
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical sensing in two dimensional porous covalent organic nanosheets†
Gobinda
Das‡
a,
Bishnu P.
Biswal‡
ad,
Sharath
Kandambeth‡
ad,
V.
Venkatesh
b,
Gagandeep
Kaur
b,
Matthew
Addicoat
c,
Thomas
Heine
c,
Sandeep
Verma
b and
Rahul
Banerjee
*ad
aPhysical and Materials Chemistry Division, CSIR-National Chemical Laboratory, Dr HomiBhabha Road, Pune-411008, India. E-mail: r.banerjee@ncl.res.in; Fax: +91-20-25902636; Tel: +91-20-25902535
bDST-Thematic Unit of Excellence on Soft Nanofabrication, Indian Institute of Technology Kanpur, Kanpur-208016, UP, India
cCentre for Functional Nanomaterials, Engineering and Science, Jacobs University Bremen, Research III, Room 61, Campus Ring 1, 28759 Bremen, Germany
dAcademy of Scientific and Innovative Research (AcSIR), New Delhi, India
First published on 29th April 2015
Abstract
Two new imide-based crystalline, porous, and chemically stable covalent organic frameworks (COFs) (TpBDH and TfpBDH) have been successfully synthesized employing solvothermal crystallization route. Furthermore, thin layered covalent organic nanosheets (CONs) were derived from these bulk COFs by the simple liquid phase exfoliation method. These 2D CONs showcase increased luminescence intensity compared to their bulk counterparts (COFs). Notably, TfpBDH-CONs showcase good selectivity and prominent, direct visual detection towards different nitroaromatic analytes over TpBDH-CONs. Quite interestingly, TfpBDH-CONs exhibit a superior “turn-on” detection capability for 2,4,6-trinitrophenol (TNP) in the solid state, but conversely, they also show a “turn-off” detection in the dispersion state. These findings describe a new approach towards developing an efficient, promising fluorescence chemosensor material for both visual and spectroscopic detection of nitroaromatic compounds with very low [10−5 (M)] analyte concentrations.
Introduction
Chemical sensing using porous materials, especially Metal–Organic Frameworks (MOFs)1 and Porous Organic Frameworks (POFs)2 has picked up considerable attention recently. An important feature of these frameworks is their flexible and adjustable pores in which guest molecules can freely interact with the pore walls.2e–g However, the sensitivity and selectivity of detection in these materials is generally not sufficient, either due to the poor analyte–host interactions inside the amorphous porous organic frameworks, or due to the chemical instability of the crystalline metal–organic frameworks. Hence, a chemically stable, crystalline and luminescent porous material with electronically adjustable π-conjugation is an attractive choice for acting as an efficient chemical sensor.Covalent Organic Frameworks (COFs)3 are one such class of porous materials that, like MOFs, exhibit a well-defined and predictable crystalline network. A large number of COFs have been synthesized over the past few years, but these materials have only rarely been explored as chemosensors3h owing to their chemical instability in aqueous and acidic/basic medium. With this in mind, we have synthesized two imide based COFs, each possessing a two dimensional layered structure, that exhibit porosity, crystallinity and chemical stability. Since imide functionalized linkers show good photophysical properties, one might predict that these COFs would act as efficient chemical sensors.4 However, the bulk COFs exhibit only moderate chemical sensing ability due to the aggregated π-stacked layers, poor electron mobility and ineffective interaction with analytes. In order to address these shortcomings, we exfoliated the aggregated π-stacked COF layers using the Liquid Phase Exfoliation (LPE) method5a–c to produce Covalent Organic Nanosheets (CONs). Since π–π interactions between the stacked layers are considerably weakened in these two dimensional covalent organic nanosheets (2D CONs), we anticipated that these CONs might show superior chemical sensing capabilities compared to the bulk COF. There have been a few reports on the growth of thin COF layers on 2D surafaces,5i but very little effort has been made to isolate such 2D materials in bulk quantities and to investigate their usefulness. In this paper, we show that these exfoliated CONs exhibit highly selective and visual detection of 2,4,6-trinitrophenol (TNP), over other nitroaromatic analytes such as 2,4,6-trinitrotoluene (TNT), 2,6-dinitrophenol (DNP), 2,6-dinitrotoluene (DNT), and 2-nitrophenol (NP), via a “turn-on” mechanism in the solid state and a “turn-off” fluorescence quenching in the dispersed state. Furthermore, the sensitivity of the CONs towards nitroaromatic analytes detection is ∼10 fold (turn-on) and 63% (turn-off) higher with respect to the bulk COF, even at a very low [10−5 (M)] analytes concentration.
Experimental section
General procedure for the synthesis of TpBDH, TpBDH-CONs, TfpBDH and TfpBDH-CONs
The COFs reported in this paper [TpBDH and TfpBDH] were synthesized using a standard solvothermal protocol reported previously.6 The detailed synthesis of materials and experimental methods can be found in Section S1 and S2, ESI.† In brief, 1,3,5-triformylphloroglucinol (Tp) (0.3 mmol, for TpBDH) or 1,3,5-tris(4-formylphenyl)benzene (Tfp) (for TfpBDH) and pyromellitic-N,N′-bisaminoimide (BDH) (0.45 mmol) can be allowed to react solvothermally in the presence of 1,4-dioxane, N,N′-dimethylacetamide (DMA) and 6 M acetic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:0.3) in a pyrex tube at 120 °C for 3 days. After Soxhlet purification, these COFs were dried at 180 °C under vacuum for 12 hours to give a deep red/gray colored powder in ∼80% isolated yield. To produce TpBDH and TfpBDH CONs, 50 mg of as-synthesized COFs was placed in 50 mL of isopropyl alcohol (IPA) and sonicated at room temperature for 45–60 minutes. The resulting suspension was centrifuged at 1000 rpm for 5 minutes. After complete evaporation of solvent, the residue of the material transferred from the settled solids to the solution as a result of sonication was obtained as CONs in ∼4 wt% isolated yield.
:2:0.3) in a pyrex tube at 120 °C for 3 days. After Soxhlet purification, these COFs were dried at 180 °C under vacuum for 12 hours to give a deep red/gray colored powder in ∼80% isolated yield. To produce TpBDH and TfpBDH CONs, 50 mg of as-synthesized COFs was placed in 50 mL of isopropyl alcohol (IPA) and sonicated at room temperature for 45–60 minutes. The resulting suspension was centrifuged at 1000 rpm for 5 minutes. After complete evaporation of solvent, the residue of the material transferred from the settled solids to the solution as a result of sonication was obtained as CONs in ∼4 wt% isolated yield.
