 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarvesting the photoexcited holes on a photocatalytic proton reduction metal–organic framework†
J. G.
Santaclara
a,
A. I.
Olivos-Suarez
a,
I.
du Fossé
 a,
A.
Houtepen
b,
J.
Hunger
c,
F.
Kapteijn
a,
J.
Gascon
a and
M. A.
van der Veen
*a
a,
A.
Houtepen
b,
J.
Hunger
c,
F.
Kapteijn
a,
J.
Gascon
a and
M. A.
van der Veen
*a
aCatalysis Engineering, Department of Chemical Engineering, Delft University of Technology, van der Maasweg 9, Delft, 2629 HZ, The Netherlands. E-mail: m.a.vanderveen@tudelft.nl
bOptoelectronic Materials, Department of Chemical Engineering, Delft University of Technology, van der Maasweg 9, Delft, 2629 HZ, The Netherlands
cMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz, 55128, Germany
First published on 14th March 2017
Abstract
The highly porous titanium based metal–organic framework NH2-MIL-125(Ti) has recently attracted significant attention in the field of photocatalysis as a promising material for H+ reduction. This work reveals charge transfer upon visible light illumination from this MOF to two different charge acceptors, as an alternative to sacrificial electron donors. Charge transfer is demonstrated through a combined spectroscopic study between this MOF and: (1) 2-(1H-pyrazol-3-yl)phenol, a molecule that functionally mimics the tyrosine–histidine pair, responsible for shuttling the holes to the oxygen evolving centre in natural photosynthesis, and (2) TEMPO, a well known and stable radical. Charge transfer of the holes from the MOF to these occluded molecules takes place on the picosecond time scale. This work suggests that, by coupling a stable and recyclable charge acceptor to the photogenerated holes, the charges can be utilised for oxidation reactions and, thus, link the reduction to the oxidation reactions in water splitting.
Introduction
The demand for renewable energy sources that harvest solar energy is greater than ever.1 Using the sun, the largest exploitable resource, as a power source is a sustainable and clean alternative to fossil fuels. Thus, the production of solar fuels by artificial photosynthesis has attracted a great deal of attention.2 The first example in which water was split under UV radiation using TiO2 as catalyst was reported by Fujishima and Honda in 1972.3 Using the photosynthetic blueprint, many different materials and devices have been reported since then, yet there is no efficient, cheap and stable system to achieve the desired light-driven water splitting for the production of solar fuels.4The challenge to implement water splitting is the coupling of the oxidation and the reduction half reactions. This 4-electron reaction requires the adequate light induced separation of charges to harvest light (4 photons), but also that the charges, with adequate redox potential, are transferred to the respective catalytic sites.5 Ideally, both the reduction and oxidation half reactions are spatially separated, and only interconnected via the light harvesting component to avoid the undesired back electron transfer.6 Inspired by natural photosynthesis, researchers have focussed on developing multicomponent relay systems, where different components aid the spatial separation, and electron transfer rates are adequately matched.6–9
In nature, the ratio between forward and backward reaction rates is controlled via molecular redox relays. Here, the tyrosine–histidine pair (Tyr/His in Fig. 1) is a key component of photosystem II (PS II) that mediates electron transfer between the photoexcited primary electron donor (P680˙+) and the Mn-containing oxygen evolving complex (OEC).10–12 In this pair, the one electron oxidised alcohol group is stabilised by an adequately positioned basic histidine residue. Implementation of similar recyclable charge carriers in an artificial photosynthetic system can be done via recyclable electron donors that provide the coupling between the catalytic centres for the hydrogen evolution and the water oxidation half reactions, in a Z-scheme.13–15
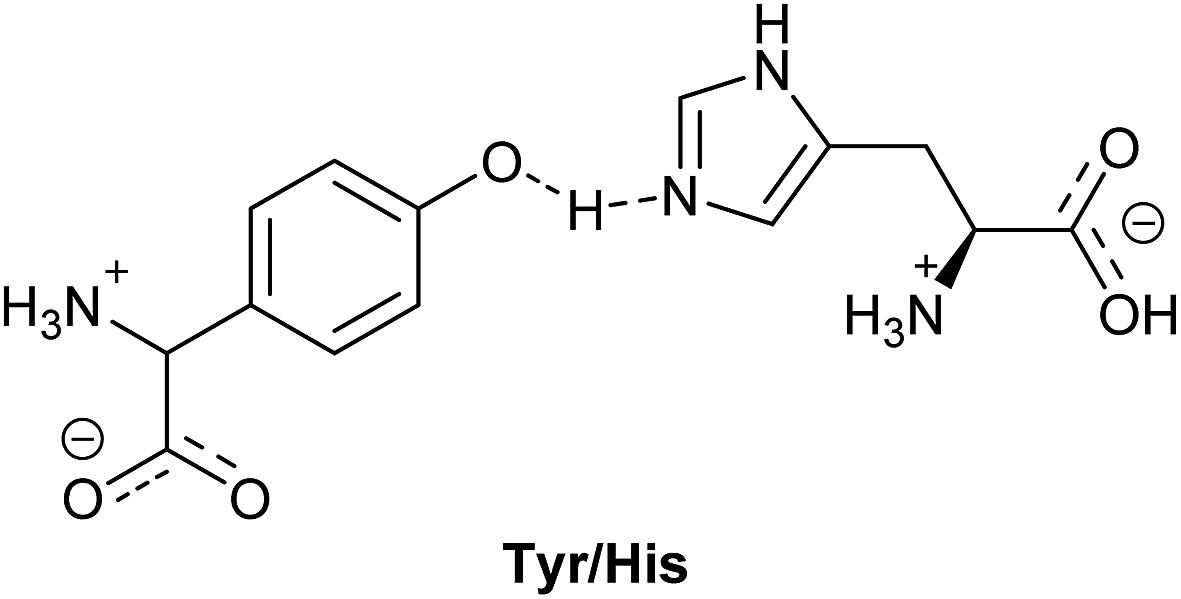 | ||
| Fig. 1 Scheme of tyrosine/histidine pair in PS II. | ||
Metal organic frameworks (MOFs) have been recently applied to study and design artificial photosynthetic systems.4 The modular structure that MOFs provide is one of the most attractive features to integrate the three fundamental steps of artificial photosynthesis into a single material: (1) light harvesting, (2) charge separation; and (3) controlled arrangement of the individual components to favour the utilization of charges in redox reactions.
Although many frameworks have been reported to be able to evolve H2, reduce CO2 and perform organic oxidations, the fundamentals behind the photocatalytic reactions are still elusive,16–18 and there has only been focus on the reductive half reaction. Recent studies of the photoexcitation processes of MIL-125(Ti) have demonstrated that functionalities, such as the amino group in NH2-MIL-125(Ti), lead to an enhancement towards visible range light absorption, and more importantly, help to separate charges (beyond 10 ns) featuring ligand-to-metal charge transfer (LMCT).19 Moreover, the comparison of photoexcitation of NH2-MIL-125(Ti) with other d0-based transition metal frameworks, such as NH2-UiO-66(Zr/Hf), highlights the importance of good overlap between metal and ligand orbitals for efficient separation of photoexcited charges (lifetimes of only 1–100 ps for Zr/Hf MOFs).20 However, the slow decay kinetics of NH2-MIL-125(Ti) alone do not guarantee highly efficient photocatalytic performance. A well-known problem in molecular artificial photosynthesis is back-electron transfer after photoinduced charge separation.21
It is not surprising, as mentioned above, that only the reduction side (H2 evolution and CO2 reduction reaction) has been reported for MOF-mediated photocatalysis, since MOFs are built up from carboxylate linkers that do not possess the oxidative power to run the water oxidation half reaction.22 This means that these MOFs need to be coupled to an adequate water oxidation catalyst via e.g. a recyclable charge carrier.
In this work, we aim to (i) improve the fundamental understanding and (ii) implement charge transfer to recyclable charge carriers. Thus, we demonstrate charge transfer from the visible-light absorbing NH2-MIL-125(Ti) framework, a hydrogen evolution reaction (HER) photocatalyst, to charge-acceptor molecules incorporated in the pores (Fig. 2). By substituting traditional sacrificial agents for more stable electron donors, we are able to mimic the charge shuttle tyrosine–histidine pair that natural photosynthesis uses. By a combined study of the multicomponent oxidation potentials and the dynamics at ultrafast time scales, we demonstrate charge transfer from the holes in NH2-MIL-125(Ti) to the occluded molecules, and highlight the importance of efficient electron transfer in stabilizing the photo-induced charge separated state.
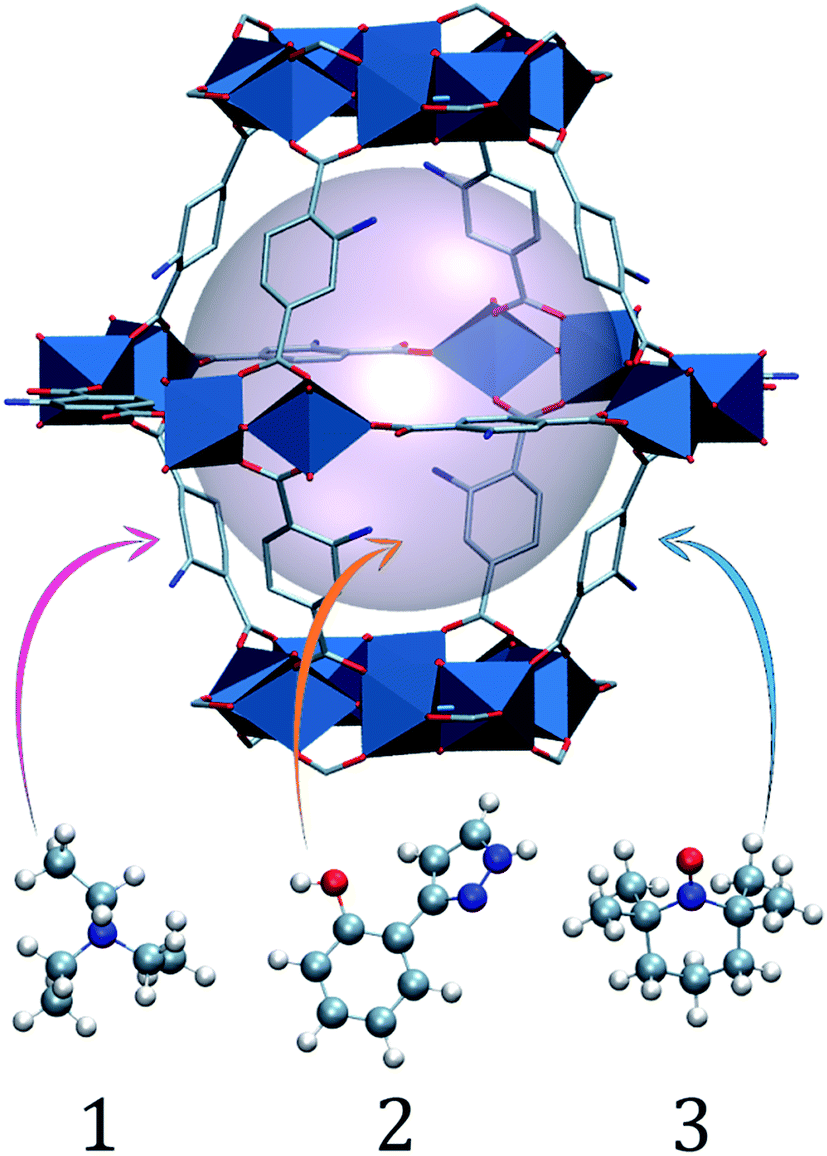 | ||
| Fig. 2 Schematic (ball and stick and polyhedra) representation of molecules 1, 2 and 3 occluded in a large NH2-MIL-125 cage. The light pink ball represents the pore volume. | ||
Experimental
Materials and reagents
All chemicals were purchased from Sigma Aldrich and used without further purification.Synthesis and characterization of NH2-MIL-125(Ti)
The MOF was synthesized using a protocol reported by Walsh and co-workers.23 In a typical synthesis, 0.5 g (2.76 mmol) 2-aminoterephthalic acid was dissolved in a mixture of 16 mL anhydrous N,N-dimethylformamide (DMF) and 4 mL anhydrous methanol at room temperature in a glove box. Then 0.55 mL of titanium isopropoxide (1.93 mmol) was added and the mixture was placed in an autoclave. The autoclave was sealed and heated for 72 h at 383 K. The obtained yellow solid was filtered, dispersed in fresh DMF and heated at 383 K overnight in order to remove residual linker. Then, the same procedure was repeated using methanol for 6 hours at 353 K. The solid was finally dried under air at 353 K. After synthesis, the following characterization techniques were used in order to determine the purity of the MOF (Fig. S1–S3†):The powder X-ray diffraction pattern of NH2-MIL-125(Ti) was recorded using a Bruker-AXS D5005 instrument with Co-Kα radiation.
Nitrogen physisorption measurements of NH2-MIL-125(Ti) were performed in a Tristar II 3020 Micromeritics unit at liquid nitrogen temperature (77 K). Prior to the experiment the samples were degassed for 16 h at 423 K under vacuum. The BET areas were calculated using intervals allowing positive BET constants.24 The total pore volumes were calculated at a relative pressure of 0.9.
Thermogravimetric analysis (TGA) of NH2-MIL-125(Ti) was performed on a Mettler Toledo TGA/SDTA851 instrument, under a synthetic air flow of 60 mL min−1 at a heating rate of 10 K min−1 up to 1073 K.
Characterization
Transmittance UV/VIS spectra were collected using a Perkin-Elmer Lambda 40 spectrophotometer. Liquid samples and composites (2@NH2-MIL-125 and 3@NH2-MIL-125) were studied in Hellma 110-QS cuvettes with a 2 mm optical path length.
The Vis-pump infrared (IR)-probe experiments were based on a regenerative Ti:sapphire amplified laser system (Spitfire Ace, Spectra Physics), providing pulses at 800 nm with a duration of 35 fs and a pulse energy of 5 mJ at a repetition rate of 1 kHz. A commercial optical parametric amplifier (TOPAS-Prime, Light Conversion) was pumped with 1.5 mJ of the 800 nm pulses. The signal and idler pulses from one TOPAS-Prime were used in a difference frequency mixing process in a silver gallium disulphide (AgGaS2) crystal, resulting in ∼8 μJ tuneable IR pulses with a FWHM of 300 cm−1. A wedged CaF2 window was used to generate a probe and a reference pulse from the IR pulse, with the reflection of the front side used as the probe pulse and the back-reflection used as a reference pulse. A second portion of the 800 nm pulses was frequency-doubled in a beta-barium borate crystal, yielding visible pump pulses with a 400 nm central wavelength. A translational stage was used to control the timing of the Vis pump-pulses relative to the IR probe-pulses. A mechanical chopper was used to block every second pump pulse, which allows for active background subtraction. The pump, probe, and reference pulses were focused into the sample and the pump and the probe pulses were spatially overlapped. The probe and the reference pulses were recollimated and spectrally dispersed with an imaging spectrograph (Horiba Triax 180). The intensities, I, were detected on two separate lines of a 2 × 32 pixel mercury–cadmium–telluride (MCT, Infrared Associates) detector. The pump-induced absorption changes were thus given as ΔA = −ln[Iprobe × Ireference,0/(Iprobe,0 × Ireference)], where subscript ‘0’ refers to the IR intensities recorded without pump excitation. For all the measurements 40 mg NH2-MIL-125(Ti) was dried overnight at 423 K, then dispersed in 800 μL solvent containing 2 (24 mg) and sonicated for 15 min.
Photocatalytic tests
Photocatalytic experiments were carried out using a home-built set-up equipped with a 500 W Xe/Hg lamp (66983, Newport). It consists of a custom-made stirred Pyrex-glass reactor, which has a volume of 42.1 mL and is equipped with a water jacket to allow for precise temperature control. Light intensity from the light source was measured with AvaSpec-3648-2-USB2 (Avantes, the Netherlands). The light emitted by the lamp passes through a lens assembly (77330, Newport) focussing the beam on the reactor window, an H2O filter (61945, Newport) and a 385 nm cut-off optical filter. A CP 9001 gas chromatograph (Chrompack) analyzes the headspace on-line, by a KSLA gas pump that is applied to ensure a sufficient mixing of gases in the headspace of the reactor and the stainless steel tubes (2.5 mL min−1 continuous operation). Every 60 min a probe of the headspace is analyzed by the GC.Results and discussion
The NH2-MIL-125(Ti) framework is capable of separating charges, featuring a LMCT transition upon visible light irradiation and producing enough reducing power for the hydrogen evolution reaction (HER). The LMCT efficiency of this system produces long-lived charge separated states (up to 10 ns).19,20 An extensive spectroscopic characterization has pictured the nature of the separated charges and pointed to a nitrogen centred localized hole and an electron that is delocalised at the Ti8O8 ring in the MOF (see Fig. 3b).19 On the acceptor side, the electron transfer to a Co-based catalyst has been evidenced by the increase of the photocatalytic HER efficiency.25 On the donor side, however, sacrificial electron donors are still a requirement to provide the necessary electrons and complete the cycle. In spite of the slow decay kinetics observed in NH2-MIL-125 that prevents fast recombination and renders the reduction half reaction, the integration of this system with an additional component that can act as electron donor and, at the same time, interconnect the charge transfer between this reductive catalytic system and an additional water oxidation catalytic system is still desired (Fig. 3a, right).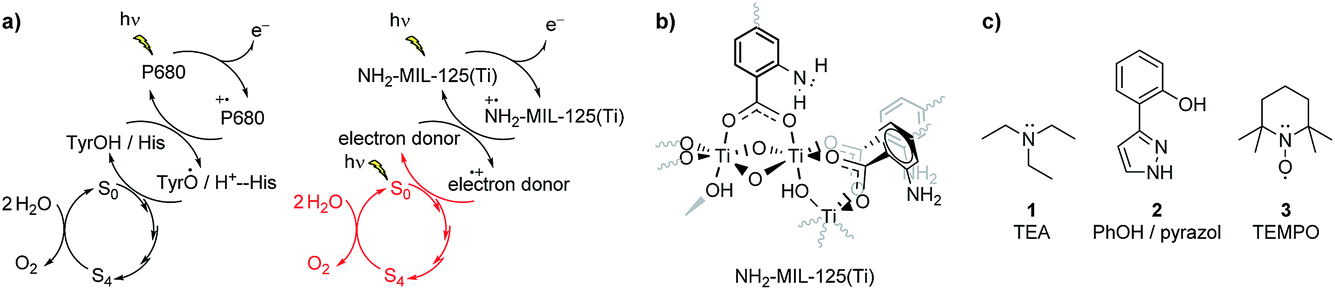 | ||
| Fig. 3 (a) Schematic representation of the electron transfer reactions in PS II and in the photocatalytic MOF system (in red is shown the desired oxidation half reaction coupled to a Z-scheme), (b) scheme of NH2-MIL-125(Ti), and (c) compounds 1–3 used as electron donors in this work. | ||
We chose three charge carrier molecules that are small enough to readily diffuse (see Section 4, ESI†) through the cages of NH2-MIL-125. NH2-MIL-125 has two types of cage, with free diameters (defined as the distance between the van der Waals surfaces of either side of the cage) of 4.7 and 10.7 Å, and it has large cage windows with a free diameter of 7 Å.26,27
Triethylamine is widely used as a hole harvester for proton reduction in order to facilitate fundamental studies of this half reaction. However, its oxidation leads to a decomposition of the positive amine radicals, making it unsustainable for technological applications.28,29 Implementation of a charge carrier with functional groups that can mimic the Tyr/His pair is attractive.30,31 Efforts have been made in protecting phenolic analogues to the Tyr/His pair with bulky groups and making them more stable as charge carriers, but this leads to bulky molecules that are incompatible with porous hosts.32 Thus, we chose the unsubstituted 2-(1H-pyrazol-3-yl)phenol (2) as a hole harvester (Fig. 4c, middle), because it contains the phenolic group and, as proton acceptor, the basic pyrazole group, thus mimicking the Tyr/His component of the PSII. In the case of molecule 2, it does not present an entirely reversible oxidation wave (Fig. 4). Moreover, from Fig. 4 we can conclude that under neutral conditions the oxidation potential of the hole at the linker in the MOF (EP = 0.85 V vs. Ag/AgCl, PO43−/HPO42− in water) has a higher oxidation potential than 2 (Ep = 0.65 V vs. Ag/AgCl, PO43−/HPO42− in water), suggesting that 2 may be a suitable candidate to carry the positive charges for the oxidation reaction, although not in a reversible fashion. Interestingly, we can also see that the oxidation potential of the MOF is lower than that of water, confirming that the MOF is incapable of the water oxidation half reaction, and that a Z-scheme would be necessary for achieving overall water splitting in this system (Fig. 3a, red).
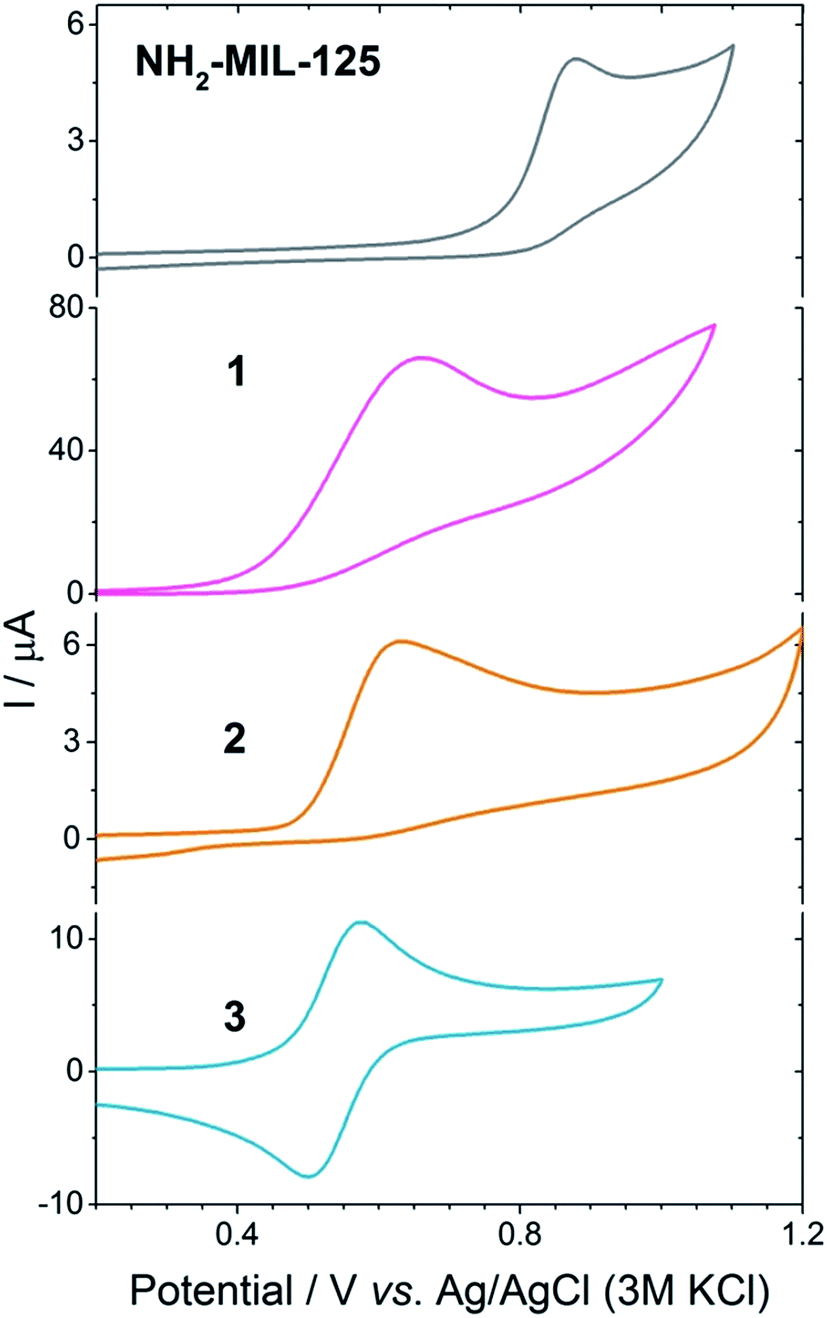 | ||
| Fig. 4 Cyclic voltammetry at 50 mV s−1 of NH2-MIL-125(Ti), 1, 2 (phosphate buffer, pH 7) and 3 (0.1 M LiClO4) in water as it is the only medium that allowed for electrochemical measurements of the MOF. | ||
TEMPO is a well-known radical scavenger that has been successfully applied as a (catalytic) oxidant in its oxidized form.33 This compound provides an oxidation potential suitable to carry the positive charge (EP = 0.57 V vs. Ag/AgCl, PO43−/HPO42− in water) and, moreover, its oxidation wave is fully reversible (see Fig. 4, bottom) to warrant the availability of oxidation power and connect the reduction with the oxidation half reactions (Fig. 3a).
Noteworthily, the amount of occluded electron donor varied for the three different composites (and was directly correlated to the diffusion limitations of each molecule in terms of size and affinity for the pore, see Section 4, ESI†). To assess whether the adsorbed molecules interact with the photoexcited MOF and have the possibility to undergo charge transfer, time-resolved photoluminescence (PL) studies were performed on the NH2-MIL-125(Ti) and composites (Fig. 5). Fig. 5 shows that, when the molecules are present, there is a faster decay (τ) in all the composites compared to the pristine MOF, with the decay of the photoluminescence being much faster for 1 and 3 than for 2 (τ1 = 0.1 ns, τ3 = 0.3 ns and τ2 = 6.1 ns, respectively, see Fig. 5). To detect if there is any influence by the solvent, we also performed all measurements in acetonitrile, observing the same trend (Fig. S8†). This means that the photoluminescence of the MOF is quenched by the occluded molecules, confirming the expected charge transfer from the MOF to the occluded molecules.25 These results point towards a different photoexcitation and/or recombination to the ground state mechanism compared to the pristine MOF. However, the time resolution of the measurements (limited to 50 ps) cannot distinguish processes that might already occur on shorter time scales. Thus, further studies with subpicosecond time resolution were carried out with transient absorption (TA) spectroscopy.
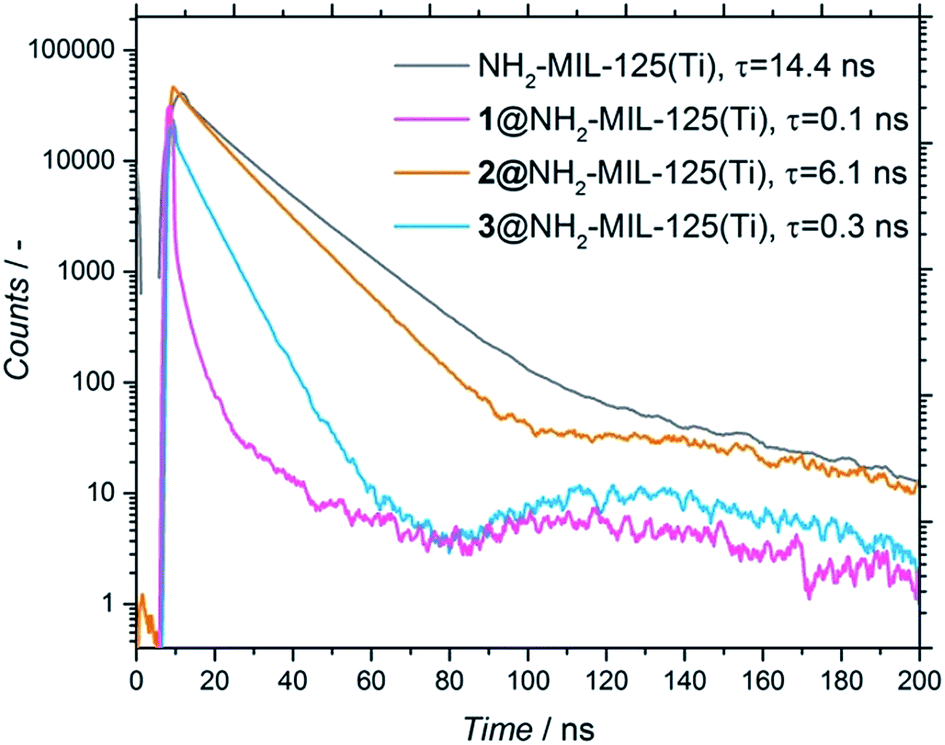 | ||
| Fig. 5 Time resolved fluorescence of NH2-MIL-125(Ti) in water (grey), 1@NH2-MIL-125(Ti) in acetonitrile (pink), 2@NH2-MIL-125(Ti) in DMF (orange) and 3@NH2-MIL-125(Ti) in water (blue). The suspensions were excited by a 404 nm laser. The decrease in fluorescence over time was measured at a wavelength of 430 nm and fitted by a sum of exponential decays (only the fastest τ component, which is the biggest contribution, is shown for clarity). | ||
In the case of the pristine MOF, the photoexcitation with a Vis-pump (400 nm) results in an intense induced absorbance (λmax = 570 nm with a shoulder at 530 nm) that can be assigned to the holes on the NH2. An additional broad induced absorbance of lower intensity is present from 500 nm, extending beyond 750 nm, which is characteristic for the electron on the Ti3+ (Fig. S9†).19 We have previously shown that the kinetics and, to a large degree, the shape of the transient absorbance spectra are solvent independent.19
Fig. 6a shows the TA spectrum of 2@NH2-MIL-125(Ti) in dimethylformamide (DMF) upon 400 nm excitation. Despite the fact that the shape of this spectrum is very similar to the original from solely the MOF (Fig. S9†), the decay of the induced absorbance attributed to the photogenerated holes (at λ = 570 nm) on the MOF and on 2@NH2-MIL-125(Ti) is remarkably different. Clearly, there is a faster decay for 2@NH2-MIL-125(Ti) compared to the pristine MOF. From Fig. 6a (inset), we see for 2@NH2-MIL-125(Ti) a decay of 25% of the band associated with the hole within 10 ps, in contrast to no decay for the pristine MOF, indicating that a fraction of the holes on the MOF are most likely transferred to compound 2. To further gain insight into the charge transfer process, we studied the structural changes of the functional organic groups during the photoexcitation of the composite by transient IR absorption spectroscopy. Whereas for the pristine MOF the transient IR-signals equilibrate after excitation,19 for 2@NH2-MIL-125(Ti) we observed transient positive and negative signals, which grow in amplitude with increasing delay time. The transient spectrum (Fig. 6b) in DMF at long delay times shows two negative ΔA peaks (spectral bleaches) with two adjacent peaks with positive ΔA (induced absorbance). Such combinations of a bleach and an induced absorption can be assigned to vibrational peaks that are red-shifted upon excitation with the visible pump pulse: the photo-conversion leads to a depletion of the starting materials (MOF or 2, see below), reflected in a reduced absorption of the molecular vibrations (spectral bleach). Simultaneously, the photo-product with red-shifted (relative to the initial bands) vibrational band is generated, which results in the observed induced absorption. Two such pairs of a spectral bleach and an adjacent induced absorption appear with increasing delay time (e.g. 1540 shifts to 1520 cm−1, and 1580 shifts to 1560 cm−1). As both the MOF and 2 have weak bands in the IR absorption spectra in this frequency range (Fig. S13†), these transient shifts may be due to photo-oxidation of 2 or reduction of the MOF. DMF was chosen as the solvent for a better comparison with the TA and PL results, however, it is not IR transparent in the region of the amino group C–N vibration in the MOF (∼1257 cm−1). Studies with more transparent solvents (tetrachloroethane, 1210–1310 cm−1 range) show similar dynamics as in DMF (new spectral features appearing, Fig. S14†). These observations mean that the N-centred holes from the MOF are transferred to compound 2, leading to spectral features arising from a photoadduct (that can be either the now negatively charged MOF or oxidised 2). In fact, the time dynamics of the appearance of these new spectral features and the faster decay detected in 2@NH2-MIL-125(Ti) by a Vis-probe (compared to the MOF, Fig. 6a, inset) are very similar (see Fig. 6c). Indeed, the transient visible spectra can be fitted to a sum of exponentials, where an initial exponential decay (with a time constant of 1 ps, attributed to vibrational cooling), is followed by a decay with a time constant of 14 ps, which is comparable to the exponential growth observed in the mid-IR-probe spectra, where a time constant of 10 ps was found (Fig. 6c), suggesting that both spectra are related to the transfer of holes.
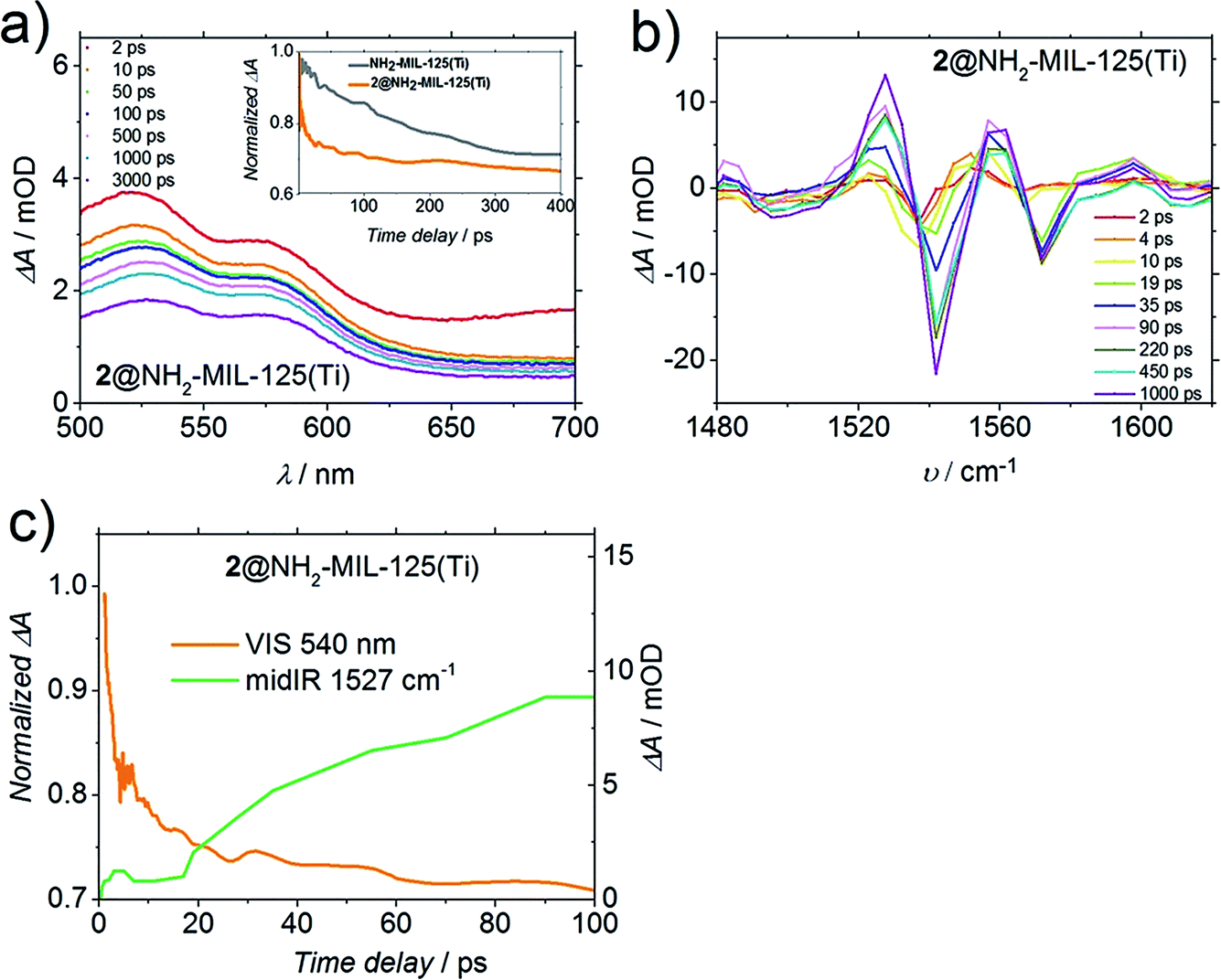 | ||
| Fig. 6 (a) Differential transient spectra of 2@NH2-MIL-125(Ti) in DMF upon excitation at 400 nm, inset: comparison of the time traces of NH2-MIL-125(Ti) and 2 occluded in NH2-MIL-125(Ti) at 540 nm in DMF, (b) differential transient absorption mid-IR spectra for 2@NH2-MIL-125(Ti) in DMF upon excitation at 400 nm, and (c) representative time traces of 2@NH2-MIL-125(Ti) upon excitation at 400 nm in DMF recorded at 1527 cm−1 (green). The orange line (corresponding to the left axis) represents the time traces at 540 nm in DMF (same as in (a), inset orange line). | ||
Even though fast charge transfer occurs between 2 and the MOF, only 48% of the signal assigned to the holes (at 570 nm, Fig. 6a) has decayed after 3 ns, the time window of the Vis-probe experiment (vs. 46% observed in the case of the pristine MOF). Intriguingly, when the TA is performed in acetonitrile (Fig. S10†), we observe decay dynamics for composite 2@NH2-MIL-125(Ti) similar to the pristine MOF, indicating that charge transfer does not take place, and it is solvent dependent. As 2 is not soluble in water, unfortunately we could not perform these experiments in water, the most relevant solvent for water splitting.
In contrast, in the transient spectra of 3@NH2-MIL-125(Ti) in water (Fig. 7b), the signal attributed to the holes (at λ = 570 nm) is not even observed. This indicates that within the time resolution of the experiment (considering that before 1 ps the spectrum is dominated by the coherent artefact) charge transfer of the holes to occluded 3 has already taken place. In acetonitrile, the charge transfer from the MOF to 3 still takes place, albeit with slower kinetics (Fig. S11†), despite the fact that the concentration of 3 in the composite 3@NH2-MIL-125(Ti) is higher in this solvent (6 and 2 molecules per large NH2-MIL-125(Ti) cage in acetonitrile and in water, respectively, see Section 4, ESI†).
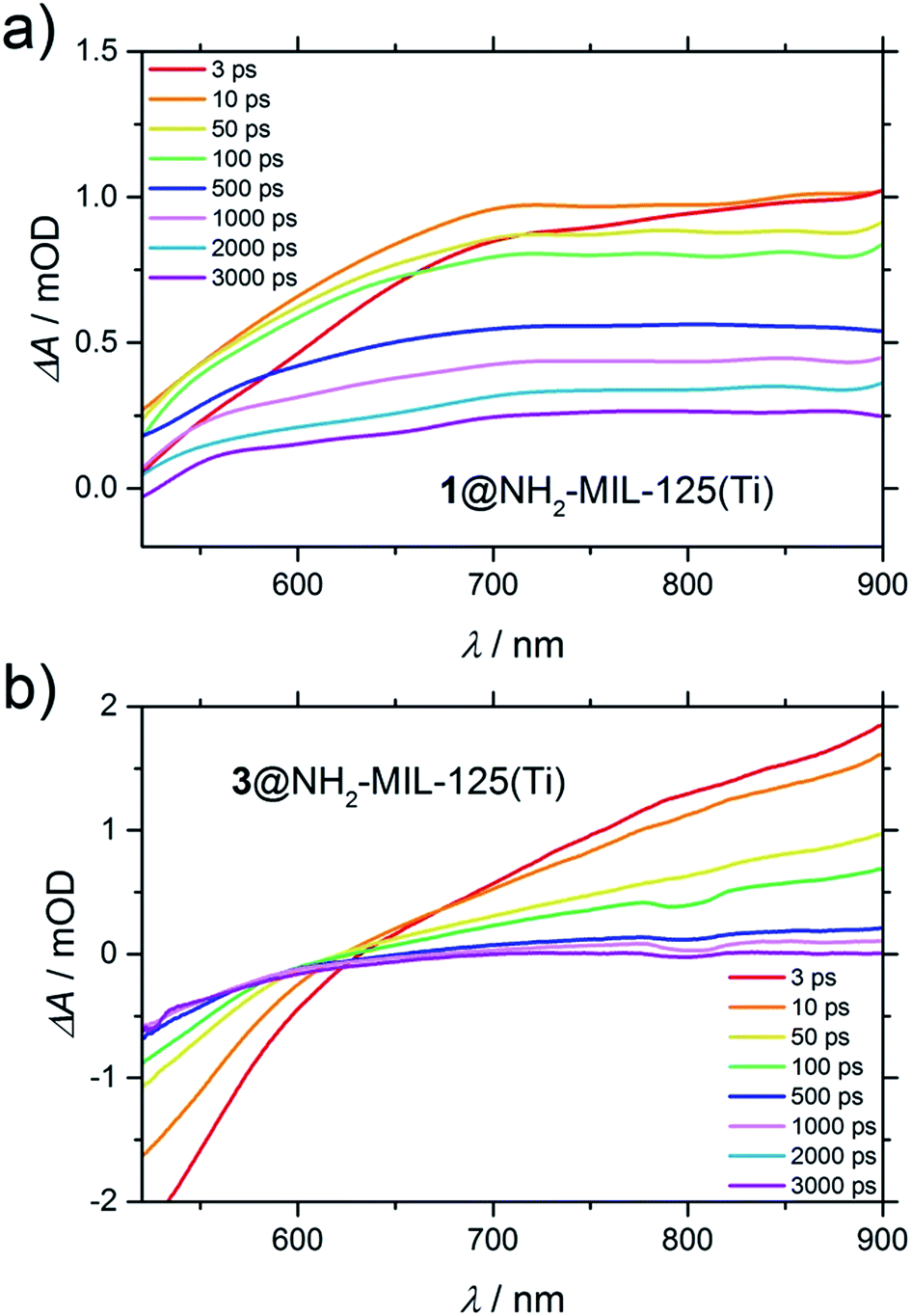 | ||
| Fig. 7 Difference transient spectra of: (a) 1@NH2-MIL-125(Ti) in acetonitrile and (b) 3@NH2-MIL-125(Ti) in water upon excitation at 400 nm. | ||
Interestingly, as in the case of 3@NH2-MIL-125(Ti) in water, the signal attributed to the holes in 1@NH2-MIL-125(Ti) in acetonitrile is also absent in the transient spectra. In both cases (Fig. 7a and b), the lower or even negative induced absorbance towards lower wavelengths is caused by stimulated emission, that is red-shifted from the ground state depletion (centred at the pump wavelength of 400 nm) that now dominates this range of the spectrum. In Fig. 7a, the stimulated emission is present as the superposition of a negative peak (up to 700 nm at 3 ps) with a very broad positive absorbance, alike in shape and wavelength to the UV generated (pump at λ = 315 nm) Ti3+ excited state in the non-functionalized MIL-125(Ti) (Fig. S15†). Since the accessibility of the photoexcited state in NH2-MIL-125(Ti) is only possible under visible light because of the participation of the amino group, and because it is necessary for this charge separation to be able to observe Ti3+, the transient spectra must be composed of the photogenerated electrons on the Ti3+ and the absence of holes due to their scavenging by the guest molecules 1 or 3.
Moreover, the fact that the electronic spectrum of the composite 3@NH2-MIL-125(Ti) is clearly a superposition of the MOF and 3 shows that there is no complexation of the two components (Fig. S17†). Additionally, the spectroelectrochemistry of 3 shows no induced absorbance in the visible range upon oxidation (Fig. S18†), and the visible TA spectrum of 3 shows also no change in absorption upon excitation at 400 nm (Fig. S19†), meaning that the broad induced absorbance extending into the near-IR observed in Fig. 7b can be attributed to the photogenerated Ti3+ on the MOF. Interestingly, the spectral band attributed to Ti3+ decays faster for 3@NH2-MIL-125(Ti) than for 1@NH2-MIL-125(Ti). In the case of 3@NH2-MIL-125(Ti), this signal fully decays after 2 ns. This is in contrast to the case of 1@NH2-MIL-125(Ti), where there is still 30% induced absorbance remaining after 3 ns. To understand the different decay of the Ti3+ spectral features observed in TA between composites 1 and 3, and its implications on the reduction half reaction of the water-splitting reaction, we performed the HER (Fig. S22†). 3@NH2-MIL-125(Ti) as the donor/catalyst pair shows no H2 evolution when using an excess of 3 (60 equivalents with respect to the expected H2 from the MOF), whereas the activity of the MOF is partially restored when using only 17 equivalents of 3. In contrast, the efficiency in the HER (μmol H2 produced during the reaction time) is significantly greater when 1 is added compared to the addition of 3 (see Table 1). In this case, a decreased concentration of the sacrificial electron donor 1 shows lower activity using 1@NH2-MIL-125(Ti) as the catalyst. An additional experiment with a mixture of the two donors (1 and 3) showed a further decrement of activity in comparison with any of the concentrations of 1 studied here (Table 1).
| Electron donor | Hydrogen evolution after 18 h μmol−1 |
|---|---|
| 600 eq. 1 | 26.47 |
| 60 eq. 1 | 7.35 |
| 60 eq. 3 | 0 |
| 17 eq. 3 | 0.27 |
| 60 eq. 1, 60 eq. 3 | 0.18 |
Based on its reduction potential, compound 3 can also be reduced and react with the photogenerated electron on Ti3+ species (see Fig. S20†). This is a reactivity that compound 1 does not show (probably due to its irreversible oxidation).28 If we combine this information with the TA spectroscopic results that showed a faster decay of the signal attributed to Ti3+ for 3@NH2-MIL-125(Ti) than for 1@NH2-MIL-125(Ti), it becomes very likely that reverse electron transfer is taking place for 3@NH2-MIL-125(Ti) (Fig. S21†).6
In summary, charge transfer is observed for the three studied compounds with notable differences in the charge transfer rates that show solvent dependency, and acetonitrile being our base solvent, only molecule 1 shows charge transfer. For molecule 2, we observed that acetonitrile was not suitable as the solvent, and we chose DMF for our studies (as 2 is not soluble in aqueous media). In the case of molecule 3, charge transfer in water was shown to be more efficient than in acetonitrile.
Due to the irreversible oxidation of 1 and 2, the availability of the holes for the oxidation reaction is hindered. In contrast, the reversible oxidation of 3 results in the availability of the positive charges. Indeed, when using benzyl alcohol oxidation as a probe reaction, we observe an increase in activity in the presence of 3 (Fig. S23†).
Conclusions
In this work, we have demonstrated charge transfer from NH2-MIL-125(Ti), a visible-light absorbing Ti-based MOF, to molecules occluded in its pores. We have confirmed by a combination of ultrafast spectroscopic techniques, fast charge transfer from the MOF to 2, a molecule that mimics the Tyr/His pair in PS II. Considering the structural changes observed in the mid-IR-probe (and in agreement with the irreversible oxidation wave of 2), we propose that the oxidation of 2 results in a chemically different species after charge transfer, thus formally quenching the holes in NH2-MIL-125(Ti), and inhibiting further the utilization of these positive charges in an oxidation reaction (similarly to the case of 1). When charge transfer occurs at higher rates to a reversible species such as compound 3, the hole is formally transferred, and it is still available for oxidation, as demonstrated by the enhanced catalytic oxidation activity of the 3@NH2-MIL-125(Ti) pair compared to the pristine MOF. We conclude that 3˙+@NH2-MIL-125(Ti3+) is effectively favouring the stabilization of the photogenerated positive charges. However, we have proved that charge transfer in the composite 3@NH2-MIL-125(Ti) leads to reverse electron transfer, a drawback that still has to be solved.Our findings suggest that combining a recyclable redox shuttle, e.g. TEMPO (3), or reversibly oxidisable molecules inspired by the photosynthetic blueprint, with an efficient H+ catalytic reduction site, might indeed enable artificial photosynthetic hydrogen production combined with a water oxidation catalyst in a Z-scheme. Finding this reversible molecule that allows for spatial separation and avoids quenching of the produced charges is still an enduring great challenge.
Acknowledgements
Ruben Abellon and Riming Wang are gratefully acknowledged for their support during the photoluminescence and electrochemical measurements, respectively. We would like to acknowledge the reviewer for helping to improve the final manuscript.References
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC
.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS PubMed
- L. Hammarström and S. Styring, Energy Environ. Sci., 2011, 4, 2379–2388 Search PubMed
- L. Hammarström, L. Sun, B. Åkermark and S. Styring, Biochim. Biophys. Acta, Bioenerg., 1998, 1365, 193–199 CrossRef
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS
- A. R. Holzwarth, M. G. Mueller, M. Reus, M. Nowaczyk, J. Sander and M. Roegner, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6895–6900 CrossRef CAS PubMed
- J. Barber, Philos. Trans. R. Soc. London, Ser. A, 2007, 365, 1007–1023 CrossRef CAS PubMed
- L. Kalman, R. LoBrutto, J. P. Allen and J. C. Williams, Nature, 1999, 402, 696–699 CrossRef
- Y. Zhao, J. R. Swierk, J. D. Megiatto, B. Sherman, W. J. Youngblood, D. Qin, D. M. Lentz, A. L. Moore, T. A. Moore, D. Gust and T. E. Mallouk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15612–15616 CrossRef CAS PubMed
- B. Sherman, M. Vaughn, J. Bergkamp, D. Gust, A. Moore and T. Moore, Photosynth. Res., 2014, 120, 59–70 CrossRef CAS PubMed
- K. Maeda, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858–5868 CrossRef CAS PubMed
- C. Wang, Z. Xie, K. E. de Krafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC
- J. G. Santaclara, M. A. Nasalevich, S. Castellanos, W. H. Evers, F. C. M. Spoor, K. Rock, L. D. A. Siebbeles, F. Kapteijn, F. Grozema, A. Houtepen, J. Gascon, J. Hunger and M. A. van der Veen, ChemSusChem, 2016, 9, 388–395 CrossRef CAS PubMed
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed
- J. D. Megiatto Jr, A. Antoniuk-Pablant, B. D. Sherman, G. Kodis, M. Gervaldo, T. A. Moore, A. L. Moore and D. Gust, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15578–15583 CrossRef PubMed
- J. G. Santaclara, F. Kapteijn, J. Gascon and M. A. van der Veen, CrystEngComm, 2017 10.1039/c7ce00006e
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D’Arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed
- M. F. De Lange, T. J. H. Vlugt, J. Gascon and F. Kapteijn, Microporous Mesoporous Mater., 2014, 200, 199–215 CrossRef CAS
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. van der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 Search PubMed
- S. Hu, M. Liu, K. Li, Y. Zuo, A. Zhang, C. Song, G. Zhang and X. Guo, CrystEngComm, 2014, 16, 9645–9650 RSC
- S. Vaesen, V. Guillerm, Q. Yang, A. D. Wiersum, B. Marszalek, B. Gil, A. Vimont, M. Daturi, T. Devic, P. L. Llewellyn, C. Serre, G. Maurin and G. De Weireld, Chem. Commun., 2013, 49, 10082–10084 RSC
- M. Masui, H. Sayo and Y. Tsuda, J. Chem. Soc. B, 1968, 973–976 RSC
- J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef PubMed
- R. D. Richardson, E. J. Holland and B. K. Carpenter, Nat. Chem., 2011, 3, 301–303 CrossRef CAS PubMed
- B. C. M. Martindale, E. Joliat, C. Bachmann, R. Alberto and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 9402–9406 CrossRef CAS PubMed
- G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466–10467 CrossRef CAS PubMed
- R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7fd00029d |
| This journal is © The Royal Society of Chemistry 2017 |