
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Verification of redox-processes as switching and retention failure mechanisms in Nb:SrTiO3/metal devices†
C.
Baeumer
 *a,
N.
Raab
a,
T.
Menke
a,
C.
Schmitz
a,
R.
Rosezin
a,
P.
Müller
a,
M.
Andrä
a,
V.
Feyer
a,
R.
Bruchhaus
b,
F.
Gunkel
ac,
C. M.
Schneider
a,
R.
Waser
ac and
R.
Dittmann
a
*a,
N.
Raab
a,
T.
Menke
a,
C.
Schmitz
a,
R.
Rosezin
a,
P.
Müller
a,
M.
Andrä
a,
V.
Feyer
a,
R.
Bruchhaus
b,
F.
Gunkel
ac,
C. M.
Schneider
a,
R.
Waser
ac and
R.
Dittmann
a
aPeter Gruenberg Institute, Forschungszentrum Juelich GmbH and JARA-FIT, 52425 Juelich, Germany. E-mail: c.baeumer@fz-juelich.de
bJuelich Centre for Neutron Science at MLZ, Forschungszentrum Juelich, Lichtenbergstr. 1, 85748 Garching, Germany
cInstitute of Materials in Electrical Engineering and Information Technology II, RWTH Aachen University, 52056 Aachen, Germany
First published on 30th March 2016
Abstract
Nanoscale redox reactions in transition metal oxides are believed to be the physical foundation of memristive devices, which present a highly scalable, low-power alternative for future non-volatile memory devices. The interface between noble metal top electrodes and Nb-doped SrTiO3 single crystals may serve as a prominent but not yet well-understood example of such memristive devices. In this report, we will present experimental evidence that nanoscale redox reactions and the associated valence change mechanism are indeed responsible for the resistance change in noble metal/Nb-doped SrTiO3 junctions with dimensions ranging from the micrometer scale down to the nanometer regime. Direct verification of the valence change mechanism is given by spectromicroscopic characterization of switching filaments. Furthermore, it is found that the resistance change over time is driven by the reoxidation of a previously oxygen-deficient region. The retention times of the low resistance states, accordingly, can be dramatically improved under vacuum conditions as well as through the insertion of a thin Al2O3 layer which prevents this reoxidation. These insights finally confirm the resistive switching mechanism at these interfaces and are therefore of significant importance for the study and application of memristive devices based on Nb-doped SrTiO3 as well as systems with similar switching mechanisms.
1. Introduction
Because of their potential use in next-generation non-volatile memory applications and neuromorphic circuits, oxide-based memristive devices have developed into one of the most active topics in materials research.1–3 The mechanism driving the resistance change in these devices is widely accepted to be a nanoscale redox reaction, induced by oxygen-ion migration.1,4 This oxygen-ion migration is accompanied by a valence change in the transition metal and a local modification of the Schottky barrier height between the active layer and a noble metal electrode. A prominent example for such an interface is the Schottky barrier between Pt top electrodes and Nb-doped SrTiO3 (Nb:SrTiO3) single crystals. Despite extensive existing knowledge about the electronic transport properties across the junction,5–7 the underlying resistive switching mechanism in these devices is not understood in detail and contradicting interpretations prevail.Many reports have connected resistive switching in Pt/Nb:SrTiO3 junctions with changes in the oxygen stoichiometry within a thin interfacial dead layer, corresponding to an electron depletion layer caused by the formation of a Schottky barrier.8–12 Some reports suggested that this interfacial dead layer may be caused by a deviation from the nominal cation stoichiometry in the near-surface region of Nb:SrTiO3,13 potentially modifying the Schottky-type interfacial space charge region.14 Typical noble metal/Nb:SrTiO3 interfaces can be SET to the low resistance state (LRS) by applying a positive voltage to the top electrode, while a negative voltage RESETs the device to a high resistance state (HRS).5–7,9–12,15,16 This switching behavior is commonly referred to as eight-wise resistive switching.17 Generally, it is observed that the low resistance states achievable in these devices have a rather volatile character with LRS retention times in the range of hundreds of seconds to several hours.10,18
Additionally, it was observed that resistive switching in Pt/Nb:SrTiO3 devices strongly depends on the partial pressure of oxygen of the surrounding atmosphere.9 The creation of additional oxygen vacancies at the interface yields improved memory characteristics15 while removal of oxygen vacancies can prevent resistive switching entirely.16 Based on capacitive measurements, it has been inferred that the resistance change occurs in a homogeneous manner for small current limits, while higher voltages have been suggested to cause a filamentary change of the resistance.10 In the latter case, the switching process is commonly interpreted as a locally confined change in the Schottky barrier height,5 while the majority of the interface remains unchanged. Indeed, there exists both theoretical19 and experimental evidence20 that oxygen vacancies in the surface layer of Nb:SrTiO3 lead to a reversible reduction of the effective Schottky barrier height.
Despite the abundant evidence of dependence on the partial pressure of oxygen and the corresponding interpretation of the resistance change in transition metal oxides as an effect caused by the movement of oxygen-ions, direct proof of oxygen migration and the associated valence change is rare, resulting in competing interpretations. While some reports try to describe the resistance change in a purely electronic picture,8,21–24 comparative studies show that both electronic effects such as trapping and detrapping of defect states as well as oxygen-ion migration can explain the electrical data.18,22 Therefore, direct evidence of one or the other mechanism is indispensable.
In this article, we will present experimental evidence that oxygen-ion migration is indeed responsible for the resistance change in noble metal/Nb:SrTiO3 junctions. Eight-wise switching was observed for junctions ranging from the micrometer scale down to the nanometer regime. A comparative study of the retention behavior of these cells under ambient conditions and exposed to vacuum clearly indicates that retention failure of the LRS is driven by reoxidation involving oxygen exchange with the environment. Therefore, retention failure can be effectively suppressed by preventing the reoxidation of the Nb:SrTiO3 surface layer through the insertion of a retention-stabilization layer with slow oxygen migration properties. Even more explicitly, spectromicroscopic evidence combined with conductive atomic force microscopy (C-AFM) verifies that a filamentary valence change from Ti4+ to Ti3+ is responsible for the resistive switching with high switching currents and sheds light on the nanoscale chemistry of the switching filament. Our results thus prove the long-suspected valence change mechanism – rather than purely electronic effects – to be responsible for the resistance change in this noble metal/Nb:SrTiO3 junctions.
2. Results
In order to clarify the resistive switching mechanism in these junctions, a set of devices was fabricated by the deposition of Au or Pt layers on (001) SrTiO3 single crystals doped with 0.5 wt% Nb and subsequent structuring of the quadratic top electrodes with dimensions ranging from 100 × 100 μm2 to circular pads with a diameter of 125 nm (Fig. 1, see Experimental section for fabrication details). For electrical characterization, 10 to 100 μm-sized devices were brought into contact with tungsten probes, while smaller devices were brought into contact via C-AFM with Pt/Ir-coated tips. In both cases, voltages were applied to the top electrode while the grounded Nb:SrTiO3 substrate served as the bottom electrode.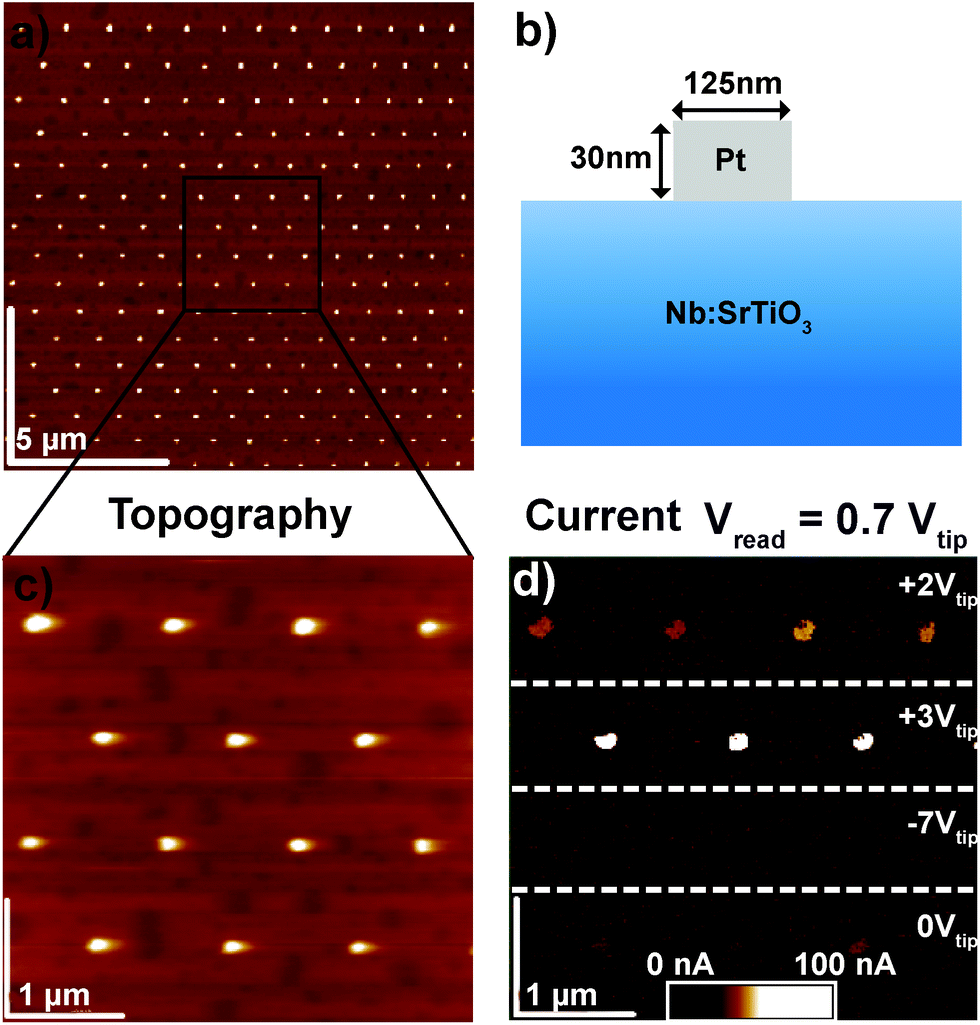 | ||
| Fig. 1 (a) AFM topography of a device array with dot diameter of 125 nm. (b) Device schematic. (c) Zoomed-in AFM topography image of the device array in (a). (d) Corresponding current image detected simultaneously. The Pt dots were previously set into different resistance states through the application of positive or negative voltages as indicated. The scan was recorded with an applied bias of +0.7 V. A previous reference measurement showed that a tip bias of +0.7 V did not change the device resistance. | ||
2.1. Spatially homogeneous resistive switching with low current limits
The Pt/Nb:SrTiO3 junctions presented in this study exhibit the typical eight-wise switching behavior. As shown in Fig. 1, multiple nanoscale-devices were addressed via C-AFM using different voltages in the range of −7 to +3 V. Subsequent scans with smaller bias (+0.7 V) were used to read out the resulting resistance states in terms of a current image (Fig. 1c and d). The devices in the first and second rows were SET to the LRS through the application of +2 V and +3 V, respectively. A negatively biased tip (−7 V) was used to obtain the HRS for the devices in the third row. The fourth row was not electrically addressed and serves as a pristine reference. In the case of devices SET to the LRS (first two rows), the READ currents showed increased conductivity. The currents flowing through pristine devices and devices in the HRS, on the other hand, are barely distinguishable from the noise level of the setup (100 pA).Following this initial characterization, devices with different top electrode sizes were investigated. Individual Pt/Nb:SrTiO3 junctions with different electrode diameters ranging from 1 μm down to 125 nm (Fig. 2a and b) were subjected to bipolar voltage sweeps with a comparably low current limit of 1 μA, yielding the typical I–V hystereses commonly observed for memristive devices (Fig. 2a). Devices of different electrode sizes show qualitatively similar I–V hystereses, which superpose each other when normalized to the junction area (Fig. 2c). Both HRS and LRS accordingly show linear scaling of the resistance with the junction area. Thus, we can conclude that the current distribution below the Pt electrode exhibits a laterally homogeneous, non-filamentary character with eight-wise switching polarity. A similar behavior was observed before for Pt/Nb:SrTiO3 junctions operated with such small current limits.10 This behavior is characteristic for Pt/Nb:SrTiO3 junctions and cannot be observed for Pt/SrTiO3/Nb:SrTiO3 devices, where only filamentary switching can be observed.
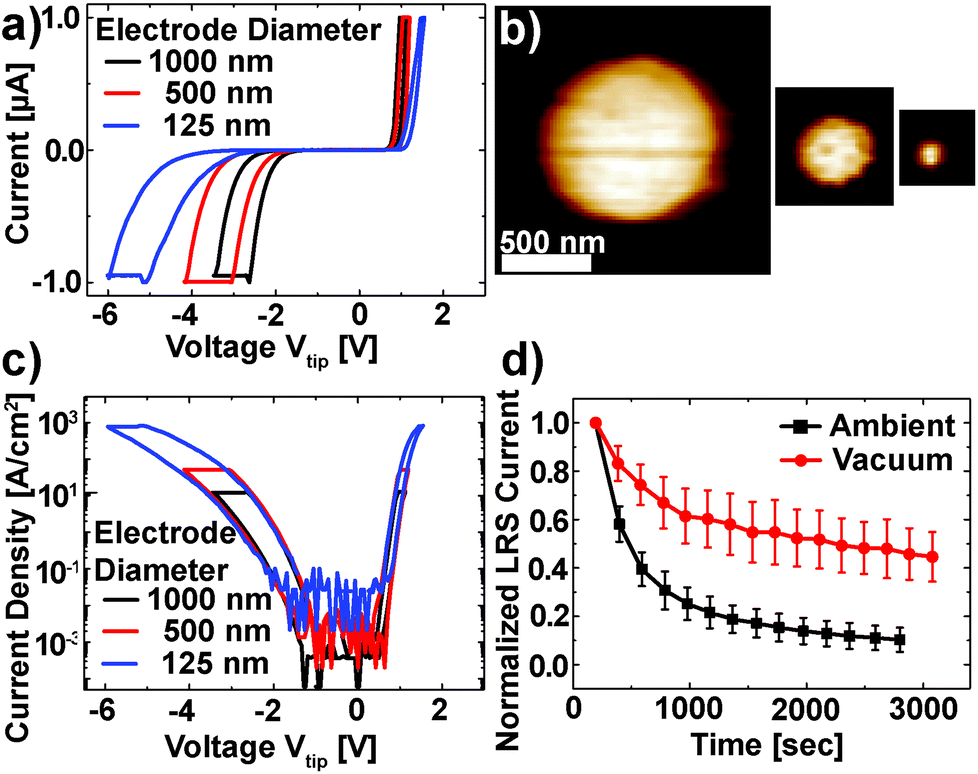 | ||
| Fig. 2 (a) I–V characteristics of circular Pt/Nb:SrTiO3 devices with different top electrode diameters. (b) The AFM topography of these devices. (c) Comparison of the current density of different electrode sizes. For each size, the current compliance was set at 1 μA. (d) Retention measurement of the LRS under ambient and vacuum conditions (black and red symbols, respectively). For each case, eleven devices were SET to the LRS and the READ current was averaged after normalization to the first READ current. Error bars indicate the standard deviation. | ||
In order to investigate the retention behavior of the low resistance states, eleven devices were SET by scanning with a positive bias (+3 V). Afterwards, READ scans were repeated every 3 minutes to track the device resistance over time. The currents of all devices were normalized to their first READ current (t = 180 s) and finally averaged over all devices (Fig. 2d). In agreement with other reports,10,18 the READ current decreased by ∼90% within 3000 s.
As discussed in the Introduction, resistive switching in these junctions has been suspected to be related to changes in oxygen stoichiometry. Consequently, we investigated the retention behavior of the LRS depending on the surrounding atmosphere to verify whether the environment has any significant effect on the Pt/Nb:SrTiO3 device resistance states (Fig. 2d). Similar to the experiment under the ambient atmosphere, eleven devices were SET to the LRS by scanning with a positive bias (+3 V) under vacuum conditions (ptot ∼ 10−3 mbar). Subsequently, the READ current was monitored over time. We observed a dramatic change in the retention behavior of these devices compared to the retention behavior under ambient conditions: within 3000 s, the READ current decreased by only 52% under vacuum conditions compared to more than 90% under ambient conditions. Hence, the devices measured under vacuum conditions show an improved stability in the LRS suggesting that the devices interact with the environment and are oxygen-sensitive.
2.2. Spectromicroscopic evidence of the valence change-driven filamentary switching
After the characterization of Pt/Nb:SrTiO3 junctions operated at low currents, we now turn our attention to higher current limits in order to investigate the suspected filamentary switching mechanism.10 For this purpose, we investigated larger devices which allow for spectromicroscopic analysis. In a first step, the low-current switching regime (<10 μA) of these devices was characterized and compared to the nanostructures described above. Again, devices of different electrode sizes show qualitatively similar I–V hystereses with eight-wise switching, which superpose each other when normalized to the junction area (Fig. 3a). Thus, we can conclude that the resistive switching of these devices also exhibits a laterally homogeneous, non-filamentary character for low current limits. Upon utilization of significantly higher current limits, however, the normalized I–V hystereses do not superpose each other. Instead, smaller devices show a higher LRS current density and a higher HRS current density than larger devices (Fig. 3b). This behavior is due to a more pronounced forming step: while the homogeneous switching for low current compliances observed above is forming free (HRS current equal to virgin current; the first SET sweep and subsequent sweeps superpose each other), higher currents in the mA range lead to a soft forming step, the first SET sweep and subsequent sweeps do not superpose each other (Fig. 3c). The difference between the first and the second SET increases with decreasing junction area for a given current limit, i.e. with increasing current density. In line with the vanishing of the area scaling of both LRS and HRS observed here, forming steps in such junctions have been proposed to yield filamentary switching.10 However, the switching polarity as well as the shape of the I–V curves do not change upon forming, indicating a similar underlying mechanism.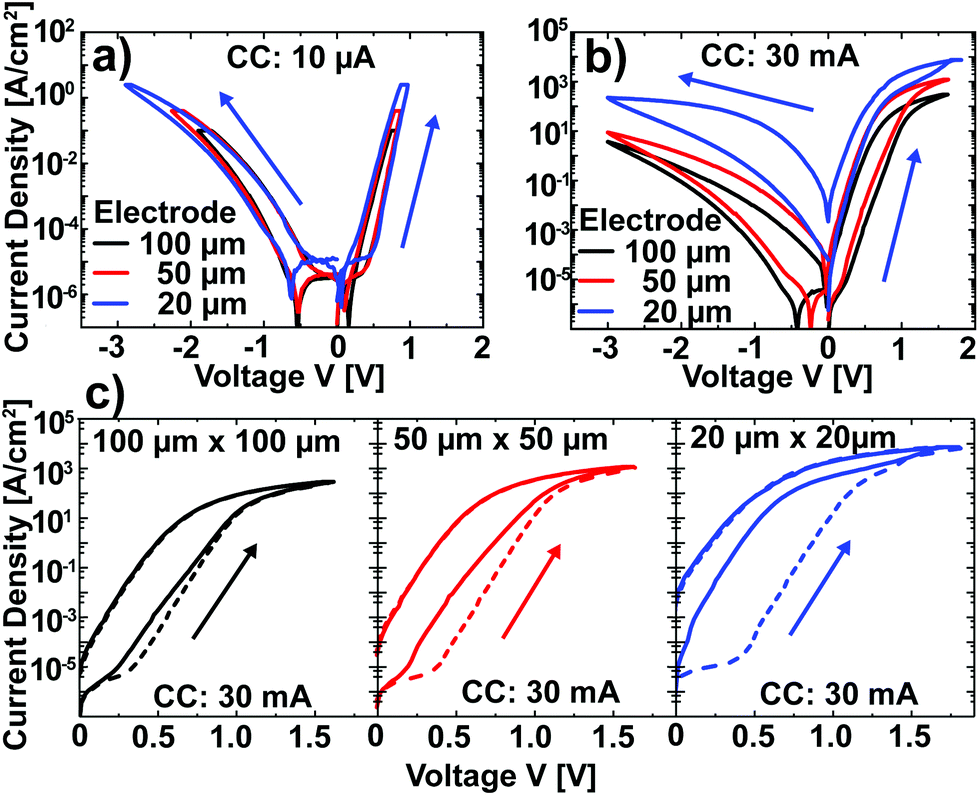 | ||
| Fig. 3 (a) I–V characteristics of Pt/Nb:SrTiO3 junctions with different electrode sizes, normalized to the junction area. In each case, the current compliance was 10 μA. (b) I–V characteristics of Pt/Nb:SrTiO3 junctions with different electrode sizes, normalized to the junction area. In each case, the current compliance was 30 mA. (c) The forming process (dashed line) and subsequent SET (solid line) for the devices presented in (b). The current compliance was 30 mA for forming and switching. | ||
To visualize switching-induced local changes of the electronic structure and chemical properties of the Nb:SrTiO3, we investigated memristive Au/Nb:SrTiO3 devices using X-ray photoemission electron microscopy (XPEEM) in absorption mode. In this case, Au top electrodes were chosen to allow for mechanical top electrode delamination before spectromicroscopic investigation. In a first step, I–V hystereses were recorded for Au/Nb:SrTiO3 junctions with 10 × 10 μm2 area using a current limit of 40 mA, yielding a stable and reversible switching between the HRS (>109 Ω) and the LRS (<104 Ω). Comparison with I–V hystereses Pt/Nb:SrTiO3 junctions of the same size confirms very similar switching characteristics, suggesting the same switching mechanism for both top electrode materials (ESI Fig. 1†).
After delamination of the top electrode (see Experimental section), XPEEM imaging of a representative device in the LRS reveals an area with darker contrast (Fig. 4a) at a photon energy of hν = 459.5 eV, which corresponds to the Ti4+ L3 absorption edge. A direct comparison of the XPEEM image with a C-AFM image of the same device reveals that this area is in fact the only area with increased conductivity (Fig. 4b), yielding direct evidence for the filamentary change of the resistance. If the valence change mechanism is responsible for this change of resistance, we consequently expect this area to exhibit a spectroscopic signature of reduced SrTiO3. A detailed analysis of the full Ti L absorption edge reveals that this switching filament in fact consists of a mixture of Ti4+ and Ti3+ states,25,26 while the insulating surrounding only exhibits Ti4+ states (Fig. 4c). The local confinement of this valence change is evident from the false color map of Ti3+ contribution (Fig. 4d, see Experimental section for details): only within the filament area does one find a significant Ti3+ contribution. While this locally confined, chemically reduced component could also be induced through the electroforming step,25 different valence states between LRS and HRS are necessary to validate that the valence change mechanism is responsible for the resistive switching in these devices. We therefore analyzed a device that was RESET to the HRS before XPEEM imaging. Although the XPEEM images indicate the presence of a locally modified area, which we ascribe to the switching filament in the HRS (i.e., a reoxidized switching filament, not shown here), no Ti3+ spectral contributions could be detected in this region (Fig. 4e). In fact, a false color mapping does not show any Ti3+ contribution for the entire device area. In consequence, we have demonstrated explicitly that a change of the Ti valence in a locally confined filament is indeed responsible for the resistive switching in Pt/Nb:SrTiO3 junctions operated at high current limits. In other words, field-driven oxygen-ion migration and the corresponding local redox reactions rather than purely electronic effects constitute the fundamental mechanism behind resistive switching in this system.
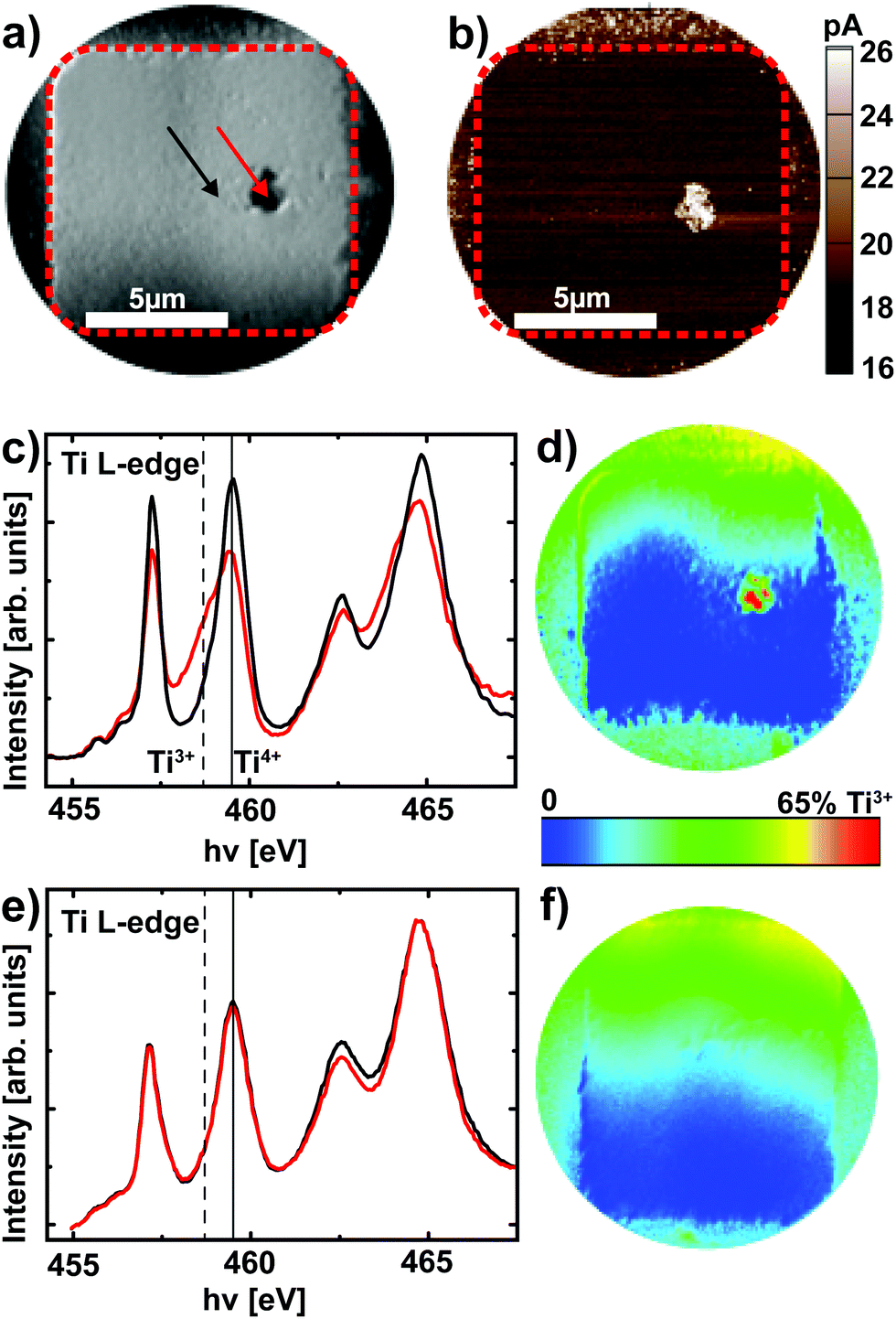 | ||
| Fig. 4 (a) The XPEEM image of a device in the LRS recorded with a photon energy of 459.5 eV. (b) C-AFM current image of the device shown in (a). (c) Ti L-edge spectra recorded from the XPEEM image stack for a region within the filament (red line) and for the surrounding device area (black line) as indicated with the arrows in (a). The solid line and the dashed line denote the eg level of the Ti L3 edge in Ti4+ and Ti3+ configurations, respectively. (d) A false color map of the Ti3+ contribution for the device in (a). The enhanced contrast surrounding the active device is caused by the reactive ion beam etching involved in the electrode structuring and is not associated with the device conductance. The apparent vertical gradient is an artefact from the uncorrected but small energy dispersion. The resulting contrast is much lower, however, than the real contrast detected for the filament. (e) Ti L-edge spectra recorded from the XPEEM image stack for a device in the HRS from a region within the suspected filament (red line) and for the surrounding device area (black line). The dotted line and the dashed line denote the eg level of the Ti L3 edge in Ti4+ and Ti3+ configurations, respectively. (d) A false color map of the Ti3+ contribution for the device in (e). No Ti3+ contribution is discernible for the entire device area. | ||
2.3. Filament chemistry and device retention
While Fig. 4 already validates the valence change mechanism, a closer inspection of the switching filament is essential to shed light on the details of the switching process itself. In order to analyze the link between conductivity and the valence state in more detail, regions with different conductivities were selected based on the C-AFM data and used as reference for the analysis of the spectromicroscopic data (Fig. 5a). For each of these regions, Ti L edge absorption spectra were recorded (Fig. 5b). Comparison of the two representative spectra recorded within the switching filament indicates an inhomogeneous distribution of Ti3+ states within the filament. Upon first sight, one may expect that areas with a high Ti3+ concentration should correspond to the areas of highest conductivity inside this switching filament. Our C-AFM data, however, indicate the opposite behavior: areas of high conductivity correspond to mediocre Ti3+ concentrations, while areas of mediocre conductivity correspond to high Ti3+ concentrations. This apparent contradiction can be explained through the consideration of the spatially resolved Sr 3d spectra recorded from the same regions as the Ti L-edge spectra (Fig. 5c). As previously observed for SrTiO3 thin film devices, we observe a local phase separation into Sr-deficient SrTiO3 with a SrO coverage of the switching filament.25,27 This SrO coverage is apparent from the high binding energy shoulder of the Sr 3d spectra. Such phase separations occur if sufficient Joule heating is provided, which is the case for the high current limits necessary for filamentary switching.25 Comparison of the Sr 3d spectra and the C-AFM data reveals that the SrO layer is responsible for the non-uniform conductivity of the switching filament: regions with high conductivity show only a slight shoulder, indicative of a thin SrO layer. Regions with lower conductivity, on the other hand, exhibit a more pronounced shoulder, indicative of a thicker SrO layer. This correlation is directly understandable in terms of the electronic structure of SrO: the filament region underneath is well conducting even for a small Ti3+ contribution. SrO, however, is an insulator and therefore presents a tunneling barrier for electronic transport into the filament. Thin SrO layers therefore do not impede electronic transport, while thicker layers lead to a non-negligible series resistance. This scenario is schematically depicted in Fig. 5d.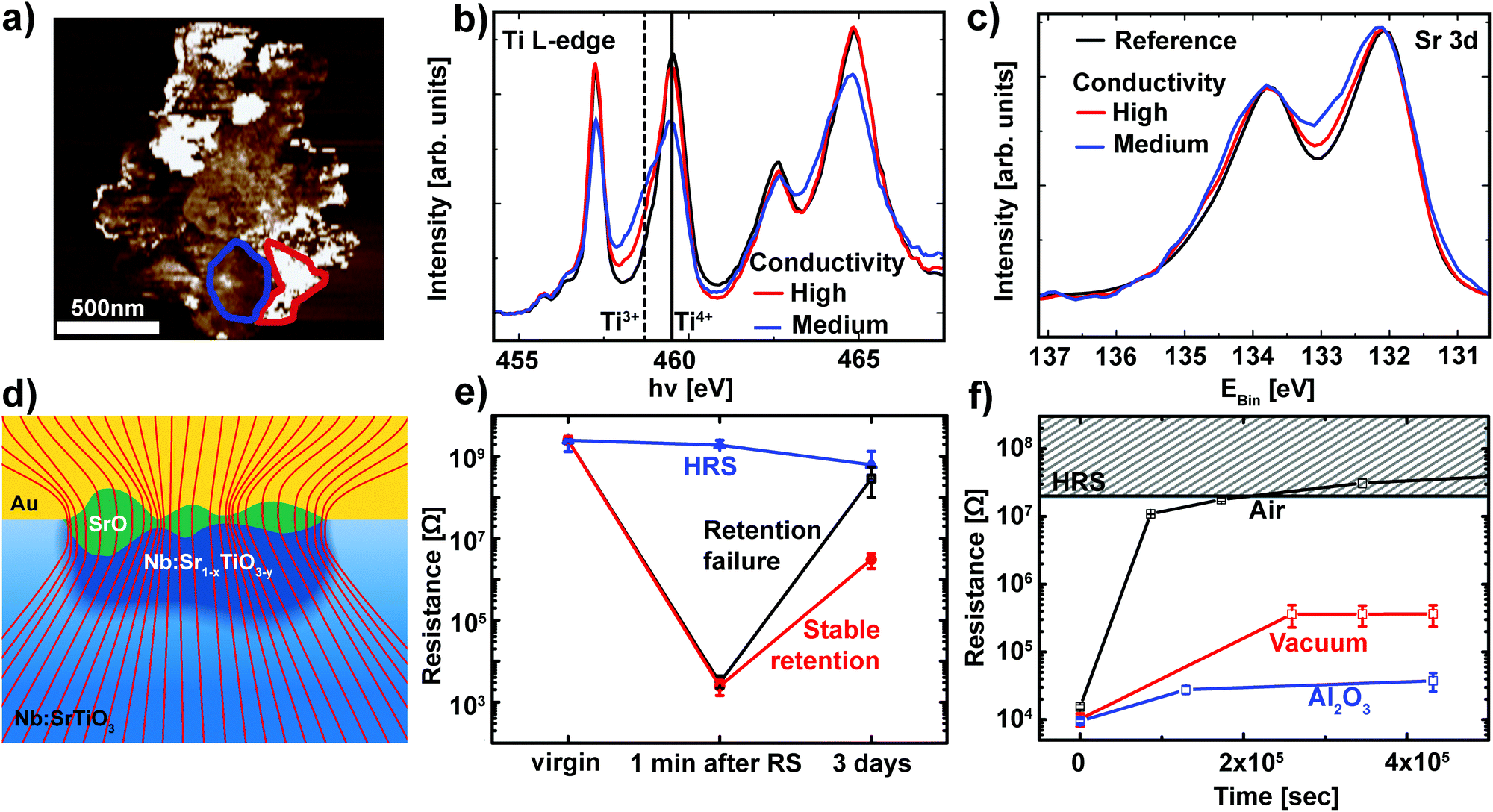 | ||
| Fig. 5 (a) A high-magnification C-AFM image of the switching filament for the LRS device in Fig. 4. Representative regions of high conductivity (red) and mediocre conductivity (blue) were chosen for spectroscopic evaluation. (b) Ti L-edge spectra recorded from the XPEEM image stack for two regions inside the filament outlined in (a) (red and blue lines) and for the surrounding device area (black line). The solid line and the dashed line denote the eg level of the Ti L3 edge in Ti4+ and Ti3+ configurations, respectively. (c) Sr 3d XPS spectra for the same regions as in (b). (d) Schematic illustration of the phase separation in the switching filament. Nb:SrTiO3 single crystal in blue with an insulating surface layer (light blue) underneath the Au top electrode. The switching filament consists of a well-conducting, Nb:Sr1−xTiO3−y region (dark blue) which penetrates the insulating surface layer. This filament is covered by a SrO layer (green) of non-uniform thickness. The thickness dependent series resistance of this SrO layer leads to non-uniform conductance, as depicted by the current density lines (red lines). The lines are drawn such that the density of lines indicates the current density. (e) Retention behavior of different devices on the same chip. Devices RESET to the HRS remain at a constant resistance over several days (blue curve). Devices that were SET to the LRS show two classes of retention behavior: stable retention and retention failure (red and black curves, respectively). For each class of retention behavior, the resistance was averaged for several representative devices. Error bars indicate the minimum and maximum values. (f) Retention measurement of the LRS for Pt/Nb:SrTiO3 devices under ambient and vacuum conditions (black and red symbols, respectively). The retention times can be further improved through the insertion of a thin Al2O3 layer (blue symbols). For each case, several devices were SET to the LRS and the READ current was averaged. Error bars indicate the minimum and maximum values. | ||
As discussed in section 2.1, the LRS of these devices can undergo a retention failure, which appears to be connected to exchange with the ambient atmosphere. Therefore, a closer inspection of the retention behavior of these Nb:SrTiO3-based devices operated at high current limits is indispensable. For this purpose, several devices were SET to the LRS and the resistance of each device was monitored over a period of several days (Fig. 5e). As we recently observed for SrTiO3 devices,27 two classes of retention characteristics of the LRS can be distinguished: for most devices in the LRS, the resistance increases by several orders of magnitude over a period of few days, indicative of retention failure. On the other hand, some devices retain a resistance below 107 Ω. In the following, these devices will be referred to as devices with retention failure and devices with stable retention, respectively.
For the XPEEM investigation of the filamentary switching mechanism described above, we selected a representative device with stable retention to allow for the characterization of the LRS state even after the unavoidable delay between switching and XPEEM investigation. Similar to our observations for SrTiO3 devices, all devices with stable retention investigated here exhibit a SrO layer on top of the reduced switching filament.27 In contrast, we did not find any SrO signature for devices with a retention failure, indicating a close connection between the appearance of the SrO layer and the LRS retention, as discussed below.
Similar to the devices operated at low current limit (section 2.1), we also investigated the retention behavior of filamentary switching under the ambient atmosphere as well as under vacuum conditions. Several devices without the previously observed phase separation were SET to the LRS and the resistance of each device was monitored over a period of several days while the devices were exposed to ambient atmosphere or stored in a vacuum (ptot ∼ 10−6 mbar, Fig. 5f). Again, we observe dramatic improvement of the retention times under vacuum conditions while the LRS resistance under ambient conditions increases by 3 orders of magnitude within one day and consequently becomes indistinguishable from the HRS, the LRS resistance under vacuum conditions remains 2 orders of magnitude lower. As is evident from the small error bars, all of the devices stored under the same atmosphere exhibited almost identical retention behavior. We can therefore conclude that no phase separation occurred in any of these devices.
3. Discussion
As demonstrated in Fig. 4, the devices with filamentary resistive switching evidently undergo a valence change between the HRS and the LRS. Within a spatially confined region, a significant amount of Ti3+ was detected, which is indicative of electrons in the conduction band, resulting in the conductive filament observed via C-AFM. Accordingly, localized redox reactions during the switching are the cause for the change of resistance. Here, we would like to note that the device in the HRS shown in Fig. 4e and f also exhibited the phase separation into a Sr-deficient filament and a SrO cover layer (ESI Fig. 2†), confirming that the difference in the Ti valence rather than the existence or absence of a phase separation is responsible for the resistance change. This finding also helps us to explain the observed retention characteristics: previously, simulations28 and experimental observation27 revealed that the LRS retention failure of valence change memory is the result of reoxidation of the switching filament. As an oxygen deficient switching filament is also present in our devices, the retention failure under the ambient atmosphere may be attributed to reoxidation. In this scenario, the temporal increase of the resistance can then be understood by oxygen incorporation and diffusion at the interface.27,29 This reoxidation process, however, depends on the environmental conditions: with decreasing oxygen partial pressure pO2, the reincorporation rate of oxygen decreases.30 For devices stored in a vacuum it therefore appears likely that the retention times are improved because the partial pressure of oxygen is too low to completely reoxidize the samples within the measurement time.For the area-dependent resistive switching in devices operated at low current limits, the switching polarity, the shape of the I–V curves and the retention characteristics are very similar to the devices operated at high current limit. This implies that the basic switching mechanism of the devices is independent of the lateral extension of the switching event. We therefore suspect that the observed pO2-dependent retention characteristics for devices switched with low current limit are also indicative of retention failure through oxygen incorporation and diffusion.
Finally, the finding that retention failure is caused by reoxidation of the switching filament is also responsible for the retention-stabilization effect of the SrO layer observed for the switching filament in Fig. 4 and 5. We demonstrated recently that SrO exhibits very slow oxygen migration properties.27 The SrO layer covering the switching filament therefore inhibits reoxidation of the switching filament – similar to operation under vacuum conditions as described above. The phase separation into Sr-deficient SrTiO3 and a SrO surface layer therefore leads to improved retention times.
At the same time, this phase separation is a rather unpredictable process: for most devices, we did not observe the phase separation and for the device shown in Fig. 4 and 5, the SrO layer does not exhibit a uniform thickness. These aspects are certainly undesirable for the operation of memristive devices, as the inherent non-uniformity and device-to-device variation complicates any application and results in a small fraction of devices with sufficient retention. As we demonstrated for SrTiO3 thin film devices, the reoxidation-driven retention failure can also be prevented through the intentional and uniform incorporation of a retention-stabilization layer with similarly slow oxygen-migration properties. In the case of our Nb:SrTiO3-based devices, we substantiate this hypothesis through the fabrication and characterization of memristive devices with a 1 nm Al2O3 interlayer between the Pt top electrode and the Nb:SrTiO3 substrate. Al2O3 exhibits extremely slow oxygen migration with a diffusion coefficient of 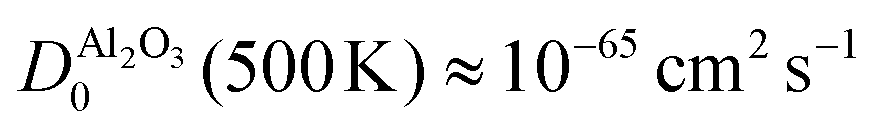 .27,31 Regarding the fast switching speed required for memory applications, one might question whether the incorporation of such layers for retention stabilization through slow oxygen migration might not lead to very slow switching times simultaneously. The extreme nonlinearity of oxygen migration with respect to the applied electric field and temperature, however, can yield sufficiently fast switching times even for large oxygen-migration activation energies due to Joule heating.32 Therefore, the insertion of this thin retention-stabilization layer does not impede or retard the resistive switching of the active layer; we observe similar I–V characteristics as for devices without an interlayer.
.27,31 Regarding the fast switching speed required for memory applications, one might question whether the incorporation of such layers for retention stabilization through slow oxygen migration might not lead to very slow switching times simultaneously. The extreme nonlinearity of oxygen migration with respect to the applied electric field and temperature, however, can yield sufficiently fast switching times even for large oxygen-migration activation energies due to Joule heating.32 Therefore, the insertion of this thin retention-stabilization layer does not impede or retard the resistive switching of the active layer; we observe similar I–V characteristics as for devices without an interlayer.
As expected, the LRS retention times of Pt/Al2O3/Nb:SrTiO3 devices are prolonged extensively compared to Pt/Nb:SrTiO3 devices (Fig. 5f). In fact, the Pt/Al2O3/Nb:SrTiO3 devices stored in air retain an even lower resistance than the Pt/Nb:SrTiO3 devices stored in a vacuum despite an almost identical LRS resistance immediately after switching. We presume that this further improvement through the insertion of an Al2O3 layer is caused by a more effective suppression of reoxidation directly after switching, as our Pt/Nb:SrTiO3 devices were switched in air and evacuated to 10−6 mbar only after switching. As demonstrated in Fig. 5f, the resistance increase is fastest immediately after switching, which makes it reasonable to assume that the Pt/Nb:SrTiO3 devices were partially reoxidized before the vacuum was sufficient to limit this reoxidation process. Regardless of these subtle difference, the dramatic improvement of the LRS retention times through the insertion of a thin Al2O3 as well as through storage under vacuum conditions or through the creation of a SrO coverage of the switching filament verifies that reoxidation of a previously oxygen-deficient Nb:SrTiO3 surface is responsible for the retention behavior of Pt/Nb:SrTiO3 cells. In combination with the direct spectromicroscopic observation of the valence change mechanism, these results therefore indicate that oxygen-ion migration rather than trapping and detrapping of defect states is responsible for the resistive switching in these junctions.
In the case of homogeneous switching, we also observe significantly increased retention in Pt/Al2O3/Nb:SrTiO3 devices (ESI Fig. 3†). As both filamentary and homogeneous switching and retention failure are obviously connected to oxygen exchange, the question remains how the Pt/Nb:SrTiO3 junction can interact with the surrounding atmosphere despite the covering Pt layer. Considering just the devices switched at low current limits, we did not observe any scalability of the retention failure rate (decay time of the LRS current) with the electrode-size. Since these devices exhibit a homogeneous current distribution, we can conclude that the whole area of the Pt electrode must also be involved during the reoxidation effect. Therefore, it appears likely that the reoxidation occurs via oxygen transport through the top electrode (most likely along Pt grain boundaries) rather than at the electrode edge. Oxygen migration through the Pt layers has already been observed many years ago33,34 and accordingly presents a reasonable explanation for the reoxidation-driven LRS retention failure.
In fact, oxygen exchange with the environment assuming permeability of Pt to oxygen species has also been suggested as a resistive switching mechanism of Pt/Nb:SrTiO3 devices as well as other valence change memories.9,10,35–38 In this scenario, positive voltages applied to the top electrode attract the negatively charged oxygen-ions and induce the oxygen excorporation reaction. Negative voltages, on the other hand, attract oxygen vacancies, which can be annihilated at the Nb:SrTiO3 surface through oxygen incorporation, similar to the reoxidation reaction giving rise to the retention failure described above. These processes can therefore account for the observed eight-wise switching polarity, which is difficult to describe in terms of pure oxygen vacancy rearrangement under applied bias. While direct evidence for these processes is challenging to obtain, numerous reports on noble metal/Nb:SrTiO3 junctions and similar systems invoke this scenario as the most likely explanation for the observed switching and retention characteristics.9,10,35–39 For example, Buzio et al. presented oxygen excorporation and incorporation as the only reasonable explanation for the observed pO2-dependent switching characteristics in Au/Nb:SrTiO3 junctions.9 Gao et al. even presented direct evidence from transmission electron microscopy that resistive switching can be driven by the excorporation and incorporation of oxygen from the crystal lattice through the Au top electrodes.38
While direct evidence for the exact mechanism is still absent, we assume that oxygen exchange with the environment through the permeable noble metal top electrode is the most likely process underlying the valence change in these devices. For both spatially homogeneous switching and filamentary switching, we observe the same switching polarity in line with oxygen excorporation leading to the LRS, while reincorporation of oxygen under an applied negative bias RESETs the device. Similarly, reoxidation over time leads to a resistance increase – i.e. LRS retention failure – for both spatial extends. While switching polarity and the shape of the I–V curves remain similar, a substantial difference observed in these two cases is that the off/on ratio for devices operated under filamentary switching mechanism is several orders of magnitude larger. This can be understood as the filamentary switching mechanism which leads to considerable Joule heating (higher currents confined to a much smaller area), enabling thermally assisted ion migration. On the one hand, this process can lead to the observed drastic change in resistance due to the excorporation of a large amount of oxygen, as is obvious from the Ti3+ filament presented here. On the other hand, Joule heating can be so extreme that phase separations occur. The difference between spatially homogeneous switching and filamentary switching, accordingly, does not lie in the fundamental switching mechanism but in the presence or absence of Joule heating, which can assist in the field-driven ion migration and therefore lead to a more pronounced resistance change.
4. Conclusion
In conclusion, our experimental data verify that nanoscale redox reactions are responsible for the resistance change in noble metal/Nb:SrTiO3 junctions. Analysis of the retention behavior of Pt/Nb:SrTiO3 junctions indicates that reoxidation of previously oxygen-deficient areas is responsible for the resistance change over time for devices operated at all current limits investigated here. The switching mechanism for these different current limits, however, can be differentiated between spatially homogeneous switching for low current limits and filamentary switching with high off/on ratios for higher current limits. More importantly, we were able to detect direct spectromicroscopic evidence that the resistance change for high current limits is caused by a valence change within a spatially confined filament. This valence change, in turn, is caused by nanoscale redox reactions. Evaluation of the local chemistry within the switching filament allows for the prediction of retention characteristics and reveals a similar design rule as recently demonstrated for SrTiO3 devices: incorporation of thin layers which impede reoxidation extends the retention times of valence change memories dramatically.5. Experimental
5.1. Device fabrication
Noble metal/Nb:SrTiO3 junctions were fabricated through electron beam deposition or sputter-deposition of 30 nm Pt or Au and subsequent structuring via dry etching in a Reactive Ion Beam Etching tool (Ionfab 300plus, Oxford Instruments). For the nanostructures described in section 2.1, a sputter-deposited Pt layer was structured by UV nanoimprint lithography employing a custom made quartz mold.40 The imprint process is performed using a Nanonex2000 imprint system with an aircushion technique. Details regarding the imprint process can be found in ref. 41.For the microstructures described in sections 2.2 and 2.3, an electron-beam evaporated Pt layer was structured with regular photolithography. The devices were brought into contact either directly with a tungsten tip or with a Pt lead connecting the device and the tungsten tip.
For the microstructures used for spectromicroscopic analysis in sections 2.2 and 2.3, a sputter-deposited Au layer was structured with regular photolithography. The devices were brought into contact directly with a tungsten tip.
5.2 Top electrode delamination
For spectromicroscopic analysis, the top electrodes were delaminated under ultra-high vacuum conditions with an adhesive copper tape after sputter deposition of a homogeneous 30 nm Au layer onto the entire sample. The delaminated sample was transferred into the XPEEM chamber under ultra-high vacuum conditions.5.3 Spectromicroscopy
All XPEEM experiments have been performed at the NanoESCA beamline at Elettra synchrotron laboratory (Trieste, Italy) using the endstation described elsewhere.42 The total energy resolution determined from both the spectrometer broadening (the pass energy 50 eV and the entrance slit 1 mm) and the photon bandwidth was 200 meV for photoemission spectra. Various series of images were taken at increasing electron kinetic energies with a step size of 0.1 eV; the photon energy for Sr 3d XPS was 200 eV. Core level photoemission spectra were recorded from the resulting image stack in different regions of interest. Similarly, image stacks with increasing photon energy were recorded for absorption spectra, using a step size of 0.05 eV for the Ti L-edge and 0.2 eV for the O K-edge. XAS spectra were acquired in partial electron yield (PEY) collecting only electrons close to the secondary electron cutoff at an energy of E − EF = ϕ + Ekin = 4.475 eV. The image stacks were analyzed and spectra were recorded using the IGOR Pro software.5.4 Atomic force microscopy
All atomic force microscopy experiments were performed using a Jeol 4213 AFM (Akishima, Japan) or a Cypher AFM by Asylum Research (Santa Barbara, USA) in contact mode. The substrate was brought into contact with the ground sample holder using silver paste, while the voltage was applied via a Pt/Ir-coated cantilever.Author contribution
C. B., T. M. and R. D. conceived and designed the experiments. C. B., N. R., T. M., R. R. and P. M. fabricated the samples. C. B., N. R., T. M., P. M., C. S., and V. F. performed the experiments. C. B., N. R., T. M., P. M. and M. A. analyzed the retention data. C. B. analyzed the XPEEM data. C. B. and N. R. wrote the manuscript. R. B., F. G., C. M. S., R. W. and R. D. supervised the research. All authors discussed the results and commented on the manuscript.Acknowledgements
Funding from the DFG (German Science Foundation) within the collaborative research center SFB 917 ‘Nanoswitches’ is gratefully acknowledged. C. B. and R. D. also acknowledge funding from the W2/W3 program of the Helmholtz association.Notes and references
- R. Waser, R. Dittmann, G. Staikov and K. Szot, Adv. Mater., 2009, 21, 2632 CrossRef CAS.
- A. Sawa, Mater. Today, 2008, 11, 28 CrossRef CAS.
- H.-S. P. Wong, H.-Y. Lee, S. Yu, Y.-S. Chen, Y. Wu, P.-S. Chen, B. Lee, F. T. Chen and M.-J. Tsai, Proc. IEEE, 2012, 100, 1951 CrossRef CAS.
- S. Menzel, M. Salinga, U. Böttger and M. Wimmer, Adv. Funct. Mater., 2015, 25, 6306–6325 CrossRef CAS.
- E. Lee, M. Gwon, D. W. Kim and H. Kim, Appl. Phys. Lett., 2011, 98, 132905 CrossRef.
- C. Park, Y. Seo, J. Jung and D. Kim, J. Appl. Phys., 2008, 103, 54106 CrossRef.
- D. S. Shang, L. D. Chen, Q. Wang, W. D. Yu, X. M. Li, J. R. Sun and B. G. Shen, J. Appl. Phys., 2009, 105, 63511 CrossRef.
- A. Ohtomo and H. Y. Hwang, Appl. Phys. Lett., 2004, 84, 1716 CrossRef CAS.
- R. Buzio, A. Gerbi, A. Gadaleta, L. Anghinolfi, F. Bisio, E. Bellingeri, A. S. Siri and D. Marrè, Appl. Phys. Lett., 2012, 101, 243505 CrossRef.
- M. Yang, L. Z. Ren, Y. J. Wang, F. M. Yu, M. Meng, W. Q. Zhou, S. X. Wu and S. W. Li, J. Appl. Phys., 2014, 115, 134505 CrossRef.
- J. Park, D.-H. Kwon, H. Park, C. U. Jung and M. Kim, Appl. Phys. Lett., 2014, 105, 183103 CrossRef.
- S. Lee, J. S. Lee, J.-B. Park, Y. K. Kyoung, M.-J. Lee and T. W. Noh, APMIS, 2014, 2, 066103 Search PubMed.
- C. Rodenbücher, W. Speier, G. Bihlmayer, U. Breuer, R. Waser and K. Szot, New J. Phys., 2013, 15, 103017 CrossRef.
- A. Marchewka, D. Cooper, C. Lenser, S. Menzel, H. Du, R. Dittmann, R. E. Dunin-Borkowski and R. Waser, Sci. Rep., 2014, 4, 6975 CrossRef CAS PubMed.
- D. J. Seong, M. Jo, D. Lee and H. Hwang, Electrochem. Solid-State Lett., 2007, 10, H168 CrossRef CAS.
- Y. H. Wang, K. H. Zhao, X. L. Shi, G. L. Xie, S. Y. Huang and L. W. Zhang, Appl. Phys. Lett., 2013, 103, 031601 CrossRef.
- R. Muenstermann, T. Menke, R. Dittmann and R. Waser, Adv. Mater., 2010, 22, 4819 CrossRef CAS PubMed.
- M. Gwon, E. Lee, A. Sohn, E. M. Bourim and D. W. Kim, J. Korean Phys. Soc., 2010, 57, 1432 CrossRef CAS.
- S. Ho Jeon, B. Ho Park, J. Lee, B. Lee and S. Han, Appl. Phys. Lett., 2006, 89, 42904 CrossRef.
- R. Schafranek, S. Payan, M. Maglione and A. Klein, Phys. Rev. B: Condens. Matter, 2008, 77, 195310 CrossRef.
- E. Mikheev, B. D. Hoskins, D. B. Strukov and S. Stemmer, Nat. Commun., 2014, 5, 3990 CAS.
- A. Sawa, T. Fujii, M. Kawasaki and Y. Tokura, Appl. Phys. Lett., 2005, 86, 112508 CrossRef.
- T. Fujii, M. Kawasaki, A. Sawa, Y. Kawazoe, H. Akoh and Y. Tokura, Phys. Rev. B: Condens. Matter, 2007, 75, 165101 CrossRef.
- X. Sun, G. Li, L. Chen, Z. Shi and W. Zhang, Nanoscale Res. Lett., 2011, 6, 599 CrossRef PubMed.
- C. Lenser, M. Patt, S. Menzel, A. Köhl, C. Wiemann, C. M. Schneider, R. Waser and R. Dittmann, Adv. Funct. Mater., 2014, 24, 4466 CrossRef CAS.
- M. Abbate, F. M. F. de Groot, J. C. Fuggle, A. Fujimori, Y. Tokura, Y. Fujishima, O. Strebel, M. Domke, G. Kaindl, J. van Elp, B. T. Thole, G. A. Sawatzky, M. Sacchi and N. Tsuda, Phys. Rev. B: Condens. Matter, 1991, 44, 5419 CrossRef CAS.
- C. Baeumer, C. Schmitz, A. H. H. Ramadan, H. Du, K. Skaja, V. Feyer, P. Muller, B. Arndt, C. Jia, J. Mayer, R. A. De Souza, C. Michael Schneider, R. Waser and R. Dittmann, Nat. Commun., 2015, 6, 8610 CrossRef CAS PubMed.
- B. Gao, S. Yu, N. Xu, L. F. Liu, B. Sun, X. Y. Liu, R. Q. Han, J. F. Kang, B. Yu and Y. Y. Wang, IEEE International Electron Devices Meeting 2008, 2008, 563.
- M. Andrä, F. Gunkel, C. Baeumer, C. Xu, R. Dittmann and R. Waser, Nanoscale, 2015, 7, 14351 RSC.
- R. Merkle and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 3874 CrossRef CAS PubMed.
- A. Heuer, J. Eur. Ceram. Soc., 2008, 28, 1495 CrossRef CAS.
- S. Menzel, M. Waters, A. Marchewka, U. Böttger, R. Dittmann and R. Waser, Adv. Funct. Mater., 2011, 21, 4487 CrossRef CAS.
- R. Schmiedl, V. Demuth, P. Lahnor, H. Godehardt, Y. Bodschwinna, C. Harder, L. Hammer, H. P. Strunk, M. Schulz and K. Heinz, Appl. Phys. A, 1996, 62, 223 CrossRef.
- A. Grill, W. Kane, J. Viggiano, M. Brady and R. Laibowitz, J. Mater. Res., 1992, 7, 3260 CrossRef CAS.
- R. Ortega-Hernandez, M. Coll, J. Gonzalez-Rosillo, A. Palau, X. Obradors, E. Miranda, T. Puig and J. Sune, Microelectron. Eng., 2015, 147, 37 CrossRef CAS.
- R. Yang, K. Terabe, T. Tsuruoka, T. Hasegawa and M. Aono, Appl. Phys. Lett., 2012, 100, 231603 CrossRef.
- L. Goux, P. Czarnecki, Y. Y. Chen, L. Pantisano, X. P. Wang, R. Degraeve, B. Govoreanu, M. Jurczak, D. J. Wouters and L. Altimime, Appl. Phys. Lett., 2010, 97, 243509 CrossRef.
- P. Gao, Z. Wang, W. Fu, Z. Liao, K. Liu, W. Wang, X. Bai and E. Wang, Micron, 2010, 41, 301 CrossRef CAS PubMed.
- T. Menke, R. Dittmann, P. Meuffels, K. Szot and R. Waser, J. Appl. Phys., 2009, 106, 114507 CrossRef.
- R. Rosezin, M. Meier, S. Trellenkamp, C. Kügeler and R. Waser, Microelectron. Eng., 2010, 87, 1531 CrossRef CAS.
- M. Meier, S. Gilles, R. Rosezin, C. Schindler, S. Trellenkamp, A. Ruediger, D. Mayer, C. Kuegeler and R. Waser, Microelectron. Eng., 2009, 86, 1060 CrossRef CAS.
- C. Wiemann, M. Patt, I. P. Krug, N. B. Weber, M. Escher, M. Merkel and C. M. Schneider, e-J. Surf. Sci. Nanotechnol., 2011, 9, 395 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr00824k |
| This journal is © The Royal Society of Chemistry 2016 |