Description of turn off and turn on sensing experiments
Different concentrations, [0 to 5.4 × 10−5 (M)] of analyte (TNP, TNT, DNP, DNT and NP) were added to a homogeneous dispersion of CONs (1 mg in 3 mL of IPA). The fluorescence intensity of the CONs decreased with the increasing concentration of TNP; however, a slight decrease was observed for the other analytes. Secondly, the turn on sensing ability of these isolated CONs was ascertained by filter paper strip assay experiments.7 Test strips were prepared by drop casting the homogeneous suspension of CONs in IPA (1 mg in 5 mL) on a strip of filter paper (2 cm × 2 cm), then dried under vacuum at room temperature to obtain the fluorescent paper sensor. Upon addition of 200 μL [1 mg in 100 mL, 5.4 × 10−5 (M)] of TNP in IPA onto the test strip, the color of the strip immediately changed from blue to yellow (under 365 nm UV lamp), which can also be visually observed with the naked eye. The same experiment was performed with the other nitroaromatic compounds; TNT, DNP, DNT and NP, but unlike TNP, they did not show any pronounced sensing response.Results and discussion
The PXRD patterns of TpBDH and TfpBDH showed intense peaks at 2θ ∼3.3°and ∼3.8°, which correspond to the reflection from (100) planes (Fig. 1b and c). In addition to that, TpBDH exhibited two minor peaks at 2θ ∼5.8° and ∼8.7°; which could be ascribed to (110) and (200) plane reflections respectively. A broad diffuse peak appears at 2θ ∼27.3° for TpBDH and between 2θ ∼15.3° to 27° for TfpBDH. This broad peak is mainly due to the reflection from the (001) plane. In order to elucidate the probable structures of these imide based COFs, all possible 2D eclipsed (AA) and staggered (AB and ABC) models were built using AuToGraFS and optimized by the SCC-DFTB method.8 The best fitting of the simulated and experimental PXRD determined the probable structure of these imide based COFs. The two COFs adopt a P1 space group with unit cell parameters, a = 44.00 Å, b = 44.02 Å, c = 6.6472 Å (for TfpBDH) and a = 29.9 Å, b = 27.9 Å, c = 3.3 Å (for TpBDH) (Section S3, ESI†). The π–π stacking distance between two successive COF layers was found to be ∼3.4 Å and ∼3.3 Å respectively for TpBDH and TfpBDH. The experimental PXRD pattern of the TpBDH is in good agreement with the simulated pattern from the eclipsed (AA) stacking model. However, TfpBDH showed an unusually poor PXRD pattern unlike other hexagonal COFs reported so far.6 All possible models related to stacking arrangements were built for TfpBDH COF to establish the stable structure via rigorous structural modelling and Pawley refinement on the AA, AB and ABC orientations, correlating the experimental and calculated PXRD profiles (Fig. 1 and 2). As a result, we found that the experimental PXRD profile of TfpBDH roughly fits with the simulated ABC stacking mode only and not with the AA and AB or any slipped AA or AB structures. Similarly the refinement values [Rp = 2.57%, Rwp = 3.08% and Rwp (w/o bck) = 3.85%] are found to be minimum for ABC compared to AA or AB model. The results of all refinement and the statistics for the various stacking arrangements are elaboratated given in Section S3, ESI.† On the other hand the pore size distribution peak maxima obtained for TfpBDH (1.6 nm) from the N2 adsorption experiment is close to the theoretical (ABC) pore diameter (∼1.2 nm) and that further support the predicted ABC stacking model rather than AA model (∼3.4 nm) and AB (∼2.0 nm). Although, the first experimental PXRD peak (2θ ∼ 3.8°) of TfpBDH matches well with the simulated ABC stacking mode, but the peaks at 2θ ∼8°, ∼15° and at higher angles (27.5–32.5°) are unidentified and appear as a broad hump. From the theoretical calculations it was found that AA is the most thermally stable form based on stacking energy (Table S4, ESI†). However, the pore size given by the N2 adsorption experiment better supports assignment of the ABC structural model. Notably, owing to the very poor crystallinity of TfpBDH, our prediction of ABC model for TfpBDH is speculative and therefore it can also be termed as a two dimensional porous polymer. Needless to mention we have tried different solvent combinations to synthesize TfpBDH but even after several attempts we were unable to improve the crystallinity (Fig. S3, ESI†).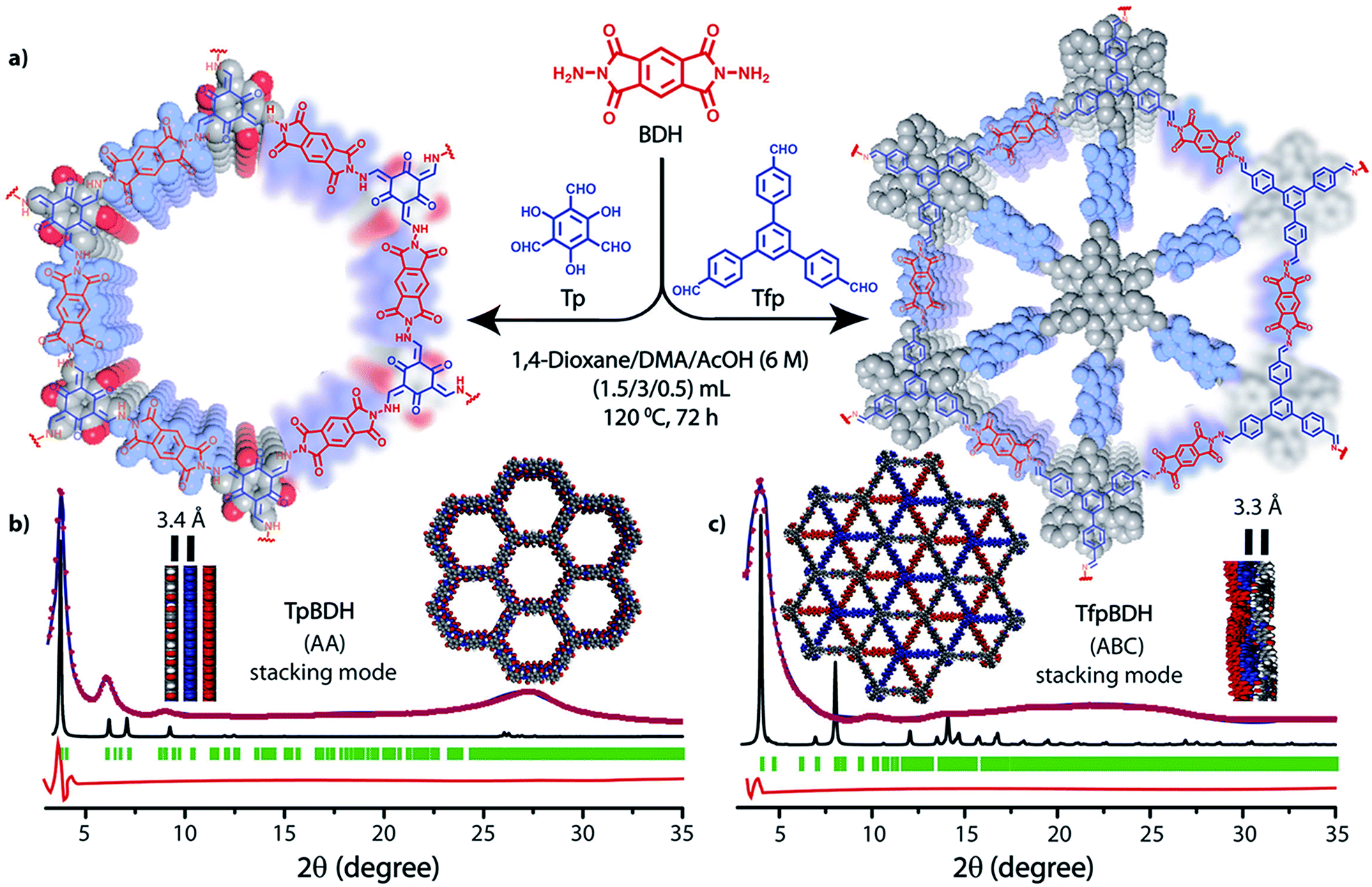 | ||
| Fig. 1 (a) Schematic representation for the synthesis of TpBDH and TfpBDH [one unit of a space filling model of TpBDH and TfpBDH is shown in the inset of the chemical drawing model]; (b and c) The experimental PXRD patterns (blue) compared with simulated (eclipsed; black) and Pawley refined difference between experimental and calculated data (red) [Rp = 1.94%, Rwp = 4.6% and Rwp (w/o bck) = 4.33% for TpBDH and Rp = 2.57%, Rwp = 3.08% and Rwp (w/o bck) = 3.85% for TfpBDH]; for TpBDH and TfpBDH respectively [inset models showing the eclipsed AA and staggered ABC stacking of consecutive 2D layers of TpBDH and TfpBDH6c respectively]. | ||
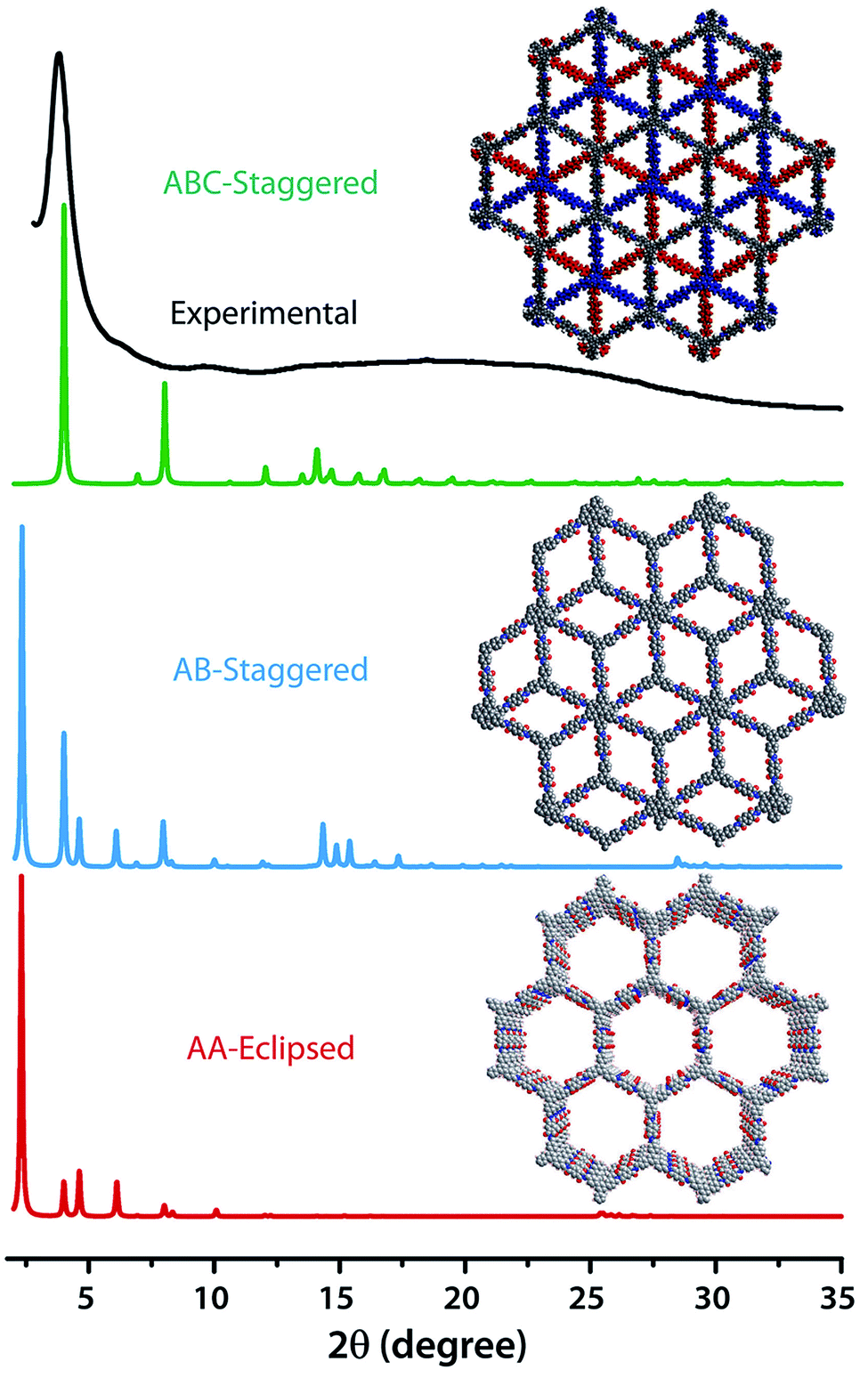 | ||
| Fig. 2 The comparison of PXRD patterns between possible AA, AB and ABC staking models of TfpBDH.6c | ||
The FT-IR spectra of TpBDH and TfpBDH indicate the absence of the –NH stretching band of BDH at ∼3339 cm−1, the carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching band of Tp/Tfp at 1639 cm−1 and at 1689 cm−1 respectively. This indicates the complete consumption of the starting materials, as well as the formation of new bonds (Section S5, ESI†). A strong peak for –CC at ∼1583 cm−1 for TpBDH appears as a result of enol to keto tautomerism. This peak is merged with the –CO stretching frequency (∼1609 cm−1) of the keto group present in the framework. The appearance of two peaks at ∼1445 and ∼1256 cm−1 correspond to the aromatic –CC and –C–N bond in the keto form of TpBDH. However, the band at ∼1604 cm−1 for the FT-IR spectra of TfpBDH is due to the presence of the –CN bond, which further confirms the covalent bond formation between the aldehyde group of Tfp and the amino group of BDH. As evidenced from FT-IR spectroscopy (Fig. S4 and S4a, ESI†), TpBDH exists in the keto-enamine form resembling our previously reported COFs6 where as TfpBDH exists purely in the imine form.
O) stretching band of Tp/Tfp at 1639 cm−1 and at 1689 cm−1 respectively. This indicates the complete consumption of the starting materials, as well as the formation of new bonds (Section S5, ESI†). A strong peak for –CC at ∼1583 cm−1 for TpBDH appears as a result of enol to keto tautomerism. This peak is merged with the –CO stretching frequency (∼1609 cm−1) of the keto group present in the framework. The appearance of two peaks at ∼1445 and ∼1256 cm−1 correspond to the aromatic –CC and –C–N bond in the keto form of TpBDH. However, the band at ∼1604 cm−1 for the FT-IR spectra of TfpBDH is due to the presence of the –CN bond, which further confirms the covalent bond formation between the aldehyde group of Tfp and the amino group of BDH. As evidenced from FT-IR spectroscopy (Fig. S4 and S4a, ESI†), TpBDH exists in the keto-enamine form resembling our previously reported COFs6 where as TfpBDH exists purely in the imine form.
Further support for the local structure of these imide based COFs is given by 13C CP-MAS solid state NMR spectroscopy. TpBDH showed a clear NMR signal at 182.6 ppm corresponding to the carbonyl (–CO) carbons of the keto form (Fig. S5, ESI†). However, the resonance of carbonyl carbons of the BDH linker unit appears at 163.8 ppm. The peak at 152 ppm confirms the presence of the –C–N bond, instead of the –CN bond (∼165 ppm), which would have been a signature peak if TpBDH had been existing in the enol form. For TfpBDH, the presence of the –CN bond was established by a characteristic resonance signal at 158.7 ppm. As depicted in Fig. S6, ESI;†TfpBDH showed a down field resonance at 162.7 ppm for the carbonyl carbon (–CO) which confirms the presence of the imide (BDH) linker unit. The assignment of the peak patterns of both TpBDH and TfpBDH is consistent with their corresponding monomer compounds made for comparison (Fig. S5 and S6, ESI†).
The external morphology of these imide based COFs has been verified using SEM and TEM techniques. The SEM images of TpBDH indicated that several COF layers agglomerate to construct a flower-like morphology (Fig. S10(a), ESI†), while for TfpBDH, agglomeration of irregular shaped layers with rough surface was observed, (Fig. S10(b), ESI†). We have performed the TGA analysis under the flow of N2 gas to check the weight loss (%) of these COFs at high temperature. As observed, the TpBDH COF retain the plateau up to ∼394 °C, whereas TfpBDH showed similar behavior up to ∼386 °C (Fig. S9, ESI†). N2 adsorption isotherms were collected to determine the architectural rigidity and permanent porosity of these COFs at 77 K. Both TpBDH and TfpBDH showed type II isotherms with reversible adsorption and desorption profiles. The Brunauer–Emmett–Teller (BET) surface areas were found to be 642 and 738 m2 g−1 (Fig. 3c and d) with pore-size distributions peak maxima at 2.4 and 1.6 nm for TpBDH and TfpBDH respectively calculated on the basis of nonlocal density functional theory (NLDFT) method (Fig. S7 and S8, ESI†). Although the pore size distribution peak maxima obtained for TfpBDH (∼1.6 nm) is slightly higher than the theoretical pore size value (1.2 nm), that further support the predicted ABC staking model rather than AA model (∼3.4 nm). The relative PXRD peak intensity and peak positions of TpBDH and TfpBDH remained identical after 7 days of water and acid (3 N HCl) treatment, indicating the high chemical stability of these COFs (Fig. 3a–d). The existence of enol-to-keto tautomerization provides the chemical stability for TpBDH, whereas the chemical stability in TfpBDH is possibly due to the presence of imide functionalities near the imine bonds, which decreases the electrophilicity of –CN bonds through the mesomeric effect.11 The surface area of acid treated TpBDH decreases slightly (642 to 517 m2 g−1), whereas the water treated sample holds the usual surface area (642 to 627 m2 g−1). However, for TfpBDH, the values decrease significantly for both water and acid treated samples [from 738 to 541 m2 g−1 (for water) to 375 m2 g−1 (for acid)]. This could be due to the partial degradation, pore blockage or delamination when exposed to acid (3 N HCl) and water for a longer period (7 days).
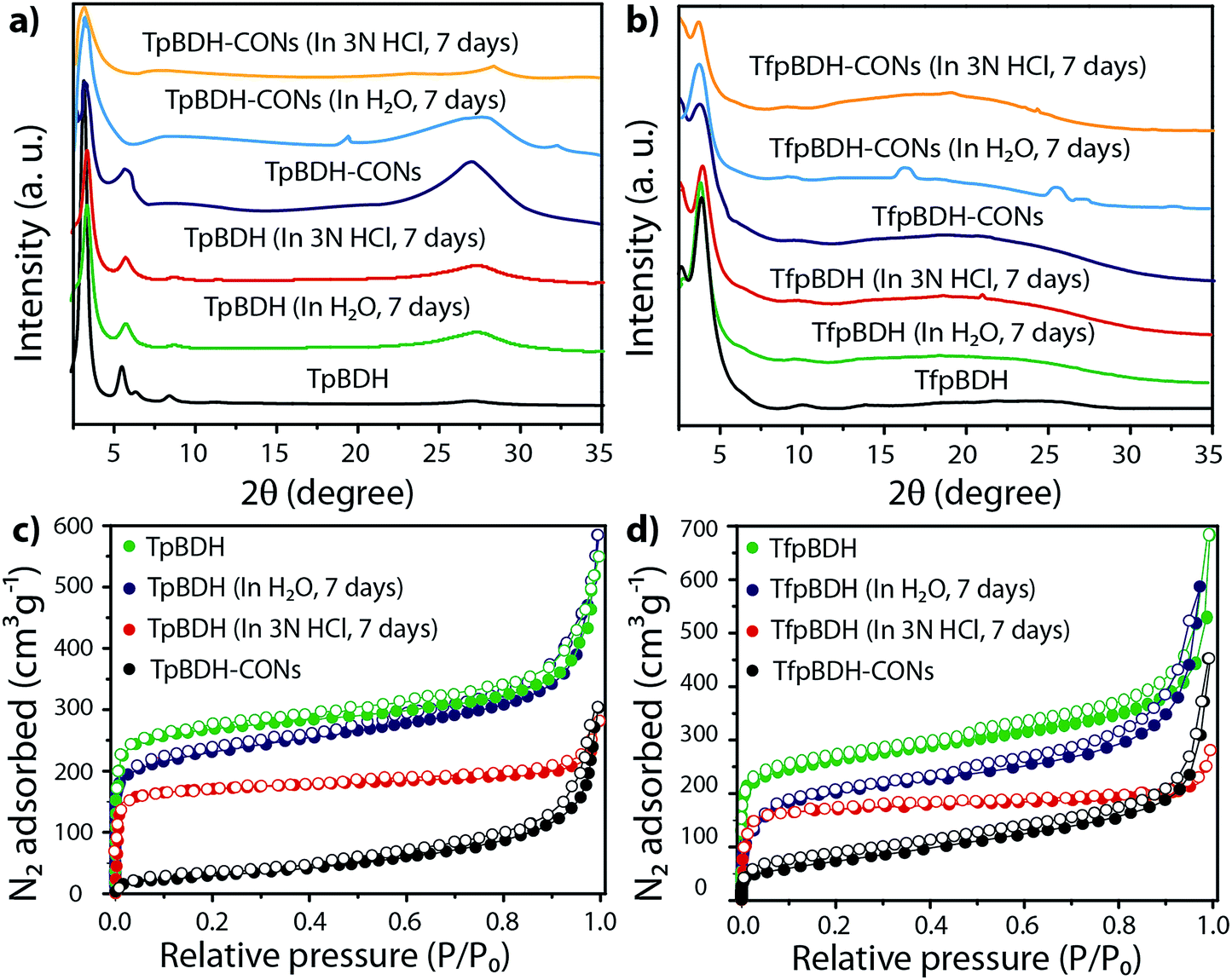 | ||
| Fig. 3 (a and b) PXRD patterns before and after chemical stability tests of TpBDH, TpBDH-CONs; TfpBDH, and TfpBDH-CONs respectively; (c and d) N2 sorption isotherms; shows the chemical stability of TpBDH, TfpBDH in water and 3 N HCl for 7 days. | ||
Since TpBDH and TfpBDH contain electron rich triphenyl/keto (donor) and electron deficient imide (acceptor) moieties, we decided to check their nitroaromatic analyte sensing ability. However, the sensitivity and selectivity was found to be quite low for bulk COFs, due to the extensive aggregation of layers, which reduces the available electrons for analyte-COF interaction. Hence, we decided to exfoliate these COFs into 2D CONs to minimize the aggregation and maximize the availability of electron density among the layers.5 One-step liquid phase exfoliation (LPE) was undertaken in isopropyl alcohol (IPA) at room temperature to synthesize thin 2D CONs (Fig. 4a). HR-TEM and AFM analyses confirm their typical flat thin 2D-nanosheet structures (Fig. 4b–g). Both TpBDH and TfpBDH CONs exhibit wrinkles and folding with well-resolved 0.45 nm periodic lattice fringes (Fig. S13 and S14, ESI†). AFM analysis indicates that the heights of TpBDH-CONs and TfpBDH-CONs are in the range of ∼1.5–5.1 nm, indicating that these CONs are composed of ∼5–15 stacked COF layers (Fig. 4f, g and S15, ESI†). Furthermore, the PXRD patterns of these CONs are identical to that of the parent COFs with a decrease in intensity of the first peak (100) and a broadening of peak at the wide angles (001) (Fig. 3a and b). Such peak broadening and reduced intensity phenomenon has previously been observed for thin layered COF materials and CONs.5d,6b Further, due to exfoliation, the long-range pore structure of the COFs is disturbed and only shallow pores remain accessible for N2 sorption resulting in only moderate surface area values for these CONs [270 m2 g−1 for TfpBDH and 112 m2 g−1 for TpBDH] (Fig. 3c and d). FT-IR and TGA also confirms the retention of structural integrity and thermal behaviour (TpBDH-CONs and TfpBDH-CONs are stable above 380 °C) of these CONs similar to their bulk counterparts (Fig. S9, ESI†). As anticipated, these CONs were found to be highly stable in water, acid (3 N HCl) and even in various organic solvents for 7 days (Fig. 3, S17 and S18 ESI†).
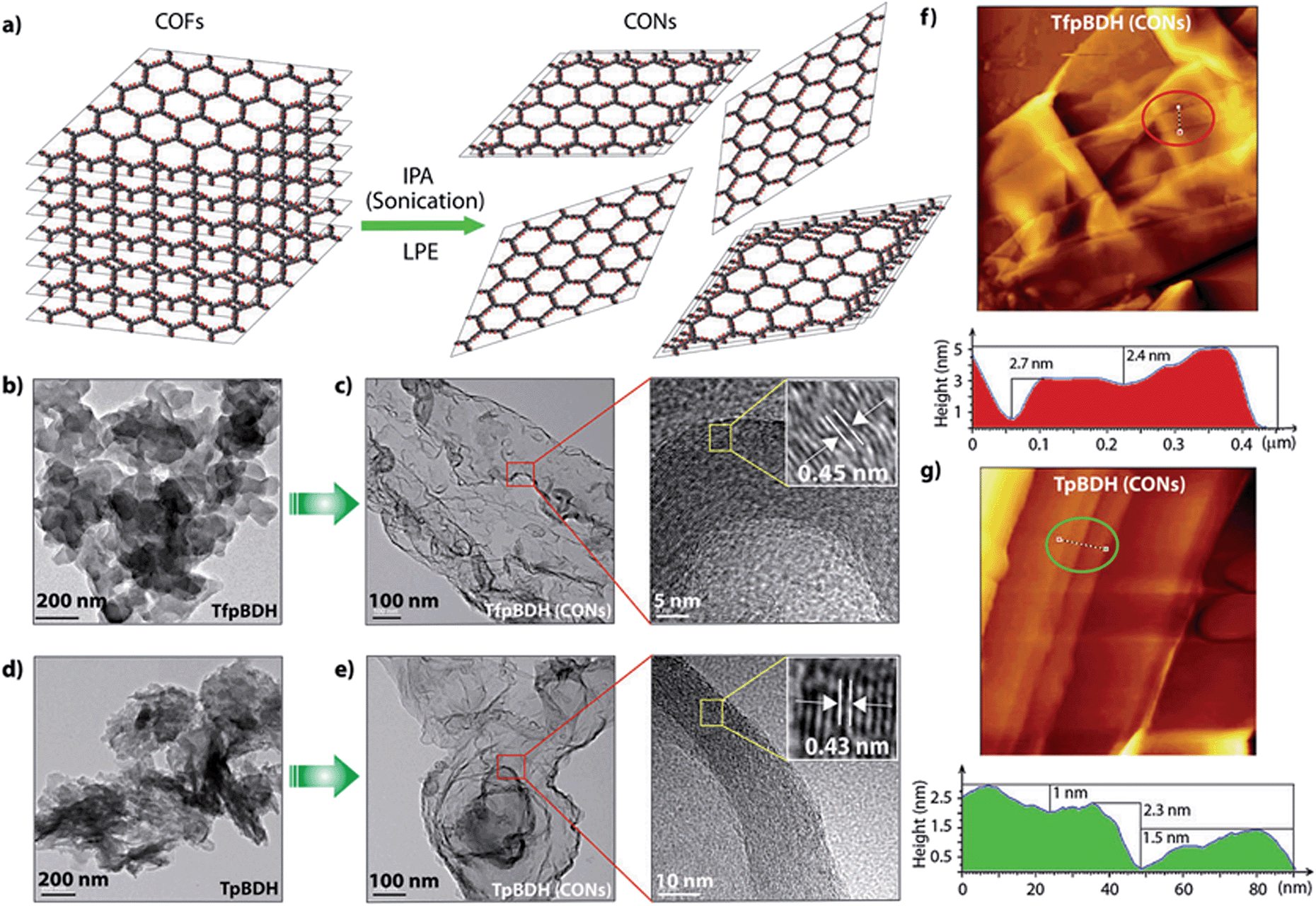 | ||
| Fig. 4 (a) Schematic representation of COFs to CONs formation; HR-TEM images of (b and d) TfpBDH and TpBDH; and (c and e) TfpBDH-CONs and TpBDH-CONs; (f and g) AFM images of TfpBDH-CONs and TpBDH-CONs with corresponding height profiles (the height profiles, colour-filled red and green, are measured along the corresponding tracks inside the circle shown in the AFM images). | ||
We examined the photoluminescence (PL) spectra of these COFs and CONs reported in this paper with different nitro aromatic analytes i.e. TNP, TNT, DNP, DNT and NP. Surprisingly, TpBDH and TpBDH-CONs do not showed any PL activity, as in both TpBDH and TpBDH-CONs there exist enol–keto tautomerization6 which leads to the disturbance in π-conjugation and “switches off” the fluorescence.
However, due to the presence of extended π-conjugation and donor–acceptor charge transfer, TfpBDH and TfpBDH-CONs, showcase enhanced luminescence compared to TpBDH and TpBDH-CONs. Further, we found that TfpBDH-CONs exhibit an intense PL peak (∼6–10 times intensity) in both solution as well as solid state compared to the bulk COFs (Section S13, ESI†). TfpBDH-CONs showed a slight blue shift (∼20 nm) in the PL spectra with reference to the bulk COF samples.9a,b The HOMO–LUMO gap of the exfoliated TfpBDH-CONs (ΔE = 1.71 eV, calculated from the UV-visible spectra) is found to be larger than that of bulk COF samples (ΔE = 1.14 eV), which clearly justifies the blue shift in the emission maxima (Fig. S22, ESI†). Further, these nanosheets facilitate efficient electron transfer between the HOMO and LUMO energy levels, thus TfpBDH-CONs exhibit enhanced luminescence with respect to the bulk COF. Considering these features, we selected TfpBDH-CONs only to study in detail of sensing behavior of the following different nitroaromatic analytes: TNP, TNT, DNP, DNT and NP. Solid TfpBDH-CONs were suspended in IPA. Upon excitation at 365 nm, this suspension displayed emission maxima at ∼447 nm which originates from the aromatic triphenyl chromophore units and a weak shoulder at ∼541 nm due to the charge transfer from the aromatic chromophore to the bisimide fragments. Luminescence spectrometric titration experiments were performed with above mentioned nitroaromatics (TNP, TNT, DNP, DNT and NP) at 5.4 × 10−5 (M) concentration in IPA. Fluorescence emission intensity was significantly quenched upon addition of increasing amounts of analyte solution. It was found that TfpBDH-CONs are highly sensitive and exhibit maximum ca. 63% quenching efficiency towards TNP at 5.4 × 10−5 (M) concentration in IPA over other nitroaromatic explosives (TNT: 31%, DNT: 3%, DNP: 23%, and NP: 4%) (Fig. 5b and S20, ESI†). The standard linear curve-fitting in the Stern–Volmer (SV) equation was employed to understand the quenching phenomena (Fig. 5c and S21, ESI†). The quenching constant for TNP was found to be 2.6 × 104 M−1 and are in the order TNP ≫ TNT > DNP > NP > DNT. Although both TNP and TNT have the same standard reduction potential, the quenching efficiency of TNP is ca. 63% higher than that of TNT (31%) in the dispersed state. This result can be attributed to the electronic charge transfer between the picrate anion (TNP−) to the π-electronic cloud of the protonated TfpBDH-CONs. Furthermore, theoretical calculations (Fig. S27, ESI†) indicate that the ground-state electronic charge transfer takes place from the HOMO of picrate anion (TNP−) to the LUMO of the protonated TfpBDH-CONs and as a result, luminescence quenching of TfpBDH-CONs was observed. On the other hand, the HOMO energy level (−7.749 eV) of TNT is much lower than the LUMO energy level (−4.861 eV) of the TfpBDH-CONs. Therefore in this case, the electron transfer is not favorable and no such quenching phenomenon occurs (Fig. S28 and S29, ESI†). The photo-induced energy transfer from TNP− to TfpBDH-CONs was further supported by fluorescence decay studies. Fluorescence decay was collected at 448 nm with an excitation wavelength of 365 nm in IPA at 25 °C. The fluorescence decay profile of TfpBDH-CONs (1 mg in 5 mL of IPA) at λex = 390 nm exhibited biexponential decay with a lifetime of 0.75 ns, which reduced further to 0.35 ns in the presence of TNP (5.4 × 10−5 M). Reduction in the average lifetime upon addition of TNP indicates efficient energy transfer. The lifetime reduced sharply with increasing TNP concentration, which clearly suggests that the quenching is dynamic in nature (Fig. 5d). Further, we have investigated the change in thicknesses of CONs with respect to sonication time (samples collected at 0, 20, 30 and 60 minutes respectively). As expected, we found that the thickness of these CONs decreases gradually with aforementioned sonication time and monitored via AFM imaging with corresponding height profiles at (Fig. S16, ESI†). We have also attempted to understand the change in florescence intensity of these CONs recovered with different sonication time intervals (0, 20, 30 and 60 minutes).
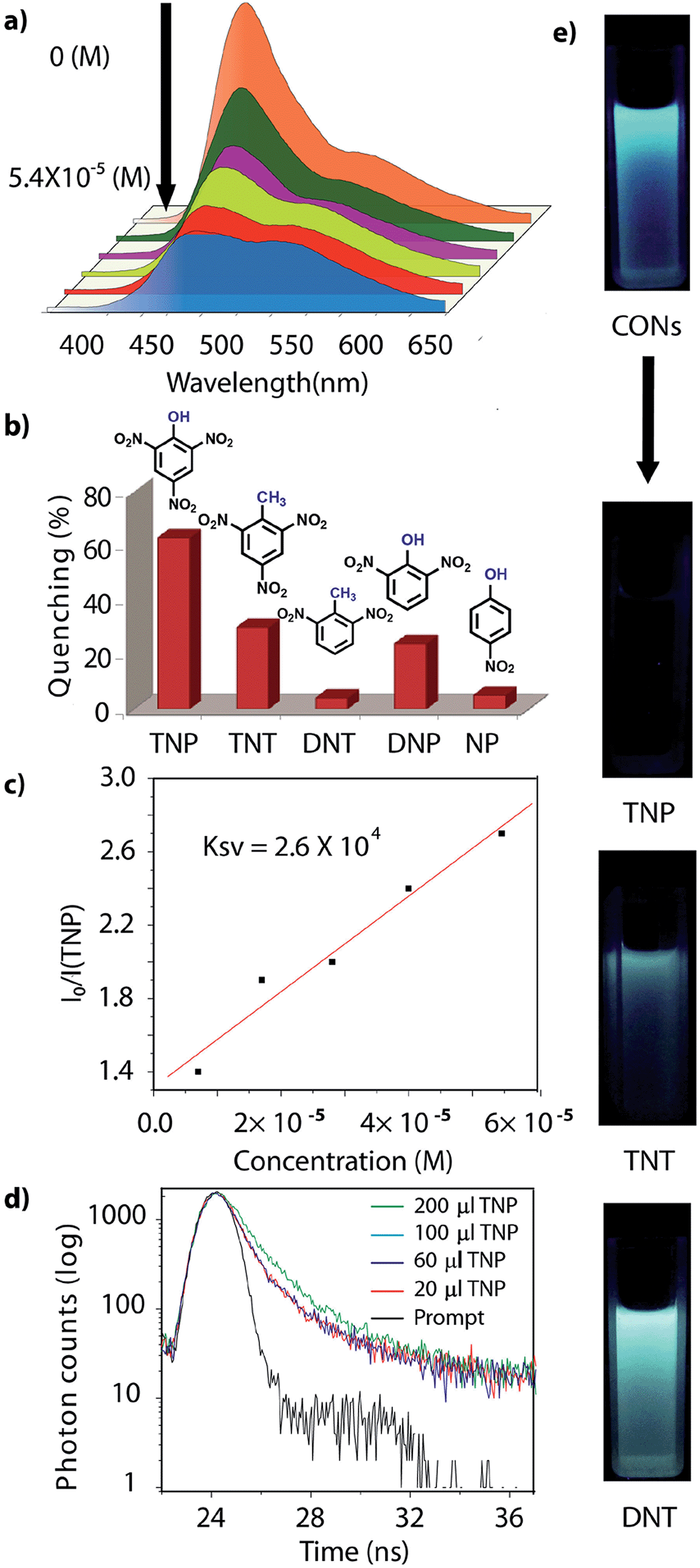 | ||
| Fig. 5 (a) Turn off sensing (PL quenching); (b) degree of fluorescence quenching of TfpBDH-CONs with analytes; (c) Stern–Volmer plots for the quenching of CONs by TNP; (d) fluorescence decay profile (λex = 365 nm and λem = 447 nm) of TfpBDH-CONs, in the presence of different analytes; (e) digital photographs of the turn off sensing of analytes under UV light (λex = 365 nm). | ||
It was observed that with increase in the sonication time, CONs exhibits a stronger PL peak almost ∼90 times intense compared to that of bulk COF samples (Fig. S24a, ESI†). Similarly, a notable effect was also seen in the UV excitation wavelength, gradual and obvious blue shift in excitation wavelength which varies significantly with CONs recovered at higher sonication time (60 minutes) that could be due to the decrease of stacking layers (Fig. S24b, ESI†). Further, curiously we have also monitored the effect of size using dynamic light scattering (DLS) experiment by collecting CONs samples at different time intervals (0, 20, 30 and 60 minutes respectively). It has been found from the DLS experiment that the average size of the CONs decreases as the sonication time increases (Fig. S31 and S32, ESI†). Such behavior has already been observed for other reported nanosheets.5g,h
Besides solution phase detection, solid state luminescence experiments were also performed with TfpBDH-CONs in the presence of the same nitroaromatic analytes mentioned above. For this purpose, CONs-coated paper strips were used, which served as a simple, effective, fast, and low-cost support for detection of nitroaromatic analytes. Solid TfpBDH-CONs powder (10 mg in 50 mL of IPA) was deposited on the paper strip and displayed weak blue fluorescence under an UV lamp at an excitation wavelength of 365 nm. When only 200 μL [1 × 10−3 (M)] of TNP solution in IPA was deposited on the paper strip surface, a distinct colour change from blue to bright-yellow was immediately observed (Fig. 6c). The quantitative fluorescence spectral changes of the TfpBDH-CONs were also monitored in the solid state, by varying the TNP concentrations in IPA (0 to 1 × 10−3 M). Interestingly, the PL emission maxima in solid state (Fig. 6b) get red shifted and the intensity is enhanced (10 times higher than that of the pristine TfpBDH-CONs), with increasing TNP concentration (Fig. S23, ESI†). However, other nitroaromatic analytes did not show any significant effects on TfpBDH-CONs in the solid state.10a This “turn on” sensing behaviour in the solid state could be due to the proton transfer from TNP to the basic nitrogen atom of the imine (–CN) bond. We speculate that the protonation occurs predominantly on the surface of the CONs layers as these TfpBDH-CONs are being aggregated in the solid state. In order to understand the interaction between the acidic phenolic (–OH) groups of TNP with the imine bonds of TfpBDH-CONs and to check the reversibility of chemosensing in the solid state, we performed a simple experiment using the concept of acid–base interaction. In the presence of amine (TEA) vapour, the yellow colored paper strip of TfpBDH-CONs returns back to its initial gray colour within a few seconds (Fig. 6d and e). Such reversible protonation–deprotonation of the –CN bond of the imine linkage with the phenolic –OH group is known for other imine bonded macrocyclic and polyazomethine compounds.10a The above observations suggest that the CONs based chemosensor is highly selective, sensitive and reversible towards TNP over other explosives of the same family.10b
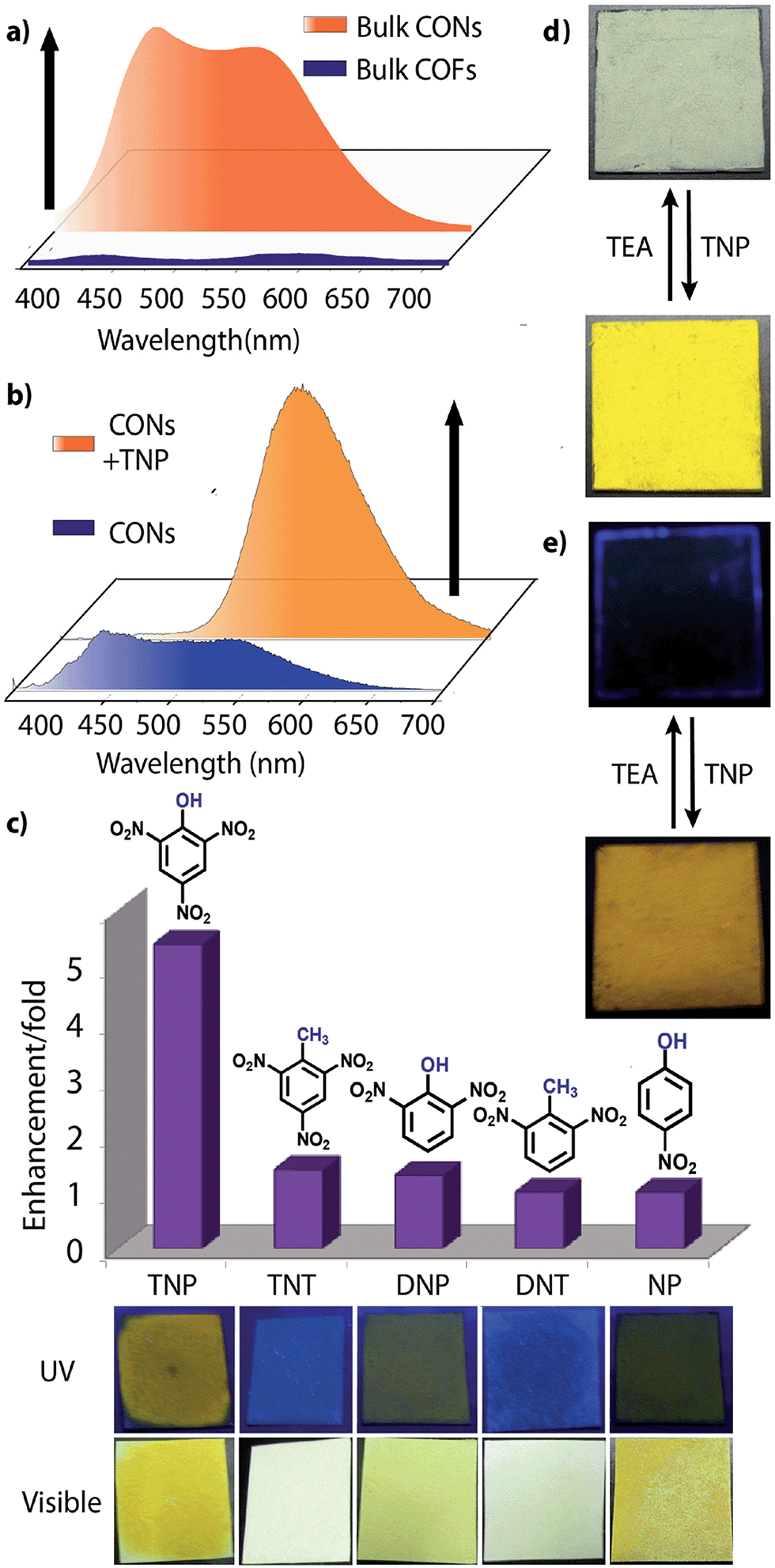 | ||
| Fig. 6 (a) Photoluminescent spectra of bulk TfpBDH and TfpBDH-CONs; (b) photoluminescent spectra showing turn on (PL enhancement) of bulk TfpBDH-CONs and with TNP; (c) photoluminescence enhancement plots of different analytes [at 5.4 × 10−3 (M)] with TfpBDH-CONs; (d and e) solid state sensing (turn on) by TfpBDH-CONs in presence of TNP under visible and UV light (365 nm) which also exhibits the reversible color change from blue to yellow and vice versa upon TEA treatment. | ||
Conclusions
In summary, we have successfully synthesized two imide based covalent organic frameworks by using simple Schiff-base reactions. These COFs are crystalline, porous and showed good hydrolytic and acidic stability. Thin layered covalent organic nanosheets were derived from these bulk COF materials via the liquid phase exfoliation method. Quite surprisingly, TfpBDH-CONs showed superior luminescent property under UV light, whereas the TpBDH-CONs are almost non-emissive. TfpBDH-CONs exhibit chemical sensing of nitroaromatic analytes, showing highly selective luminescence in both “turn on” (∼10 fold) and “turn off” (63%) modes. In the bulk aggregate state, the TfpBDH-CONs interact with the TNP molecule and show reversible “turn-on” sensing maximum lower concentration up to [1 × 10−3 (M)], which is also visually detectable by the naked eye. Once dispersed, TfpBDH-CONs showed luminescent quenching behavior under the influence of the same analytes. To the best of our knowledge, this is the first report where 2D-CONs have been engaged for fast and highly selective detection of nitroaromatic analytes via both turn-on/off sensing mechanisms. Although the quick and selective detection of TNT is highly desirable than any other nitroaromatics but we believe that the methodology presented in this work will open up new opportunities for the design and synthesis of two dimensional analytical platforms for the detection of highly explosive compounds like TNT in near future.Acknowledgements
B. P. B and S. K acknowledge UGC and CSIR, New Delhi, India, for fellowship. G. D and R. B acknowledge CSIR's Five Year Plan (CSC0122 and CSC0102) and DST-fast Track Scheme (SB/FT/CS-120/2013, GAP-306126) for funding. Financial assistance from DST (SB/S1/IC-32/2013) is also acknowledged. We acknowledge Dr T. G. Ajithkumar and Mr Srikant Kunjir for providing NMR, Dr C. Ramesh and Mr Yogesh Marathe for PXRD and Dr Amitava Das, Dr S. K. Asha, Ms Prajitha K. and Mr Hridesh Agarwal for UV-PL spectroscopy facility.Notes and references
- (a) H. Hayashi, A. P. Côté, H. Furukawa, M. O'keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501 CrossRef CAS PubMed; (b) Y. Hwang, H. Sohn, A. Phan, O. M. Yaghi and R. N. Candler, Nano Lett., 2013, 13, 5271 CrossRef CAS PubMed; (c) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (d) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153 CrossRef CAS PubMed; (e) D. Ma, B. Li, X. Zhou, Q. Zhou, K. Liu, G. Zeng, G. Li, Z. Shi and S. Feng, Chem. Commun., 2013, 49, 8964 RSC; (f) J.-S. Qin, S.-J. Bao, P. Li, W. Xie, D.-Y. Du, L. Zhao, Y.-Q. Lan and Z.-M. Su, Chem.–Asian J., 2014, 9, 749 CrossRef CAS PubMed; (g) L. V. Meyer, F. Schönfeld and K. M. Buschbaum, Chem. Commun., 2014, 50, 8093 RSC; (h) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC; (i) A. Lan, K. Li, H. Wu, H. D. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334 CrossRef CAS PubMed; (j) Y. Li, S. Zhang and D. Song, Angew. Chem., Int. Ed., 2013, 52, 710 CrossRef CAS PubMed; (k) N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dinca, J. Am. Chem. Soc., 2013, 135, 13326 CrossRef CAS PubMed.
- (a) J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423 CrossRef CAS; (b) J. Liu, K.-K. Yee, K. K.-W. Lo, K. Y. Zhang, W.-P. To and C.-M. Che, J. Am. Chem. Soc., 2014, 136, 2818 CrossRef CAS PubMed; (c) D. Gopalakrishnan and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 8357 CrossRef CAS PubMed; (d) Y. Yuan, R. Hao, F. Sun, X. Jing, K. Cai, X. Zhao, Y. Wang, Y. Wei and G. Zhu, J. Mater. Chem., 2012, 22, 24558 RSC; (e) E. L. Spitler, B. T. Koo, J. L. Novotney, J. W. Colson, F. J. Uribe-Romo, G. D. Gutierrez, P. Clancy and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 19416 CrossRef CAS PubMed; (f) J. F. Eubank, L. Wojtas, M. R. Hight, T. Bousquet, V. C. Kravtsov and M. Eddaoudi, J. Am. Chem. Soc., 2011, 133, 17532 CrossRef CAS PubMed; (g) O. K. Farha, A. M. Shultz, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 5652 CrossRef CAS PubMed.
- (a) A. P. Côté, A. I. Benin, N. W. Ockwig, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed; (b) C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235 CrossRef CAS PubMed; (c) X. Feng, X. S. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC; (d) E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672 CrossRef CAS PubMed; (e) A. P. Cote, A. I. Benin, N. W. Ockwig, M. O. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS PubMed; (f) S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC; (g) H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875 CrossRef CAS PubMed; (h) S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS PubMed; (i) S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, J. Am. Chem. Soc., 2008, 130, 11580 CrossRef CAS PubMed; (j) D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952 CrossRef CAS PubMed; (k) S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Son, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816 CrossRef CAS PubMed; (l) J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS PubMed.
- (a) S. Jin, K. Furukawa, M. Addicoat, L. Chen, T. Seiya, S. Irle, T. Nakamura and D. Jiang, Chem. Sci., 2013, 4, 4505 RSC; (b) S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. E. Khouly, T. Nakamura, S. Irle, S. Fukuzumi, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2013, 52, 2017 CrossRef CAS PubMed.
- (a) D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952 CrossRef CAS PubMed; (b) I. Berlanga, R. M. Balleste and F. Zamora, Chem. Commun., 2012, 48, 7976 RSC; (c) I. Berlanga, M. L. R. González, J. M. G. Calbet, J. L. G. Fierro, R. M. Ballesté and F. Zamora, Small, 2011, 7, 1207 CrossRef CAS PubMed; (d) S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853 CrossRef CAS PubMed; (e) Y. Zhang, M. Tan, H. Li, Y. Zheng, S. Gao, H. Zhang and J. Y. Ying, Chem. Commun., 2011, 47, 7365 RSC; (f) H. Zhu, L. Gao, X. Jiang, R. Liu, Y. Wei, Y. Wang, Y. Zhao, Z. Chai and X. Gao, Chem. Commun., 2014, 50(3), 695 Search PubMed; (g) Y. Qi, N. Wang, Q. Xu, H. Li, P. Zhou, X. Lu and G. Zhao, Chem. Commun., 2015, 51, 6726 RSC; (h) L.-S. Lin, Z.-X. Cong, J. Li, K.-M. Ke, S.-S. Guo, H.-H. Yang and G.-N. Chen, J. Mater. Chem. B, 2014, 2, 1031 RSC; (i) J. W. Colson, A. R. Woll, A. Mukherjee, M. P. Levendorf, E. L. Spitler, V. B. Shields, M. G. Spencer, J. Park and W. R. Dichtel, Science, 2011, 332, 228 CrossRef CAS PubMed.
- (a) S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524 CrossRef CAS PubMed; (b) B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328 CrossRef CAS PubMed; (c) Notably, TfpBDH material can also be called as a 2D porous polymer due to very poor crystallinity. However, based on pore size analysis and rough PXRD fitting we have speculatively predicted the ABC structure.
- (a) K. K. Kartha, S. S. Babu, S. Srinivasan and A. Ajayaghosh, J. Am. Chem. Soc., 2012, 134, 4834 CrossRef CAS PubMed; (b) Y. Ma, H. Li, S. Peng and L. Wang, Anal. Chem., 2012, 84, 8415 CrossRef CAS PubMed; (c) V. Vij, V. Bhalla and M. Kumar, ACS Appl. Mater. Interfaces, 2013, 5, 5373 CrossRef CAS PubMed.
- (a) M. A. Addicoat, D. E. Coupry and T. Heine, J. Phys. Chem. A, 2014, 118, 9607 CrossRef CAS PubMed; (b) B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678 CrossRef CAS PubMed.
- (a) X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18 CrossRef CAS PubMed; (b) S. J. Woltornist, A. J. Oyer, J.-M. Y. Carrillo, A. V. Dobrynin and D. H. Adamson, ACS Nano, 2013, 7, 7062 CrossRef CAS PubMed; (c) F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, J. Am. Chem. Soc., 2011, 133, 11478 CrossRef CAS PubMed; (d) Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630 CrossRef CAS.
- (a) H.-T. Feng and Y.-S. Zheng, Chem.–Eur. J., 2014, 20, 195 CrossRef CAS PubMed; (b) It is important to note that the turn on mechanism showed by TfpBDH-CONs in the solid state is only selective among the nitroaromatic compounds mentioned in this paper. We also observed that TfpBDH-CONs showed similar colour change [“turn on” fluorescence sensing] as that observed for TNP when exposed to trifluoroacetic acid (TFA) and hydrochloric acid (HCl) vapour. This further prove the concept of proton transfer mechanism which is most likely playing the key role for such colour change in TfpBDH-CONs upon expose to acidic environment.
- V. V. Korskak, S. V. Vinogradova, Y. S. Vygodskii, Z. V. Gerashchenko and N. I. Lushkina, Polym. Sci. U.S.S.R., 1972, 14, 2153 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5sc00512d |
| ‡ G.D, B.P.B and S.K contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |