
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitrogen fixation catalyzed by ferrocene-substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes: unique behavior of ferrocene moiety as redox active site†
Shogo
Kuriyama
a,
Kazuya
Arashiba
a,
Kazunari
Nakajima
a,
Hiromasa
Tanaka
b,
Kazunari
Yoshizawa
*bc and
Yoshiaki
Nishibayashi
*a
aInstitute of Engineering Innovation, School of Engineering, The University of Tokyo, Yayoi, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ynishiba@sogo.t.u-tokyo.ac.jp
bElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Nishikyo-ku, Kyoto 615-8520, Japan
cInstitute for Materials Chemistry and Engineering and International Research Center for Molecular System, Kyushu University, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: kazunari@ms.ifoc.kyoshu-u.ac.jp
First published on 20th April 2015
Abstract
A series of dinitrogen-bridged dimolybdenum–dinitrogen complexes bearing metallocene-substituted PNP-pincer ligands is synthesized by the reduction of the corresponding monomeric molybdenum–trichloride complexes under 1 atm of molecular dinitrogen. Introduction of ferrocene as a redox-active moiety to the pyridine ring of the PNP-pincer ligand increases the catalytic activity for the formation of ammonia from molecular dinitrogen, up to 45 equiv. of ammonia being formed based on the catalyst (22 equiv. of ammonia based on each molybdenum atom of the catalyst). The time profile for the catalytic reaction reveals that the presence of the ferrocene unit in the catalyst increases the rate of ammonia formation. Electrochemical measurement and theoretical studies indicate that an interaction between the Fe atom of the ferrocene moiety and the Mo atom in the catalyst may play an important role to achieve a high catalytic activity.
1. Introduction
Activation and transformation of molecular dinitrogen using transition metal–dinitrogen complexes have been extensively studied to achieve novel nitrogen fixation systems under mild reaction conditions.1,2 Although a variety of stoichiometric reactivities of transition metal–dinitrogen complexes have been reported upto now, the catalytic conversion of molecular dinitrogen under ambient reaction conditions using these complexes as catalysts has so far been limited to only a few cases.3 The first successful example of the catalytic transformation of molecular dinitrogen into ammonia under ambient reaction conditions in the presence of a transition metal–dinitrogen complex was reported by Schrock and co-workers in 2003, where a molybdenum–dinitrogen complex bearing a triamidoamine ligand worked as a catalyst to produce 8 equiv. of ammonia based on the catalyst.4 In 2010, we reported another successful example of ammonia formation from molecular dinitrogen by using a dinitrogen-bridged dimolybdenum–dinitrogen complex bearing PNP-pincer ligands [Mo(N2)2(PNP)]2(μ-N2) (1a; PNP = 2,6-bis(di-tert-butylphosphinomethyl)pyridine) (Scheme 1) as a catalyst.5 More recently, Peters and co-workers have found the third successful example by using well-designed iron–dinitrogen complexes as catalysts.6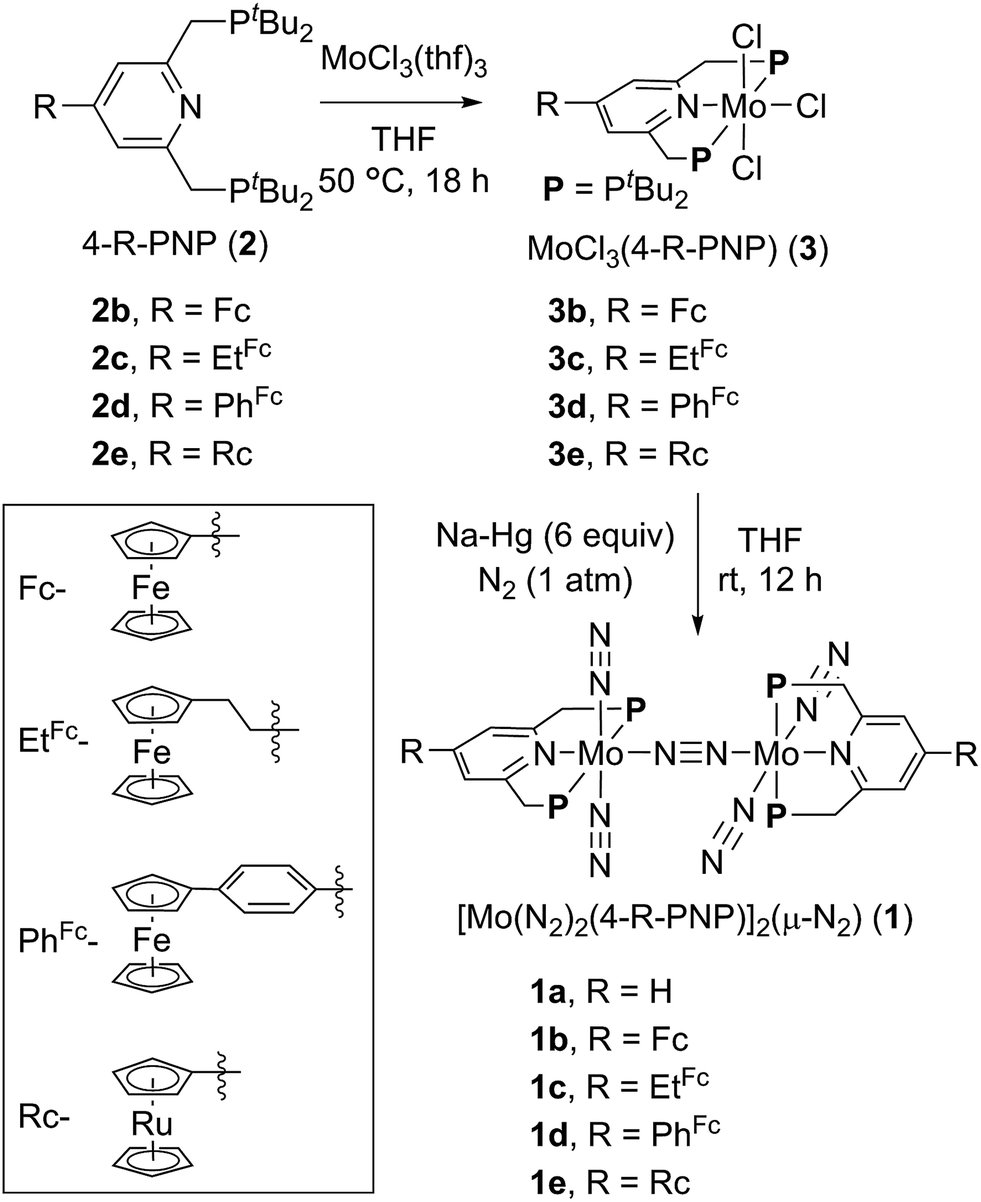 | ||
| Scheme 1 Synthesis of dinitrogen-bridged dimolybdenum–dinitrogen complexes bearing metallocene-substituted PNP-pincer ligands 1. | ||
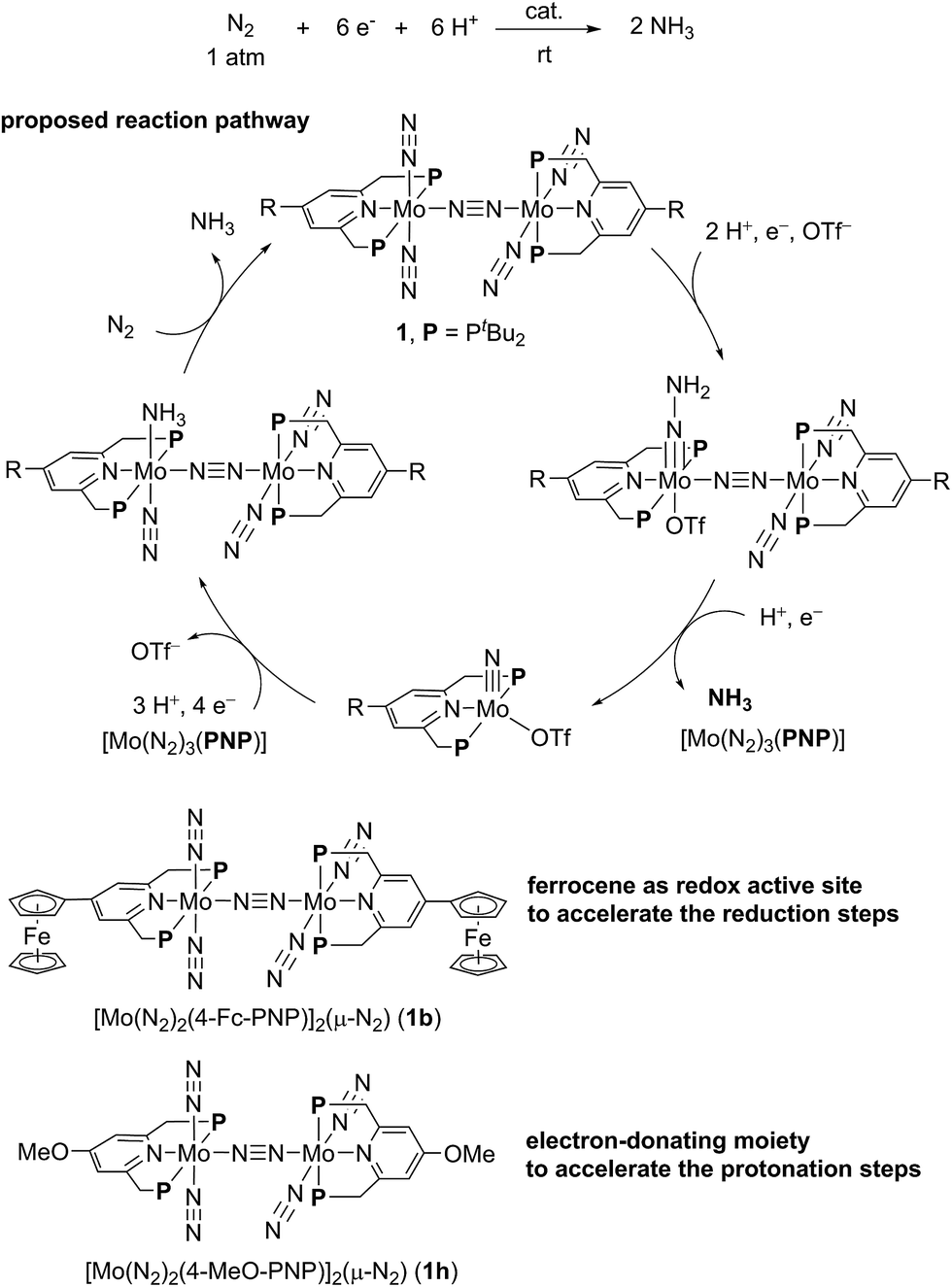
In our reaction system, a theoretical study revealed that a synergistic effect between two Mo moieties of the dinitrogen-bridged dimolybdenum–dinitrogen complex plays an important role to achieve the catalytic activity.5,7,8 An electron transfer from a Mo core to the active site of the other core via the bridging dinitrogen ligand contributes to the protonation steps of the coordinated nitrogenous ligands on the Mo atom. On the basis of the proposed catalytic pathway, we have succeeded in the development of a more effective catalyst by tuning the electronic property of the PNP-pincer ligand, where up to 52 equiv. of ammonia based on the catalyst (26 equiv. based on the Mo atom of the catalyst) were produced by using catalysts bearing an electron-donating group at the 4-position of the pyridine ring of the PNP-pincer ligand.9 Mechanistic studies suggested that the introduction of an electron-donating group to the PNP-pincer ligand enables a facile protonation on the terminal dinitrogen ligand on the Mo atom and also the suppression of the formation of a side product, molecular dihydrogen.9 However, not expectedly, the introduction of an electron-donating group such as methoxy group to the PNP-pincer ligand decreased the rate of ammonia formation probably due to the difficulty in reduction steps involved in the catalytic reaction. These results prompted us to develop a more efficient reaction system, where the reduction steps in the catalytic reaction can also be accelerated by suitably modifying the catalyst.
In biological nitrogen fixation, nitrogenase enzymes which are composed of two proteins named the MoFe protein and Fe protein have been known to catalyze ammonia formation from molecular dinitrogen under ambient reaction conditions (Fig. 1).10 The active site of the nitrogenase,11 known as FeMo cofactor, receives electrons successively from P- and 4Fe4S-clusters, which are located adjacent to FeMo cofactor, to facilitate the multi-electron reduction of molecular dinitrogen into ammonia. By considering the so-far proposed reaction system of nitrogenase, we have envisaged the introduction of a ferrocene unit as a redox-active moiety to the PNP-pincer ligand of the dinitrogen-bridged dimolybdenum–dinitrogen complex, hoping to accelerate the reduction steps involved in the catalytic reaction via an intramolecular electron transfer from the Fe atom of ferrocene to the active site of the Mo atom in the complex.12 In fact, several research groups have already reported enzyme model complexes bearing a ferrocene unit as a redox-active moiety13 because ferrocene is known to be one of the most popular redox-active compounds due to its reversible one-electron redox behavior.14,15 As a typical example, Camara and Rauchfuss reported that a thiolate-bridged diiron complex bearing a ferrocenylphosphine as a redox-active ligand acted as a [FeFe]hydrogenase model complex (Fig. 2), where an intramolecular electron transfer from the Fe atom in the thiolated-bridged diiron core to the Fe atom of the ferrocene unit plays a critical role to promote the catalytic oxidation of molecular dihydrogen under ambient reaction conditions.13a
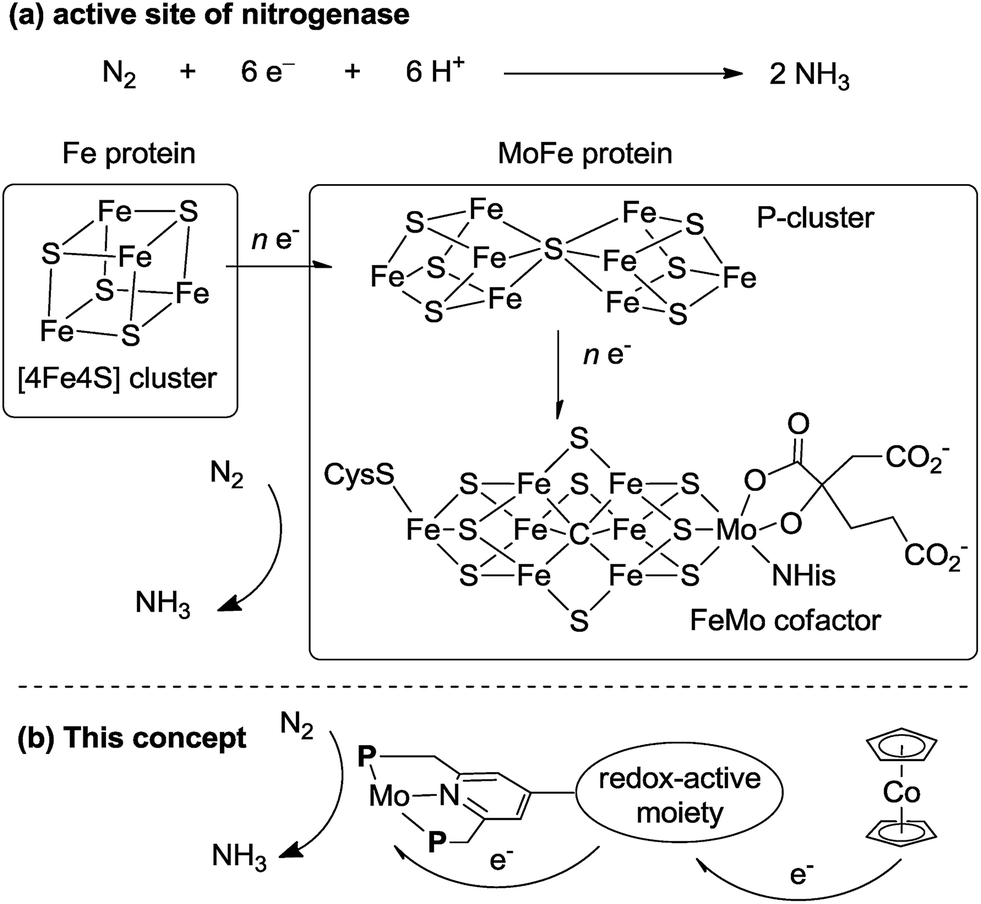 | ||
| Fig. 1 (a) Structure of the 4Fe4S cluster, P-cluster and FeMo cofactor of nitrogenase and an electron flow toward FeMo cofactor. (b) Our concept for a new reaction system by using Mo complexes bearing a redox-active moiety. | ||
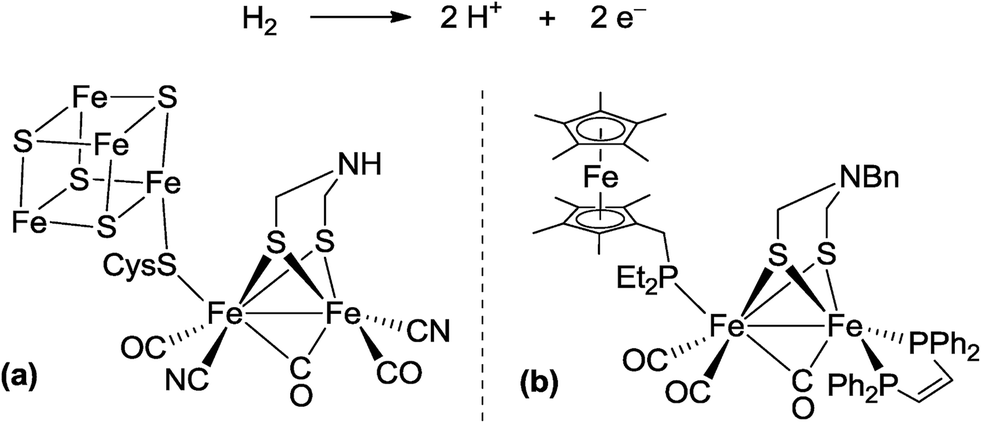 | ||
| Fig. 2 Reaction system of hydrogenase for oxidation of molecular dihydrogen. (a) Active site of [FeFe]hydrogenase. (b) Thiolate-bridged diiron complex bearing a ferrocenylmethylphosphine as a redox-active ligand. | ||
In this article, we describe the preparation and catalytic activity of a series of dinitrogen-bridged dimolybdenum–dinitrogen complexes bearing a metallocene unit such as ferrocene and ruthenocene (1) (Scheme 1). The dinitrogen-bridged dimolybdenum–dinitrogen complexes bearing ferrocene-substituted PNP-pincer ligands have been found to work as efficient catalysts, where up to 45 equiv. of ammonia are produced based on the catalyst (22 equiv. of ammonia based on each Mo atom of the catalyst). The unique behavior of the newly introduced ferrocene unit as a redox-active moiety in the complexes was discussed by means of experimental and theoretical methods.
2. Results and discussion
2.1. Synthesis of molybdenum complexes bearing metallocene-substituted PNP-pincer ligands
A series of metallocene-substituted PNP-pincer ligands 4-R-PNP (2; 4-R-PNP = 4-substituted 2,6-bis(di-tert-butylphosphinomethyl)pyridine) was newly prepared (Scheme 1) and the detailed experimental procedure is described in the ESI.† A variety of substituents such as ferrocenyl (Fc), 2-ferrocenylethyl (EtFc), 4-ferrocenylphenyl (PhFc), and ruthenocenyl (Rc) groups were introduced to the 4-position of pyridine ring of the PNP-pincer ligand. Treatment of [MoCl3(thf)3] (thf = tetrahydrofuran) with 1 equiv. of 4-R-PNP 2 in tetrahydrofuran (THF) at 50 °C for 18 h gave the corresponding molybdenum–trichloride complexes bearing the corresponding PNP-pincer ligands [MoCl3(4-R-PNP)] (3) in high yields. The molecular structures of 3 were confirmed by X-ray crystallography (see the ESI in Fig. S3–S5†).Reduction of 3 with 6 equiv. of Na–Hg in THF at room temperature for 12 h under an atmospheric pressure of molecular dinitrogen gave the corresponding metallocene-substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes [Mo(N2)2(4-R-PNP)]2(μ-N2) (1) in good yields. These new dinitrogen-bridged dimolybdenum–dinitrogen complexes were characterized spectroscopically. Detailed molecular structures of [Mo(N2)2(4-Fc-PNP)]2(μ-N2) (1b) and [Mo(N2)2(4-Rc-PNP)]2(μ-N2) (1e) were determined by X-ray crystallography. ORTEP drawings of 1b and 1e are shown in Fig. 3. No distinct differences of bond distances and angles were observed in these complexes (Table S9†).
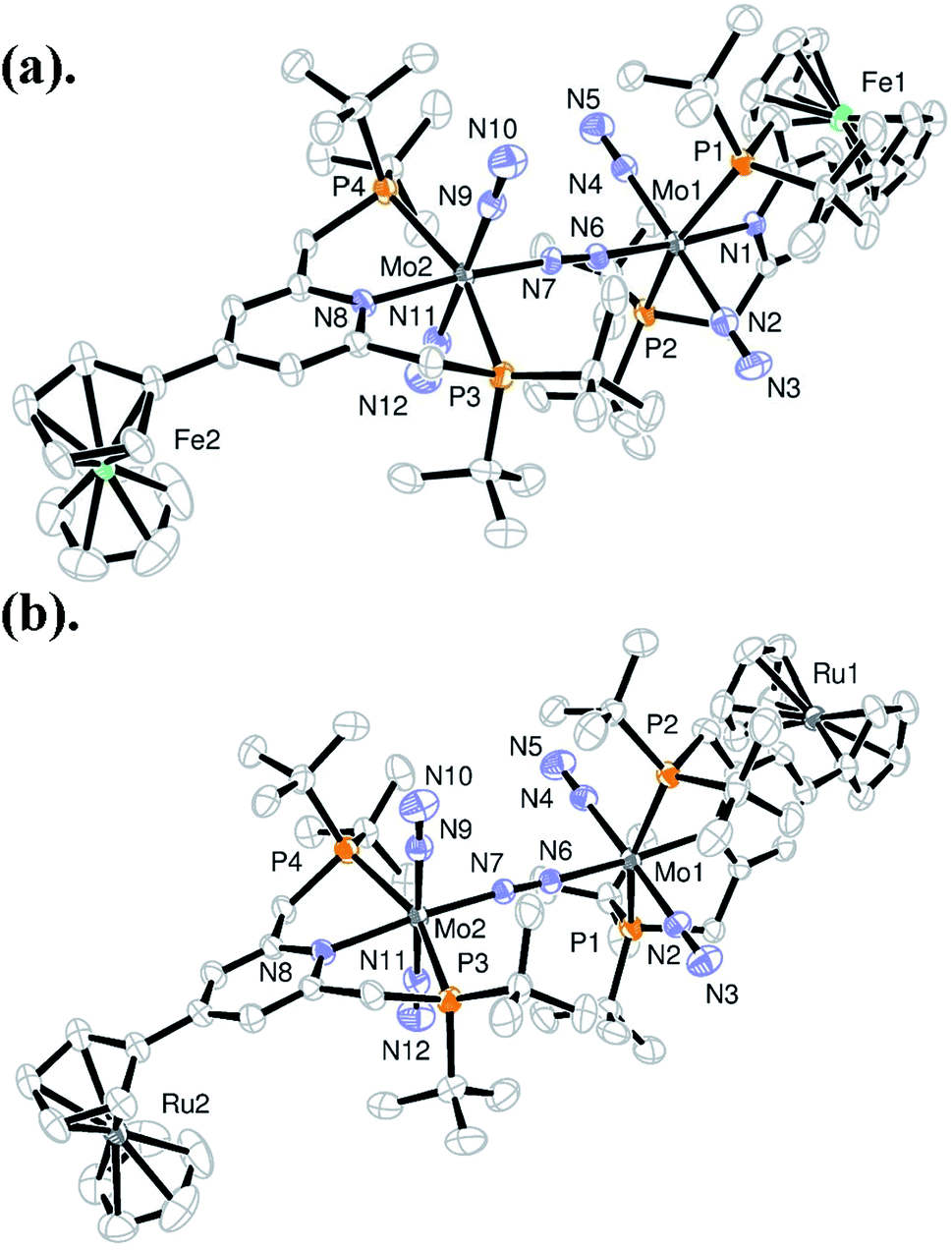 | ||
| Fig. 3 ORTEP drawings of 1b (a) and 1e (b). Hydrogen atoms and solvent molecules are omitted for clarity. | ||
The nature of metallocene as substituent was examined by IR spectroscopy because the electronic nature of substituent at the 4-position in pyridine ring of the PNP pincer-ligand is known to affect absorptions derived from the corresponding terminal dinitrogen ligand.9 Typical results are shown in Table 1. The presence of Fc and Rc groups at the 4-position in pyridine ring of the PNP-pincer ligands in 1 did not change the N≡N stretching frequencies (1944 cm−1 (1b) and 1944 cm−1 (1e), respectively). These results indicate that the introduction of these metallocene moieties to the pyridine ring of the PNP-pincer ligand would not affect the electronic density on the molybdenum atom in the complex 1. On the other hand, the N≡N stretching frequency of [Mo(N2)2(4-EtFc-PNP)]2(μ-N2) (1c) (1939 cm−1) is smaller than that of 1a and the same as that of [Mo(N2)2(4-Me-PNP)]2(μ-N2) (1f, 1939 cm−1).9 This result indicates that the EtFc group has an electron-donating ability as strong as the methyl group in the complex. In contrast, the N≡N frequency of [Mo(N2)2(4-PhFc-PNP)]2(μ-N2) (1d) is larger than that of 1a, and the same as that of [Mo(N2)2(4-Ph-PNP)]2(μ-N2) (1g, 1950 cm−1), indicating that the PhFc group acts as an electron-withdrawing substituent in the complex.
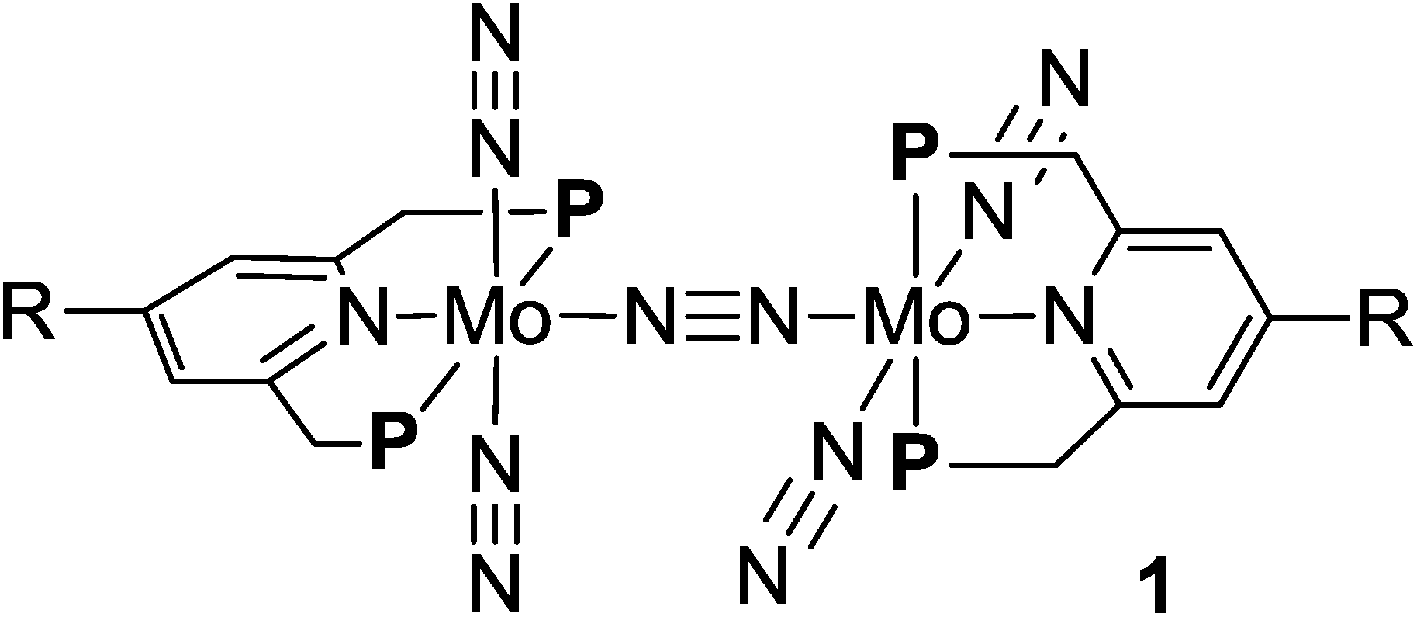
2.2. Electrochemical property of molybdenum complexes bearing metallocene-substituted PNP-pincer ligands
Cyclic voltammograms (CVs) of PNP-pincer ligands 2b–e in THF with [NBu4]BArF4 (ArF = 3,5-(CF3)2C6H3) as supporting electrolyte showed oxidation processes assigned to metallocene moieties. Typical results are shown in Table 2. The PNP-pincer ligand bearing a ferrocene moiety, 2b, showed a reversible one-electron oxidation wave derived from the Fe(II/III) couple at E1/2 = +0.10 V vs. FeCp20/+ (Fig. 4). The PNP-pincer ligands bearing ferrocene units such as EtFc and PhFc groups, 2c and 2d, were oxidized at E1/2 = −0.03 V (2c) and +0.04 V (2d), respectively. On the other hand, the PNP-pincer ligand bearing a Rc moiety, 2e, showed one irreversible oxidation wave at Ea = +0.79 V.16| Compd. | R | Mo(III/IV)/V | Fe(II/III)/V | ΔE(Fe)b/V |
|---|---|---|---|---|
| a Measured by cyclic voltammetry in THF at 0.1 V s−1 with [NBu4]BArF4 as supporting electrolyte. Potentials are indicated as E1/2 values vs. FeCp20/+. b ΔE(Fe) is the difference of the Fe(II/III) potential between 2 and 3. c E pa value of ruthenocene oxidation. d Measured by differential pulse voltammetry (DPV). e Oxidation of ruthenocene was not observed in the electrochemical window. | ||||
| 2b | Fc | — | +0.10 | — |
| 2c | EtFc | — | −0.03 | — |
| 2d | PhFc | — | +0.04 | — |
| 2e | Rc | — | +0.79c | — |
| 3a | H | +0.14 | — | — |
| 3b | Fc | +0.10 | +0.42 | +0.32 |
| 3c | EtFc | +0.05 | +0.19 | +0.22 |
| 3d | PhFc | +0.06d | +0.13d | +0.09 |
| 3e | Rc | +0.09e | —e | — |
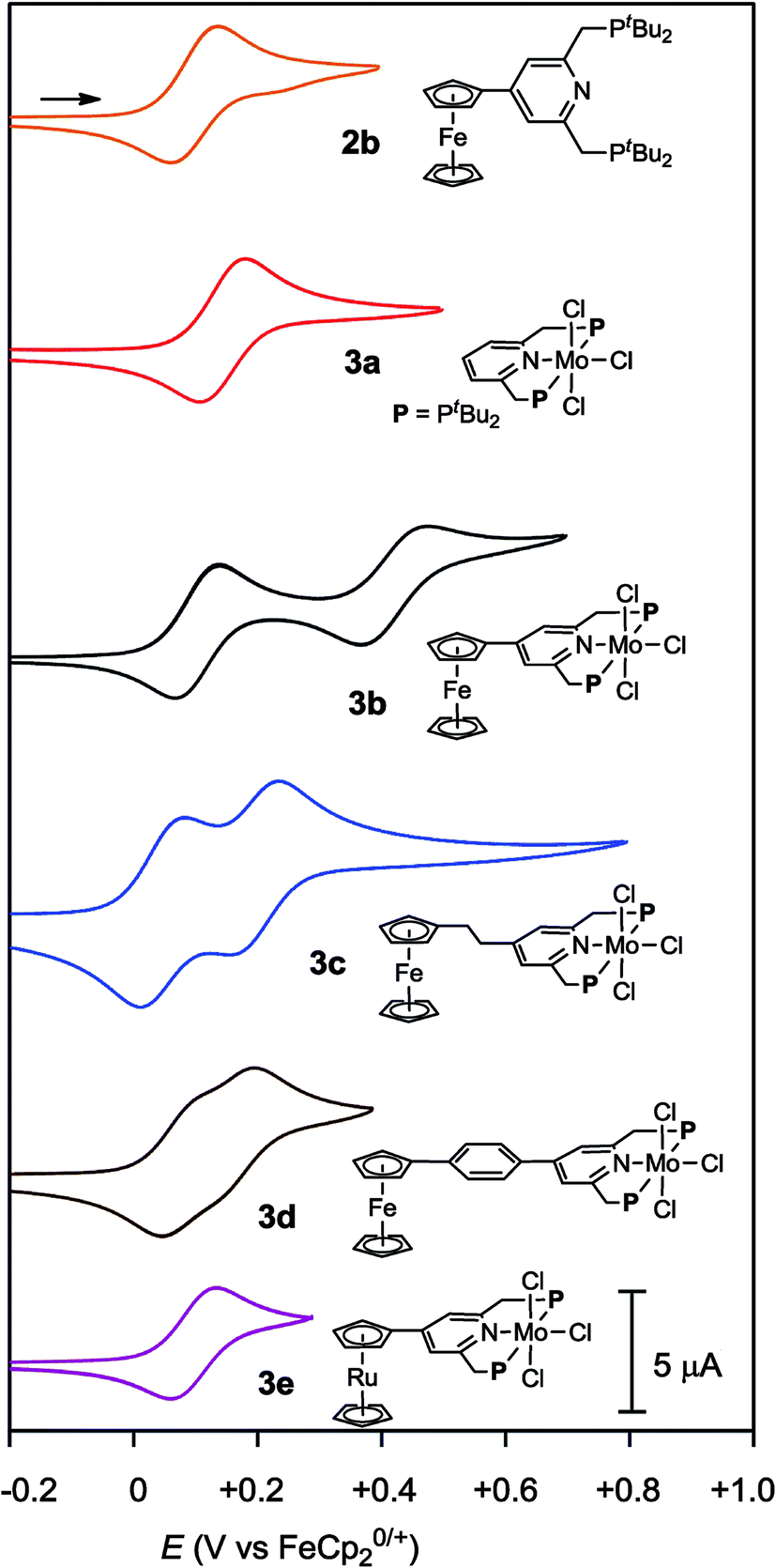 | ||
| Fig. 4 Cyclic voltammograms of 2b and 3a–e. | ||
Electrochemical studies were also carried out on the molybdenum–trichloride complexes bearing a substituted PNP-pincer ligand 3. Typical results are shown in Table 2 and the CVs of 3 are shown in Fig. 4. Non-substituted complex [MoCl3(PNP)] (3a) has one reversible wave at E1/2 = +0.14 V assignable to Mo(III/IV), while the CV of molybdenum–trichloride complex bearing a ferrocene-substituted PNP-pincer ligand, 3b, showed two successive reversible one-electron waves at +0.10 and +0.42 V. We have performed density functional theory (DFT) calculations to assign the observed oxidation waves. The electronic ground state of the cation of 3b ([3b]+) is a triplet state, where the Mulliken spin density is highly localized at the Mo atom (1.85). The removal of an electron from 3b changes the formal charge of the Mo atom from +3 (d3) to +4 (d2), and thus the oxidation at E1/2 = +0.10 V is assignable to Mo(III/IV). On the other hand, the dication of 3b ([3b]2+) adopts a quartet state as the ground spin state, where the spin densities at the Mo and Fe atoms are calculated to be 1.82 and 1.27, respectively. This result suggests that the second oxidation should occur at the Fe atom and the oxidation at E1/2 = +0.42 V is assignable to Fe(II/III). In the molybdenum–trichloride complex 3b, the E1/2 value of Fe(II/III) of the ferrocene moiety shifts by +0.32 V upon coordination. This is evidence of the electronic interaction between the Mo and Fe centers in the complex 3b. In fact, similar shifts of the E1/2 values of Fe(II/III) of the ferrocene moiety were observed in several transition metal complexes bearing 4-ferrocenylpyridine and 4′-ferrocenyl-2,2′:6′,2′′-terpyridine as ligands, indicating an electronic interaction between the metal and Fe centers in these complexes.17,18
The electronic interaction between the Mo and Fe centers in 3b is also supported by the results of spectroelectrochemical measurements. A weak absorption appeared in the near-IR region at around 800–1800 nm when 3b was oxidized to [3b]+ (Fig. S8 and S9†). A time-dependent BP86 calculation of [3b]+ predicted three electron transitions at 3188, 2276 and 1073 nm. Fig. 5 depicts electron density difference maps (EDDMs) for these transitions, where EDDMs present isosurface plots of loss (light green) and gain (purple) of electron density for the corresponding electron transitions. Although the observed absorption of [3b]+ in the near IR region was weak, the calculated EDDMs clearly show that these low-energy electron transitions were assignable to the metal-to-metal charge transfer (MMCT) from Fe(II) of the ferrocene to Mo(IV). The relationship between electronic couplings and intramolecular MMCTs has already been observed in some transition-metal complexes bearing a 4-ferrocenylpyridine scaffold as a ligand.18 In fact, intramolecular MMCTs between the Fe atom in the ferrocene moiety and the Ru atom were observed in ruthenium complexes bearing 4-ferrocenylpyridines18a,b and/or 4′-ferrocenyl-2,2′:6′,2′′-terpyridine as ligands.18c
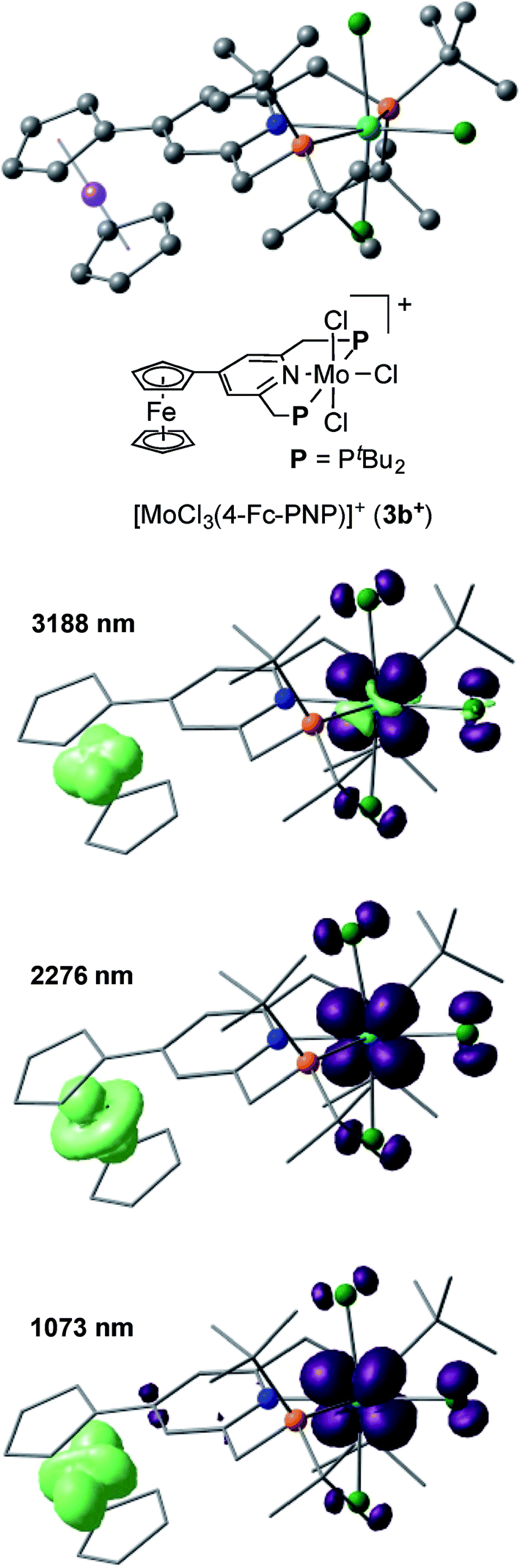 | ||
| Fig. 5 Electron density difference maps (EDDMs) of the electron transitions at 3188, 2276, 1073 nm of [3b]+ calculated with a time-dependent BP86 method. Isosurface plots of loss and gain of electron density are presented in light green and purple, respectively. | ||
The CVs of the molybdenum–trichloride complexes bearing EtFc and PhFc groups, 3c and 3d, showed similar two merged reversible waves. On the basis of the result of the CV of 3b, we assigned the former waves (E1/2: +0.05 and +0.06 V) to Mo(III/IV) and the latter (E1/2: +0.19 and +0.13 V) to Fe(II/III) in the CVs of 3c and 3d (Table 2). In both cases, oxidation of the Mo atom at first may occur in the anodic scan and then oxidation of the Fe atom of the ferrocene unit in the PNP-pincer ligand may proceed sequentially. The E1/2 values of Fe(II/III) of the ferrocene units shift by +0.22 and +0.09 V, respectively, upon coordination. These results indicate that the electronic communication is weakened by the introduction of ethylene or phenylene group into the linkage between the ferrocene and the pyridine of the PNP-pincer ligand. Similar phenomena have already been observed in a rhodium complex bearing 4-(4-ferrocenylphenyl)pyridine as a ligand17c and diruthenium complexes bearing a bridging dipyridine ligand.19 In the latter case, the peak to peak separation in the CVs of the dinuclear ruthenium complexes decreased with the increase of the metal separation of the two Ru centers in the complex.19
The CV of the molybdenum–trichloride complex bearing a Rc-substituted PNP-pincer ligand, 3e, showed only one reversible wave assignable to Mo(III/IV) in the electrochemical window, indicating that the Rc group in 3e did not act as a redox active moiety, in contrast to the ferrocene units in 3b, 3c and 3d (vide supra).
2.3. Evaluation of electronic interaction between Fe and Mo atoms in ammonia formation
The electrochemical and theoretical studies revealed the presence of electronic interaction between the Mo and Fe atoms in the series of molybdenum–trichloride complexes bearing a ferrocene-substituted PNP-pincer ligand. Such interactions could enhance the catalytic ability of the dinitrogen-bridged dimolybdenum complexes via an electron transfer from the Fe atom in the ferrocene moiety to the Mo atom of the complexes bearing nitrogenous ligands that are transformed into ammonia. Among these molybdenum complexes proposed as reactive intermediates in the catalytic cycle in our previous study,7 we have found that a dinitrogen-bridged dimolybdenum–hydrazidium (–NNH3) complex met our hypothesis. In the reaction pathway starting from 1a, dinitrogen-bridged dimolybdenum–hydrazide(2−) complex [Mo(PNP)(OTf)(NNH2)–N≡N–Mo(PNP)(N2)2] (Ia) is protonated to yield the corresponding dinitrogen-bridged dimolybdenum–hydrazidium complex [Mo(PNP)(OTf)(NNH3)–N≡N–Mo(PNP)(N2)2]+ (IIa), which is then reduced with cobaltocene to afford ammonia and the dinitrogen-bridged dimolybdenum–nitride complex [Mo(PNP)(OTf)(N)–N≡N–Mo(PNP)(N2)2] (IIIa) (Scheme 2). The protonation of Ia is slightly exothermic (ΔE = −1.6 kcal mol−1) with very low activation energy of 1.9 kcal mol−1, while the reduction of IIa followed by the N–N bond cleavage is extremely exothermic (ΔE = −156.1 kcal mol−1).7 We have now carried out DFT calculations to examine the impact of the introduction of ferrocene to the pyridine ring of the PNP-pincer ligand. Fig. 6(a) presents an optimized structure of the ferrocene-substituted dinitrogen-bridged dimolybdenum–hydrazidium complex [Mo(4-Fc-PNP)(OTf)(NNH3)–N≡N–Mo(4-Fc-PNP)(N2)2]+ (IIb). For electronic transitions of IIb calculated by the TD-BP86 method (Fig. 6(b)), a low-energy transition at 1174 nm is assignable to the electron transfer from the Fe atom of the ferrocene moiety to the Mo atom. Fig. 6(c) and (d) describe the EDDM of the transition at 1174 nm and molecular orbitals responsible for the transition (HOMO−2 and LUMO). This can be assigned to a MMCT transition from the Fe atom to the Mo atom. It should also be noted that the LUMO of IIb has antibonding character between the two N atoms of the NNH3 ligand. Thus, the N–N bond of the NNH3 ligand in IIb will be more activated, once an electron occupied the LUMO. As presented in our previous report, a neutral species formed by the reduction of the hydrazidium complex, IIa, was easily transformed to a pair of the nitride complex IIIa and ammonia via a spontaneous N–N bond cleavage.7 If the ferrocene-substituted PNP-pincer ligand enables the Mo center of the hydrazidium complex IIb to utilize an intramolecular MMCT, the formation of ammonia would be promoted by the reduction of the Mo–NNH3 moiety. As a result, we expect that the acceleration of the reduction step from the hydrazidium complex to the nitride complex provides an advantage for the catalytic reaction, because the protonation of the hydrazide complex into the hydrazidium complex is an isoenergetic process with a very small activation barrier, and the backward reaction (proton detachment) of the hydrazidium complex into the hydrazide complex is also likely to occur before the reduction of the hydrazidium complex with cobaltocene, producing the nitride complex and ammonia. At present, we can not exclude another possibility such as the acceleration of other steps and the stabilization of some reactive intermediates in the catalytic cycle by the introduction of the ferrocene moiety to the catalyst.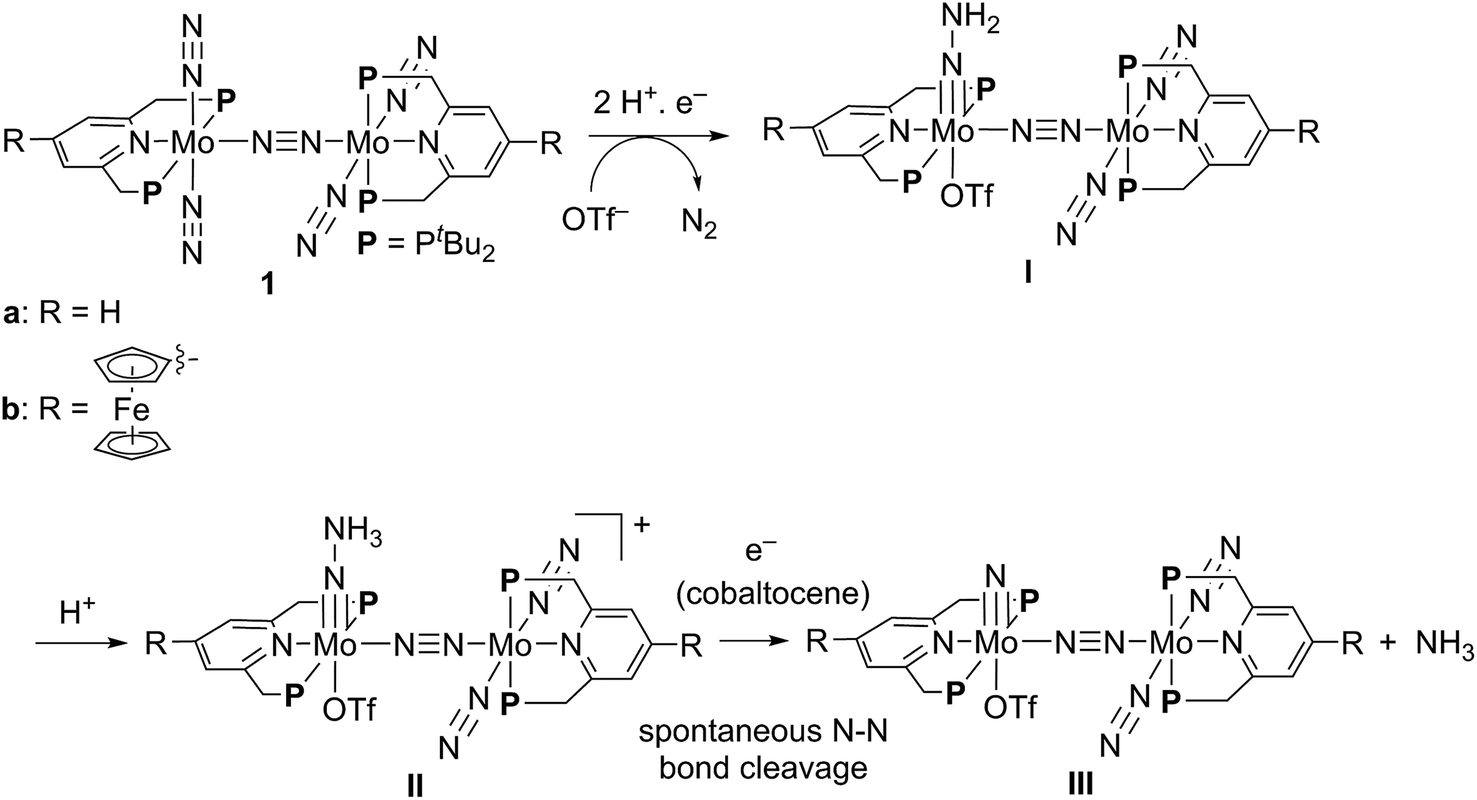 | ||
| Scheme 2 Reaction pathway from dinitrogen-bridged dimolybdenum–dinitrogen complex 1 into dinitrogen-bridged dimolybdenum–nitride complex IIIvia dinitrogen-bridged dimolybdenum–hydrazide and –hydrazidium complexes I and II. | ||
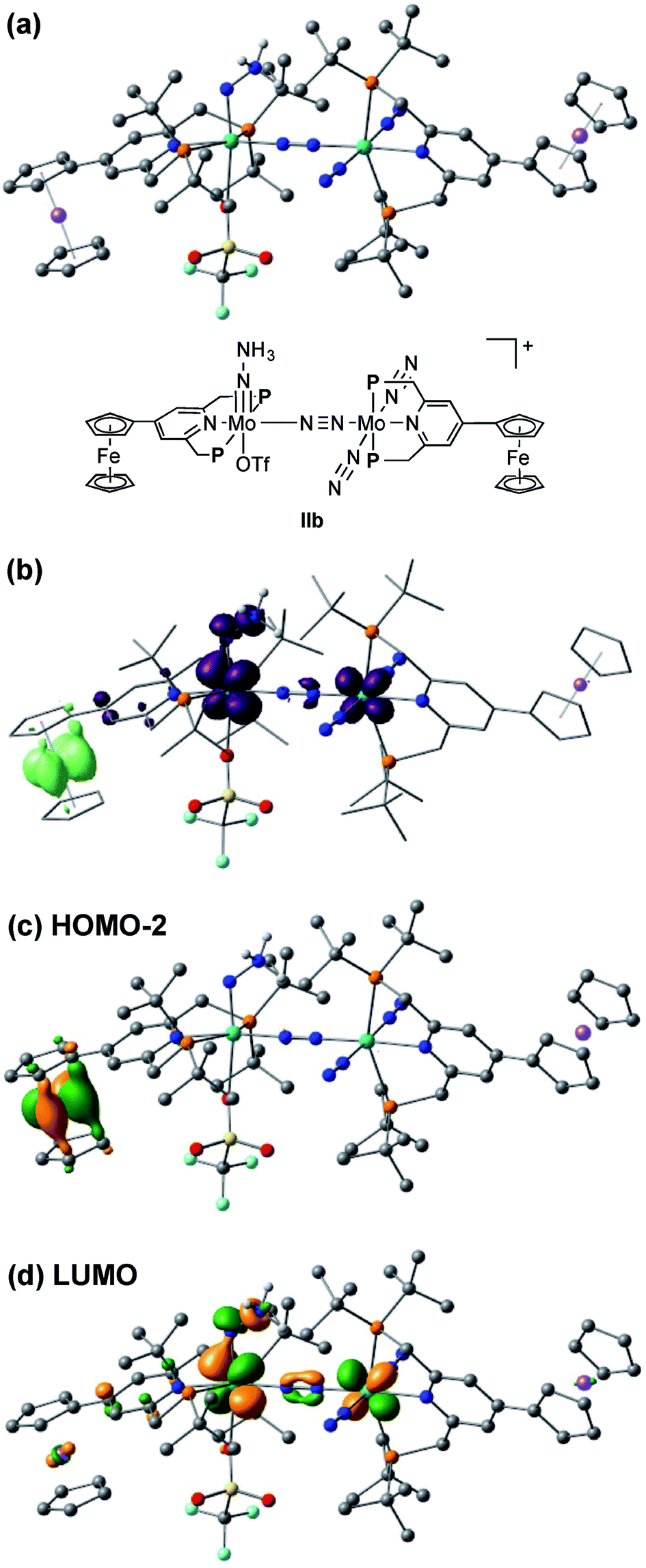 | ||
| Fig. 6 (a) Optimized structure of the ferrocene-substituted dinitrogen-bridged dimolybdenum–hydrazidium complex [Mo(4-Fc-PNP)(OTf)(NNH3)–N≡N–Mo(4-Fc-PNP)(N2)2]+ (IIb). (b) EDDM of the electron transition at 1174 nm based on the time-dependent BP86 result. (c) Molecular orbitals responsible for the transition at 1174 nm. | ||
2.4. Catalytic formation of ammonia from molecular dinitrogen
|
|
||||||
|---|---|---|---|---|---|---|
| Run | Cat. | R | NH3b (equiv.) | NH3c (%) | H2b (equiv.) | H2c (%) |
| a To a mixture of the catalyst (1: 0.010 mmol) and [LutH]OTf (288 equiv. to the catalyst) as proton source in toluene (1.0 mL) was added a solution of CoCp2 (216 equiv. to catalyst) as a reductant in toluene (4.0 mL) at room temperature over a period of 1 h, followed by stirring the resulting mixture at room temperature for another 19 h under an atmospheric pressure of dinitrogen. b Mol equiv. relative to the catalyst. Averages of multiple runs are shown. c Yield based on CoCp2. d Ref. 9. e FeCp2 (2 equiv. to 1a) was added. f Variation of ±2 equiv. between experiments. g Variation of ±3 equiv. between experiments. h Variation of ±1 equiv. between experiments. | ||||||
| 1 | 1b | Fc | 37f | 51 | 36h | 33 |
| 2d | 1a | H | 23 | 31 | 46 | 43 |
| 3e | 1a | H | 21g | 29 | 44h | 41 |
| 4 | 1c | EtFc | 30f | 41 | 35f | 32 |
| 5 | 1d | PhFc | 10f | 14 | 50g | 46 |
| 6 | 1e | Rc | 25g | 35 | 38h | 36 |
| 7d | 1h | MeO | 34 | 47 | 36 | 33 |
The reaction by use of 1b as a catalyst gave 37 equiv. of ammonia and 36 equiv. of molecular dihydrogen, respectively, based on the catalyst (Table 3, run 1).21,22 The amount of produced ammonia was higher than that obtained by using 1a as a catalyst under the same reaction conditions (Table 3, run 2). For comparison, we carried out the catalytic reaction by using 1a as a catalyst in the presence of 2 equiv. of ferrocene to 1a, where 21 equiv. of ammonia were produced based on the catalyst (Table 3, run 3). These results indicated that the ferrocene moiety directly connected to the pyridine ring of the PNP-pincer ligand plays an important role to promote the catalytic reaction more efficiently. As shown in the previous section, the electronic density of the Mo atom of 1b is almost the same with that of 1a (Table 1). The presence of ferrocene at the PNP-pincer ligand in 1 did not affect the nature of the coordinated terminal dinitrogen ligand in 1.9 Thus, the ferrocene moiety in 1b may not act as an electron-donating group but rather as a redox active group for assisting to promote the reduction steps of the coordinated nitrogenous ligands on the Mo atom more smoothly.
Next, we carried out the catalytic ammonia formation by using other ferrocene-substituted dinitrogen-bridged dimolybdenum–dinitrogen complexes as catalysts. In the reactions using 1c and 1d as catalysts, lesser amounts of ammonia compared to that by use of 1b was formed (30 equiv. and 10 equiv.23 of ammonia based on the catalysts, respectively) (Table 3, runs 4 and 5). The catalytic activity of 1 decreased with the decrease of the electrochemical interaction between the Mo atom and the Fe atom of the ferrocene unit in the PNP-pincer ligands (Table 4), and actually a good relationship between ΔE(Fe) [the difference of the Fe(II/III) potential (E1/2) between 2 and 3] and ΔNH3 [the difference of the catalytic activity of 1] was observed as shown in Fig. 7. This result also shows that the complex having a larger ΔE(Fe) value has a higher catalytic activity for ammonia formation.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Run | Complex bearing ferrocene moiety | Reference complexa | ΔNH3b (equiv.) | ΔE(Fe)c/V | ||||
| Complex | R | NH3b (equiv.) | Complex | R | NH3b (equiv.) | |||
| a Ref. 9. b The amount of ammonia based on the catalyst. c Difference of E1/2 value of the Fe(II/III) couple of the ferrocene moiety between the corresponding PNP-pincer ligand 2 and the molybdenum–trichloride complex 3. See Table 2. d Difference between the amount of ammonia catalyzed by 1b and that by 1a. e Difference between the amount of ammonia catalyzed by 1c and that by 1f. f Difference between the amount of ammonia catalyzed by 1d and that by 1g. | ||||||||
| 1 | 1b | Fc | 37 | 1a | H | 23 | 14d | 0.32 |
| 2 | 1c | EtFc | 30 | 1f | Me | 31 | −1e | 0.22 |
| 3 | 1d | PhFc | 10 | 1g | Ph | 21 | −11f | 0.09 |
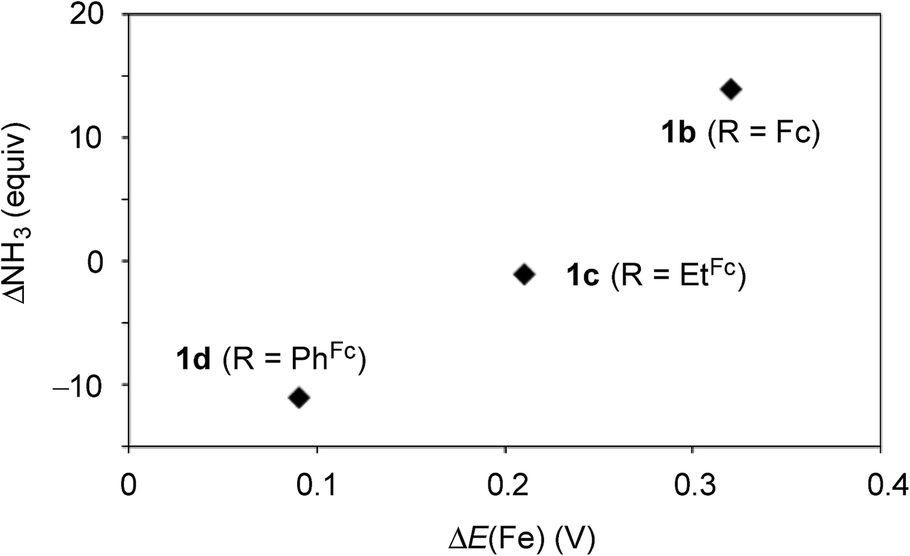 | ||
| Fig. 7 Relationship between the difference E1/2 value of Fe(II/III) of the ferrocene unit in the PNP-pincer ligand 2 on the coordination as 3 and the difference of the catalytic activity of 1. See Table 4 in details. | ||
The reaction using the ruthenocene-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex 1e as a catalyst under the same reaction conditions gave only 25 equiv. of ammonia based on the catalyst (Table 3, run 6). The catalytic activity of 1e was almost the same with that of 1a and lower than that of 1b. This result indicates that the ruthenocene moiety in 3e did not act as a redox active group in the present reaction system.
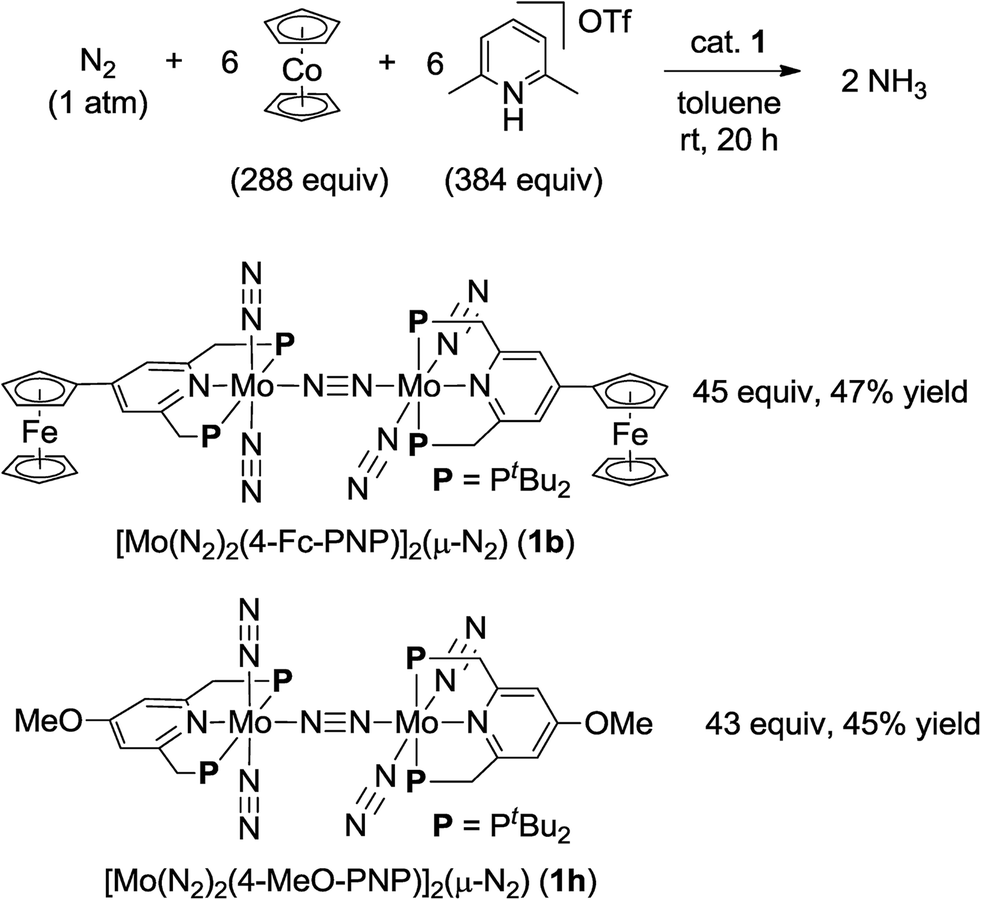 | ||
| Scheme 3 Catalytic formation of ammonia from molecular dinitrogen in the presence of 1b and 1h as catalysts by using larger amounts of cobaltocene and proton source. | ||
To obtain more information on the catalytic behavior of 1b, we monitored the time profile of catalytic reactions by using 1b as a catalyst. Typical results are shown in Fig. 8 together with the time profiles of catalytic reactions by using 1a and 1h as catalysts. The catalytic conversion of molecular dinitrogen into ammonia in the presence of a catalytic amount of 1b proceeded more rapidly than that with 1a and 1h, and the turnover frequency (TOF) for ammonia, which was determined by mols of ammonia produced in initial 1 h per catalyst, by 1b as a catalyst was 23 h−1. Typical results are shown in Table 5. On the other hand, the catalytic formation of ammonia in the presence of catalytic amounts of 1a and 1h proceeded more slowly than that of 1b under the same reaction conditions.9 In fact, the TOFs for ammonia formation using 1a and 1h as catalysts, 17 h−1 and 7 h−1, respectively, were lower than that using 1b.9 These results indicate that the ferrocene-substituted dinitrogen-bridged dimolybdenum–dinitrogen complex 1b has the highest performance from the viewpoint of both the catalytic ability and the rate for ammonia formation.
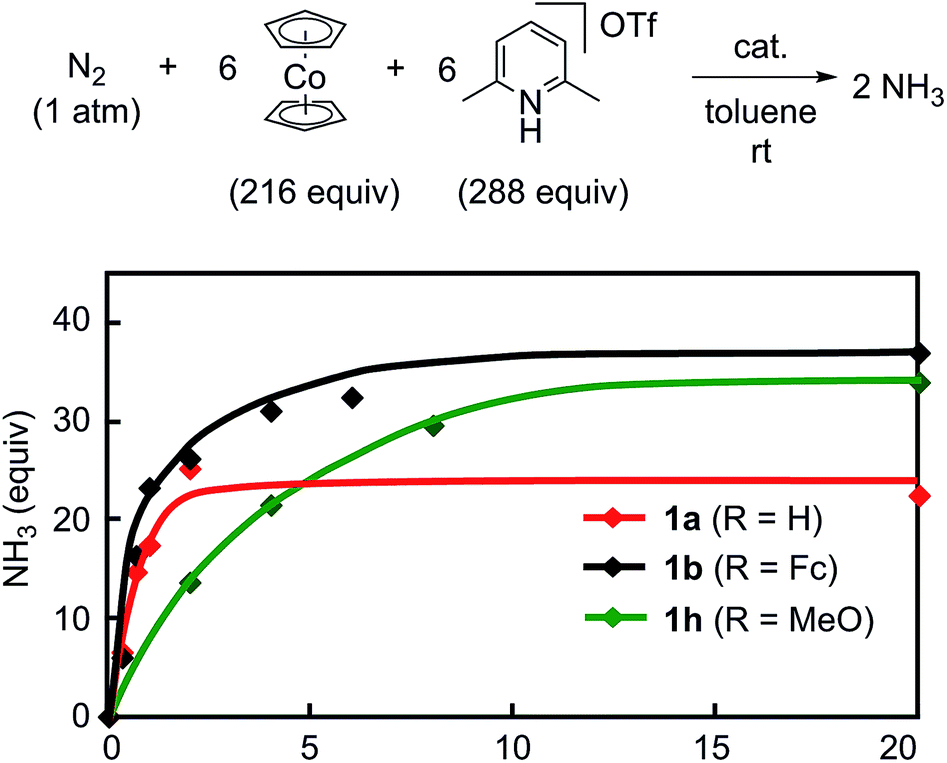 | ||
| Fig. 8 Time profiles of ammonia formation with 1a (red), 1b (black) and 1h (green) as catalysts. | ||
|
|
|||||
|---|---|---|---|---|---|
| Complex | R | TOF (NH3)/h−1 | TOF (H2)/h−1 | TON (NH3) | TON (H2) |
| a TOF and TON are determined based on the catalyst. Average of multiple runs is shown. See Table 3 for reaction conditions. Values of 1a and 1h are from ref. 9. b Variation of ±1 equiv. between experiments. c Variation of ±2 equiv. between experiments. | |||||
| 1b | Fc | 23b | 17c | 37c | 36b |
| 1a | H | 17 | 23 | 23 | 46 |
| 1h | MeO | 7 | 5 | 34 | 36 |
Previously we disclosed that the formation of ammonia and molecular dihydrogen is complementary in the present reaction system.9 To obtain more information on the catalytic activity of 1b toward the formation of molecular dihydrogen, we monitored it in the catalytic reaction using 1b as a catalyst. Typical results are shown in Fig. 9. The formation of molecular dihydrogen proceeded simultaneously with ammonia formation, where the TOF for molecular dihydrogen using 1b as a catalyst is 17 h−1 (Table 5). As shown in the previous paper, the TOF for molecular dihydrogen using 1a as a catalyst is 23 h−1.9 Thus, the TOF of 1b for molecular dihydrogen is lower than that of 1a. These results indicate that 1b can promote the formation of ammonia and, on the other hand, suppress the formation of molecular dihydrogen.
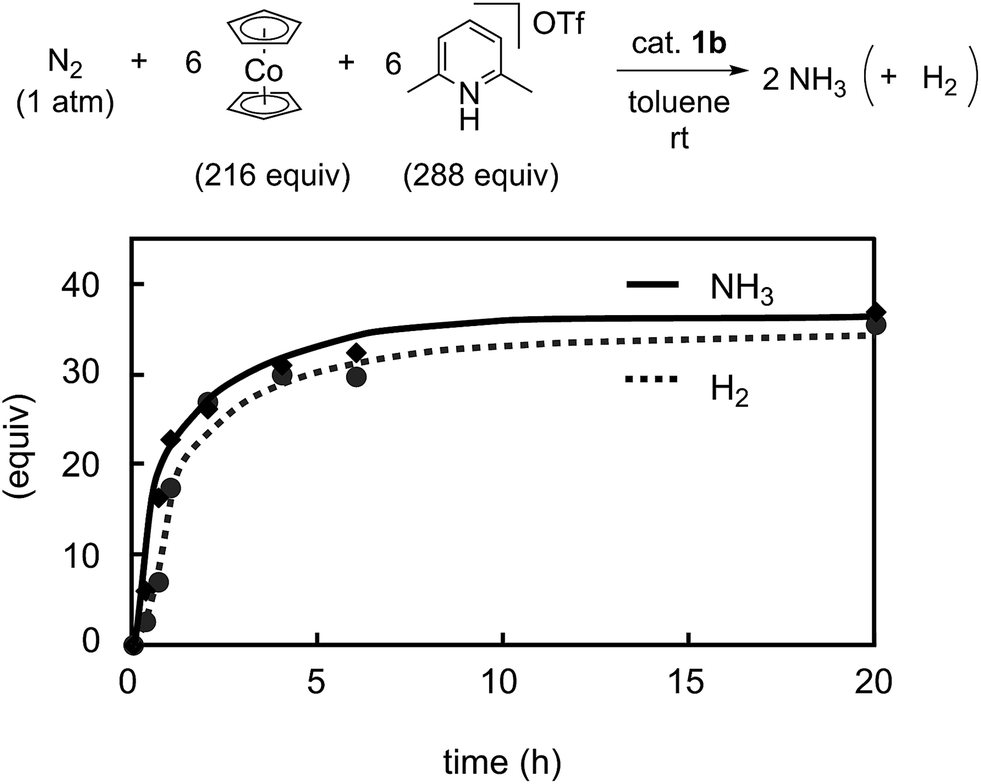 | ||
| Fig. 9 Time profiles of the formation of ammonia (solid line) and molecular dihydrogen (dashed line) with 1b as a catalyst. | ||
Based on the unique behavior and the DFT calculation result of 1b, we consider that the presence of the ferrocene moiety directly connected to the pyridine ring of the PNP-pincer ligand in the catalyst increases the rate of the reduction steps of the coordinated nitrogenous ligand on the Mo atom. In this reaction system, the electronic interaction between the Fe atom of the ferrocene moiety and the Mo atom of the complex plays an important role to promote the catalytic formation of ammonia more smoothly, where the ferrocene moiety works as an intramolecular reductant, clarified by the reversible one-electron redox behavior, toward the high-oxidation state of the Mo complexes such as hydrazidium complexes. The observed effect of direct introduction of a ferrocene moiety to the pyridine ring of the PNP-pincer ligand is in sharp contrast to the effect observed by introduction of an electron-donating group such as methoxy group to the pyridine ring of the PNP-pincer ligand. In the latter case, the electron-donating group accelerates the protonation step of the coordinated nitrogenous ligands on the Mo atom of the complexes in the catalytic reaction (Fig. 10), while a ferrocene moiety increases the rate of the reduction step.9 At present, we can not exclude another possibility of the role of the ferrocene moiety in the catalyst and further work is necessary to clarify the exact role of the ferrocene moiety in the catalyst. However, the result described in the present paper provides the first successful example of the substantial improvement of the catalytic reactivity by the presence of the ferrocene moiety as a redox active site in the catalytic reduction of molecular dinitrogen into ammonia under ambient reaction conditions.24
 | ||
| Fig. 10 The role of substituent to the PNP-pincer ligand toward catalytic ammonia formation from molecular dinitrogen. | ||
The electron transfer from the Fe atom of the ferrocene moiety to the active site of the Mo atom in the complex plays an important role to promote catalytic ammonia formation effectively. As presented in the introduction part, a similar electron transfer may occur at the active site of nitrogenase enzyme in biological nitrogen fixation (Fig. 1). We believe that the mechanistic insight obtained in the present paper may provide valuable information to understand the mechanism of biological nitrogen fixation by nitrogenase.
3. Conclusion
We have prepared and characterized a series of dinitrogen-bridged dimolybdenum–dinitrogen complexes bearing metallocene-substituted PNP-pincer ligands spectroscopically and theoretically. The complex bearing a ferrocene group on the pyridine ring of the PNP-pincer ligand 1b has been found to work as the most efficient catalyst toward the catalytic formation of ammonia from molecular dinitrogen, where up to 45 equiv. of ammonia were produced based on the catalyst (22 equiv. of ammonia based on each Mo atom of the catalyst). The catalytic activity of 1b is almost the same with that of 1h, which was previously reported to work as the most effective catalyst toward ammonia formation under ambient reaction conditions.9 Mechanistic study indicated that the presence of ferrocene as a redox active moiety in the PNP-pincer ligand increases the rates of the reduction step involved in the catalytic process of ammonia formation via an intramolecular electron transfer from the Fe atom of the ferrocene to the active site of the Mo atom in the complex. The present result is another useful innovation to the previous finding that the introduction of an electron-donating group to the pyridine ring of the PNP-pincer ligand accelerated the protonation step involved in the catalytic reaction.9 We believe that the result described in this article provides useful information to design a more effective catalyst toward the catalytic formation of ammonia from molecular dinitrogen under ambient reaction conditions. Further work is currently in progress to develop a more effective reaction system.25Acknowledgements
The authors are grateful to M. Takahashi and Prof. H. Houjou in The University of Tokyo for spectroelectrochemical analysis. Y.N. thanks Grants-in-Aid for Scientific Research (nos 26288044, 26620075 and 26105708) from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT). K.Y. thanks Grants-in-Aid for Scientific Research (nos 22245028 and 24109014) from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) and the MEXT Projects of “Integrated Research on Chemical Synthesis” and “Elements Strategy Initiative to Form Core Research Center”. H.T. thanks a Grant-in-Aid for Research Activity Start-up (nos 26888008) from the Japan Society for the Promotion of Science (JSPS). S.K. is a recipient of the JSPS Predoctoral Fellowships for Young Scientists. We also thank the Research Hub for Advanced Nano Characterization at The University of Tokyo for X-ray analysis.Notes and references
- For selected recent reviews, see: (a) K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559 CrossRef CAS PubMed; (b) M. D. Fryzuk, Chem. Commun., 2013, 49, 4866 RSC; (c) H. Broda, S. Hinrichsen and F. Tuczek, Coord. Chem. Rev., 2013, 257, 587 CrossRef CAS PubMed; (d) Y. Tanabe and Y. Nishibayashi, Coord. Chem. Rev., 2013, 257, 2551 CrossRef CAS PubMed; (e) H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547 RSC; (f) C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183 RSC; (g) C. Köthe and C. Limberg, Z. Anorg. Allg. Chem., 2015, 641, 18 CrossRef PubMed; (h) N. Khoenkhoen, B. de Bruin, J. N. H. Reek and W. I. Dzik, Eur. J. Inorg. Chem., 2015, 567 CrossRef CAS PubMed.
- For selected recent examples see: (a) T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549 CrossRef CAS PubMed; (b) S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 11373 CrossRef CAS PubMed; (c) M. T. Mock, S. Chen, M. O'Hagan, R. Rousseau, W. G. Dougherty, W. S. Kassel and R. M. Bullock, J. Am. Chem. Soc., 2013, 135, 11493 CrossRef CAS PubMed; (d) I. Klopsch, M. Finger, C. Würtele, B. Milde, D. B. Werz and S. Schneider, J. Am. Chem. Soc., 2014, 136, 6881 CrossRef CAS PubMed; (e) A. J. Keane, B. L. Yonke, M. Hirotsu, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2014, 136, 9906 CrossRef CAS PubMed; (f) K. C. MacLeod, D. J. Vinyard and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 10226 CrossRef CAS PubMed; (g) Y. Ishida and H. Kawaguchi, J. Am. Chem. Soc., 2014, 136, 16990 CrossRef CAS PubMed; (h) T. Miyazaki, H. Tanaka, Y. Tanabe, M. Yuki, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2014, 53, 11488 CrossRef CAS PubMed; (i) G. W. Margulieux, Z. R. Turner and P. J. Chirik, Angew. Chem., Int. Ed., 2014, 53, 14211 CrossRef CAS PubMed; (j) C. Gradert, N. Stucke, J. Krahmer, C. Näther and F. Tuczek, Chem.–Eur. J., 2015, 21, 1130 CrossRef CAS PubMed; (k) Y. Lee, F. T. Sloane, G. Blondin, K. A. Abboud, R. García-Serres and L. J. Murray, Angew. Chem., Int. Ed., 2015, 54, 1499 CrossRef CAS PubMed.
- (a) T. A. Bazhenova and A. E. Shilov, Coord. Chem. Rev., 1995, 144, 69 CrossRef CAS; (b) A. E. Shilov, Russ. Chem. Bull., 2003, 52, 2555 CrossRef CAS; (c) K. Shiina, J. Am. Chem. Soc., 1972, 94, 9266 CrossRef CAS; (d) K. Komori, H. Oshita, Y. Mizobe and M. Hidai, J. Am. Chem. Soc., 1989, 111, 1939 CrossRef CAS; (e) K. Komori, S. Sugiura, Y. Mizobe, M. Yamada and M. Hidai, Bull. Chem. Soc. Jpn., 1989, 62, 2953 CrossRef CAS; (f) H. Oshita, Y. Mizobe and M. Hidai, J. Organomet. Chem., 1993, 456, 213 CrossRef CAS; (g) M. Mori, J. Organomet. Chem., 2004, 689, 4210 CrossRef CAS PubMed; (h) H. Tanaka, A. Sasada, T. Kouno, M. Yuki, Y. Miyake, H. Nakanishi, Y. Nishibayashi and K. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 3498 CrossRef CAS PubMed; (i) M. Yuki, H. Tanaka, K. Sasaki, Y. Miyake, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2012, 3, 1254 CrossRef PubMed; (j) Q. Liao, N. Saffon-Merceron and N. Mézailles, Angew. Chem., Int. Ed., 2014, 53, 14206 CrossRef CAS PubMed.
- (a) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76 CrossRef CAS PubMed; (b) V. Ritleng, D. V. Yandulov, W. W. Weare, R. R. Schrock, A. S. Hock and W. M. Davis, J. Am. Chem. Soc., 2004, 126, 6150 CrossRef CAS PubMed; (c) R. R. Schrock, Acc. Chem. Res., 2005, 38, 955 CrossRef CAS PubMed; (d) W. W. Weare, X. Dai, M. J. Byrnes, J. M. Chin, R. R. Schrock and P. Müller, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17099 CrossRef CAS PubMed; (e) R. R. Schrock, Angew. Chem., Int. Ed., 2008, 47, 5512 CrossRef CAS PubMed.
- (a) K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120 CrossRef CAS PubMed; (b) E. Kinoshita, K. Arashiba, S. Kuriyama, Y. Miyaka, R. Shimazaki, H. Nakanishi and Y. Nishibayashi, Organometallics, 2012, 31, 8437 CrossRef CAS.
- (a) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef CAS PubMed; (b) J. Rittle and J. C. Peters, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15898 CrossRef CAS PubMed; (c) S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105 CrossRef CAS PubMed; (d) J. Rittle, C. C. L. McCrory and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13853 CrossRef CAS PubMed; (e) G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532 CAS.
- H. Tanaka, K. Arashiba, S. Kuriyama, A. Sasada, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2014, 5, 3737 Search PubMed.
- Independently, Batista and co-workers have recently reported a theoretical study on our reaction pathway, and they also concluded bimetallic complexes as reactive intermediates: Y.-H. Tian, A. W. Pierpont and E. R. Batista, Inorg. Chem., 2014, 53, 4177 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, K. Nakajima, H. Tanaka, N. Kamaru, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2014, 136, 9719 CrossRef CAS PubMed.
- For a recent review, see; B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041 CrossRef CAS PubMed.
- (a) T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed; (b) K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974 CrossRef CAS PubMed; (c) K. M. Lancaster, Y. Hu, U. Bergmann, M. W. Ribbe and S. DeBeer, J. Am. Chem. Soc., 2013, 135, 610 CAS; (d) J. A. Wiig, Y. Hu, C. C. Lee and M. W. Ribbe, Science, 2012, 337, 1672 CrossRef CAS PubMed.
- For recent reviews on redox-active ligands, see: (a) V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228 CrossRef CAS PubMed; (b) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC.
- (a) J. M. Camara and T. B. Rauchfuss, Nat. Chem., 2011, 4, 26 CrossRef PubMed; (b) S. Samanta, K. Mittra, K. Sengupta, S. Chatterjee and A. Dey, Inorg. Chem., 2013, 52, 1443 CrossRef CAS PubMed; (c) S. Ghosh, G. Hogarth, N. Hollingsworth, K. B. Holt, S. E. Kabir and B. E. Sanchez, Chem. Commun., 2014, 50, 945 RSC; (d) C. Hüttinger, C. Förster and K. Heinze, Chem. Commun., 2014, 50, 4285 RSC; (e) S. Roy, S. K. S. Mazinani, T. L. Groy, L. Gan, P. Tarakeshwar, V. Mujica and A. K. Jones, Inorg. Chem., 2014, 53, 8919 CrossRef CAS PubMed.
- (a) Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Stepnicka, John Wiley and Sons, Chichester, U.K., 2008 Search PubMed; (b) E. S. Philips, Ferrocenes: Compounds, Properties 6 Applications (Chemical Engineering Methods and Technology), Nova Science Publishers, New York, 2011 Search PubMed.
- Catalytic reactions by using ferrocene unit as a redox-active moiety in the catalyst, see: (a) C. S. Slone, C. A. Mirkin, G. P. A. Yap, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 1997, 119, 10743 CrossRef CAS; (b) M. Süßner and H. Plenio, Angew. Chem., Int. Ed., 2005, 44, 6885 CrossRef PubMed; (c) C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410 CrossRef CAS PubMed; (d) G. Liu, H. He and J. Wang, Adv. Synth. Catal., 2009, 351, 1610 CrossRef CAS PubMed; (e) A. G. Tennyson, V. M. Lynch and C. W. Bielawski, J. Am. Chem. Soc., 2010, 132, 9420 CrossRef CAS PubMed; (f) E. M. Broderick, N. Guo, T. Wu, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, T. Cantat and P. L. Diaconescu, Chem. Commun., 2011, 47, 9897 RSC; (g) E. M. Broderick, N. Guo, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278 CrossRef CAS PubMed; (h) C. D. Varnado Jr, E. L. Rosen, M. S. Collins, V. M. Lynch and C. W. Bielawski, Dalton Trans., 2013, 42, 13251 RSC; (i) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264 CrossRef CAS PubMed; (j) P. Neumann, H. Dib, A.-M. Caminade and E. Hey-Howkins, Angew. Chem., Int. Ed., 2015, 54, 311 CrossRef CAS PubMed.
- It is reported that the electrochemical oxidation of ruthenocene is an irreversible process: J. C. Swarts, A. Nafady, J. H. Roudebush, S. Trupia and W. E. Geiger, Inorg. Chem., 2009, 48, 2156 CrossRef CAS PubMed.
- (a) C. D. Nunes, T. M. Santos, H. M. Carapuça, A. Hazell, M. Pillinger, J. Madureira, W.-M. Xue, F. E. Kühn and I. S. Gonçalves, New J. Chem., 2002, 26, 1384 RSC; (b) T.-Y. Dong, M.-C. Lin, M. Y.-N. Chiang and J.-Y. Wu, Organometallics, 2004, 23, 3921 CrossRef CAS; (c) J. Rajput, A. T. Hutton, J. R. Moss, H. Su and C. Imrie, J. Organomet. Chem., 2006, 691, 4573 CrossRef CAS PubMed; (d) K.-Q. Wu, J. Guo, J.-F. Yan, L.-L. Xie, F.-B. Xu, S. Bai, P. Nockemann and Y.-F. Yuan, Organometallics, 2011, 30, 3504 CrossRef CAS.
- (a) T.-Y. Liu, Y. J. Chen, C.-C. Tai and K. S. Kwan, Inorg. Chem., 1999, 38, 674 CrossRef CAS; (b) Y. J. Chen, C.-H. Kao, S. J. Lin, C.-C. Tai and K. S. Kwan, Inorg. Chem., 2000, 39, 189 CrossRef CAS; (c) S.-H. Wu, J.-J. Shen, J. Yao and Y.-W. Zhong, Chem.–Asian J., 2013, 8, 138 CrossRef CAS PubMed.
- (a) D. E. Richardson and H. Taube, Coord. Chem. Rev., 1984, 60, 107 CrossRef CAS; (b) J. E. Sutton and H. Taube, Inorg. Chem., 1981, 20, 3125 CrossRef CAS; (c) Y. Kim and C. M. Lieber, Inorg. Chem., 1989, 28, 3990 CrossRef CAS.
- M. W. Weatherburn, Anal. Chem., 1967, 39, 971 CrossRef CAS.
- Separately, we confirmed the direct conversion of molecular dinitrogen into ammonia by using 15N2 gas in place of normal 14N2 gas (see the ESI† for details).
- Separately, we also investigated catalytic reactions by using 1b as a catalyst with another reductant such as CrCp*2 (Cp* = η5-C5Me5) having a higher reducing ability and proton sources such as [2-PicH]OTf (2-Pic = 2-picoline) and [ColH]OTf (Col = 2,4,6-collidine) under the same reaction conditions. In all cases, however, the yield of ammonia was lower compared with the result using CoCp2 and [LutH]OTf as a reductant and a proton source, respectively. See the ESI† for details.
- By comparison of the IR absorbance of the terminal dinitrogen ligand in 1a with that in 1d (vide supra), the PhFc-moiety in 1d is considered to act as an electron-withdrawing group, suppressing the rate of catalytic formation. This may be the reason why a lower amount of ammonia was produced under the same reaction conditions.
- (a) Previously, George and Tisdale found a substantial improvement of the stoichiometric formation of ammonia by the protonation of a molybdenum–dinitrogen complex bearing a ferrocenyldiphosphine as an auxiliary ligand.24b More recently, we found that a molybdenum–dinitrogen complex bearing two ferrocenyldiphosphines as auxiliary ligands worked as an effective catalyst toward the formation of silylamine from molecular dinitrogen under ambient reaction conditions.3h Electronic interaction between the Fe atom of the ferrocenyldiphosphine and the Mo atom was not observed in both cases; (b) T. A. George and R. C. Tisdale, J. Am. Chem. Soc., 1985, 107, 5157 CrossRef CAS.
- (a) Y. Tanabe, S. Kuriyama, K. Arashiba, Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 9290 RSC; (b) T. Miyazaki, Y. Tanabe, M. Yuki, Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem.–Eur. J., 2013, 19, 11874 CrossRef CAS PubMed; (c) K. Arashiba, K. Nakajima and Y. Nishibayashi, Z. Anorg. Allg. Chem., 2015, 641, 100 CrossRef CAS PubMed; (d) quite recently, Rebreyend and de Bruin have highlighted the photolytic N2 splitting2h as a road to sustainable NH3 production: C. Rebreyend and B. de Bruin, Angew. Chem., Int. Ed., 2015, 54, 42 CrossRef CAS PubMed; (e) E. Kinoshita, K. Arashiba, S. Kuriyama, A. Eizawa, K. Nakajima and Y. Nishibayashi, Eur. J. Inorg. Chem., 2015, 1789 CrossRef CAS PubMed; (f) K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 5666 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1048952–1048956. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00545k |
| This journal is © The Royal Society of Chemistry 2015 |