DOI:
10.1039/C7QO00285H
(Review Article)
Org. Chem. Front., 2017,
4, 1876-1890
Lu's [3 + 2] cycloaddition of allenes with electrophiles: discovery, development and synthetic application
Received
15th May 2017
, Accepted 19th June 2017
First published on 20th June 2017
Abstract
Lu's [3 + 2] cycloaddition reaction has experienced explosive development due to its versatile and powerful ability to access highly functionalized carbo- and heterocycles. After the first example reported by Prof. Xiyan Lu's group, many research groups expanded its substrate scope, and developed its asymmetric variants, and demonstrated its synthetic applications as well. This review briefly introduces the history of Lu's [3 + 2] cycloaddition reaction and highlights the important developments and synthetic applications with respect to Lu's [3 + 2] cycloaddition reaction.
 Yin Wei | Dr Yin Wei received her PhD from Ludwig-Maximilians-Universität of München (Germany) in 2009 under the direction of Professor Hendrik Zipse. Subsequently she joined Professor Min Shi's group at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC, CAS), and was promoted as an associate professor in 2012. She has authored over 100 scientific publications, and is mainly working on the theoretical studies of organocatalysis. |
 Min Shi | Prof. Dr Min Shi is the vice director of the State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC, CAS). Having obtained a PhD degree in 1991 at Osaka University, Japan, he gained postdoctoral research experience working with Prof. Kenneth M. Nicholas at the University of Oklahoma (1995–6) and worked as an ERATO Researcher in Japan Science and Technology Corporation (JST) (1996–8). Then, he was appointed as a full professor at SIOC in 1998. Professor Shi has authored over 500 scientific publications, and his current research interests include photochemistry, total synthesis of natural products, asymmetric synthesis, Morita–Baylis–Hillman reaction and fixation of CO2 using a transition metal catalyst. |
1. Introduction
Phosphines have been commonly used in organic synthesis, in which they are employed as ligands in transition-metal-catalyzed reactions, and are also utilized as reagents in the Wittig reaction. From the beginning of the 21st century, phosphines have frequently been utilized as catalysts in a wide range of reactions, and organophosphorus catalysis has experienced explosive development to a highly dynamic chemical research area due to its wide applicability in organic synthesis, which has drawn remarkable growing research interest from a number of research groups.1 Among a large number of phosphine-catalyzed reactions, the so-called “Lu reaction” is highly versatile and powerful to access functionalized cyclopentenes and dihydropyrroles. The “Lu reaction” can be generally defined as the phosphine-catalyzed [3 + 2] cycloaddition of allenes with electrophiles such as alkenes, alkynes, and imines. In 1995, Prof. Xiyan Lu and co-workers at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences (SIOC) (Fig. 1) reported the first example of phosphine-catalyzed [3 + 2] cycloaddition of allenes and alkenes to give functionalized cyclopentenes.2 Since then, Lu's [3 + 2] cycloaddition has been developed and demonstrated as one of the few types of annulation processes where an allene acts as a three-carbon synthon.3 Currently, there are more than 20 research groups that are developing and applying Lu's [3 + 2] cycloaddition reaction to the syntheses of natural products and biologically active molecules. In this review, the following sections will be discussed: (1) a brief review of the history of Lu's [3 + 2] cycloaddition reaction, (2) a highlight of the selected examples for the development of Lu's [3 + 2] cycloaddition reaction after 2000, (3) mechanistic studies for Lu's [3 + 2] cycloaddition reaction and (4) applications of Lu's [3 + 2] cycloaddition reaction in organic synthesis.
 |
| | Fig. 1 Prof. Xiyan Lu at SIOC. | |
2. Discovery of Lu's [3 + 2] cycloaddition
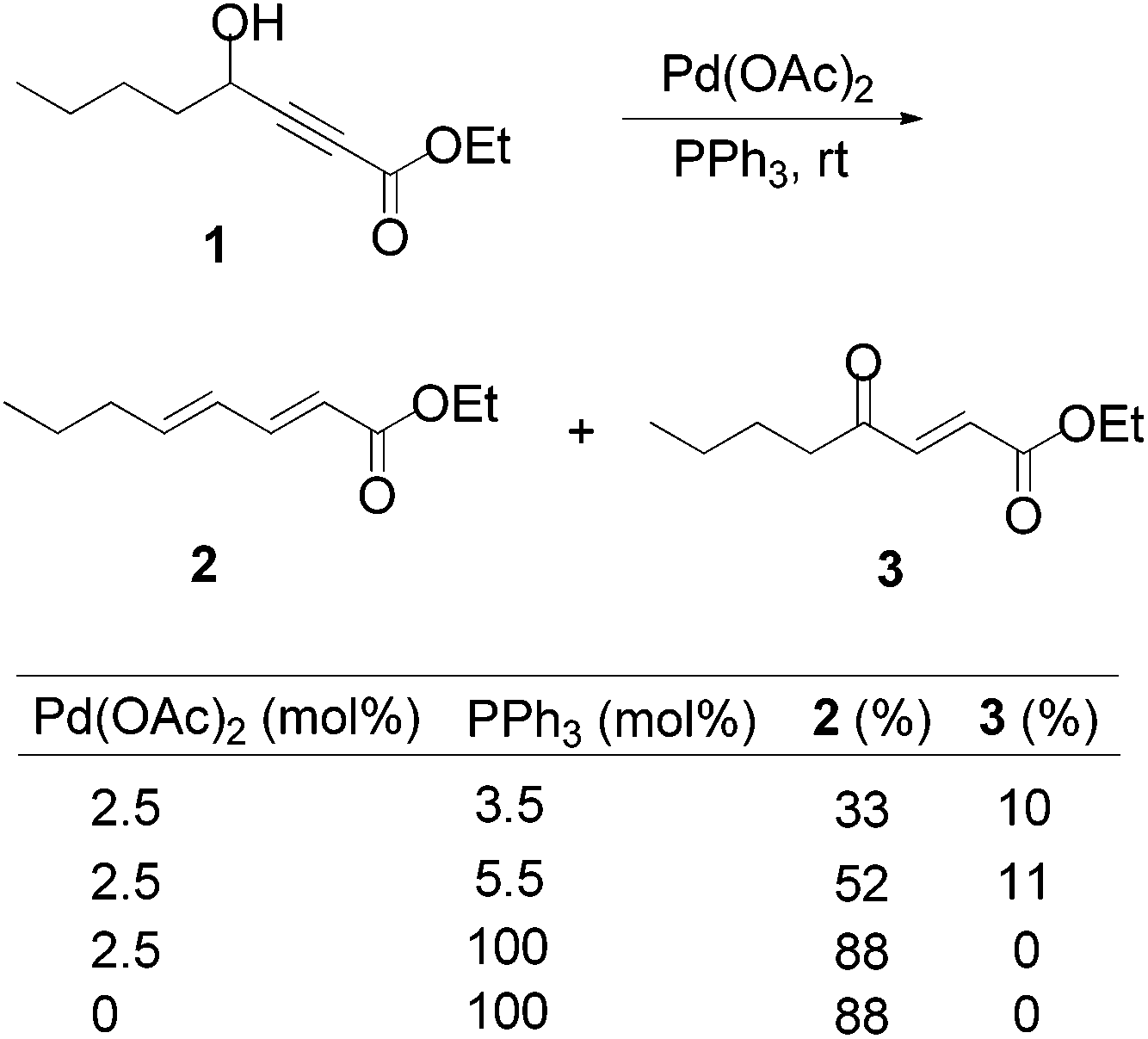
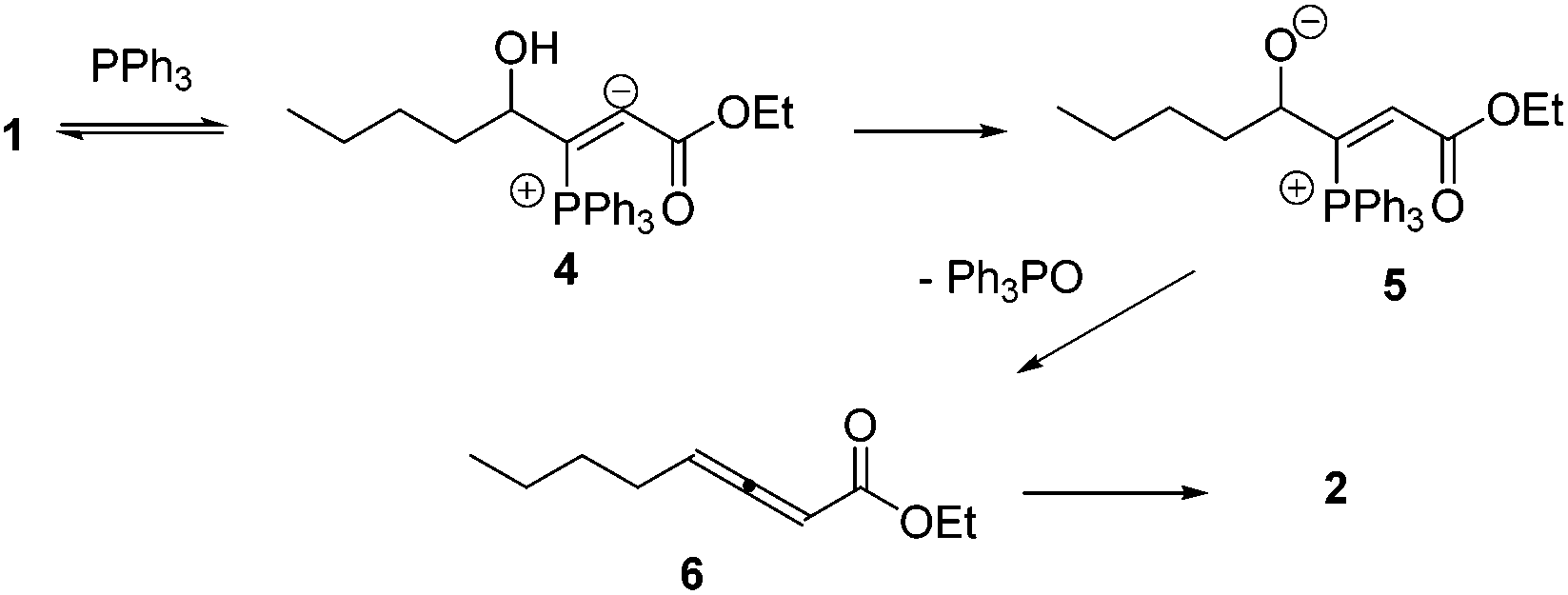
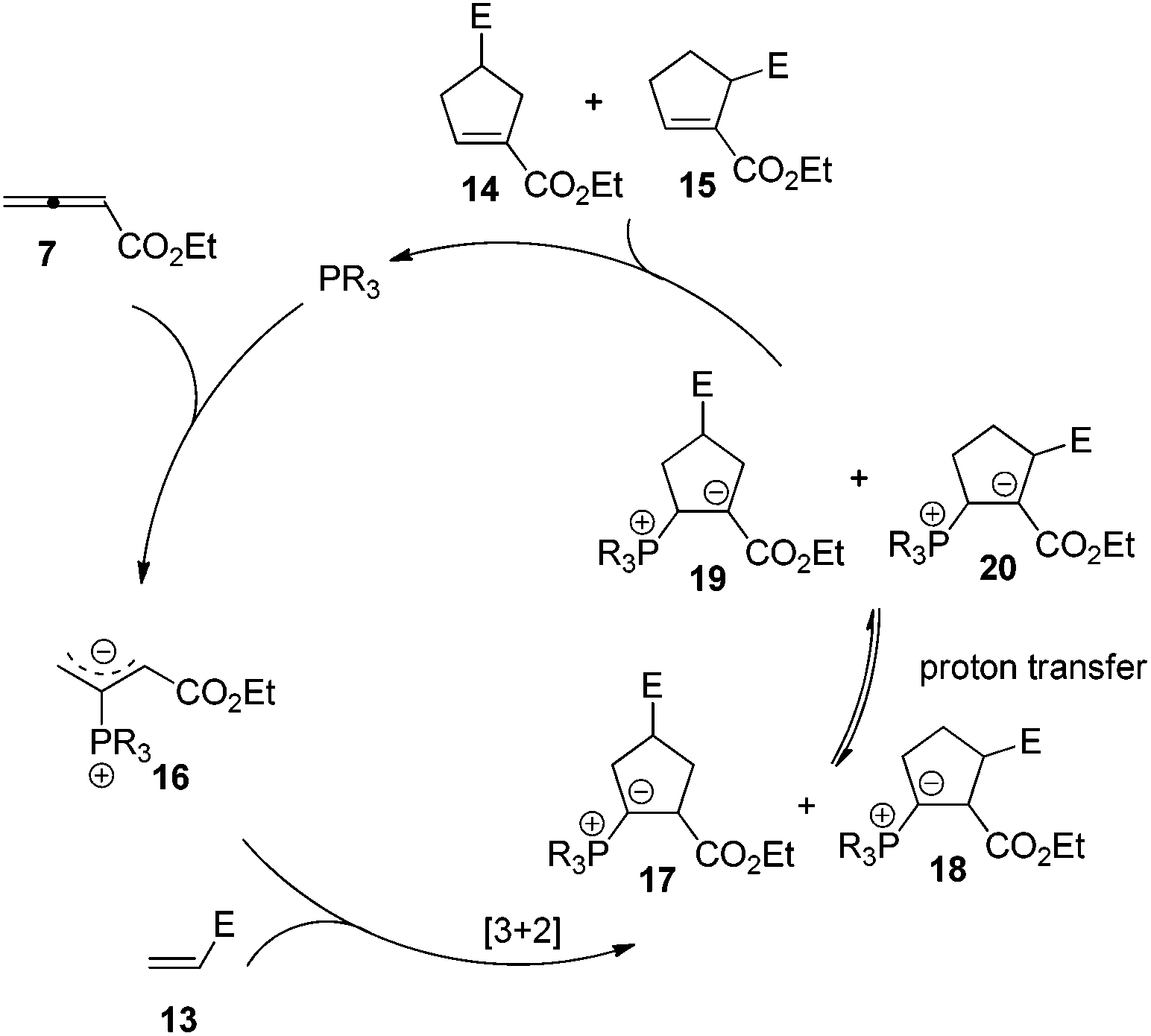
Lu's group published a series of reports about the isomerization of alkynes to dienes by catalysis of transition metals with phosphine ligands.4 Trost5 and Inoue6 also reported similar reactions, respectively. When Lu and co-workers extended their methodology to synthesize ene-dicarbonyls via isomerisation of 4-hydroxy-2-ynoic ester 1 catalyzed by a Pd(OAc)2–PPh3 complex, they surprisingly found that the unpredicted dienoate 2 was obtained as a major product instead of the desired product 3; they further achieved the dienoate 2 in good yields in the absence of Pd(OAc)2 (Scheme 1).7 Then, they found that Pd(OAc)2 did not play a catalytic role in this reaction; however, PPh3 was critical for this reaction; thus, they proposed a plausible reaction mechanism as shown in Scheme 2. The nucleophilic addition of PPh3 to substrate 1 generates the intermediate 4 which undergoes the proton shift to give intermediate 5; it then undergoes a deoxygenation reaction to yield an allene intermediate 6, which is isomerised to dienoate 2. In further investigation, Lu's group and Trost's group found that the isomerisation reactions could take place smoothly in the presence of a catalytic amount of tertiary phosphine in the absence of transition metal catalysts, respectively.8,9 The research results for the isomerisation reactions of acetylenic derivatives found that the nucleophilic phosphines could be utilized as catalysts for this type of reaction to synthesize polyenyl carbonyl compounds, which also provide a solid foundation for the discovery of Lu's [3 + 2] cycloaddition reaction.
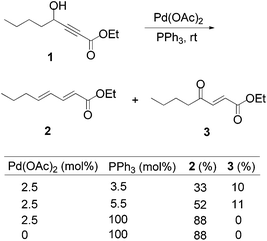 |
| | Scheme 1 Isomerization of 4-hydroxy-2-ynoic ester. | |
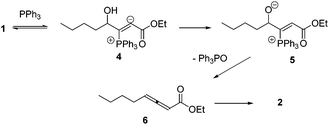 |
| | Scheme 2 Proposed mechanism. | |
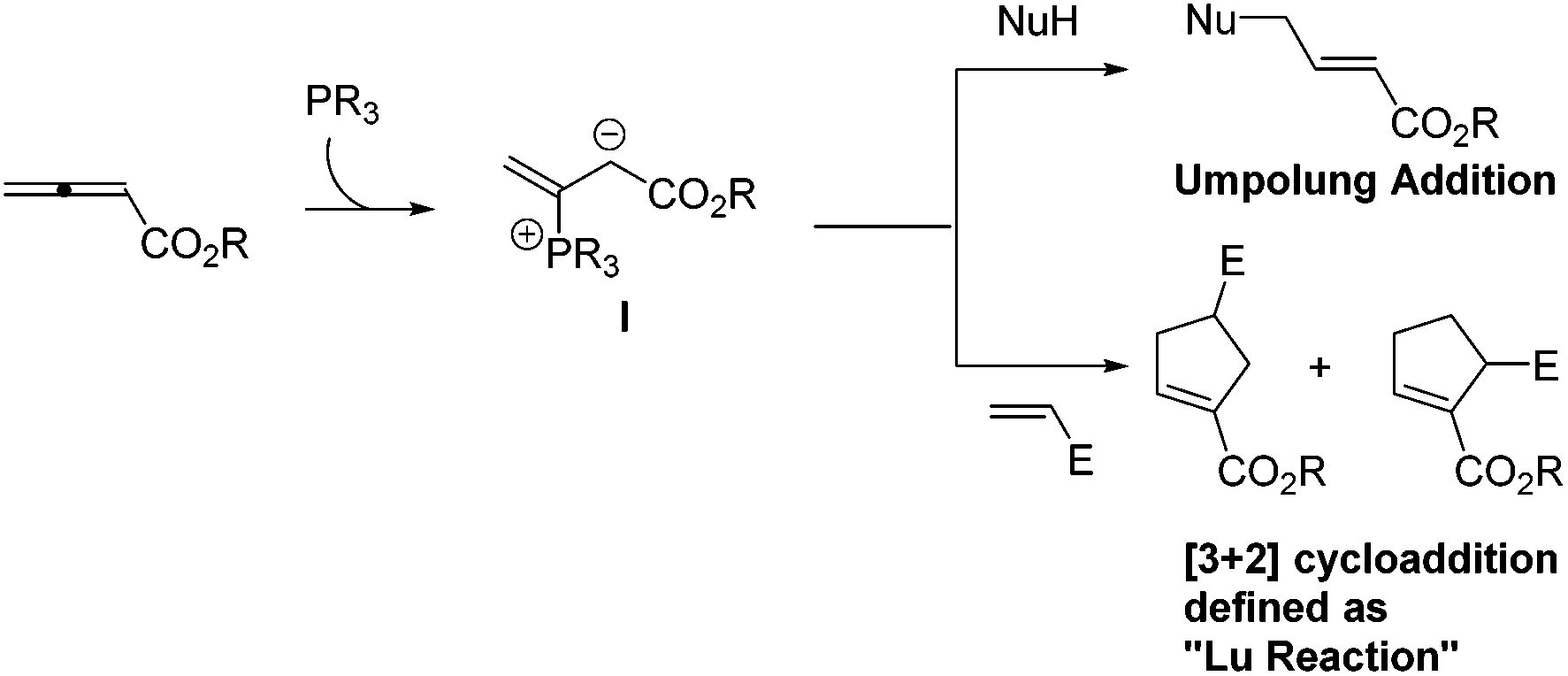
Lu's group subsequently hypothesized that using terminal allenoates as substrates, the generated zwitterionic intermediates I from the addition of tertiary phosphine to terminal allenoates could be trapped by nucleophiles or reacted with other electrophiles (Scheme 3). Based on this working hypothesis, they developed umpolung reactions10 and [3 + 2] cycloaddition reactions2,11 of allenes with electron-deficient alkenes and imines which are currently defined as Lu's [3 + 2] cycloaddition reaction.
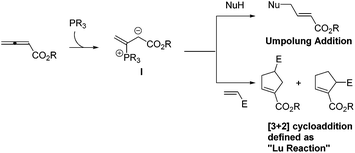 |
| | Scheme 3 Working hypothesis by Lu's group. | |
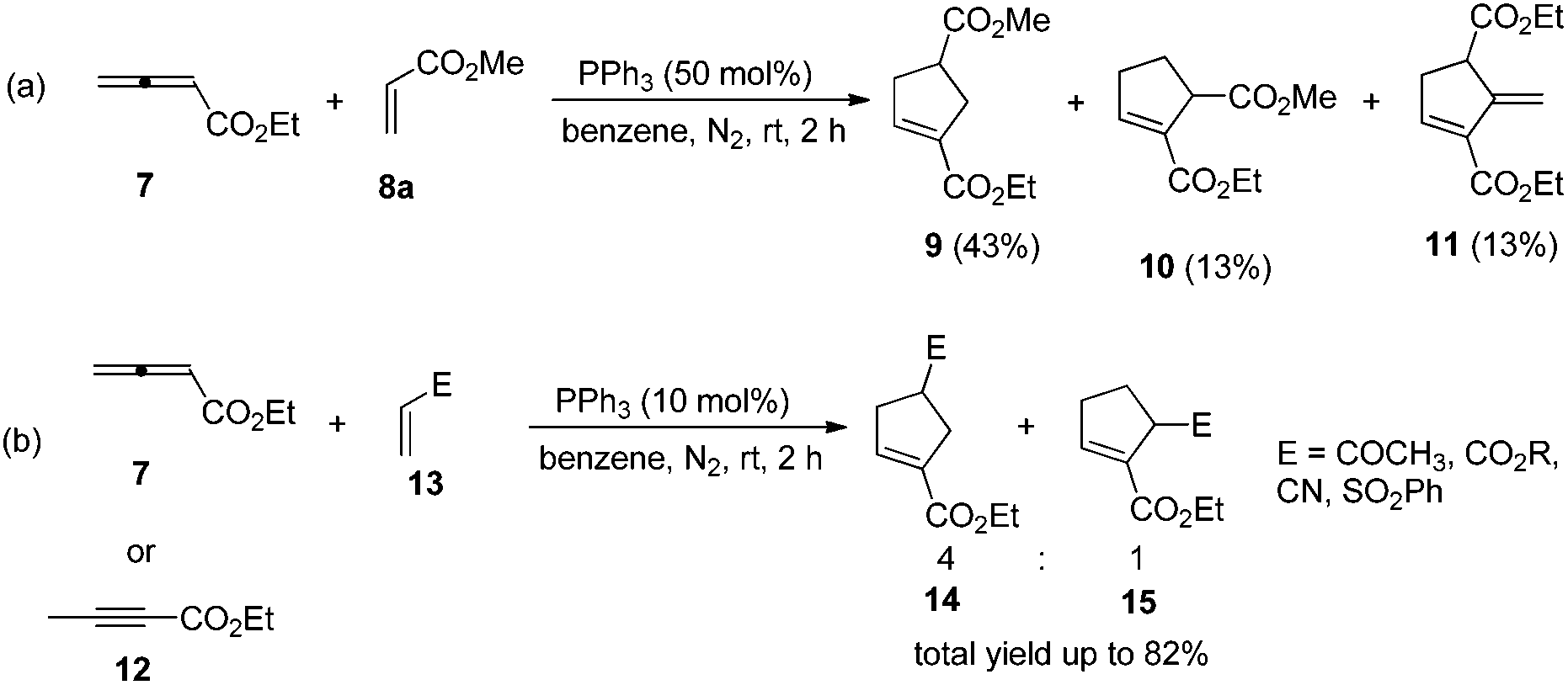
The first seminal report of the Lu reaction was published in 1995.2 They initially explored the reaction of ethyl 2,3-butadienoate 7 with methyl acrylate 8a in the presence of triphenylphosphine (50 mol%) in dry benzene at room temperature, and three products 9–11 were isolated after 2 h (Scheme 4a). Products 9 and 10 were the cycloadducts of 7 and 8, and product 11 was the self-cycloaddition product of 7. They further extended the substrates to 2-butynoate 12 and a series of electron-deficient alkenes 13 including esters, ketones, and nitriles, giving the corresponding cycloadducts 14 and 15 in moderate to good yields (Scheme 4b). They also proposed a plausible mechanism for this reaction as shown in Scheme 5. In the proposed mechanism, the zwitterionic intermediate 16 is generated readily through the addition of phosphine to the ethyl 2,3-butadienoate 7. The zwitterionic intermediate 16 undergoes a [3 + 2] cycloaddition with an electron-deficient alkene to give phosphorous ylides 17 and 18. Then an intramolecular [1,2] proton transfer occurs to convert the phosphorus ylides to intermediates 19 and 20, which, upon elimination of the phosphine catalyst, afford the final cycloadducts 14 and 15.
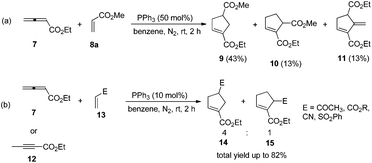 |
| | Scheme 4 Phosphine-catalyzed [3 + 2] cycloadditions. | |
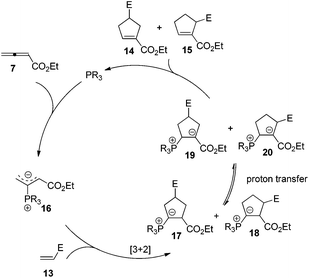 |
| | Scheme 5 Proposed mechanism for Lu's [3 + 2] cycloaddition. | |
3. Development of Lu's [3 + 2] cycloaddition
Since the discovery of Lu's [3 + 2] cycloaddition reaction, many research groups have developed this reaction, as it has been a highly versatile and powerful tool for syntheses of functionalized cyclopentenes and dihydropyrroles.
3.1 Lu's [3 + 2] cycloaddition of allenes with activated alkenes
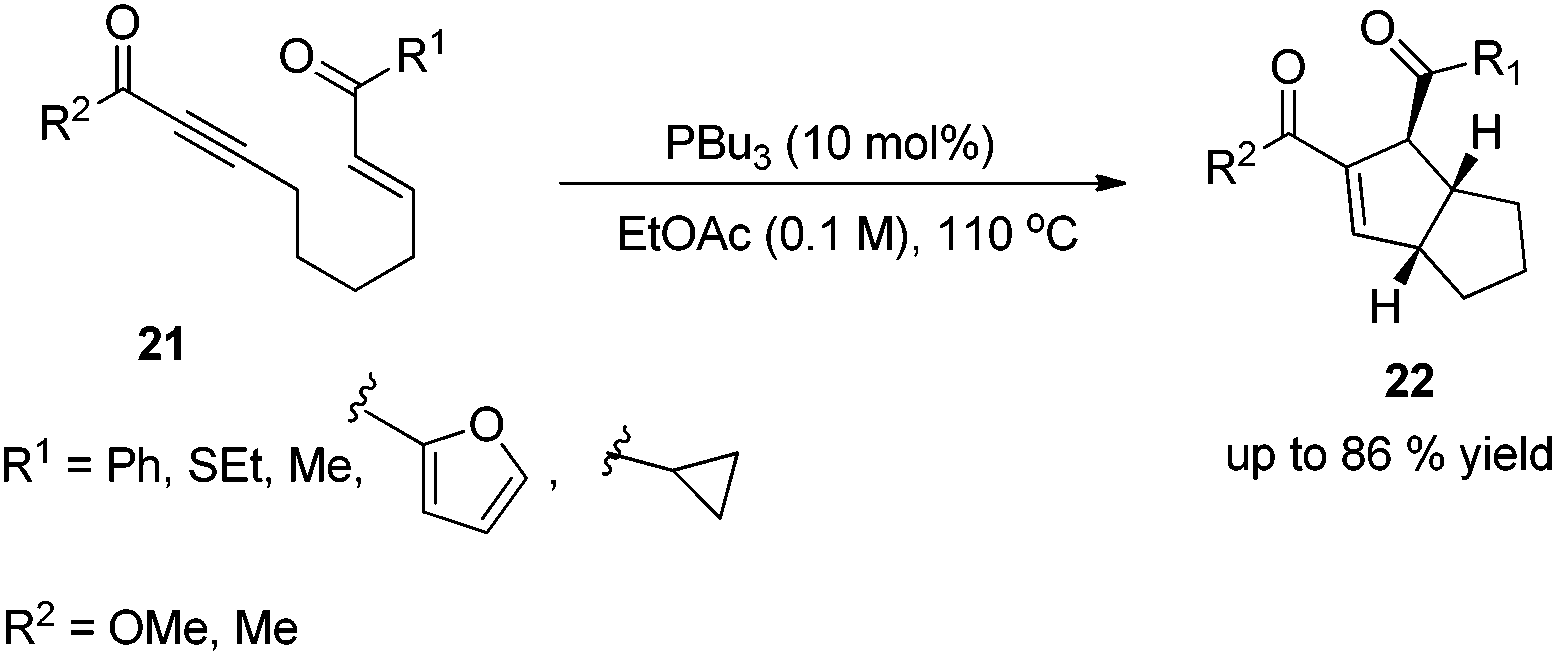
In 2003, Krische and co-workers developed Lu's [3 + 2] cycloaddition reaction to an intramolecular version for the synthesis of highly functionalized diquinanes.12 A range of electron-deficient 1,7-enynes 21 underwent this intramolecular phosphine-catalyzed [3 + 2] cycloaddition smoothly, affording the corresponding cycloadducts 22 in good yields (Scheme 6). This intramolecular phosphine-catalyzed [3 + 2] cycloaddition of electron-deficient 1,7-enynes is highly diastereoselective, which represents a robust method for building bicylo[3.3.0] ring systems, enabling the diastereoselective formation of three contiguous stereogenic centers in a single operation.
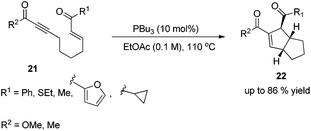 |
| | Scheme 6 Synthesis of diquinanes through intramolecular Lu's [3 + 2] cycloaddition. | |
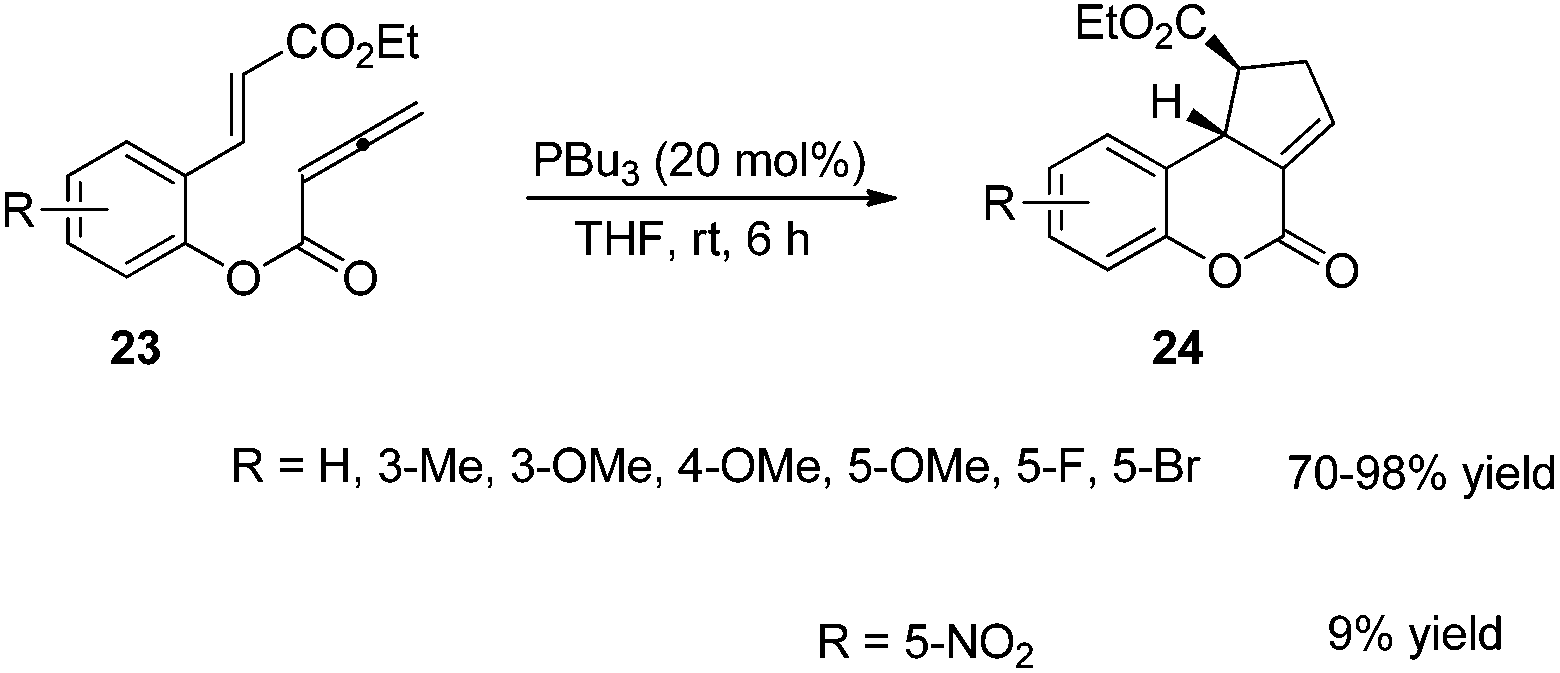
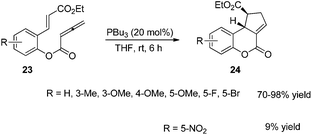
Kwon and co-worker reported another intramolecular [3 + 2] cycloaddition of 2-styrenyl allenoates 23 to produce cyclopentene-fused dihydrocoumarins 24 in excellent to good yields with exclusive diastereoselectivity (Scheme 7).13 This method provides an efficient way to construct highly functionalized coumarins which are often found in natural products and used widely in medicinal compounds. Substrates 23 containing both electron-withdrawing and -donating substituents on the benzene ring underwent this reaction smoothly under mild reaction conditions. However, using substrates having a nitro substituent only gave a trace amount of the product, probably, due to the ready hydrolysis of its allenoate ester moiety in this case.
 |
| | Scheme 7 Synthesis of dihydrocoumarins through intramolecular Lu's [3 + 2] cycloaddition. | |
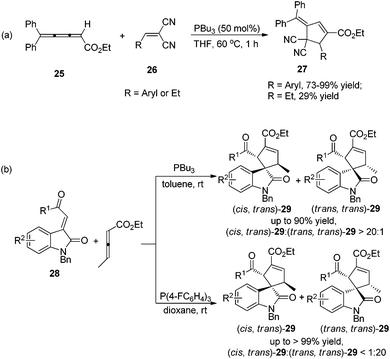
In 2009, Shi's group developed Lu's [3 + 2] cycloaddition using 2,3,4-pentatrienoate 25 and arylidenemalononitriles 26, furnishing an easy access to a variety of novel polysubstituted cyclopentenes 27.14a The 2,3,4-pentatrienoate 25 still served as a three-carbon synthon in this reaction, and a normal [3 + 2] cycloaddition reaction took place to afford polysubstituted cyclopentenes. A relatively high loading (50 mol%) of PBu3 was necessary for this reaction to acquire good efficiency. Both electron-donating and electron-withdrawing functionalities on arylidenemalononitrile were well accommodated, giving the desired products in high yields; when a less-reactive alkylidenemalononitrile was employed, the product yield dramatically decreased (Scheme 8, eqn (a)). Subsequently, Shi's group developed a highly diastereoselective [3 + 2] cycloaddition employing ethyl 2,3-pentadienoate and isatin-derived electron-deficient alkenes 28, affording the functionalized spirocyclic products 29 in good to excellent yields, with high regioselectivities and diastereoselectivities (Scheme 8, eqn (b)).14b Interestingly, they found that the phosphine catalyst influenced the diastereoselectivity. Using the highly nucleophilic phosphine PBu3 as a catalyst, (cis, trans)-29 was obtained as the major product; however, employing a weaker nucleophilic phosphine P(F-C6H4)3 as a catalyst, the major product was switched to (trans,trans)-29.
 |
| | Scheme 8 Synthesis of polysubstituted cyclopentenes and spirocyclic products through Lu's [3 + 2] cycloaddition developed by Shi's group. | |
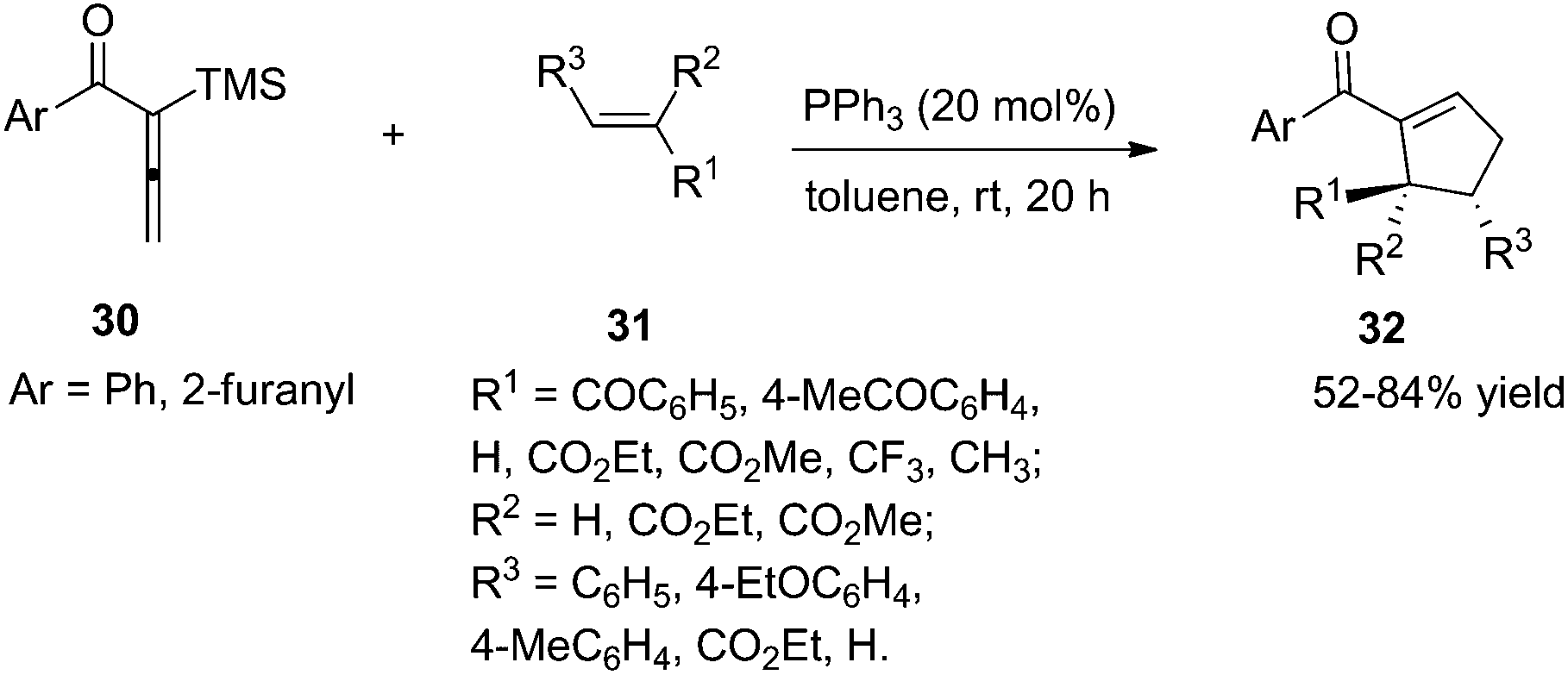
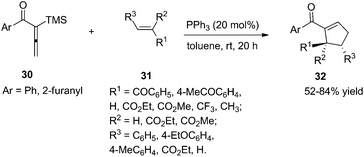
As per Lu's seminal report, self-cycloaddition of 7 could occur to give a side product 11. In order to inhibit this undesired pathway, Loh and co-workers introduced a trimethylsilyl (TMS) group at the α-position of allenone to prohibit the dimerization.15 Highly reactive phenyl allenones 30 having an α-TMS moiety underwent the desired phosphine catalyzed [3 + 2] cycloaddition smoothly with a range of activated alkenes 31, affording highly functionalized cyclopentenes 32 in moderate to good yields (Scheme 9). The α-TMS was hydrolyzed during the reaction. Probably due to the steric congestion at the α-position, trans γ-addition products were exclusively obtained with excellent diastereoselectivity.
 |
| | Scheme 9 Synthesis of polysubstituted cyclopentenes through Lu's [3 + 2] cycloaddition using aryl allenones. | |
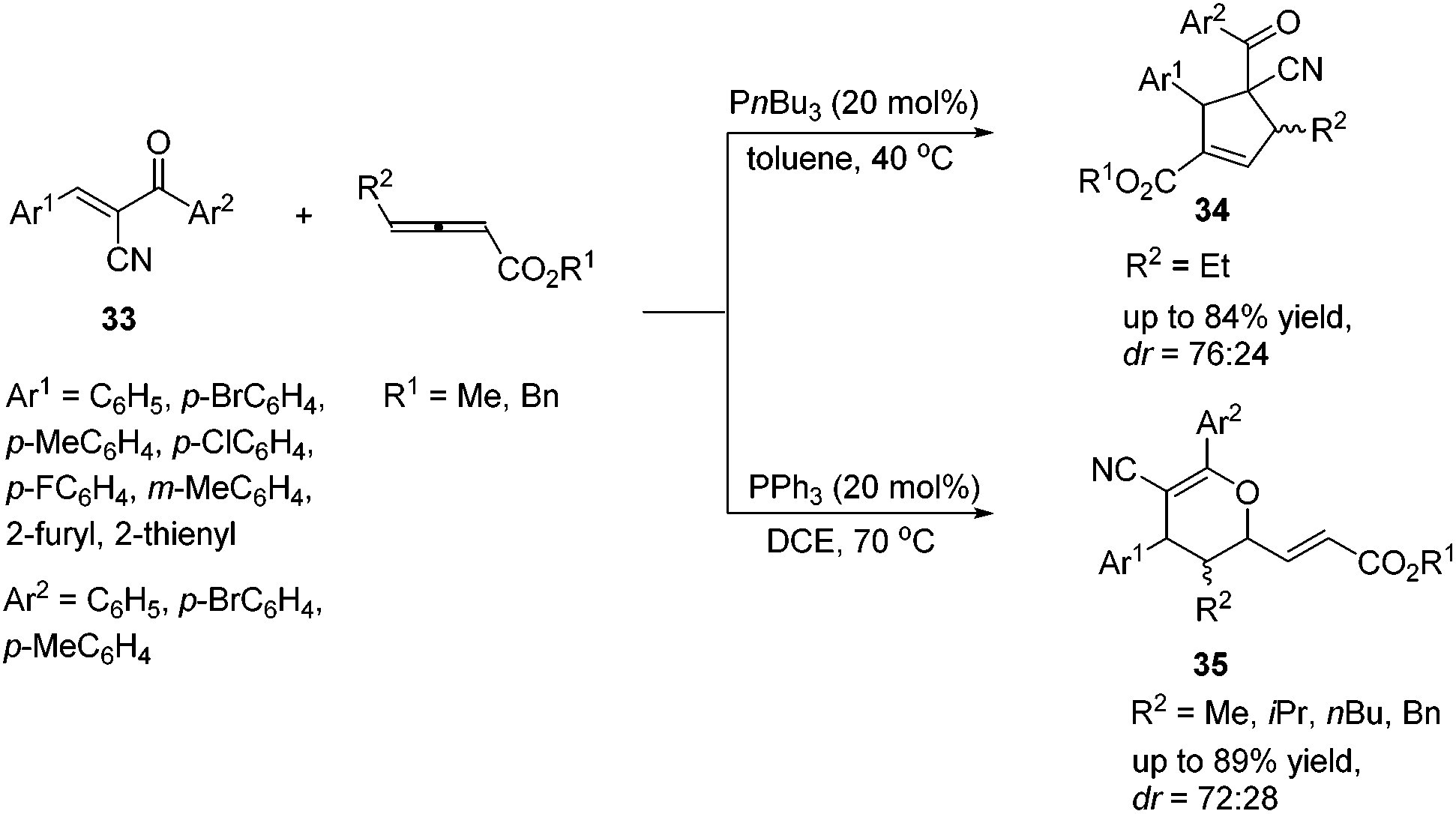
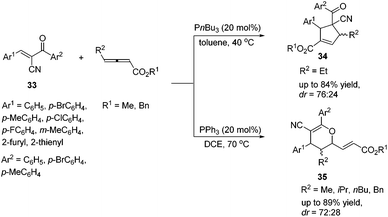
In 2015, Huang's group demonstrated a P(nBu)3 catalyzed [3 + 2] cycloaddition reaction of γ-substituted allenoates and α-cyano-α,β-unsaturated ketones 33, affording multifunctional cyclopentene products 34 in moderate to good yields with moderate diastereoselectivities.16 Lu's [3 + 2] cycloaddition reactions of a wide range of α-cyano-α,β-unsaturated ketones 33 with γ-substituted allenoates proceeded smoothly (Scheme 10). In general, aromatic ketones having electron-withdrawing substituents furnished higher yields than those with electron-donating substituents. They also found that changing P(nBu)3 to PPh3 or replacing an ethyl γ-substituent with a methyl, isopropyl, or n-butyl group afforded [4 + 2] products 35.
 |
| | Scheme 10 Phosphine-catalyzed cycloaddition reactions of γ-substituted allenoates and α-cyano-α,β-unsaturated ketones. | |
3.2 Lu's [3 + 2] cycloaddition of allenes with imines
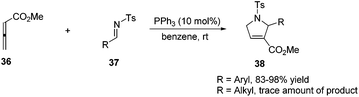
Lu's group expanded the [3 + 2] cycloaddition of allenes and alkenes to an allene–imine variant in 1997, shortly after their first report, providing functionalized dihydropyrroles.11a In a similar manner, the phosphine-catalyzed [3 + 2] cycloaddition of allenes 36 with aryl N-tosylimines 37 took place smoothly, however, affording the corresponding cycloadducts 38 with only one regioisomer in high yields (Scheme 11). Alkyl N-tosylimine could not undergo this reaction, probably due to the ready hydrolysis of alkyl aldimines.
 |
| | Scheme 11 Synthesis of dihydropyrroles through Lu's [3 + 2] cycloaddition. | |
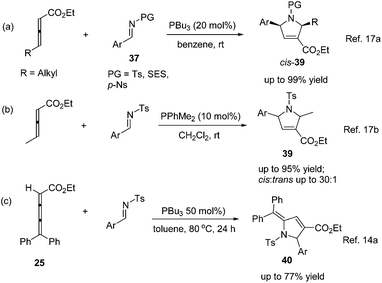
In 2005, Kwon and Shi independently reported Lu's [3 + 2] cycloaddition of activated allenes with imines catalyzed by phosphine to produce pyrrolidine derivatives.17 Kwon and co-workers employed γ-substituted allenoates with a series of imines 37 having different protecting groups catalyzed by PPh3 in benzene to afford 1,2,3,5-tetrafunctionalized dihydropyrroles 39 in good yields with high diastereoselectivity (Scheme 12, eqn (a)).17a Shi and co-workers reported the cycloadditions of ethyl penta-2,3-dienoate and aryl N-tosylimines catalyzed by PPhMe2 in CH2Cl2, affording dihydropyrroles 39 in moderate to good yields with good diastereoselectivity (Scheme 12, eqn (b)).17b In the following research, they reported Lu's [3 + 2] cycloaddition of 2,3,4-pentatrienoate 25 and aryl N-tosylimines catalyzed by PBu3 in toluene, giving the corresponding [3 + 2] cycloaddition products 40 in moderate to good yields (Scheme 12, eqn (c)).14a Switching the activated allenes to alkynyl ketones, Lu's [3 + 2] cycloaddition could also carry on smoothly with imines catalyzed by phosphine, affording highly functionalized pyrrolidines.18
 |
| | Scheme 12 Synthesis of highly functionalized pyrrolidines through Lu's [3 + 2] cycloaddition. | |
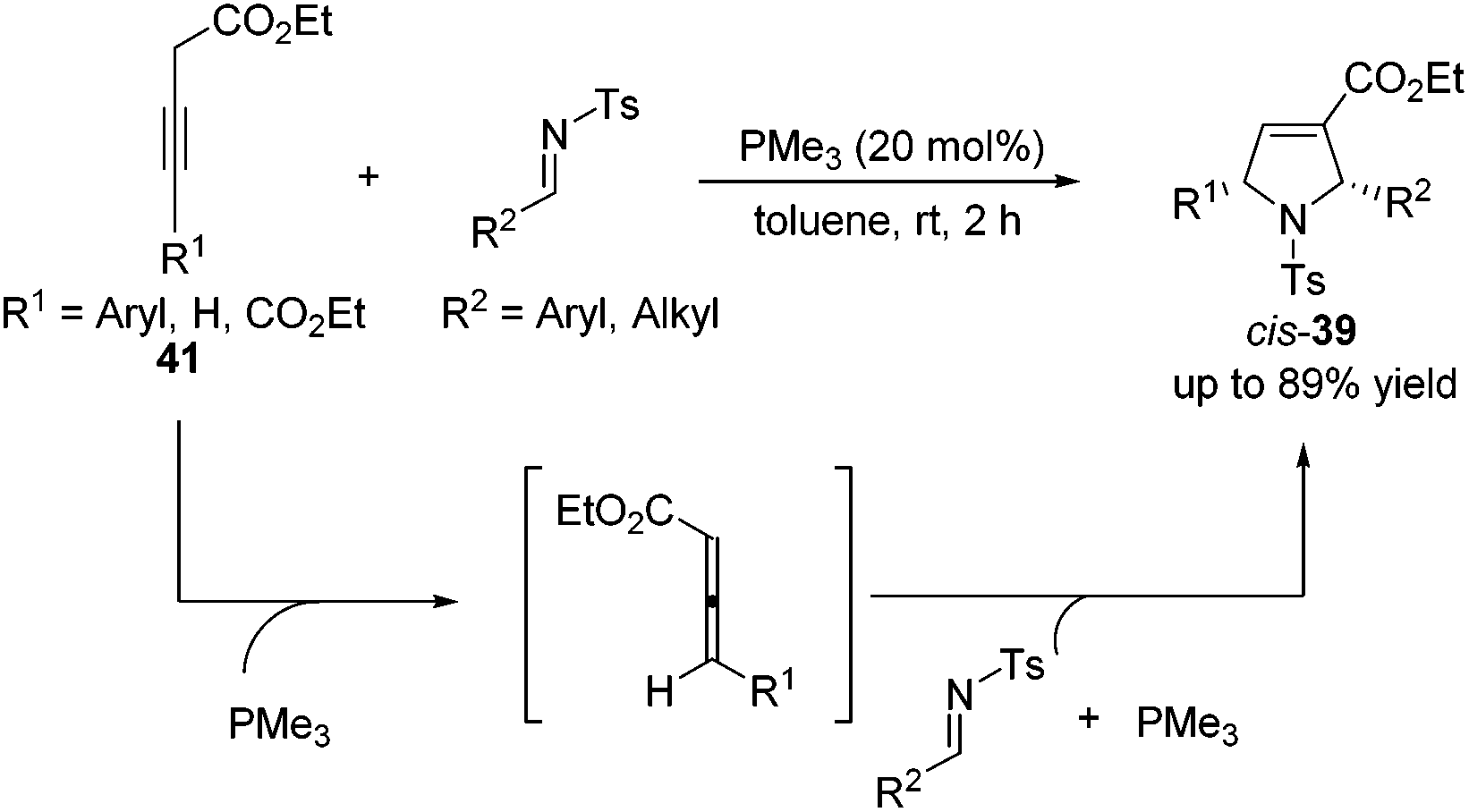
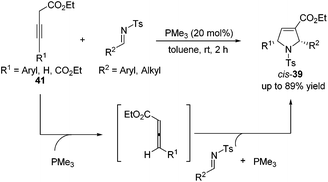
Although high yields with good diastereoselectivities were achieved by Lu's [3 + 2] cycloaddition of allenes with imines, the range of imines was limited to aryl imines. It was difficult to acquire satisfactory yields for this reaction when employing alkyl imines, mainly because of their decomposition through rapid hydrolysis. In 2011, Loh reported the first examples of the formation of highly efficient dihydropyrroles from various alkyl N-tosylimines.19 Unlike previous reports, Loh's group employed 3-alkynoates 41 as substrates, which were in situ isomerized to allenoates in the presence of PMe3, to carry on this [3 + 2] cycloaddition reaction (Scheme 13). They found that a highly nucleophilic trimethylphosphine catalyst was necessary and both aryl and alkyl imines with various substitution patterns could be tolerated for this reaction.
 |
| | Scheme 13 Synthesis of dihydropyrroles through Lu's [3 + 2] cycloaddition employing alkynoates and imines. | |
3.3 Asymmetric variants
3.3.1 Catalytic asymmetric Lu's [3 + 2] cycloaddition of allenes with activated alkenes.
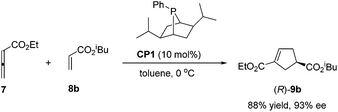
The pioneering work of catalytic asymmetric Lu's [3 + 2] cycloaddition of allenes with activated alkenes was reported by Zhang in 1997.20 A bicyclic monodentate chiral phosphine CP1 was designed and found to be an efficient catalyst for the [3 + 2] cycloaddition reaction. The best example was the reaction of allenoate 7 and isobutyl acrylate 8b conducted at 0 °C in toluene using 10 mol% chiral phosphine catalyst CP1, affording a cyclopentene product (R)-9b as a single regioisomer in 88% yield with 93% ee (Scheme 14). However, low regio- and enantioselectivities were observed in reactions using acrylates with smaller ester substituents (e.g., Me and Et). Although good enantioselectivity has been achieved for some substrates, the range of activated alkenes is limited to unsubstituted acrylate esters.
 |
| | Scheme 14 The first example of catalytic asymmetric Lu's [3 + 2] cycloaddition. | |
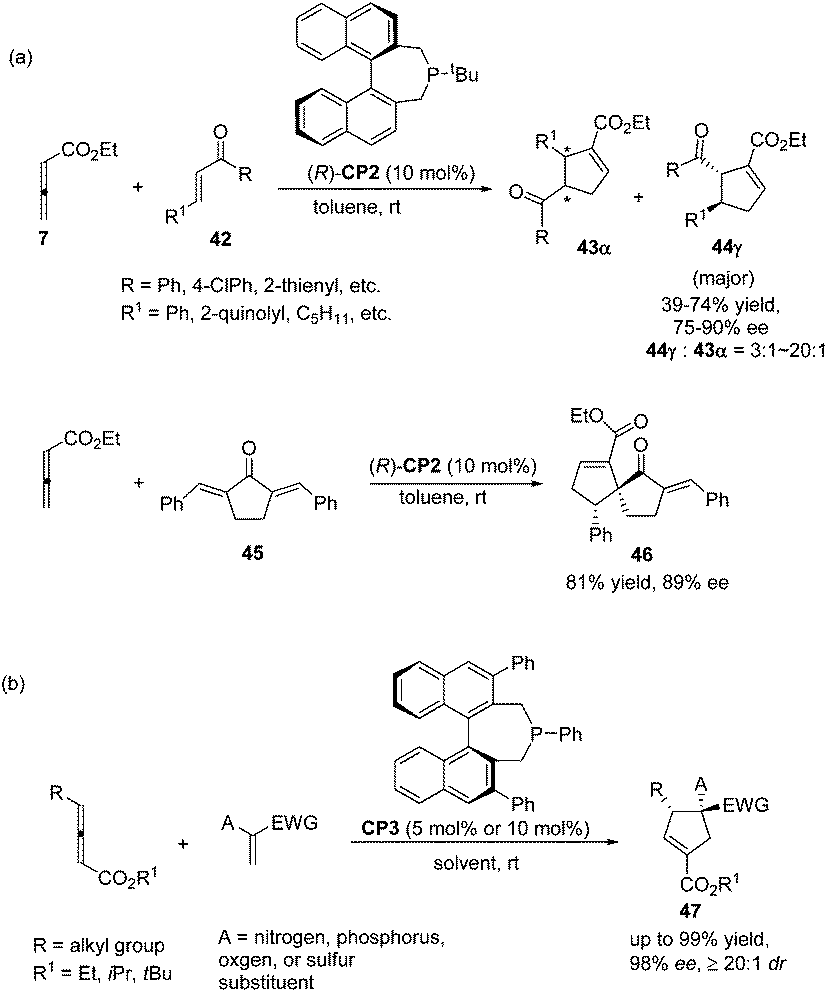
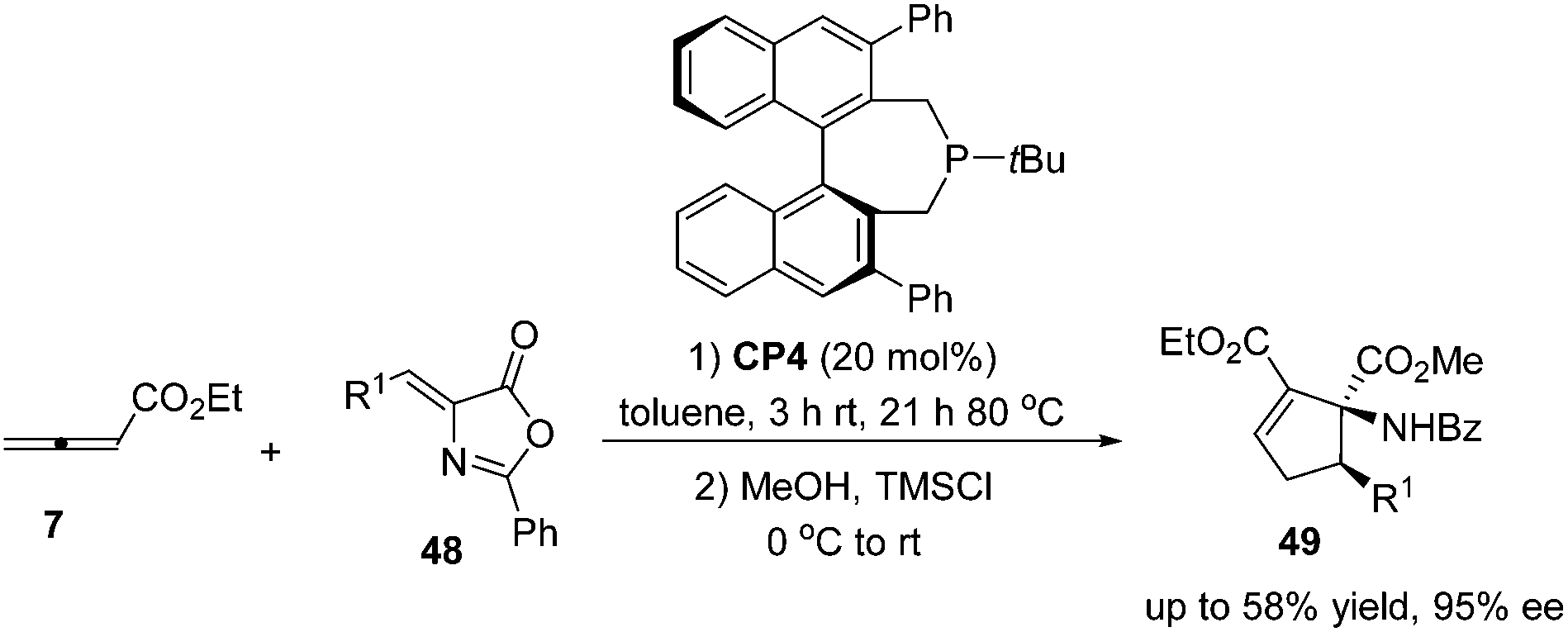
Despite this pioneering work reported in 1997, no further progress was reported on the development of catalytic asymmetric Lu's [3 + 2] cycloaddition for about a decade. In the recent decade, catalytic asymmetric Lu's [3 + 2] cycloaddition has experienced an explosive development with the development of chiral phosphine catalysts. Fu's group designed and synthesized binaphthyl-derived chiral phosphine (R)-CP2,21a which was originally designed as a ligand; however, chiral phosphine (R)-CP2 was identified as an efficient catalyst for enantioselective Lu's [3 + 2] cycloaddition of β-substituted enones and allenoates.21b The cycloaddition of 7 with chalcone substrates 42 catalyzed by chiral phosphine (R)-CP2 furnished cyclopentene products 43 and 44 with two contiguous stereogenic centers in good yields (39–74%) with high regio- (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 44:43) and good enantioselectivity (up to 90% ee) (Scheme 15, eqn (a)). They also demonstrated that dienone 45 could also undergo a highly enantioselective cycloaddition to give a spirocyclic product 46 in 81% yield with 89% ee using chiral phosphine (R)-CP2. Fu's report pointed out that the axial chirality adjacent to the phosphine center played a critical role in inducing high enantioselectivity (Scheme 15, eqn (a)). Later, Fu's group demonstrated that the chiral phosphine CP3 was an efficient catalyst in the asymmetric [3 + 2] cycloaddition of a wide range of racemic γ-substituted allenes with activated alkenes, providing cyclopentenes 47 bearing nitrogen-, phosphorus-, oxygen-, and sulfur-substituted quaternary stereocenters in high yields with good enantioselectivity, diastereoselectivity, and regioselectivity (Scheme 15, eqn (b)).21c The addition of the phosphine catalyst to the allene was identified as the turnover limiting step of the catalytic cycle through their mechanistic studies. In 2012, Jørgensen and co-workers employed chiral phosphine CP4 developed by Fu's group21a in a highly enantioselective [3 + 2] cycloaddition of 7 with olefinic azalactones 48, which was followed by a ring opening of the azlactone moiety to achieve one-pot synthesis of cyclic α-amino esters 49 in good overall yields and high enantioselectivities (up to 95% ee) (Scheme 16)22 and they also showed that products 49 could be easily transformed to amino acids or α-hydroxy-β-ketoesters.
:1, 44:43) and good enantioselectivity (up to 90% ee) (Scheme 15, eqn (a)). They also demonstrated that dienone 45 could also undergo a highly enantioselective cycloaddition to give a spirocyclic product 46 in 81% yield with 89% ee using chiral phosphine (R)-CP2. Fu's report pointed out that the axial chirality adjacent to the phosphine center played a critical role in inducing high enantioselectivity (Scheme 15, eqn (a)). Later, Fu's group demonstrated that the chiral phosphine CP3 was an efficient catalyst in the asymmetric [3 + 2] cycloaddition of a wide range of racemic γ-substituted allenes with activated alkenes, providing cyclopentenes 47 bearing nitrogen-, phosphorus-, oxygen-, and sulfur-substituted quaternary stereocenters in high yields with good enantioselectivity, diastereoselectivity, and regioselectivity (Scheme 15, eqn (b)).21c The addition of the phosphine catalyst to the allene was identified as the turnover limiting step of the catalytic cycle through their mechanistic studies. In 2012, Jørgensen and co-workers employed chiral phosphine CP4 developed by Fu's group21a in a highly enantioselective [3 + 2] cycloaddition of 7 with olefinic azalactones 48, which was followed by a ring opening of the azlactone moiety to achieve one-pot synthesis of cyclic α-amino esters 49 in good overall yields and high enantioselectivities (up to 95% ee) (Scheme 16)22 and they also showed that products 49 could be easily transformed to amino acids or α-hydroxy-β-ketoesters.
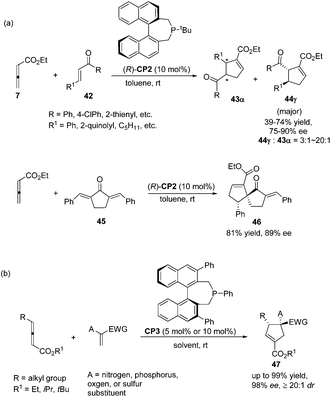 |
| | Scheme 15 The catalytic asymmetric Lu's [3 + 2] cycloadditions reported by Fu's group. | |
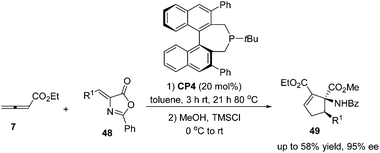 |
| | Scheme 16 The catalytic asymmetric Lu's [3 + 2] cycloadditions of allenoates with olefinic azalactones. | |
Marinettis's group has done a lot of work for the development of chiral phosphine catalyzed asymmetric [3 + 2] cycloadditions of a variety of allenes and activated alkenes. They reported a stereoselective synthetic approach to prepare a planar chiral 2-phospha[3]ferrocenophane CP5, which displayed good air-stability, and they applied it to promote enantioselective [3 + 2]-cycloadditions between allenoates and enones.23a The planar chiral phosphine CP5 demonstrated the robust ability to catalyze the [3 + 2]-cycloadditions of allenoate with various electron-deficient alkenes, affording the desired products 50 and 51 in high yields with good regio- and enantioselectivities (Scheme 17). Subsequently, they found that the chiral phosphine CP5 could be recovered after the completion of catalytic reactions and reused for a further run, giving comparable ratios of regioisomers and enantiomeric excess.23b The chiral phosphine CP5 also proved to be a remarkably efficient catalyst for enantioselective [3 + 2]-cycloadditions of allenic phosphonates and enones in terms of both product selectivity and enantioselectivity, furnishing a variety of aryl- and heteroaryl-substituted cyclopentenylphosphonates 52 in high yields with good enantioselectivities (Scheme 17).23c Based on the preliminary DFT calculations, they accounted for the chiral induction role by CP5 in these enantioselective [3 + 2]-cycloaddition reactions.23c In 2010, Marinetti's group reported that enantiomerically enriched aryl-substituted dicyanocyclopentenes could be easily accessed through the enantioselective [3 + 2]-cycloaddition of allenoates and 2-aryl-1,1-dicyanoethylenes.23e The use of (S)-t-butyl-binepine (S)-CP2 as the chiral organocatalyst allows the synthesis of functionalized cyclopentenes 53 with both aryl and heteroaryl substituents on the stereogenic carbon, in high yields with up to 95% ee (Scheme 17). This work was the first enantioselective variant of the phosphine-promoted [3 + 2] cycloaddition reaction between allenoates and 2-aryl-1,1-dicyanoethylenes, which expanded the scope of the enantioselective [3 + 2] cycloaddition reaction. In 2008 and 2009, Marinetti's group reported that the [3 + 2] cycloadditions of allenic esters and β-substituted exocyclic enones 54 catalyzed by CP5 afforded spirocyclic derivatives 55 in moderate yields with good enantioselectivities (Scheme 17).23a,b In addition, they switched substrates to 3-alkylideneindolin-2-ones 56 and the highly enantioselective [3 + 2] cycloadditions could also be achieved, giving a range of spirocyclic oxindolic cyclopentanes 57 and 58 in good yields with high regioselectivities and excellent enantioselectivities (Scheme 17).23d It should be mentioned here that the reaction of allenoate with tricyclic indolinone 59 produced the unusual spirocyclic alkaloid scaffold 60 in an acceptable isolated yield (59%) and 94% ee (Scheme 17).23d The spirocyclic moiety of 60 constitutes the core unit of known natural products such as cyclopiamine B, which demonstrates the potential synthetic applications of these enantioselective [3 + 2] cycloadditions. In 2011, Marinetti's group also demonstrated the example of an enantioselective desymmetrization process via the [3 + 2] cycloadditions of activated allenes with 4-substituted 2,6-diarylidenecyclohexanones 61 or 2,4-diarylidenebicyclo[3.1.0]hexan-3-ones 62 catalyzed by CP5 or (S)-CP2, producing the spirocyclic compounds 63, as well as the unique spiranic scaffolds 64 involving the stereocontrolled production of up to five stereogenic carbon centers in a single process (Scheme 17).23f This work extended Fu and Marinetti's previous work21b,23d on the asymmetric synthesis of a spirocyclic compound. Subsequently, the synthetic approach to access cyclopentene-fused coumarin based on phosphine-promoted [3 + 2] cycloadditions was demonstrated by Marinetti's group.23g Enantiomerically enriched compounds 65 and 66 with unprecedented substitution patterns and functionalization were obtained in good yields (Scheme 17).
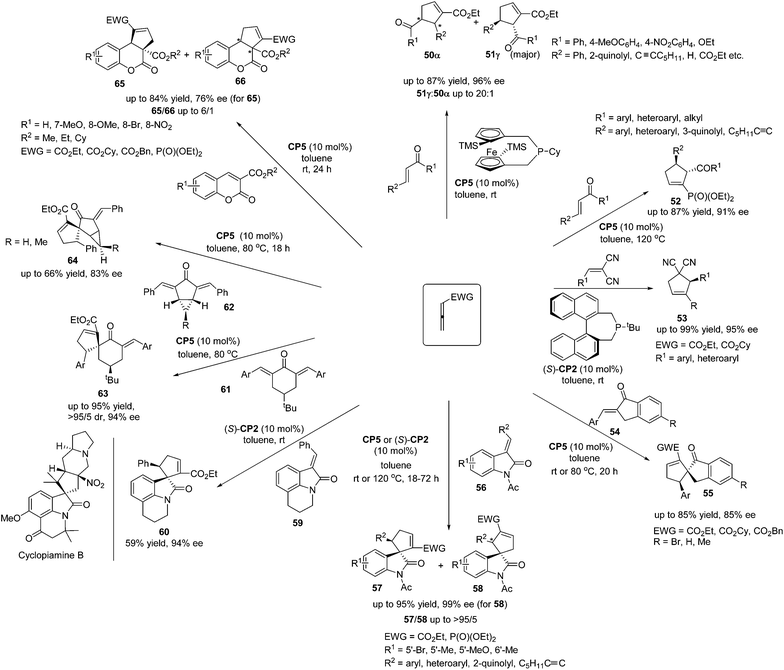 |
| | Scheme 17 The catalytic asymmetric Lu's [3 + 2] cycloaddition developed by Marinetti's group. | |
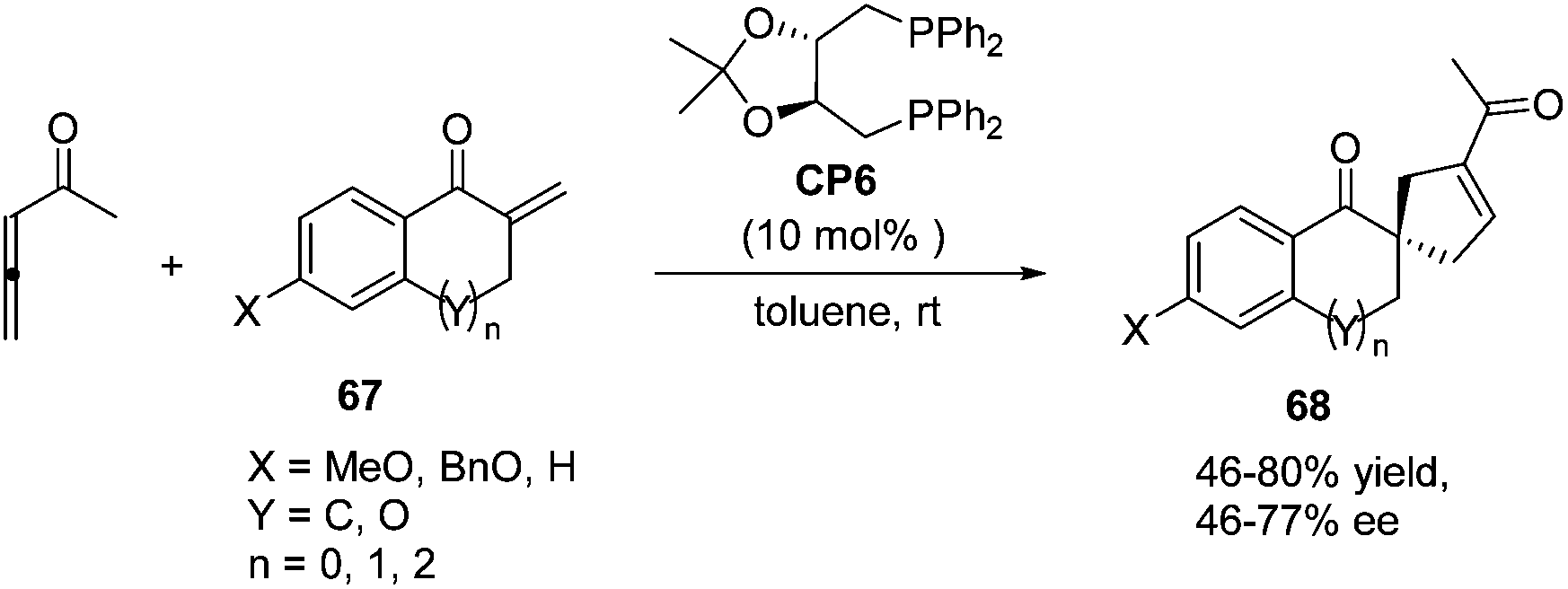
In 2007, Wallace and co-workers first applied commercially available bidentate DIOP (CP6) as a chiral phosphine catalyst in the enantioselective [3 + 2] cycloaddition of allenyl methyl ketone and substituted enones 67.24 Spirocyclic ketones 68 were acquired in moderate to good yields with moderate to good enantiomeric excess (Scheme 18).
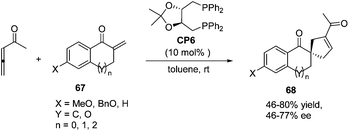 |
| | Scheme 18 The catalytic asymmetric Lu's [3 + 2] cycloadditions of allenyl methyl ketone with substituted exocyclic enones. | |
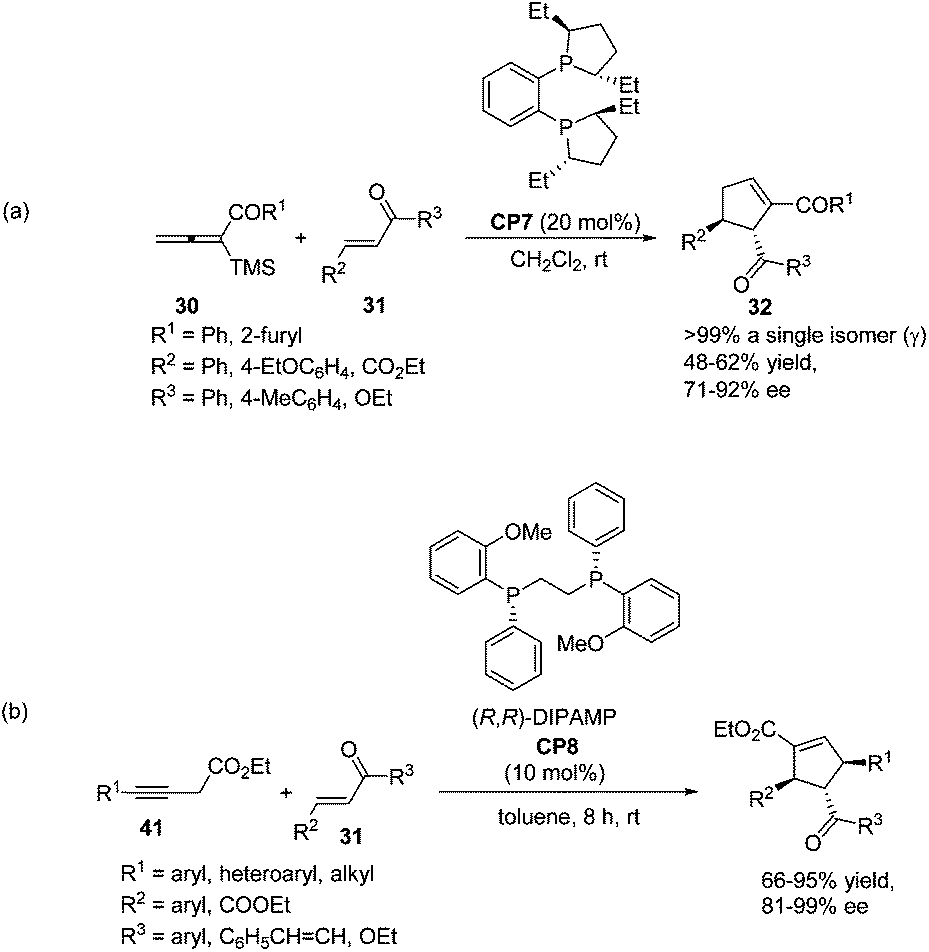
In 2009, Loh achieved the asymmetric variant of the [3 + 2] cycloadditions of aryl allenic ketones with acyclic, electron-poor olefins using commercially available chiral phosphine (S,S)-Et-DuPHOS (CP7) as a catalyst.15 The highly enantioselective products 32 were obtained in moderate yields (Scheme 19, eqn (a)). Later, a highly enantio- and regio-selective one-pot [3 + 2] cycloaddition reaction via isomerization of 3-butynoates to allenoates was also reported by Loh, in which the commercially available chiral phosphine (R,R)-DIPAMP CP8 was employed as a catalyst (Scheme 19, eqn (b)).25 Their control experiments indicated that 3-butynoates (41) were first in situ isomerised to allenoates and the chirality of the allenoates did not play a significant role in the asymmetric induction of the reaction.
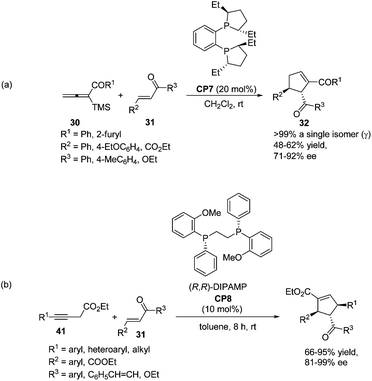 |
| | Scheme 19 The catalytic asymmetric Lu's [3 + 2] cycloadditions developed by Loh's group. | |
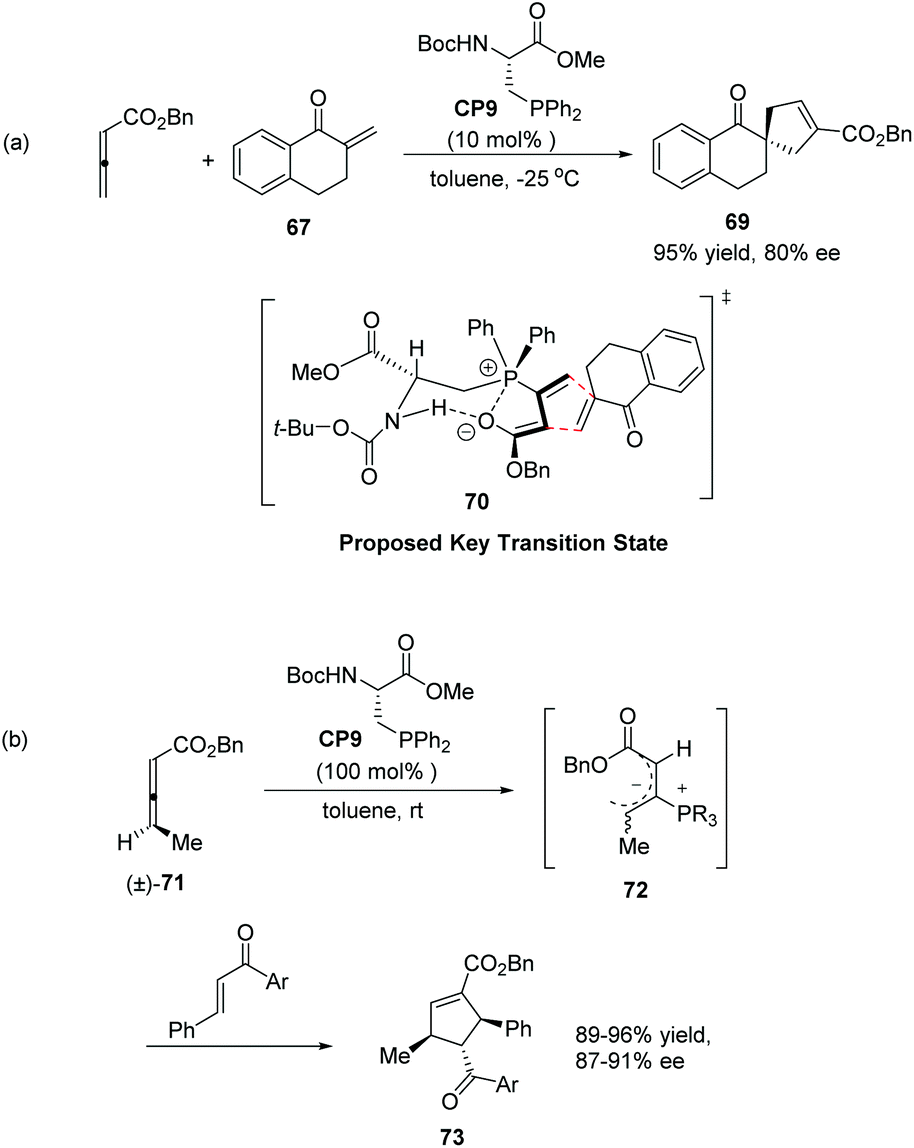
Inspired by Gilbertson's work on phosphine-embedded peptides,26 Miller's group synthesized an amino acid-based phosphine catalyst CP9 which has a chiral center separated by a methylene group from the phosphine atom for enantioselective allenoate–enone cycloadditions in 2007.27 The reaction of allenic ester with tetralone-derived exomethylene substrate 67 was smooth in the presence of 10 mol% CP9 at −25 °C in toluene to afford the desired product 69 as a single regioisomer in 95% yield with 80% ee (Scheme 20, eqn (a)). These reactions proved efficient for a variety of cyclic and acyclic exomethylenes. The origin of enantio-induction was rationalized by the intramolecular hydrogen interaction and Coulombic P+⋯O− interaction in the proposed key transition state 70. In this system, the substrate approaches the zwitterionic intermediate from the bottom face, opposite to a phenyl substituent on the phosphine catalyst. Furthermore, they developed a “dynamic kinetic asymmetric transformation” using γ-substituted racemic allene substrates (±)-71, based on the fact that the addition of a phosphine to an allene gives a zwitterionic intermediate, such as 72 which erases the element of planar chirality (Scheme 20, eqn (b)).27 They found that a stoichiometric amount of the catalyst could promote the reaction to full conversion, affording highly substituted cycloadducts 73 in excellent yields as single regio- and diastereomers (Scheme 20, eqn (b)). Decreasing catalyst loading to 20 mol%, the high enantioselectivity was retained; however, the yield was significantly decreased. These examples provided unique examples of allenoate deracemizations via chiral phosphine-catalyzed [3 + 2] cycloadditions.
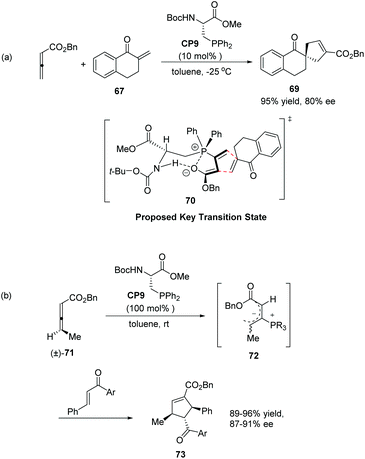 |
| | Scheme 20 The catalytic asymmetric Lu's [3 + 2] cycloadditions developed by Miller's group. | |
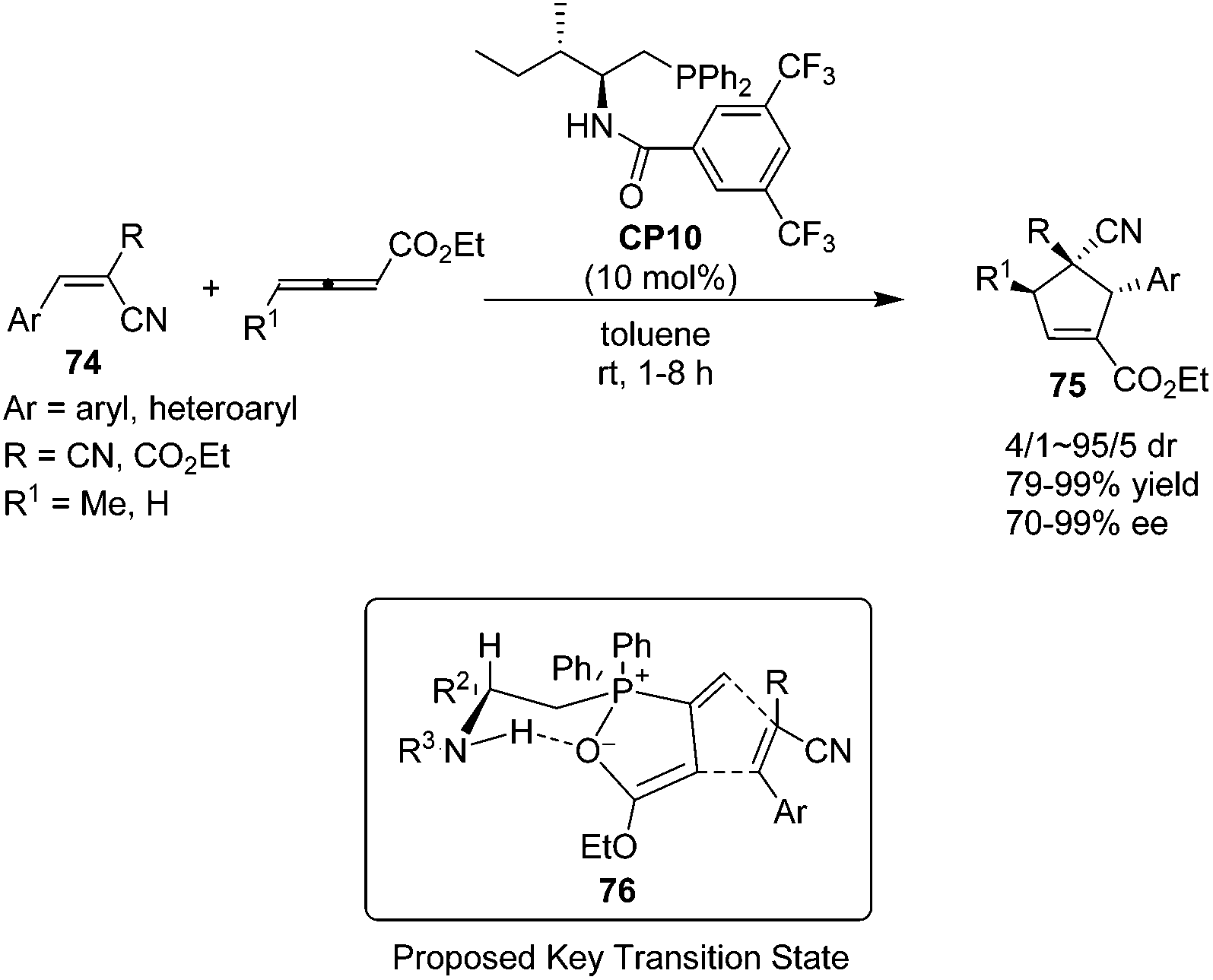
In 2010, Zhao's group prepared a novel bifunctional N-acyl amino phosphine CP10 and found that it was an effective catalyst for the asymmetric [3 + 2] cycloadditions of allenoates and activated alkenes.28 The first asymmetric organocatalytic [3 + 2] cycloaddition of allenoates with dual activated alkenes 74 using a novel bifunctional N-acyl aminophosphine catalyst was achieved, providing a series of chiral cyclopentenes 75 in high yields (79–99%) with good to excellent enantioselectivities (70–99%) (Scheme 21). Unfortunately, the activated alkenes containing aliphatic substituents were not suitable for this reaction. Compared to Marinetti's work (see Scheme 17),23e a wider scope of the substrates and a great improvement of the yields and enantioselectivities had been achieved by Zhao's work. A possible key transition state 76 similar to that suggested by Miller27 may account for the stereochemical results of their reaction. The catalyst assembles the allenoate by a synergistic action of its two functional groups to form a zwitterion. Then the dipolarophile may approach the zwitterion preferentially from the Si face to minimize the steric repulsion from the R2 and phenyl groups of the catalyst.
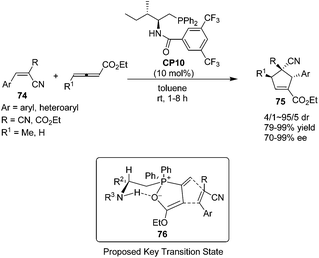 |
| | Scheme 21 The catalytic asymmetric Lu's [3 + 2] cycloadditions developed by Zhao's group. | |
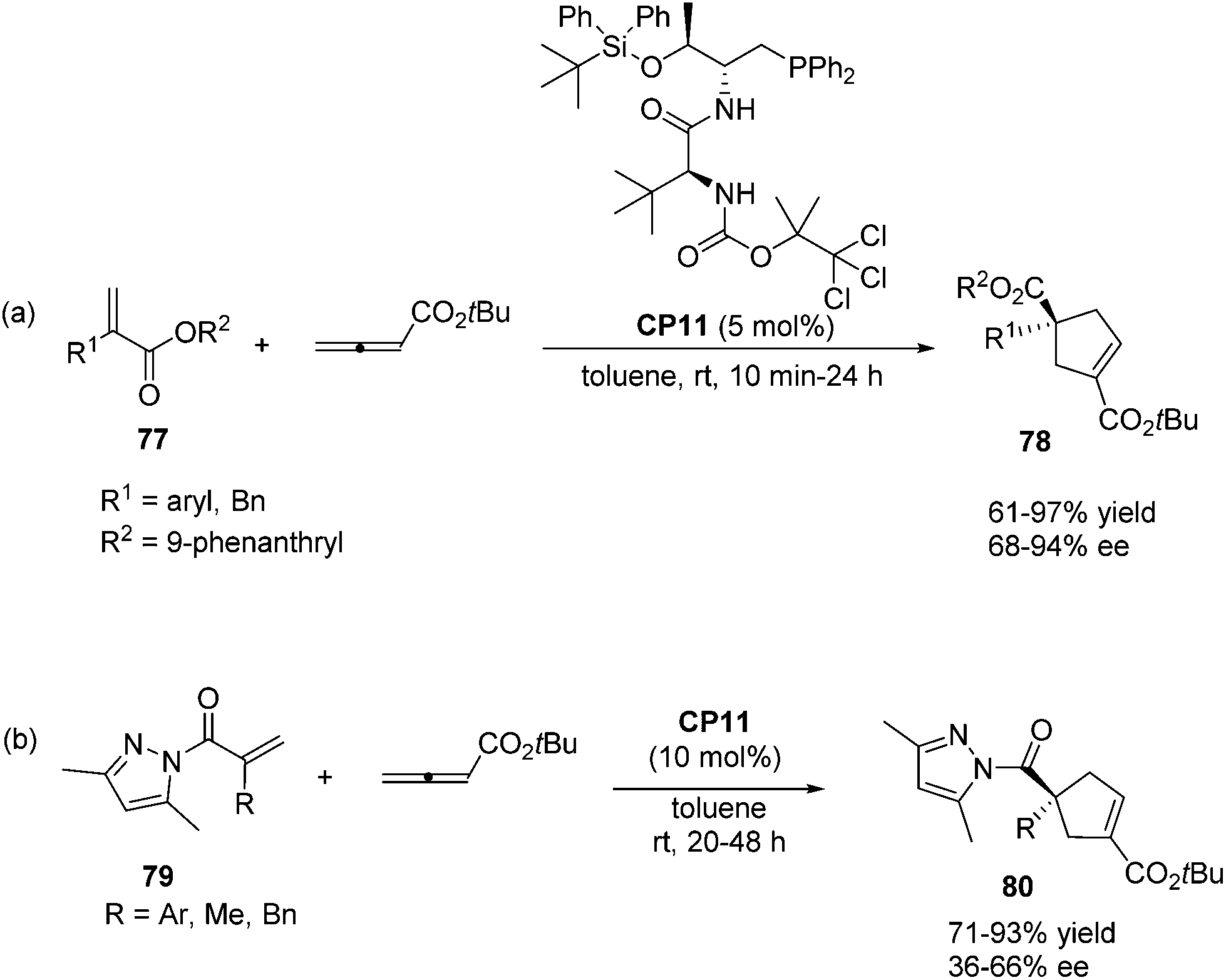
In 2011, Lu's group designed and synthesized a new family of dipeptide-based chiral phosphines.29a They first reported that the enantioselective [3 + 2] cycloaddition reactions of α-substituted acrylates 77 with tert-butyl buta-2,3-dienoate could be catalyzed by Thr-L-tert-Leu-based phosphine CP11 efficiently, affording functionalized cyclopentenes 78 containing quaternary stereocenters in 61–97% yields and 68–94% ee (Scheme 22, eqn (a)). More sterically hindered substituents, such as 9-phenanthryl and t-Bu, are necessary for higher diastereoselectivity and enantioselectivity for the formation of 78. A plausible mechanism and a key transition state model to rationalize the enantioselectivity for this reaction are presented in Scheme 23. The hydrogen-bonding interactions of the acrylate substrate and the dipeptidic backbone of the catalyst are proposed to play an important role in stereoselectivity. They propose that the nucleophilic addition of the phosphine catalyst to the allene generates the phosphonium enolate intermediate, which approaches the acrylate from its Re face to afford the major stereoisomer. The high regioselectivity is probably due to the unfavorable steric hindrance of the bulky tert-butyl group in the acrylate substrate and the sterically hindered carbamate moiety in the catalyst inhibited the generation of the γ-regioisomer. Subsequently, Lu's group first reported that acrylamides 79 derived from 3,5-dimethyl-1H-pyrazole were also suitable for the asymmetric [3 + 2] cycloaddition with tert-butyl buta-2,3-dienoate.29b Thr-L-tert-Leu-based phosphine CP11 is also proven to be the most effective catalyst for these reactions, affording regiospecific [3 + 2]-cycloaddition products 80 in excellent yields with moderate enantioselectivities (Scheme 22, eqn (b)).
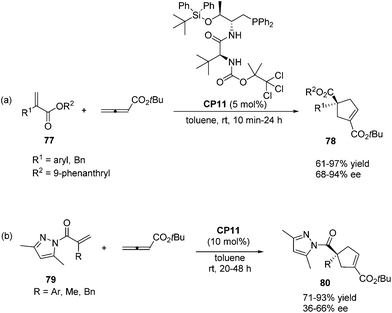 |
| | Scheme 22 The catalytic asymmetric Lu's [3 + 2] cycloadditions developed by Lu's group. | |
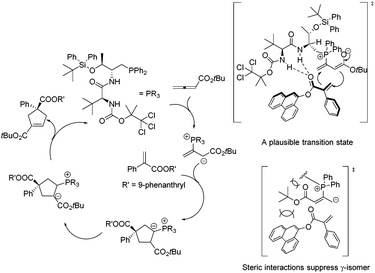 |
| | Scheme 23 The proposed mechanism and a key transition state to account for the enantioselectivity. | |
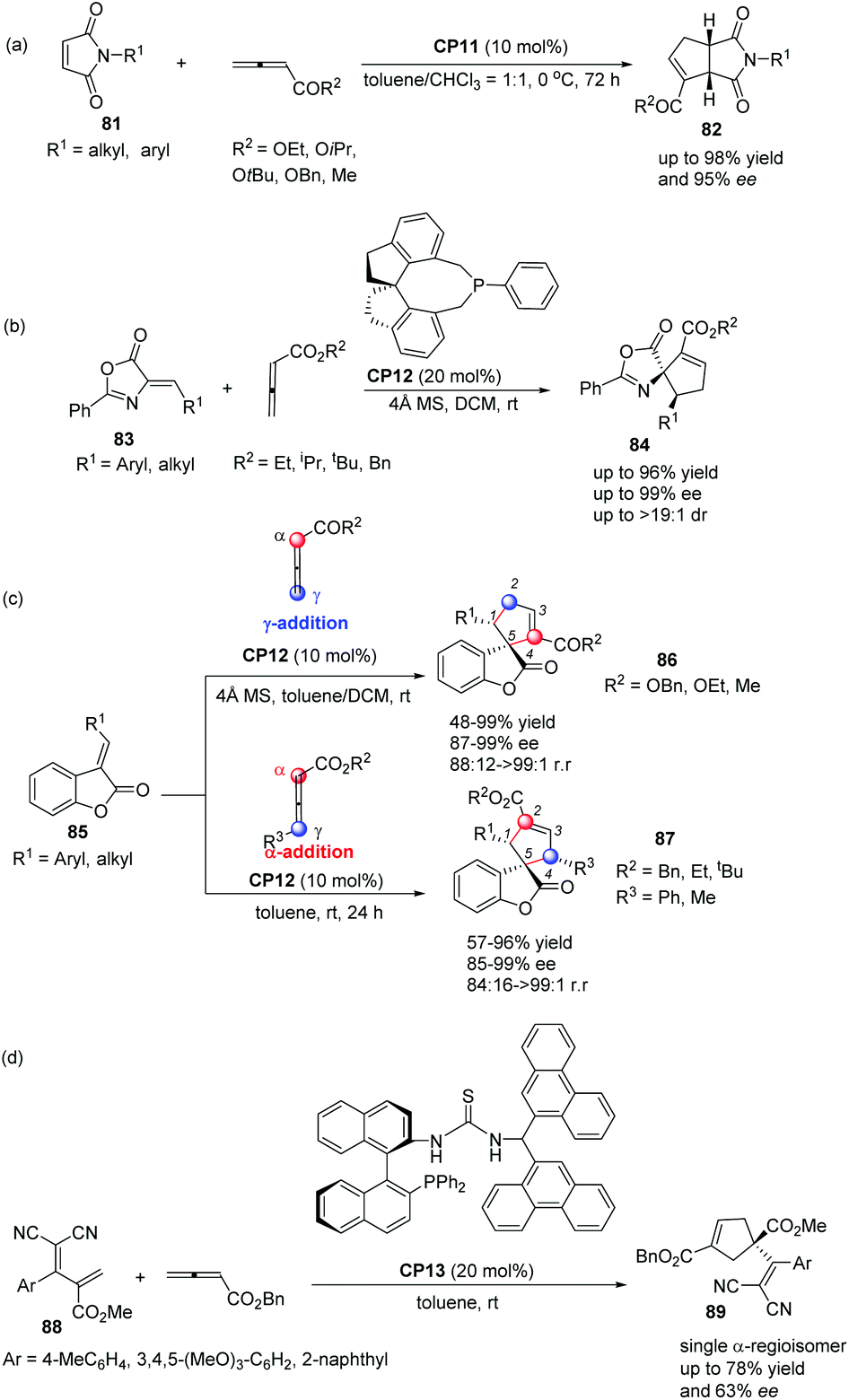
In 2012, Shi's group also applied Thr-L-tert-Leu-based phosphine CP11 in a highly enantioselective [3 + 2] cycloaddition of maleimides with allenes.30a In the catalysis of CP11, a variety of maleimides 81 and electron deficient allenes underwent the asymmetric [3 + 2] cycloaddition reaction smoothly, affording the corresponding functionalized bicyclic cyclopentenes 82 containing two tertiary stereogenic centers in moderate to good yields along with good to high enantioselectivities (Scheme 24, eqn (a)). Subsequently, they reported an interesting chiral phosphine catalyzed asymmetric [3 + 2] cycloaddition of allenoates with alkylidene azlactones 83 using chiral phosphine (R)-SITCP CP12 as the catalyst,31 furnishing the spiro cycloadducts 84 in good yields with excellent diastereo- and enantioselectivities (Scheme 22, eqn (b)).30b Later on, they again applied the chiral phosphine (R)-SITCP CP12 as the catalyst in a highly enantioselective [3 + 2] cycloaddition of benzofuranone-derived olefins 85 with allenoates and substituted allenoates.30c Interestingly, they found that the γ-substituent in the allenoate strongly affected the regioselectivity. Using allenoates without γ-substituents, γ-addition products 86 were the main products; however, employing allenoates having γ-substituents, the main products were switched to α-addition products 87 (Scheme 24, eqn (c)). In 2013, Shi's group also successfully applied their own multifunctional chiral thiourea-phosphine CP13 having an axially chiral binaphthyl scaffold in the asymmetric [3 + 2] cycloaddition of allenoates with α,α-dicyanoolefin-substituted acrylates.30d In the catalysis of CP13, α,α-dicyanoolefin-substituted acrylates 88 and benzyl allenoate underwent the [3 + 2] cycloaddition reaction, affording the single α-regioisomer 89 in good yields with moderate enantioselectivities (Scheme 24, eqn (d)). This was the first report to employ α,α-dicyanoolefin-substituted acrylates as substrates in the [3 + 2] cycloaddition of allenoates with electron-deficient alkenes, which extended the reaction scope of the asymmetric [3 + 2] cycloaddition reaction.
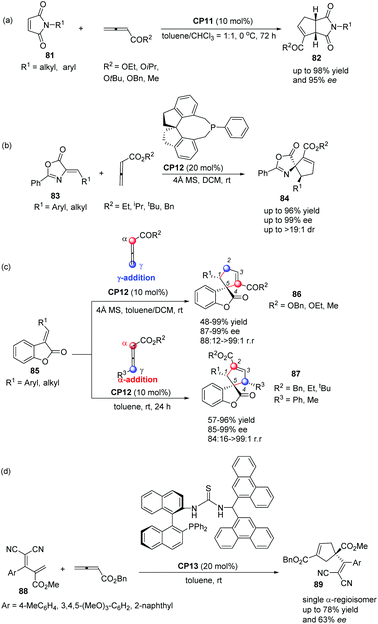 |
| | Scheme 24 The catalytic asymmetric Lu's [3 + 2] cycloaddition developed by Shi's group. | |
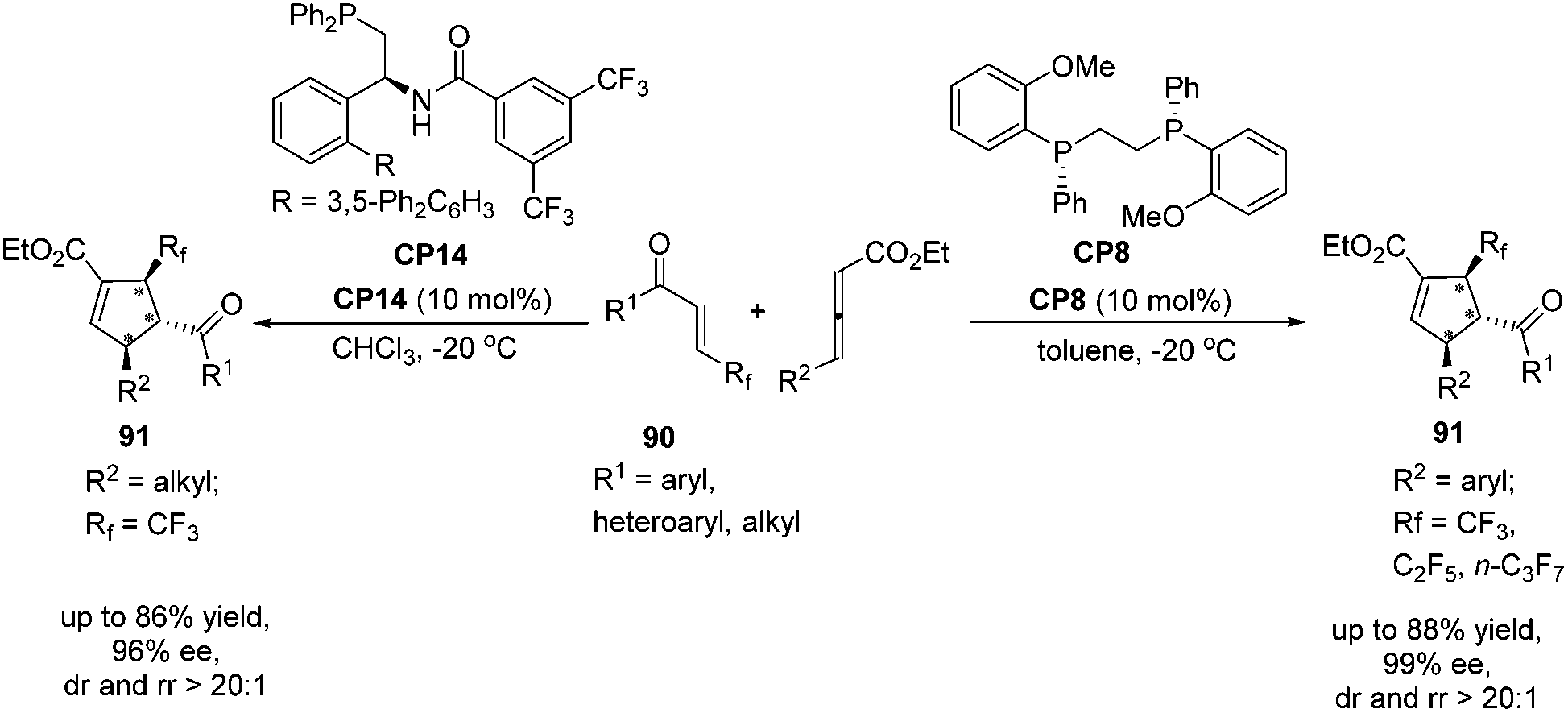
Very recently, Zhang's group reported a highly regio-, diastereo- and enantioselective [3 + 2] cycloaddition of γ-substituted allenoates with β-perfluoroalkyl enones catalyzed by (R,R)-DIPAMP CP8 or multifunctional phosphine CP14 developed by Zhang's group as a catalyst, furnishing a wide range of densely functionalized perfluoroalkylated cyclopentenes containing three contiguous chiral stereocenters.32 In the catalysis of CP8, a wide range of β-trifluoromethyl substituted enones 90 containing different electron nature functional groups with γ-aryl substituted allenoates underwent the enantioselective [3 + 2] cycloaddition smoothly, affording a series of highly regioselective α-addition tri-fluromethylated cyclopentenes 91 in good yields with high diastereo- and enantioselectivities (Scheme 25). It was noteworthy that both β-pentafluoroethyl and β-heptafluoropropyl enone were also tolerated in this asymmetric [3 + 2] cycloaddition reaction. In the catalysis of (R,R)-DIPAMP CP8, the cycloaddition of γ-alkyl substituted allenoates with β-perfluoroalkyl enones did not proceed very well. Thus, they utilized multifunctional phosphine CP14 as a catalyst to achieve a highly regio-, diastereo- and enantioselective [3 + 2] cycloaddition of γ-alkyl substituted allenoates with β-perfluoroalkyl enones (Scheme 25).
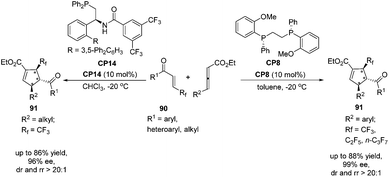 |
| | Scheme 25 The catalytic asymmetric Lu's [3 + 2] cycloaddition developed by Zhang's group. | |
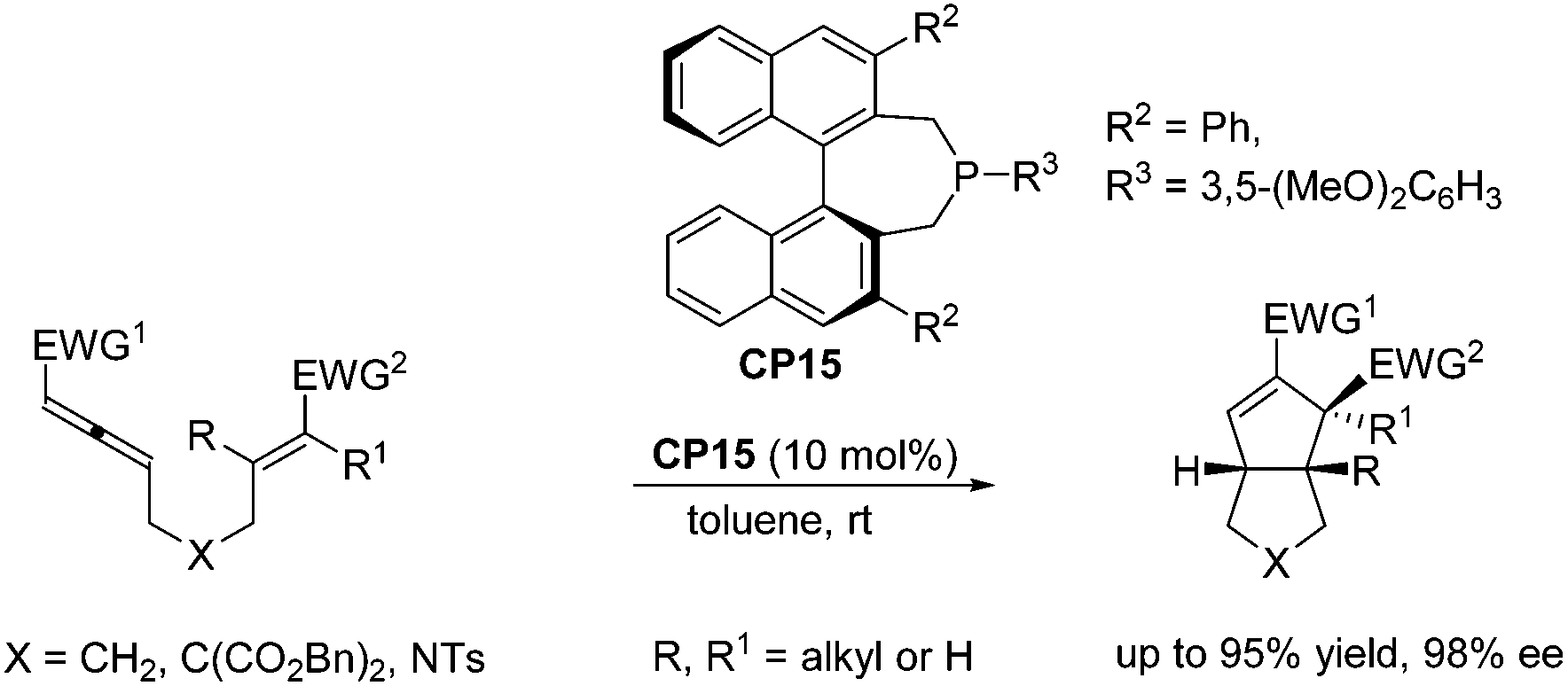
Despite the fast development of asymmetric catalysis of the intermolecular Lu's [3 + 2] cycloaddition, there has been no corresponding progress with respect to the enantioselective intramolecular Lu's [3 + 2] cycloaddition reaction. Until 2015, Fu and co-workers achieved highly enantioselective intramolecular Lu's [3 + 2] cycloaddition reactions, delivering functionalized, fused bicyclic ring systems that bear multiple contiguous stereocenters.33 Using chiral phosphine CP15 as a catalyst, a wide range of substrates underwent the intramolecular Lu's [3 + 2] cycloaddition reaction smoothly, affording a series of diquinane and quinolin-2-one derivatives in good yields with high enantioselectivies (Scheme 26), which provided an access to useful scaffolds that are found in bioactive compounds.
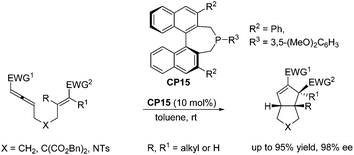 |
| | Scheme 26 The catalytic asymmetric intramolecular Lu's [3 + 2] cycloaddition reported by Fu's group. | |
3.3.2 Catalytic asymmetric Lu's [3 + 2] cycloaddition of allenes with imines.
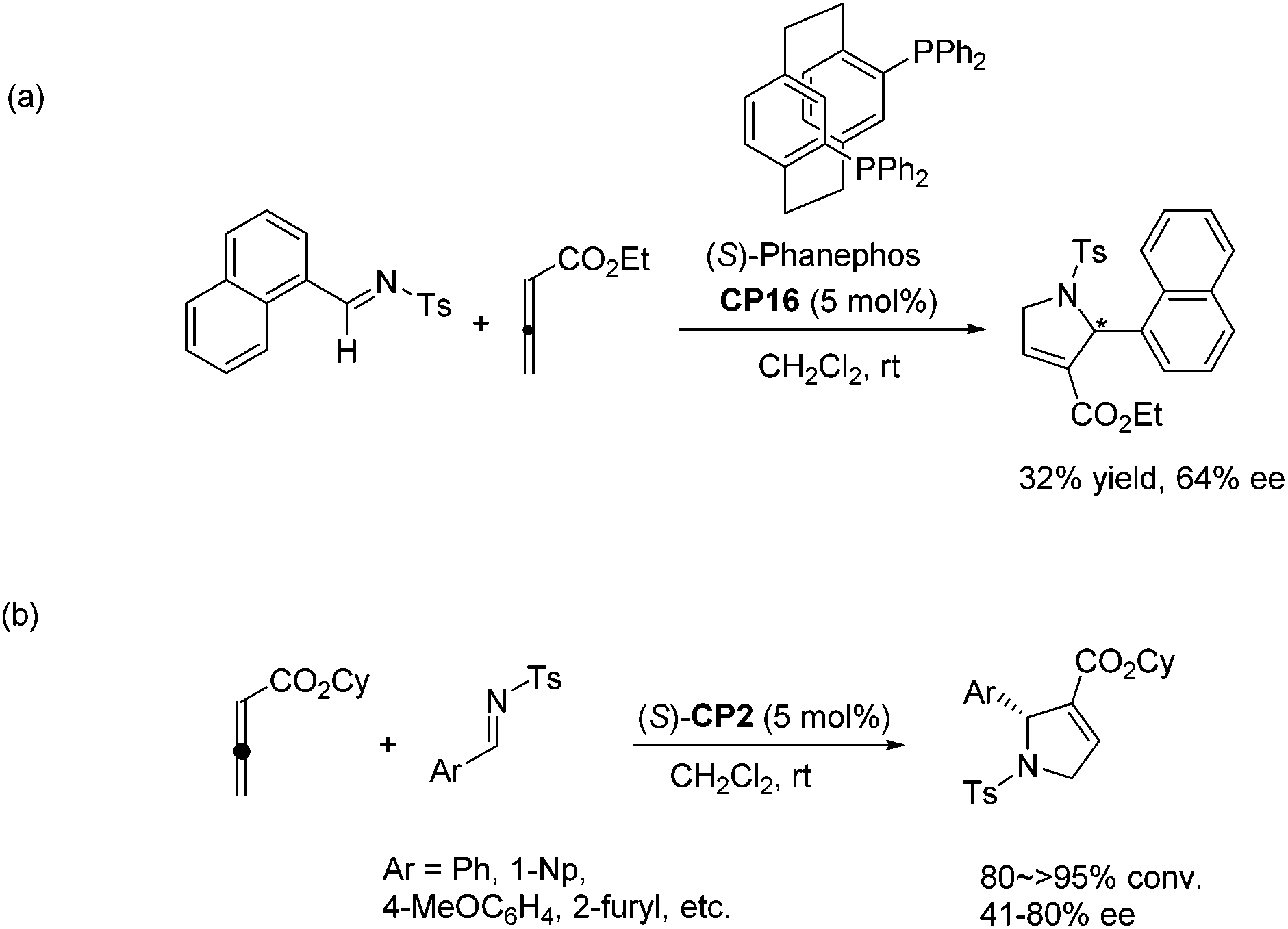
In 2006, Marinetti and co-workers first reported an asymmetric [3 + 2] cycloaddition of 2,3-butadienoates with aryl imines.34a They systematically screened a series of commercially available chiral phosphines, and identified bidentate chiral (S)-phosphine CP16 as an efficient catalyst for this asymmetric cycloaddition reaction. However, only moderate yield and enantioselectivity were achieved in their studies (Scheme 27, eqn (a)).34a Although they subsequently improved the enantioselectivity of the reaction by increasing the size of the ester group of the allenoates and employed the binaphthyl-derived monodentate chiral phosphine (S)-CP2 as the catalyst (Scheme 27, eqn (b)), high enantioselectivity was still not achieved and the substrates were limited.34b
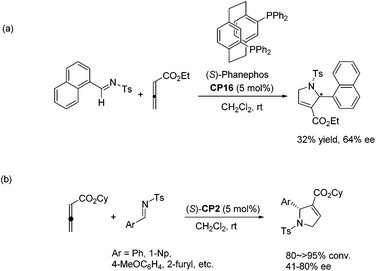 |
| | Scheme 27 The catalytic asymmetric Lu's [3 + 2] cycloaddition of allenoates with imines developed by Marinetti's group. | |
Jacobsen and co-workers made a great improvement to the [3 + 2] cycloaddition of 2,3-butadienoates with aryl imines.35 Employing N-diphenylphosphinoyl (DPP) imines 92 as substrates combined with a specifically designed chiral bifunctional thiourea-phosphine catalyst CP17 they achieved high enantioselectivity up to 98% ee to access pyrrolines 93 (Scheme 28, eqn (a)). They proposed a model to rationalize stereoinduction based on a proposed key transition state 94, in which the imine may associate with the thiourea moiety in the catalyst through hydrogen bonding. The zwitterionic enolate probably adds to the imine from the Re face in an intramolecular manner, providing the favoured enantiomer of the cycloadduct. However, high enantioselectivity did not only rely on the hydrogen bonding interaction between the chiral catalyst and the substrate, since Marinetti's group also achieved high enantioselectivity for the same reaction using chiral phosphine (S)-CP2 that did not have any hydrogen bonding interaction with the substrate.34c They employed N-DPP-substituted imines 92 as substrates and BINEPINE (S)-CP2 as the catalyst, giving pyrrolines 93 in good yields with a high enantiomeric excess (up to 92% ee) (Scheme 28, eqn (b)). In this particular case, the increased chiral induction probably resulted from the increased steric constraints owing to the bulky N-DPP substituent. Later on, Lu and co-workers achieved a highly enantioselective [3 + 2] cycloaddition of allenoates and imines by using their own amino-acid-based bifunctional phosphine CP18.36 In the catalysis of CP18, the asymmetric [3 + 2] cycloaddition of N-phosphinylimines and allenoates occurred smoothly, affording the corresponding pyrrolines 95 in high yields with excellent enantioselectivities (Scheme 28, eqn (c)). They extended the substrate scope from aryl imines to alkyl imines and proposed the key transition state 96 to account for stereoinduction. The hydrogen-bonding interactions formed between the amide and carbamate protons of the catalyst CP18 and the oxygen center of N-phosphinylimine were suggested to play a crucial role in the high enantioselectivity.
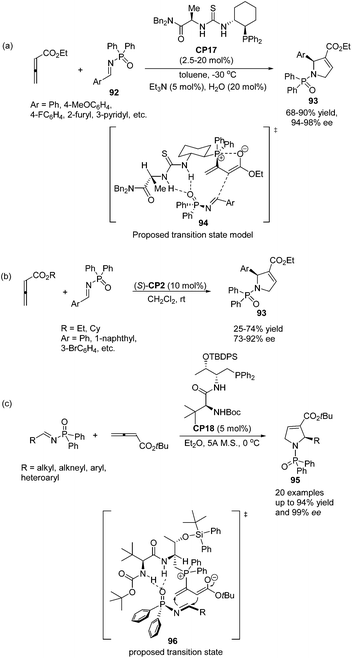 |
| | Scheme 28 Enantioselective thiourea-catalyzed [3 + 2] cycloadditions of DPP-imines with allenoates. | |
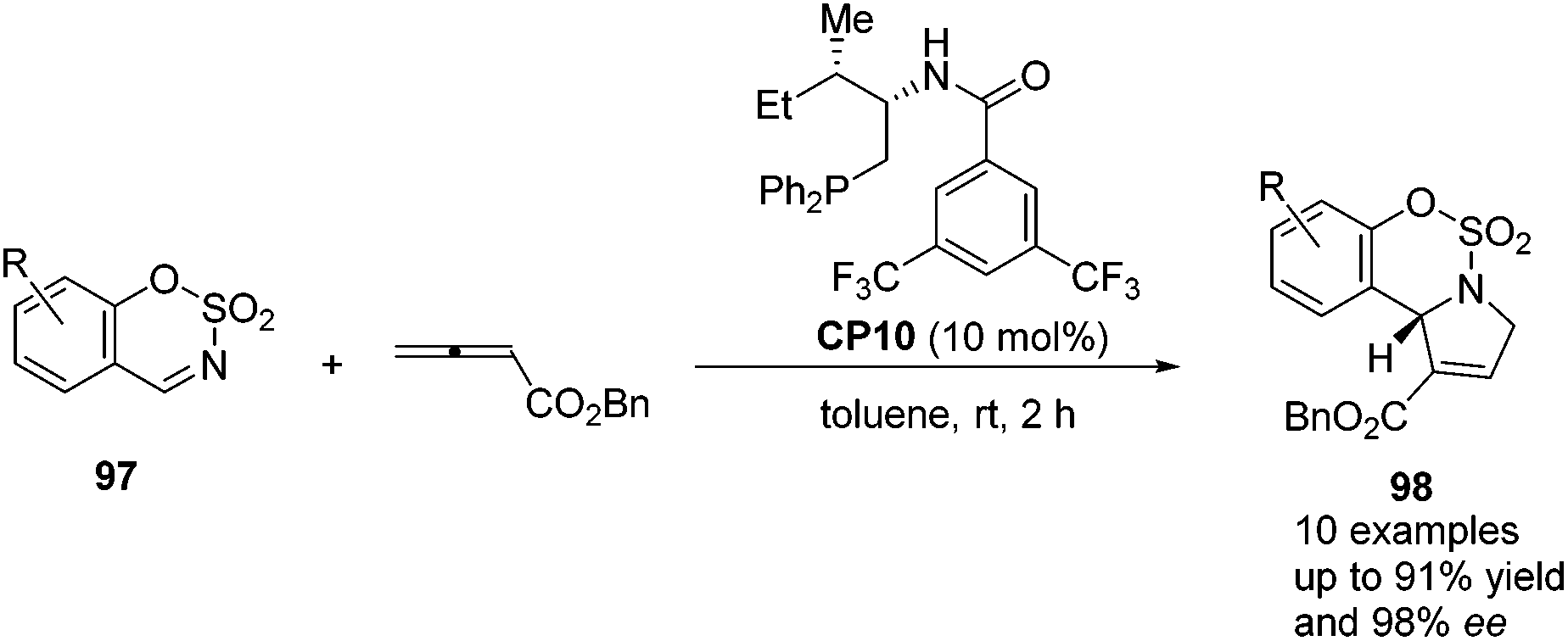
Guo's group screened a series of thiourea-based and amino acid-based bifunctional chiral phosphines to observe their performance in the asymmetric [3 + 2] cycloaddition of allenoates with sulfamate-derived cyclic imines.37 In the presence of 10 mol% amino acid-based bifunctional chiral phosphine CP10, which was first developed by Zhao's group,28 a variety of sulfamate-derived cyclic imines 97 were employed in this asymmetric [3 + 2] cycloaddition reaction, affording enantiomerically enriched sulfamate-fused dihydropyrroles 98 in good yields with moderate to excellent enantiomeric excess (Scheme 29). They also demonstrated that this reaction could be performed on a gram scale and the product 98 could undergo further transformations to provide various hetereocycles and pharmaceutically attractive compounds. Wang and co-workers almost simultaneously developed the same efficient phosphine-catalyzed [3 + 2] cycloaddition of allenoates with sulfamate-derived cyclic imines.38 Although they tried to develop an asymmetric variant by using commonly used chiral phosphines, highly enantioselective reactions were not achieved.
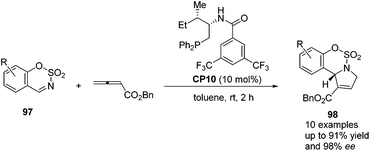 |
| | Scheme 29 Enantioselective phosphine-catalyzed [3 + 2] cycloadditions of sulfamate-derived cyclic imines with allenoates. | |
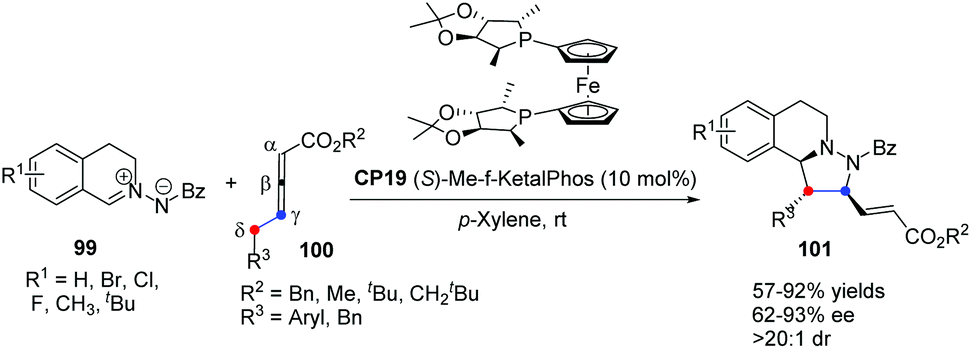
In 2014, Shi's group reported a highly enantioselective [3 + 2] cycloaddition of C,N-cyclic azomethine imines with δ-aryl-substituted allenoates.39 A wide range of C,N-cyclic azomethine imines 99 and δ-aryl-substituted allenic esters 100 underwent the γ,δ-C–C bond involving [3 + 2] cycloaddition in the catalysis of chiral phosphine CP19 (S)-Me-f-KetalPhos,40 affording functionalized tetrahydroquinolines 101 in good yields with good enantioselectivities under mild conditions (Scheme 30). This was the first time that δ-aryl-substituted allenoates were used as a C2 synthon in Lu's [3 + 2] cycloaddition, which was a new [3 + 2] reaction model and extended the substrate scope of Lu's [3 + 2] cycloadditions.
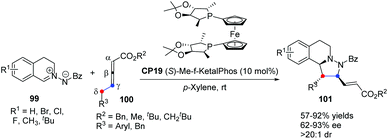 |
| | Scheme 30 Enantioselective phosphine-catalyzed [3 + 2] cycloadditions of C,N-cyclic azomethine imines with δ-aryl-substituted allenoates. | |
3.4 Mechanistic studies
Although the mechanism of Lu's [3 + 2] cycloaddition reaction was first proposed by Lu's group (see Scheme 5), the detailed mechanism was investigated through DFT calculations by Yu's group until 2007.41a Subsequently, Yu's group continued to investigate the detailed mechanism of the phosphine-catalyzed [3 + 2] cycloaddition reactions of allenoates and electron-deficient alkenes with the aid of DFT calculations and kinetic experiments.41b,c They suggested that this reaction proceeded via the following consecutive steps: (1) in situ generation of a 1,3-dipole A from the nucleophilic addition of phosphine to allenoate; (2) the first carbon–carbon bond formation to give an intermediate B and then the second carbon–carbon bond formation occurring to provide a [3 + 2] cycloaddition intermediate C, which takes place in a stepwise manner; (3) water is associated with the intermediate C to give a complex D, then a proton transfer from water to the carbon atom connected to the phosphorus atom occurs to afford a contact ion pair E which undergoes another proton transfer to give complex F; (4) elimination of water to furnish an intermediate G; (5) elimination of the phosphine catalyst to afford a product H (Scheme 31). They concluded that Lu's [3 + 2] cycloaddition is a stepwise process, and the generally accepted intramolecular [1,2] proton shift in Lu's [3 + 2] cycloaddition reaction was not possible owing to the very high activation barrier, however, the [1,2] proton shift process could take place with the assistance of trace water.
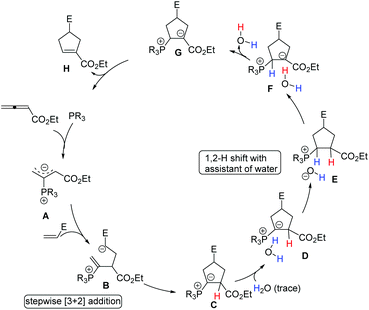 |
| | Scheme 31 The detailed mechanism for phosphine-catalyzed [3 + 2] cycloadditions proposed by Yu's group. | |
Through computational analysis at the B3LYP/6-31G(d) level of theory, Dudding and Kwon also verified that Lu's [3 + 2] annulation proceeded in a stepwise manner, and provided a rationale for the reaction regioselectivity.42 Based on the calculation results, they revealed that Lewis acid activation, strong hydrogen bonding (H-bonding), and minimization of unfavorable van der Waals contacts were the critical factors affecting regioselectivity. They also found the catalytic role of trace water, which acted as a proton-shuttle, for a proton transfer step.42b Shi's group recently rationalized the regioselectivity affected by the γ-substituent of allenoate through DFT calculations.30c
4. Synthetic applications of Lu's [3 + 2] cycloaddition
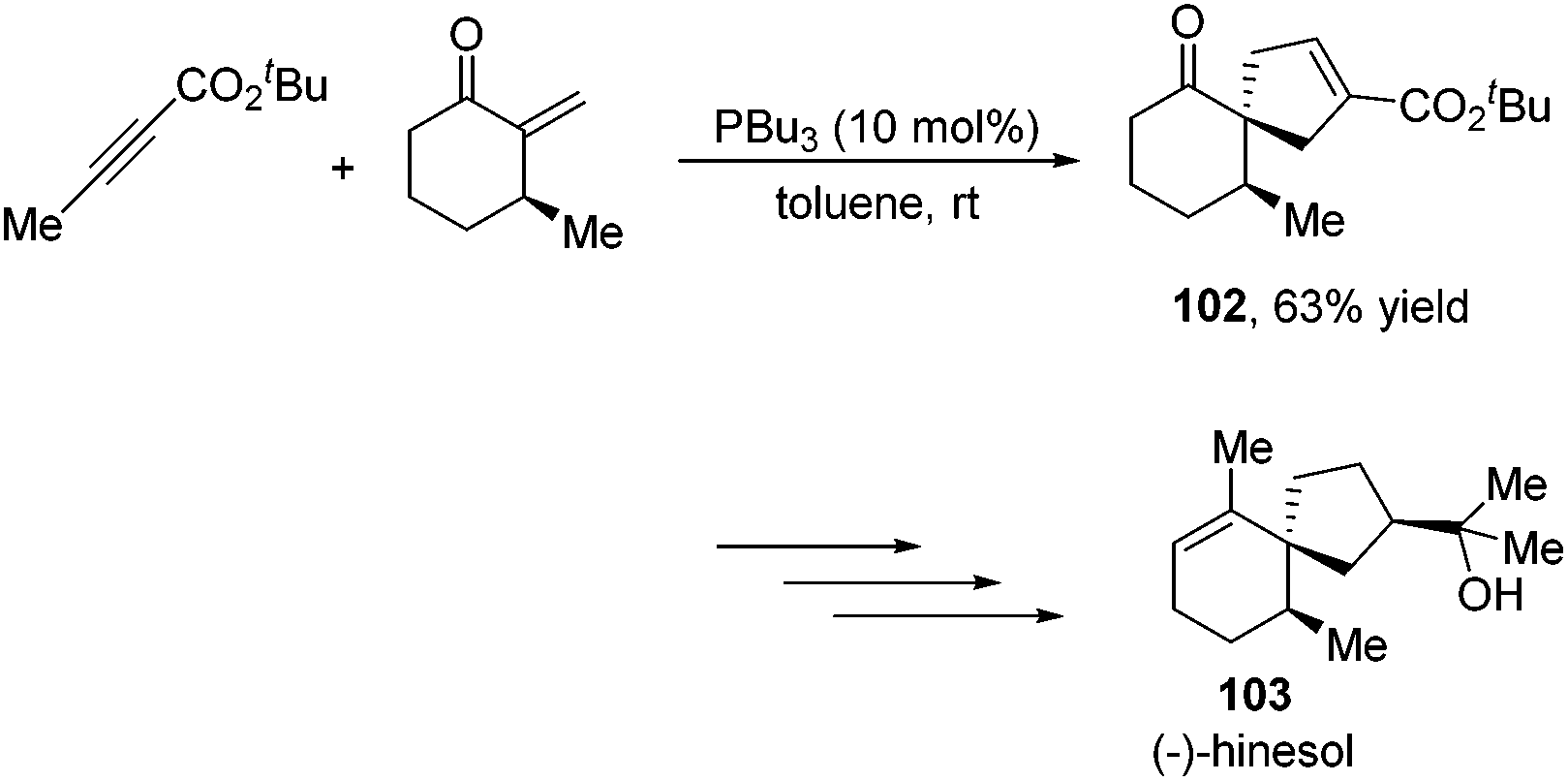
Besides the development of Lu's [3 + 2] cycloaddition reaction, a few research groups frequently applied this reaction in the total synthesis of natural products. In 2003, Lu and co-workers performed the total synthesis of (−)-hinesol 103.43 In the catalysis of PBu3, the [3 + 2] cycloaddition of 2-alkynoate with 2-methylene cyclohexenone took place to construct the spirocyclic ring skeleton 102 (Scheme 32). Further functional transformations produced (−)-hinesol 103 in good yield.
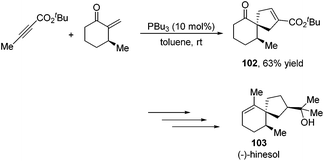 |
| | Scheme 32 Total synthesis of (−)-hinesol. | |
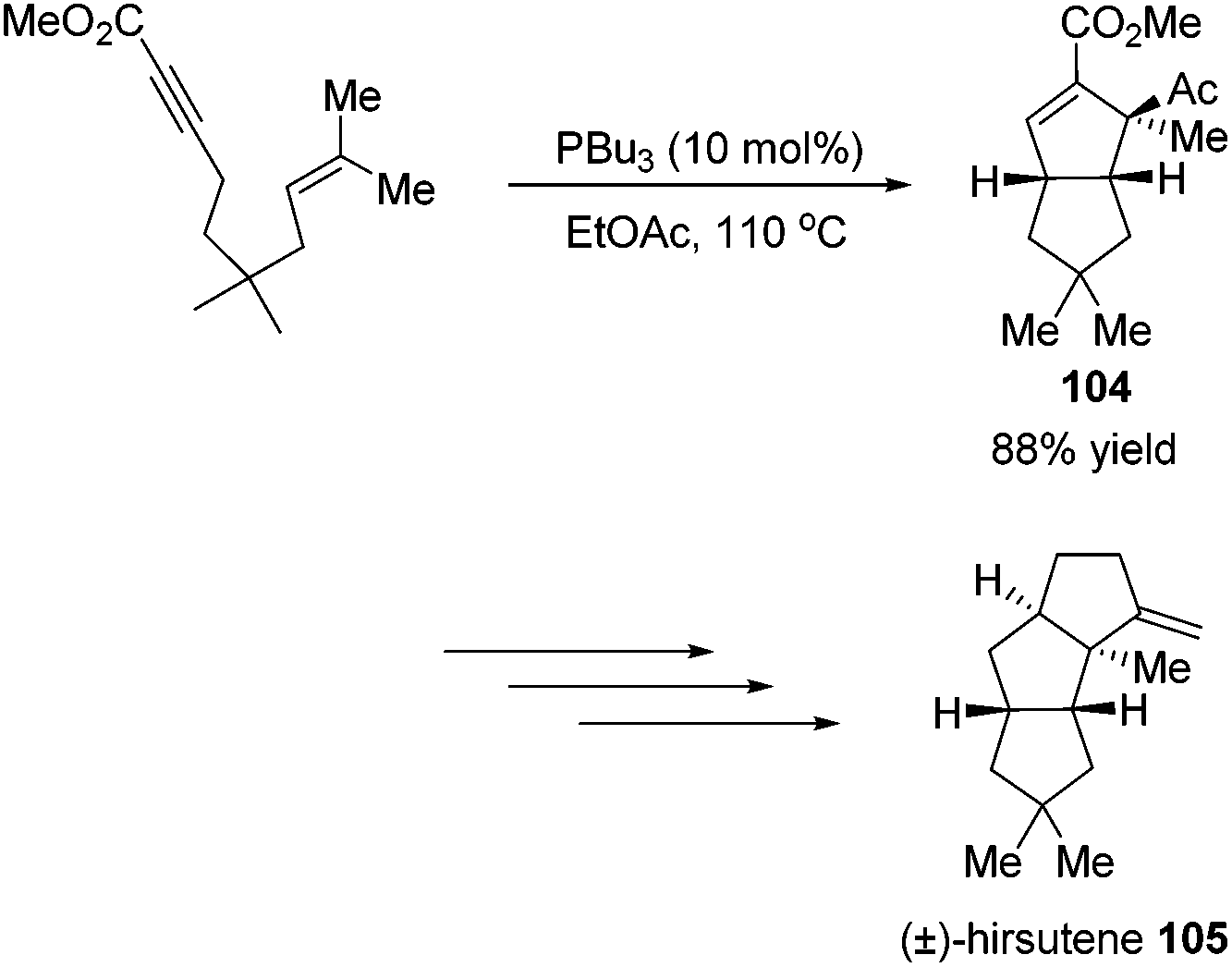
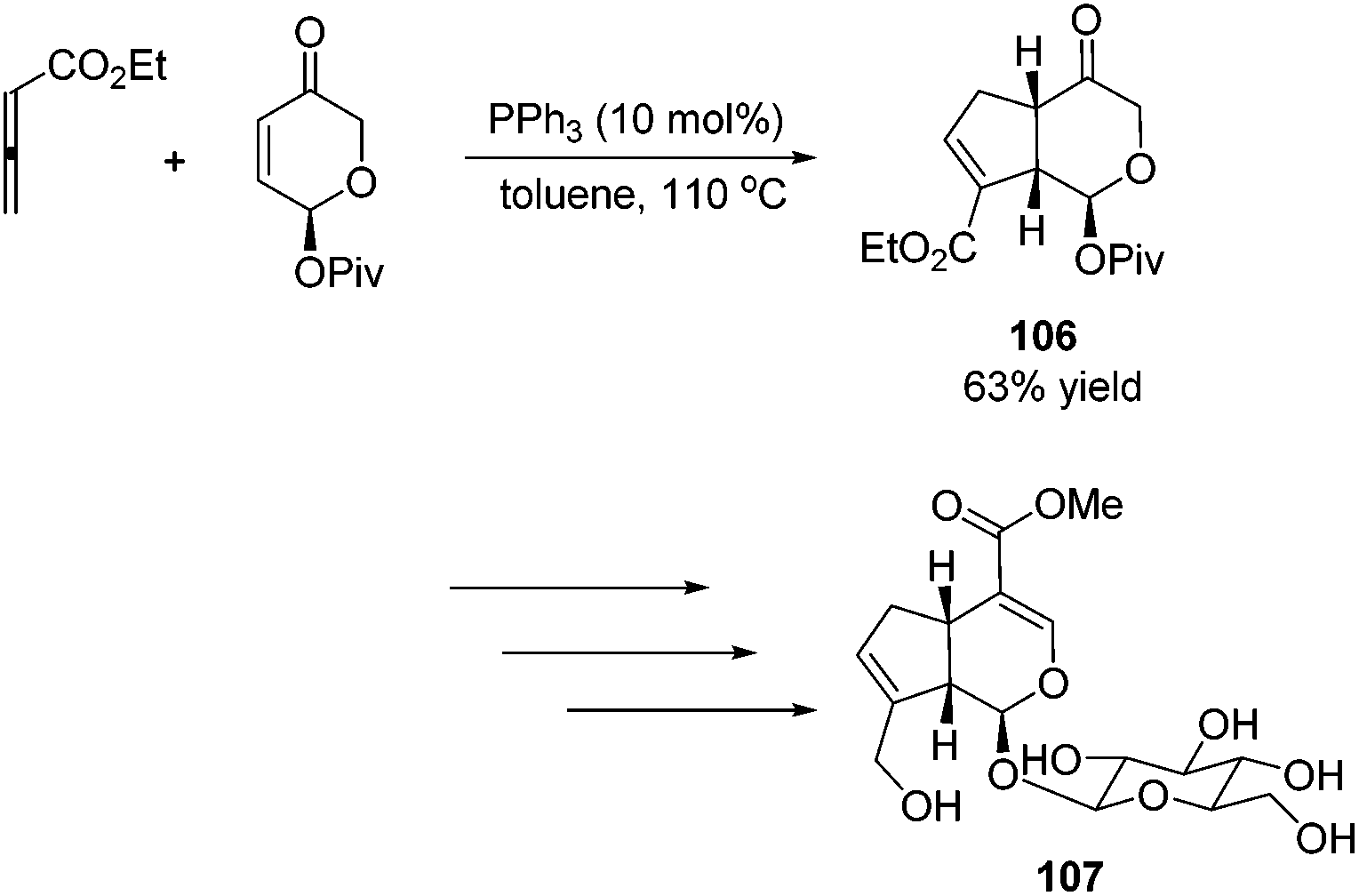
Krische's group has contributed a lot of work for the total synthesis of natural products by utilizing Lu's [3 + 2] cycloaddition reaction.44 Utilizing the intramolecular Lu's [3 + 2] cycloaddition reaction of allene and alkene, the 5,5-fused bicyclic intermediate 104 was constructed in one step, which provided two of the three rings in the target product; the following reductions and oxidations completed the total synthesis of (±)-hirsutene 105 (Scheme 33).44a Krische's group also applied Lu's [3 + 2] cycloaddition to synthesize (+)-geniposide 107 (Scheme 34).44b They constructed the core structure from Lu's [3 + 2] cycloaddition reaction of 2,3-butadienoate and enantiomerically enriched enone, furnishing a high yield of the cycloadduct 106. Functional group manipulations and the subsequent glycosidation generated (+)-geniposide 107.
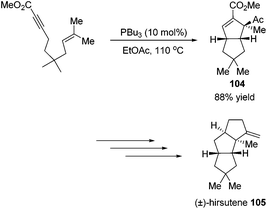 |
| | Scheme 33 Total synthesis of (±)-hirsutene. | |
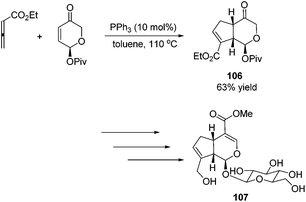 |
| | Scheme 34 Total synthesis of (+)-geniposide. | |
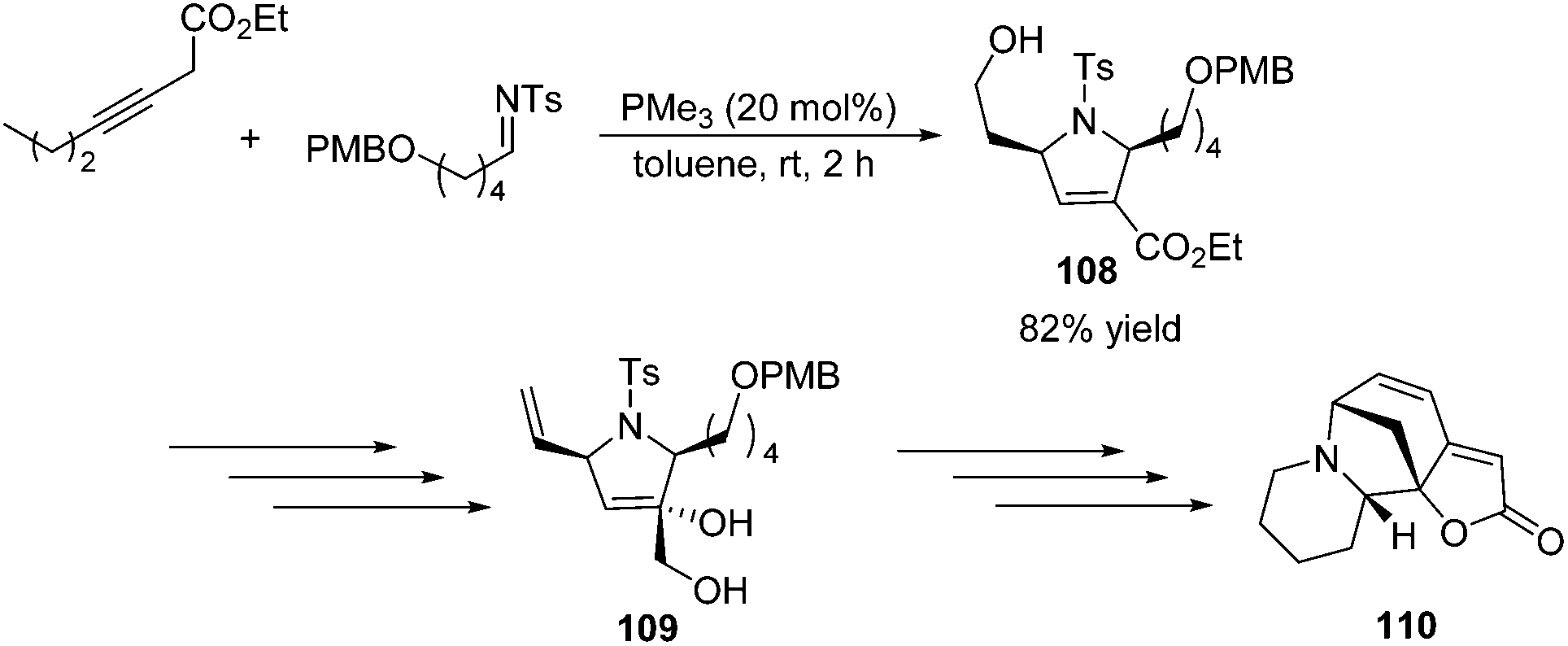
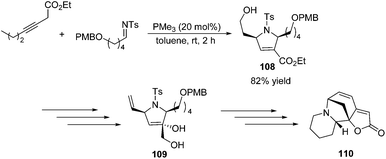
To demonstrate the utility of the [3 + 2] cycloaddition reaction of allene and alkylimine, Loh and co-workers performed the formal synthesis of (±)-allosecurinine 110 (Scheme 35).19 Remarkably, the unmasked 6-hydroxy-3-hexynoate underwent annulation with the alkylimine to produce a high yield of the desired dihydropyrroline 108 (Scheme 35). After several functional transformations, they obtained compound 109, a known synthetic intermediate of (±)-allosecurinine 110.
 |
| | Scheme 35 Formal synthesis of (±)-allosecurinine. | |
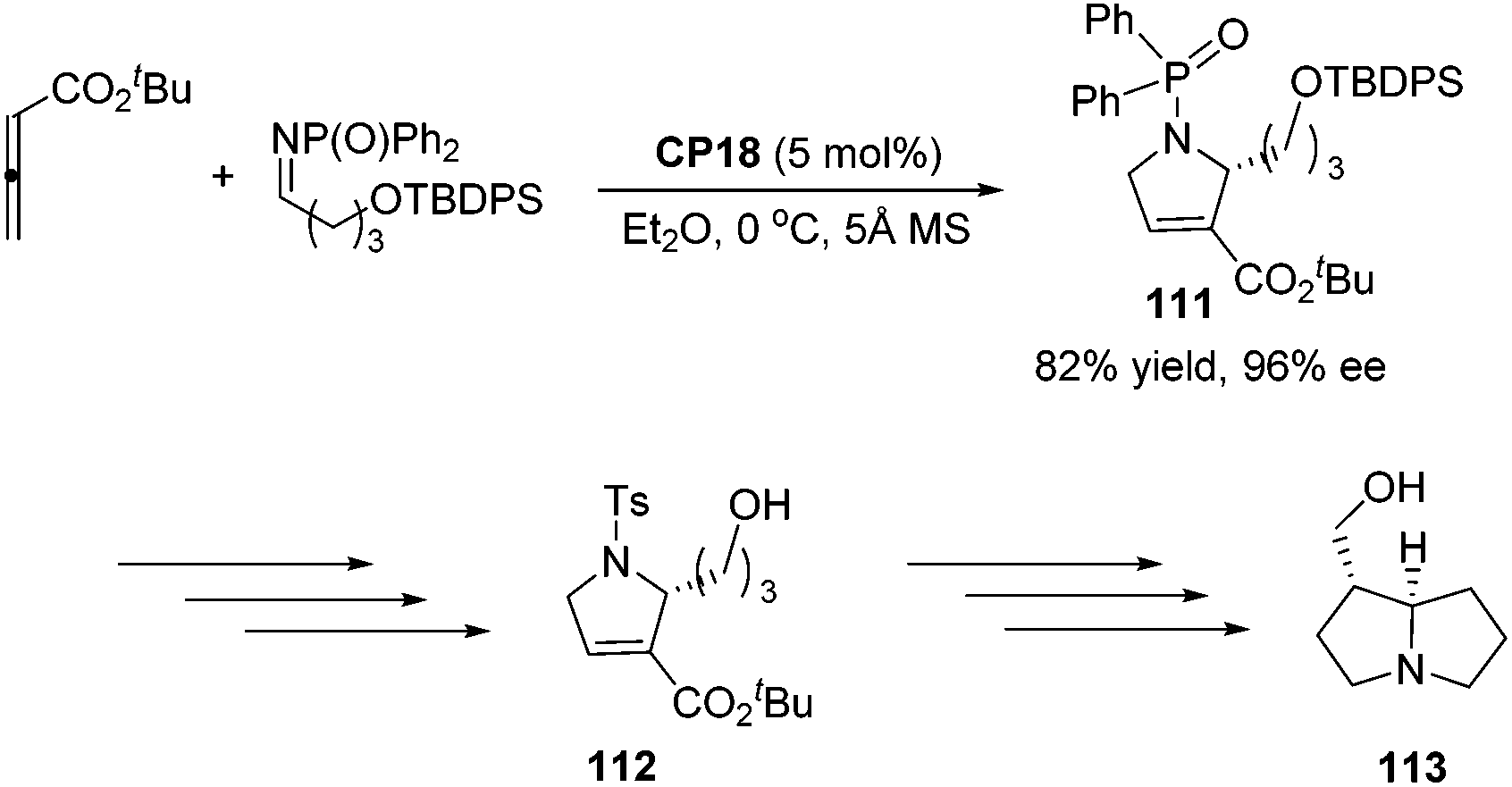
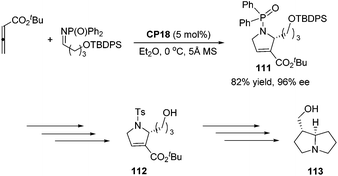
An asymmetric [3 + 2] cycloaddition of allene and alkylimine has been developed by Lu's group; they reported a concise formal asymmetric synthesis of (+)-trachelanthamidine 113 (Scheme 36).34 From the reaction of the allenoate and the alkylimine, they isolated 2-alkyl dihydropyrroline 111 in good yield and enantioselectivity. The subsequent removal of protecting groups completed the synthesis of 112, which is a known intermediate of (+)-trachelanthamidine 113.
 |
| | Scheme 36 Formal synthesis of (+)-trachelanthamidine. | |
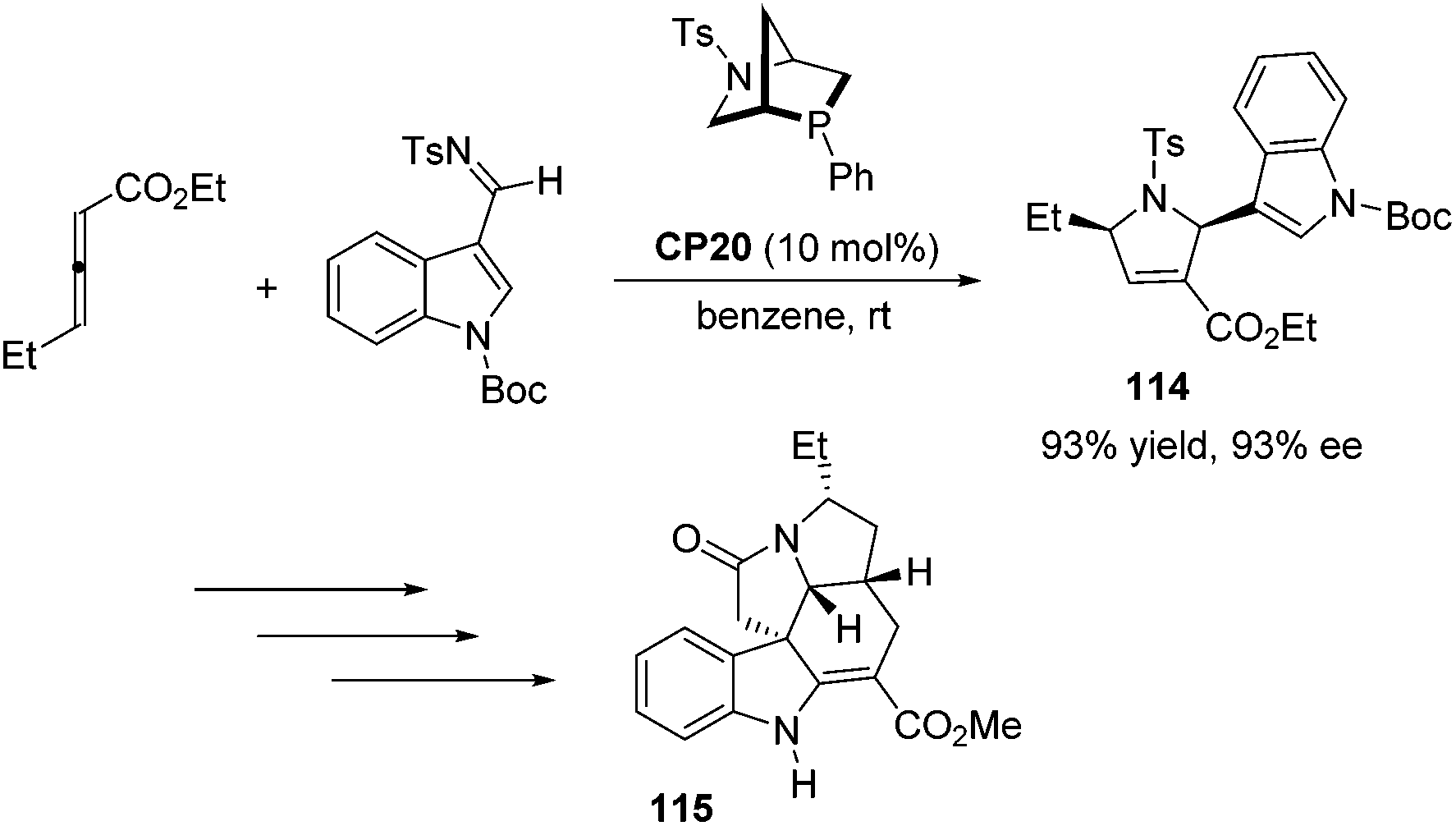
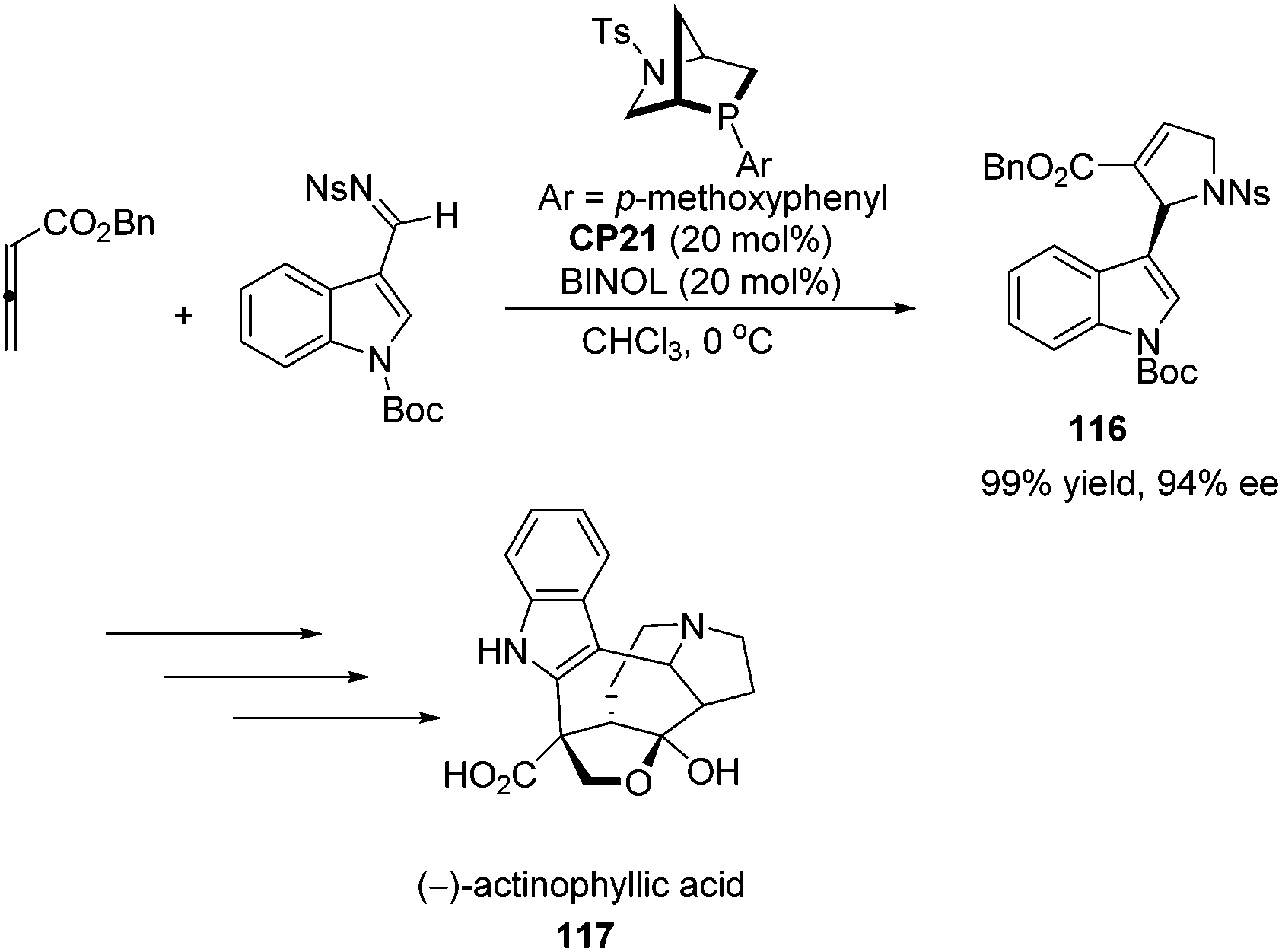
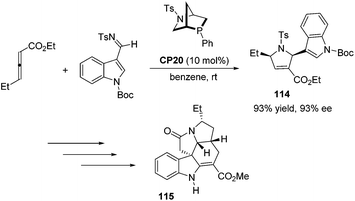
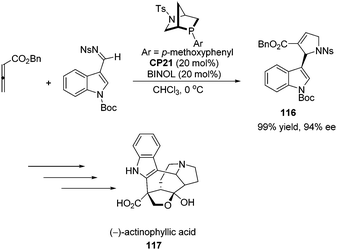
Kwon and co-workers first applied the asymmetric [3 + 2] cycloaddition of an allenoate and an indole derived imine in the total synthesis of (+)-ibophyllidine 115 (Scheme 37).45 Utilizing the readily accessible 2,3-hexadienoate and indole-3-carboxaldimine as substrates and the chiral phosphine CP20 as the catalyst, they synthesized the 2-indolyl-dihydropyrrole 114 in high yield with excellent enantioselectivity. Three of the five rings of (+)-ibophyllidine 109 were accessed in one step with high efficiency. After the formation of the remaining two rings and insertion of the functional group, the concise enantioselective synthesis of (+)-ibophyllidine 115 was complete. Very recently, they successfully performed a catalytic asymmetric total synthesis of (−)-actinophyllic acid, in which the chiral phosphine CP21 catalyzed [3 + 2] cycloaddition between an allenoate and an imine was employed to synthesize a key pyrroline intermediate 116 in 99% yield with 94% ee (Scheme 38).46 After the following CuI-catalyzed coupling between a ketoester and a 2-iodoindole to shape the tetrahydroazocine ring, intramolecular alkylative lactonization, SmI2-mediated intramolecular pinacol coupling between ketone and lactone subunits to assemble the complex skeleton of (−)-actinophyllic acid, and an unprecedented regioselective dehydroxylation, (−)-actinophyllic acid 117 was successfully obtained.
 |
| | Scheme 37 Total synthesis of (+)-ibophyllidine. | |
 |
| | Scheme 38 Total synthesis of (−)-actinophyllic acid. | |
5. Conclusions
Lu's [3 + 2] cycloaddition reaction has been developed tremendously since Prof. Xiyan Lu's group reported the first example. The substrate scope of Lu's [3 + 2] cycloaddition reaction has been dramatically expanded, affording a wide range of highly functionalized carbo- and heterocycles. With the development of chiral phosphine organocatalysts, highly enantioselective Lu's [3 + 2] cycloaddition reactions were also achieved. The detailed mechanism for Lu's [3 + 2] cycloaddition reaction has been revealed; and the synthetic applications in total synthesis have been demonstrated by a few research groups. In summary, Lu's [3 + 2] cycloaddition reaction has already become a powerful tool to synthesize highly functionalized carbo- and heterocyclic compounds. In the future, with the further development of efficient asymmetric phosphine-catalyzed transformations, new discoveries will stimulate the further development of Lu's [3 + 2] cycloaddition reaction. Last but not the least, the other related cycloaddition patterns of allenes with electrophiles, such as [4 + 2] and [4 + 1] cycloadditions have also been developing fast in recent years, which is worthy of more attention.
Acknowledgements
We acknowledge the financial support from the National Basic Research Program of China (973)-2015CB856603, the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB20000000, the National Natural Science Foundation of China (20472096, 21372241, 21572052, 20672127, 21421091, 21372250, 21121062, 21302203, and 20732008) and the Fundamental Research Funds for the Central Universities 222201717003.
References
- For reviews on phosphine-catalyzed reactions, see:
(a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed;
(b) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS;
(c) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC;
(d) Y. Wei and M. Shi, Acc. Chem. Res., 2010, 43, 1005 CrossRef CAS PubMed;
(e) Q.-Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724 RSC;
(f) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed;
(g) Y. Wei and M. Shi, Chem. – Asian J., 2014, 9, 2720 CrossRef CAS PubMed;
(h) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC;
(i) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC;
(j) A. Marinetti and A. Voituriez, Synlett, 2010, 174 CrossRef CAS;
(k) C. Gomez, J.-F. Betzer, A. Voituriez and A. Marinetti, ChemCatChem, 2013, 5, 1055 CrossRef CAS;
(l) W. Li and J. Zhang, Chem. Soc. Rev., 2016, 45, 1657 RSC;
(m) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369 CrossRef CAS PubMed.
- C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS.
- F. López and J. L. Mascareñas, Chem. – Eur. J., 2011, 17, 418 CrossRef PubMed.
-
(a) D. Ma, Y. Lin, X. Lu and Y. Yu, Tetrahedron Lett., 1988, 29, 1045 CrossRef CAS;
(b) D. Ma, Y. Yu and X. Lu, J. Org. Chem., 1989, 54, 1105 CrossRef CAS;
(c) D. Ma and X. Lu, Tetrahedron Lett., 1989, 30, 843 CrossRef CAS;
(d) D. Ma and X. Lu, Tetrahedron, 1990, 46, 3189 CrossRef CAS;
(e) D. Ma and X. Lu, J. Chem. Soc., Chem. Commun., 1989, 890 RSC;
(f) D. Ma and X. Lu, Tetrahedron Lett., 1989, 30, 2109 CrossRef CAS.
- B. M. Trost and T. A. Schmidt, J. Am. Chem. Soc., 1988, 110, 2301 CrossRef CAS.
- Y. Inoue and S. Imaizumi, J. Mol. Catal., 1988, 49, L19 CrossRef CAS.
- C. Guo and X. Lu, J. Chem. Soc., Chem. Commun., 1993, 394 RSC.
- C. Guo and X. Lu, J. Chem. Soc., Perkin Trans. 1, 1993, 1921 RSC.
- B. M. Trost and U. Kazmaier, J. Am. Chem. Soc., 1992, 114, 7933 CrossRef CAS.
- C. Zhang and X. Lu, Synlett, 1995, 645 CrossRef CAS.
-
(a) Z. Xu and X. Lu, Tetrahedron Lett., 1997, 38, 3461 CrossRef CAS;
(b) Z. Xu and X. Lu, J. Org. Chem., 1998, 63, 5031 CrossRef CAS;
(c) Z. Xu and X. Lu, Tetrahedron Lett., 1999, 40, 549 CrossRef CAS.
- J.-C. Wang, S.-S. Ng and M. J. Krische, J. Am. Chem. Soc., 2003, 42, 3682 CrossRef PubMed.
- C. E. Henry and O. Kwon, Org. Lett., 2007, 9, 3069 CrossRef CAS PubMed.
-
(a) X. Y. Guan and M. Shi, J. Org. Chem., 2009, 74, 1977 CrossRef CAS PubMed;
(b) X. C. Zhang, C. S. Cao, Y. Wei and M. Shi, Chem. Commun., 2011, 47, 1548 RSC.
- M. Sampath and T.-P. Loh, Chem. Commun., 2009, 45, 1568 RSC.
- E. Li, M. Chang, L. Ling and Y. Huang, Eur. J. Org. Chem., 2015, 710 CrossRef CAS.
-
(a) X. F. Zhu, C. E. Henry and O. Kwon, Tetrahedron, 2005, 61, 6276 CrossRef CAS;
(b) G.-L. Zhao and M. Shi, J. Org. Chem., 2005, 70, 9975 CrossRef CAS PubMed.
- L. G. Meng, P. Cai, Q. Guo and S. Xue, J. Org. Chem., 2008, 73, 8491 CrossRef CAS PubMed.
- M. Sampath, P.-Y. B. Lee and T.-P. Loh, Chem. Sci., 2011, 2, 1988 RSC.
- G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836 CrossRef CAS.
-
(a) R. P. Wurz and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 12234 CrossRef CAS PubMed;
(b) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426 CrossRef CAS PubMed;
(c) Y. Fujiwara and G. C. Fu, J. Am. Chem. Soc., 2011, 133, 12293 CrossRef CAS PubMed.
- M. Steurer, K. L. Jensen, D. Worgull and K. A. Jorgensen, Chem. – Eur. J., 2012, 18, 76 CrossRef CAS PubMed.
-
(a) A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030 CrossRef CAS PubMed;
(b) A. Voituriez, A. Panossian, N. Fleury-Brégeot, P. Retailleau and A. Marinetti, Adv. Synth. Catal., 2009, 351, 1968 CrossRef CAS;
(c) N. Pinto, M. Néel, A. Panossian, P. Retailleau, G. Frison, A. Voituriez and A. Marinetti, Chem. – Eur. J., 2010, 16, 1033 CrossRef CAS PubMed;
(d) A. Voituriez, N. Pinto, M. Néel, P. Retailleau and A. Marinetti, Chem. – Eur. J., 2010, 16, 12541 CrossRef CAS PubMed;
(e) M. Schuler, A. Voituriez and A. Marinetti, Tetrahedron: Asymmetry, 2010, 21, 1569 CrossRef CAS;
(f) N. Pinto, P. Retailleau, A. Voituriez and A. Marinetti, Chem. Commun., 2011, 47, 1015 RSC;
(g) M. Neel, J. Gouin, A. Voituriez and A. Marinetti, Synthesis, 2011, 2003 CAS.
- D. J. Wallace, R. L. Sidda and R. A. Reamer, J. Org. Chem., 2007, 72, 1051 CrossRef CAS PubMed.
- M. Sampath and T.-P. Loh, Chem. Sci., 2010, 1, 739 RSC.
-
(a) S. R. Gilbertson, S. E. Collibee and A. Agarkov, J. Am. Chem. Soc., 2000, 122, 6522 CrossRef CAS;
(b) A. Agarkov, S. Greenfield, D. Xie, R. Pawlick, G. Starkey and S. R. Gilbertson, Biopolymers, 2006, 84, 48 CrossRef CAS PubMed.
- B. J. Cowen and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 10988 CrossRef CAS PubMed.
- H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467 CrossRef CAS PubMed.
-
(a) X. Han, Y. Wang, F. Zhong and Y. Lu, J. Am. Chem. Soc., 2011, 133, 1726 CrossRef CAS PubMed;
(b) X. Han, S.-X. Wang, F. Zhong and Y. Lu, Synthesis, 2011, 1859 CAS.
-
(a) Q.-Y. Zhao, X. Han, Y. Wei, M. Shi and Y. Lu, Chem. Commun., 2012, 48, 970 RSC;
(b) D. Wang, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 2764 RSC;
(c) D. Wang, G.-P. Wang, Y.-L. Sun, S.-F. Zhu, Y. Wei, Q.-L. Zhou and M. Shi, Chem. Sci., 2015, 6, 7319 RSC;
(d) X.-N. Zhang and M. Shi, ACS Catal., 2013, 3, 507 CrossRef CAS.
-
(a) S.-F. Zhu, Y. Yang, L.-X. Wang, B. Liu and Q.-L. Zhou, Org. Lett., 2005, 7, 2333 CrossRef CAS PubMed;
(b) J.-H. Xie and Q.-L. Zhou, Acc. Chem. Res., 2008, 41, 581 CrossRef CAS PubMed;
(c) Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 2225 CrossRef CAS PubMed.
- W. Zhou, H. Wang, M. Tao, C.-Z. Zhu, T.-Y. Lin and J. L. Zhang, Chem. Sci., 2017, 8, 4660 RSC.
- S. Y. Lee, Y. Fujiwara, A. Nishiguchi, M. Kalek and G. C. Fu, J. Am. Chem. Soc., 2015, 137, 4587 CrossRef CAS PubMed.
-
(a) L. Jean and A. Marinetti, Tetrahedron Lett., 2006, 47, 2141 CrossRef CAS;
(b) N. Fleury-Brégeot, L. Jean, P. Retailleau and A. Marinetti, Tetrahedron, 2007, 63, 11920 CrossRef;
(c) N. Pinto, N. Fleury-Brégeot and A. Marinetti, Eur. J. Org. Chem., 2009, 146 CAS.
- Y.-Q. Fang and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 5660 CrossRef CAS PubMed.
- X. Han, F. Zhong, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 767 CrossRef CAS PubMed.
- H. Yu, L. Zhang, Z. Yang, Z. Li, Y. Zhao, Y. Xiao and H. Guo, J. Org. Chem., 2013, 78, 8427 CrossRef CAS PubMed.
- Y.-Q. Wang, Y. Zhang, H. Dong, J. Zhang and J. Zhao, Eur. J. Org. Chem., 2013, 3764 CrossRef.
- D. Wang, Y. Lei, Y. Wei and M. Shi, Chem. – Eur. J., 2014, 20, 15325 CrossRef CAS PubMed.
-
(a) D. Liu, W. Li and X. Zhang, Org. Lett., 2002, 4, 4471 CrossRef CAS PubMed;
(b) W. Zhang, Y. Chi and X. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS PubMed.
-
(a) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS PubMed;
(b) Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem. – Eur. J., 2008, 14, 4361 CrossRef CAS PubMed;
(c) Y. Liang, S. Liu and Z.-X. Yu, Synlett, 2009, 905 CAS.
-
(a) T. Dudding, O. Kwon and E. Mercier, Org. Lett., 2006, 8, 3643 CrossRef CAS PubMed;
(b) E. Mercier, B. Fonovic, C. Henry, O. Kwon and T. Dudding, Tetrahedron Lett., 2007, 48, 3617 CrossRef CAS.
- Y. Du and X. Lu, J. Org. Chem., 2003, 68, 6463 CrossRef CAS PubMed.
-
(a) J.-C. Wang and M. J. Krische, Angew. Chem., Int. Ed., 2003, 42, 5855 CrossRef CAS PubMed;
(b) R. A. Jones and M. J. Krische, Org. Lett., 2009, 11, 1849 CrossRef CAS PubMed.
- P. Andrews and O. Kwon, Chem. Sci., 2012, 3, 2510 RSC.
- L. Cai, K. Zhang and O. Kwon, J. Am. Chem. Soc., 2016, 138, 3298 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Min
Shi
* and
Min
Shi