AAPS J., 2017, 19, 578–586 CrossRef CAS PubMed.
- A. Ebrahimi, M. Saffari and T. Langrish, Powder Technol., 2015, 283, 171–177 CrossRef CAS.
- A. Ebrahimi, M. Saffari and T. Langrish, J. Food Eng., 2015, 156, 59–66 CrossRef CAS.
- A. Ebrahimi, M. Saffari, F. Dehghani and T. Langrish, Int. J. Pharm., 2016, 499, 217–227 CrossRef CAS PubMed.
- B. Zhang, D. Cui, M. Liu, H. Gong, Y. Huang and F. Han, Int. J. Biol. Macromol., 2012, 50, 250–256 CrossRef CAS PubMed.
- C. Wu, Z. Wang, Z. Zhi, T. Jiang, J. Zhang and S. Wang, Int. J. Pharm., 2011, 403, 162–169 CrossRef CAS PubMed.
- Z. Zhang, J. Huang, S. Jiang, Z. Liu, W. Gu, H. Yu and Y. Li, Int. J. Pharm., 2013, 444, 162–168 CrossRef CAS PubMed.
- P. R. Chang, D. Qian, D. P. Anderson and X. Ma, Carbohydr. Polym., 2012, 88, 604–608 CrossRef CAS.
- A. Dura, W. Błaszczak and C. M. Rosell, Carbohydr. Polym., 2014, 101, 837–845 CrossRef CAS PubMed.
- U. Uthumporn, I. S. M. Zaidul and A. A. Karim, Food Bioprod. Process., 2010, 88, 47–54 CrossRef CAS.
- X. Wang, H. Wang, Q. Dai, Q. Li and J. Yang, Colloids Surf., A, 2009, 346, 213–215 CrossRef CAS.
- M. T. Ali, R. Fule, A. Sav and P. Amin, AAPS PharmSciTech, 2013, 14, 919–926 CrossRef CAS PubMed.
- Y. Wang, H. Ye, C. Zhou, F. Lv, X. Bie and Z. Lu, Eur. Food Res. Technol., 2012, 234, 157–163 CrossRef CAS.
- J. Li, X. Wu, Y. Wu, Z. Tang, X. Sun, M. Pan, Y. Chen, J. Li, R. Xiao and Z. Wang, Mater. Sci. Eng., C, 2017, 77, 411–419 CrossRef CAS PubMed.
- K. Quan, G. Li, D. Luan, Q. Yuan, L. Tao and X. Wang, Colloids Surf., B, 2015, 132, 27–33 CrossRef CAS PubMed.
- D. Preisig, D. Haid, F. J. Varum, R. Bravo, R. Alles, J. Huwyler and M. Puchkov, Eur. J. Pharm. Biopharm., 2014, 87, 548–558 CrossRef CAS PubMed.
- J. Venkatesan, J. Y. Lee, D. S. Kang, S. Anil, S. K. Kim, M. S. Shim and D. G. Kim, Int. J. Biol. Macromol., 2017, 98, 515–525 CrossRef CAS PubMed.
- R. S. Byrne and P. B. Deasy, Int. J. Pharm., 2002, 246, 61–73 CrossRef CAS PubMed.
- L. Sun, S. Zhou, W. Wang, X. Li, J. Wang and J. Weng, Colloids Surf., A, 2009, 345, 173–181 CrossRef CAS.
- X. Huang, J. Gao, N. Zheng, W. Li, H. Xue and R. K. Y. Li, Colloids Surf., A, 2017, 517, 17–24 CrossRef CAS.
- X. Li, Q. Yang, J. Ouyang, H. Yang and S. Chang, Appl. Clay Sci., 2016, 126, 306–312 CrossRef CAS.
- L. Pan, Q. He, J. Liu, Y. Chen, M. Ma, L. Zhang and J. Shi, J. Am. Chem. Soc., 2012, 134, 5722–5725 CrossRef CAS PubMed.
- A. Al-Khattawi and A. R. Mohammed, Expert Opin. Drug Delivery, 2013, 10, 651–663 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |