Sulfidation of copper oxide nanoparticles and properties of resulting copper sulfide†
Rui
Ma
abh,
John
Stegemeier
ab,
Clément
Levard
bc,
James G.
Dale
bd,
Clinton W.
Noack
a,
Tiffany
Yang
ab,
Gordon E.
Brown
Jr.
efg and
Gregory V.
Lowry
*ab
aDepartment of Civil & Environmental Engineering, Carnegie Mellon University, USA. E-mail: glowry@cmu.edu
bCenter for Environmental Implications of Nanotechnology (CEINT), USA
cAix-Marseille Université, CNRS, IRD, CEREGE UM34, 13545 Aix en Provence, France
dDepartment of Geosciences, Virginia Tech, USA
eDepartment of Geological & Environmental Sciences, Stanford University, USA
fStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, USA
gDepartment of Photon Science, SLAC National Accelerator Laboratory, Menlo Park, CA, USA
hEcological Environment Institute, Chinese Academy for Environmental Planning, Beijing, China
First published on 29th April 2014
Abstract
Many nanoparticles (NPs) are transformed in the environment, and the properties of the transformed materials must be determined to accurately assess their environmental risk. Sulfidation is expected to alter the speciation and properties of CuO NPs significantly. Here, commercially available 40 nm CuO NPs were characterized and sulfidized in water by inorganic sulfide, and the properties of the resulting products were determined. X-ray absorption spectroscopy, X-ray diffraction, and transmission electron microscopy indicate that CuO (tenorite) is sulfidized by inorganic sulfide to several copper sulfide (CuxSy) species including crystalline CuS (covellite), and amorphous (CuxSy) species at ambient temperature. Some Cu(II) was reduced to Cu(I) during sulfidation, coupled with sulfide oxidation to sulfate, resulting in the formation of small amounts of several copper sulfate hydroxide species as well. The extent of sulfidation depends on the sulfide to CuO molar concentration ratio used. At the highest S/Cu molar ratio of 2.16, 100% sulfidation was not reached in 7 days, as evidenced by the persistence of small amounts of CuO in the NPs. Sulfidation increased the fraction of copper passing a 3 kDa MWCO filter representing soluble forms of Cu and any small CuxSy clusters compared to the pristine CuO NPs at environmentally relevant neutral pH. This high solubility is a result of oxidative dissolution of CuxSy, formation of relatively more soluble copper sulfate hydroxides, and the formation of small CuS nanoclusters that pass the 3 kDa MWCO filter. These findings suggest that sulfidation of CuO may increase its apparent solubility and resulting bioavailability and eco-toxicity attributed to toxic Cu2+.
Nano impactMetal and metal oxide nanomaterials that find their way into reducing environments such as wastewater treatment plants or subaquatic sediments may potentially become sulfidized. The properties of the sulfidized materials will control their fate and toxicity so those properties must be determined. This work describes the sulfidation of CuO nanoparticles, and determines the properties of the sulfidized copper product under environmental conditions that are relevant to predicting fate and toxicity, e.g. solubility. This enhances our understanding of the behavior of these nanomaterials in important environmental compartments, and better enables predictions of their interactions with natural systems. |
Introduction
Copper-based nanoparticles (NPs) are being used in products or technologies like semiconductors, heat transfer fluids, catalysts, batteries, solar cells and biocides.1–6 Copper based NPs such as CuO and elemental Cu(0) were found to be among the top five most reported metal NPs in recent (2011) nanotechnology patents, indicating the potential for an increasing use of these NPs.7 Their widespread uses will likely lead to subsequent release into the environment, and will raise concern about their potential toxicity. Nano Cu(0) was reported to have a thin oxide layer when exposed to air, where the Cu(0) is oxidized to form Cu2O and then ultimately to CuO in an aerobic environment.8 Hence, CuO NPs are an environmentally relevant form of copper for assessing the risks of nano copper products.The toxicity of CuO NPs to a variety of organisms has been studied extensively. According to a recent critical review, CuO NPs are toxic to crustaceans, algae, fish and bacteria, but typically requires higher doses than Cu2+ ions to achieve the same effects. Cu2+ was found to be more toxic to all organisms except for yeast and mammalian cells in vitro.9 Although Cu(0) NPs can catalyze the formation of reactive oxygen species (ROS), leading to toxicity.10,11 CuO dissolution to release Cu2+ accounted for most of the observed toxicity in vitro and in vivo.12 This indicates that the ability of the NPs to release Cu2+ plays a key role in copper NP toxicity.
The dissolution of CuO NPs resulting in the release of Cu2+ ions is pH dependent. The dissolution minimum is expected at near neutral pH (6–8). However, the solubility is significantly higher at lower pH (4–5). Dissolution of CuO NPs is also promoted by strong ligands such as amino acids,11,13 even at neutral pH. This behavior makes CuO NPs one of the more toxic metal oxides NP in cell culture media (containing amino acids).14
Sulfidation is an important transformation of many metal and metal oxide NPs. This is because sulfidation has been demonstrated to affect NP chemical composition,15,16 to reduce ion release,16,17 and to significantly decrease the toxicity of Ag NPs to a range of organism types.15,18 Ag, ZnO, and CuO NPs all contain class B soft metals19 and are likely to sulfidize once released into the environment. Donner et al. showed that 74–92% of the Cu is present as Cu(I) and Cu(II) sulfide (chalcocite and covellite, respectively) in fresh biosolids that had been amended with CuO NPs, but these inorganic Cu-sulfide species were transformed to Cu–organic sulfur complexes and Cu(II) sulfide in aged biosolids.20 Dimkpa et al. showed that exposure of plant roots to CuO NPs resulted in bioaccumulation of Cu2S, Cu–cysteine complexes, and CuO NPs in the plant.21 Based on these previous findings, it is important to understand the sulfidation products of CuO NPs, and the impact of sulfidation on the dissolution of these NPs to form Cu ions under environmentally relevant conditions.
Copper sulfides (CuxSy), the sulfidized product of CuO, have been successfully synthesized in the laboratory and occur naturally. An array of crystalline copper sulfide phases are stable at room temperature, including covellite (CuS), yarrowite (Cu1.12S), spionkopite (Cu1.39S), geerite (Cu1.6S), anilite (Cu1.75S), digenite (Cu1.8S), djurlite (Cu1.95S), and chalcocite (Cu2S),22 which have different optical and electrical properties.23 Controlled syntheses of copper sulfides have shown that sulfidation occurs via a Kirkendall mechanism: formation of nano CuS hollow structures (Kirkendall diffusion) by reacting Cu2O with Na2S.24,25 Luther et al. reported a mechanism of reduction of dissolved Cu2+ and formation of very small (Cu(I)S(−I)) tetrameric clusters.26 Data gaps still exist about how CuO NPs transform in the presence of sulfide (S2−/HS−) in water at ambient temperature and about the properties of the partially and fully sulfidized CuO NPs, including chemical composition, surface properties, size, morphology, and crystal structure. More information is also needed on the potential dissolution (re-oxidation) of copper sulfide species under environmental conditions (i.e. ambient temperature, neutral pH, and low ionic strength).
In this study we sulfidized commercially available CuO NPs using different S/Cu molar ratios. The pristine CuO NPs and the resulting sulfidized CuxSy/CuO NPs were extensively characterized. The objectives of this study were to (1) provide mechanistic insights about the transformation of CuO in the presence of sulfide in aqueous solution; and (2) determine the properties and solubility of the resulting sulfidized nanoparticles. The pristine and sulfidized particles were characterized using transmission electron microscopy (TEM), dynamic light scattering (DLS), thermo gravimetric analysis (TGA), X-ray diffraction (XRD), and synchrotron based X-ray absorption spectroscopy (XAS). Their dissolution rate was determined by measuring ion release by inductively coupled plasma mass spectrometry (ICP-MS).
Materials and methods
CuO NPs
The CuO NPs (40 nm) were purchased from US Research Nanomaterials, Inc. (Houston, TX). The primary particle size is ~40 nm and the particles are generally spherical. The N2-BET specific surface area provided by the manufacturer is 20 m2 g−1. The particles were used as received. The manufacturer claims that the CuO NPs contain no organic capping agent. This was confirmed by thermo gravimetric analysis (TGA) as described below. The crystal structure of the initial CuO NPs is described in the results section.Sulfidation of CuO nanoparticles
Sulfidation of CuO NPs was conducted following similar procedures as previously described for sulfidation of ZnO NPs.16 In order to minimize the oxidation of sulfide by dissolved oxygen in a typical experiment, all solutions were prepared using N2-purged DI water and N2-purged headspace. The CuO NPs were suspended in deoxygenated water by sonicating in an ice-bath with a micro tip sonicator (Branson Model 250) at a power input of 10 W for 8 minutes. The dispersed CuO NPs (100 mg L−1) in 10 mM NaNO3 electrolyte were reacted with sulfide (Na2S) in N2-purged 50 mL propylene Falcon tubes. The S/Cu molar ratio was varied from 0.21 to 2.16 to investigate the properties of NPs sulfidized to different extents. The exact S/Cu ratios used are listed in Table S1.† The initial pH of the solution during sulfidation was approximately 12 due to addition of sodium sulfide. The pH of the tubes was adjusted to 11.9 using NaOH and HCl, and the tubes were sealed and were allowed to rotate in the dark for 7 days. Then the resulting solutions were centrifuged at 4000 g for 20 min. Supernatants were carefully decanted and DI water was added. The NPs were re-suspended in sulfide-free DI water, and then the tubes were centrifuged again and the supernatants were decanted. These steps were repeated three times to remove residual sulfide in solution. Finally, the sulfidized CuO NPs were suspended in deoxygenated DI water. A portion of each of the slurries was allowed to dry and was kept for characterization as described below.Characterization of the pristine and sulfidized CuO
Characterization was performed on NPs that had been washed, dried, and re-dispersed in 10 mM NaNO3 at pH = 7.5. The sizes of the NPs were measured by dynamic light scattering (DLS) and TEM bright field imaging. DLS measurements were made using an ALV/CGS-3 compact goniometer system equipped with a 22 mW HeNe Laser (λ = 632.8 nm) at a scattering angle of 90°. TEM images were taken using a JEOL 2100 transmission electron microscope at an accelerating voltage of 200 kV. The TEM samples were mounted on lacy carbon film on 300 mesh gold grids (Ted Pella, Inc.). Samples were prepared by placing one drop of a NP suspension in ethanol using a ‘drop and wick’ technique.Thermo gravimetric analysis (TGA) was conducted using a SDT Q600 TGA (TA Instruments, New Castle, DE). Approximately 20 mg of the NPs was placed in the TGA holder. The particles were heated at a rate of 2 °C min−1 from ambient temperature up to 400 °C in air, and the weight change upon heating was recorded.
X-ray absorption spectroscopy (XAS) and X-ray diffraction (XRD) (both laboratory and synchrotron-based XRD) were used to assess structural/speciation differences among the NPs. XAS measurements were conducted at the Stanford Synchrotron Radiation Lightsource (SSRL) on beamline 11-2 to determine the speciation of pristine and sulfidized CuO NPs. Samples were pelletized after diluting with glucose to achieve an optimized absorption edge jump (Δμ) of 1 at the Cu K-edge (8988 eV). XAS spectra were collected at room temperature in transmission mode. Data were analyzed using the SixPACK software package, version 0.68.13 XAS scans were energy calibrated using a metallic Cu foil, background subtracted with E0 defined as 8988 eV, converted to frequency (k) space, and weighted by k3. Linear combination fitting (LCF) using a three copper model compounds (CuS, Cu2S & CuO) was performed on the spectra to obtain quantitative speciation information by using least-squares. To better understand the CuO to CuxSy transformation, the spectra were converted into R space via Fourier transform and the first shell was fit using two theoretical scattering paths (Cu–S & Cu–O) that were generated by IFEFFIT through Sixpack.13
Powder X-ray diffraction was first used to identify the crystal structure of the pristine CuO NPs. Diffraction patterns of these materials were collected using a laboratory-based Panalytical X-ray diffractometer, operating in the Bragg configuration using Cu Kα radiation (λ = 1.5418 Å) from 10° to 90° at a scanning rate of 0.2° min−1 for the identification of crystalline phases.
The XRD patterns for copper sulfide model compounds (CuS and Cu2S) and the sulfidized samples were collected using synchrotron-based XRD at SSRL on beamline 11-3. Incident X-rays (0.9744 Å, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 735 eV) were focused using a bent cube root I-beam Si (311) monochromator. A MAR345 area detector positioned 120 mm downstream of the sample was used to collect diffraction scans with a dwell time of 90 s. The collected images were converted into q space using Area Diffraction Machine (open source) software.
735 eV) were focused using a bent cube root I-beam Si (311) monochromator. A MAR345 area detector positioned 120 mm downstream of the sample was used to collect diffraction scans with a dwell time of 90 s. The collected images were converted into q space using Area Diffraction Machine (open source) software.
Dissolution measurements
Dissolution of CuO NPs and the sulfidized NPs was measured by quantifying the concentration of dissolved copper in solution with a known initial concentration of total Cu of ~1 mM, i.e. CuO (80 mg L−1) and CuxSy (100 mg L−1). Two sets of dissolution experiments were conducted to investigate the effect of dissolved oxygen on the rate and extent of dissolution of the sulfidized NPs. One set was performed with a dissolved oxygen (DO) concentration of 8.3 mg L−1. The other set used N2-purged deoxygenated water and was conducted in a glove box with N2 headspace.To measure the extent of dissolution of the NPs for different reaction times, the washed NPs were diluted into 200 mL serum bottles. NaNO3 was added to provide a 10 mM background concentration to maintain uniform ionic strength in all reactors. HEPES buffer (2 mM) was used to provide an initial pH of 7.4 in all reactors. The serum bottles were capped and agitated on an end-over-end rotator at 30 rpm in the dark at room temperature (20 °C).
At time points from 1 h to 2 weeks, samples were taken from the reactors. At each time point, 7 mL of solution was removed for analysis. Particles were separated using Amicon ultra-15 filters (MWCO 3 kDa). The ultra filters were centrifuged at 3000 g for 20 minutes and a 5 mL aliquot of the filtrate was collected for analysis. Control studies with 1 and 3 mg L−1 Cu2+ indicated that Cu ion (Cu(NO3)2) retention by the ultra filtration membranes can be neglected.
The samples were digested by adding concentrated HNO3 to reach 5% acid content. Each sample was analyzed by inductively coupled plasma mass spectrometry (ICP-MS) to determine the concentration of dissolved copper in solution. A multi-element calibration standard (10 mg L−1 with 5% HNO3, Agilent Technologies) was diluted with 5% HNO3 to make the desired calibration standards.
Modeling the solubility of Cu solids
Experimentally determined copper solubilities were compared to thermodynamic equilibrium for crystalline mineral phases identified by XRD using the general chemical equilibrium model MINEQL+ Version 4.5. Calculations used a constant total Cu of 1.25 mM, derived from the initial 100 mg L−1 CuO nanoparticles used in solubility experiment, assuming complete conversion to the mineral of interest. For copper-sulfide and copper-sulfate-hydroxide solids, the total S(−II) or total S(VI) was set to the stoichiometric ratio of the solid, e.g. for covellite (CuS) TOTS(−II) = 1.25 mM, for chalcocite (Cu2S) TOTS(−II) = 0.625 mM, and for brochantite (Cu4SO4(OH)6) TOTS(VI) = 0.3125 mM. Copper solubility was considered for each solid separately with each mineral of interest included in the model as dissolved and capable of precipitating. Conditions of the dissolution experiments (pH 7.4, I = 10 mM NaNO3, 20.0 °C) were fixed in the model. Oxidation–reduction reactions of copper and sulfur were ignored. Equilibrium solubility of Cu for each mineral was the resulting sum of dissolved Cu species.Results and discussion
Characterization of the pristine CuO NPs
The CuO NPs are roughly spherical particles ~30–50 nm in diameter as determined by TEM (Fig. 1). XRD data showed that the initial CuO NPs are crystalline, and the XRD pattern matches that of tenorite (Fig. 1). The relatively broad diffraction peaks are consistent with the TEM images indicating that the CuO is in the nano size range.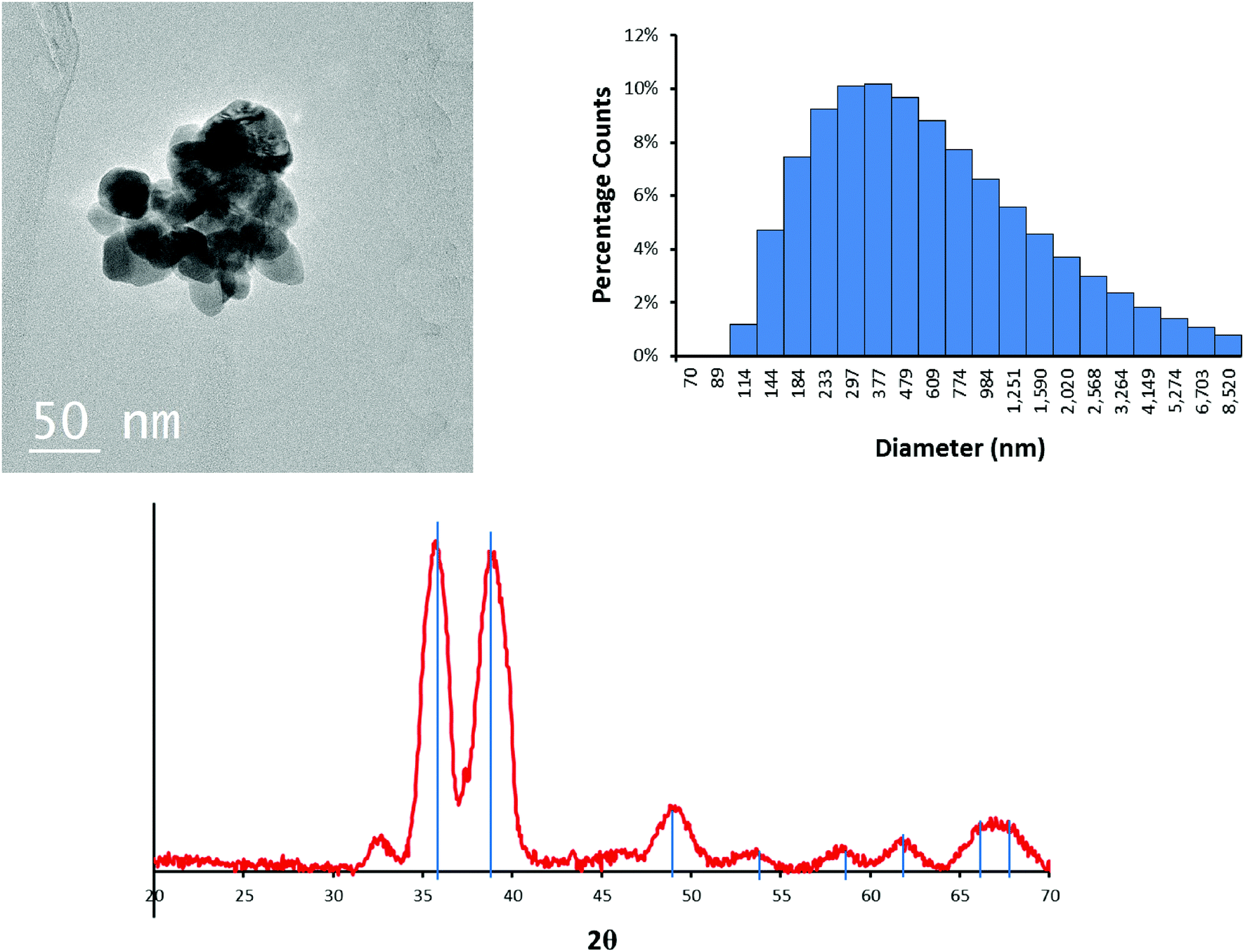 | ||
| Fig. 1 TEM image of the CuO NPs (top left), intensity averaged hydrodynamic diameter histogram (top right) of particles in 10 mM NaNO3 at pH = 7.5 measured by DLS, and the XRD pattern (bottom) of the pristine CuO NPs. Blue lines are peak matches for tenorite. | ||
The total weight loss upon heating the NPs in air to 400 °C is 3.2% (Fig. S1†). The weight loss from the initial temperature to approximately 100 °C is attributed to evaporation and removal of surface-bound water. Loss of water (before reaching 100 °C) accounted for 2.5% of the total mass. The weight loss during 100 to 200 °C was 0.74%, which is consistent with the manufacturer's claim of limited organic capping agents. Also, there was no further phase transformation of the CuO NPs upon heating to 400 °C.
Because sulfidation occurred in aqueous solution, the NPs were also characterized in water. The intensity-averaged hydrodynamic diameter of the CuO NPs in 10 mM NaNO3 at pH 7.5 is centered at 587 nm (Fig. 1). This is larger than the aggregates observed by TEM (Fig. 1), and indicates that the initial NPs were further aggregated in solution, consistent with the absence of a capping agent.
Sulfidation of CuO NPs
Copper speciation in pristine CuO NPs and the sulfidized NPs were characterized using XRD and XAS. Both lab-based (λ = 1.5418 Å) and synchrotron-based XRD (λ = 0.9744 Å) were used. To compare the XRD patterns on the same basis, the lab-based XRD spectra were converted to 2θ space corresponding to the synchrotron energy using Bragg's equation. The noisy XRD patterns collected on the lab-based instrument resulted from the lower flux of X-rays in the lab-based XRD compared to synchrotron XRD. The initial CuO NPs were identified as tenorite. Upon addition of sulfide, several new phases appeared in the sulfidized NPs with corresponding disappearance of the tenorite. The predominant phase was identified by XRD as CuS (covellite), which was confirmed by both comparison of the S/Cu 2.16 sample with a CuS model compound in Fig. 2, and by peak matching (with ICDD-no. 1074-1234, covellite CuS) from the mineral database (Fig. S2†). Sulfidation to CuS increased with increasing S/Cu ratios.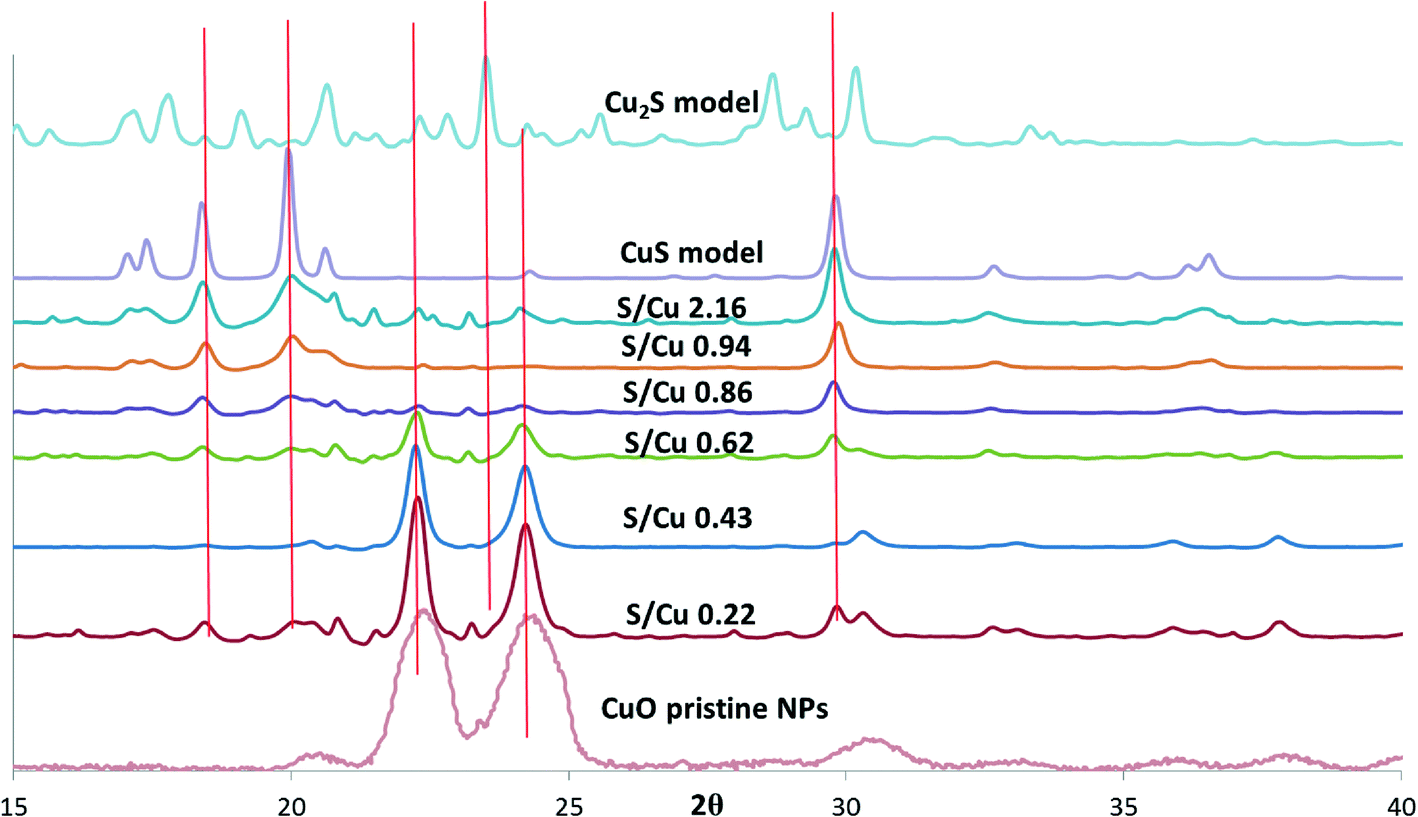 | ||
| Fig. 2 XRD patterns of CuO NPs, Cu2S, and CuS model compounds, and sulfidized particles with S/Cu ratios ranging from 0.22 to 2.16. Spectra of CuO model compounds were taken using lab-based XRD with Cu Kα incident X-rays (λ = 1.5418 Å). The spectra were converted to d spacing using Bragg's equation (λ = 2d sinθ). Then the synchrotron X-ray energy (λ = 0.9744 Å) was used to calculate the corresponding 2θ values. The intensities of the lab-based XRD patterns were scaled up by 1000 times to display on the same graph with synchrotron XRD patterns. | ||
The small peaks in the XRD pattern of the most sulfidized sample indicate the presence of other minor phases in addition to CuS. A more detailed view of the XRD pattern of the most sulfidized sample (S/Cu 2.16) is shown in Fig. 3. Besides covellite (CuS), three copper sulfate hydroxides, more or less hydrated, were identified. They are brochantite (ICSD (Inorganic Crystal Structure Database) 64688): Cu4(SO4)(OH)6, posnjakite (ICSD 100276): Cu4(SO4)(OH)6(H2O) and langite (ICSD 030724): Cu4(SO4)(OH)6(H2O). The presence of sulfate minerals indicates oxidation of some of the sulfide to sulfate in the sulfidation process, potentially during drying. Because dissolved oxygen was removed prior to the sulfidation process, Cu(II) was the likely oxidant for sulfide oxidation, resulting in formation of Cu(I). This reduction of Cu(II) to Cu(I) by sulfide has been previously observed by Luther et al.26
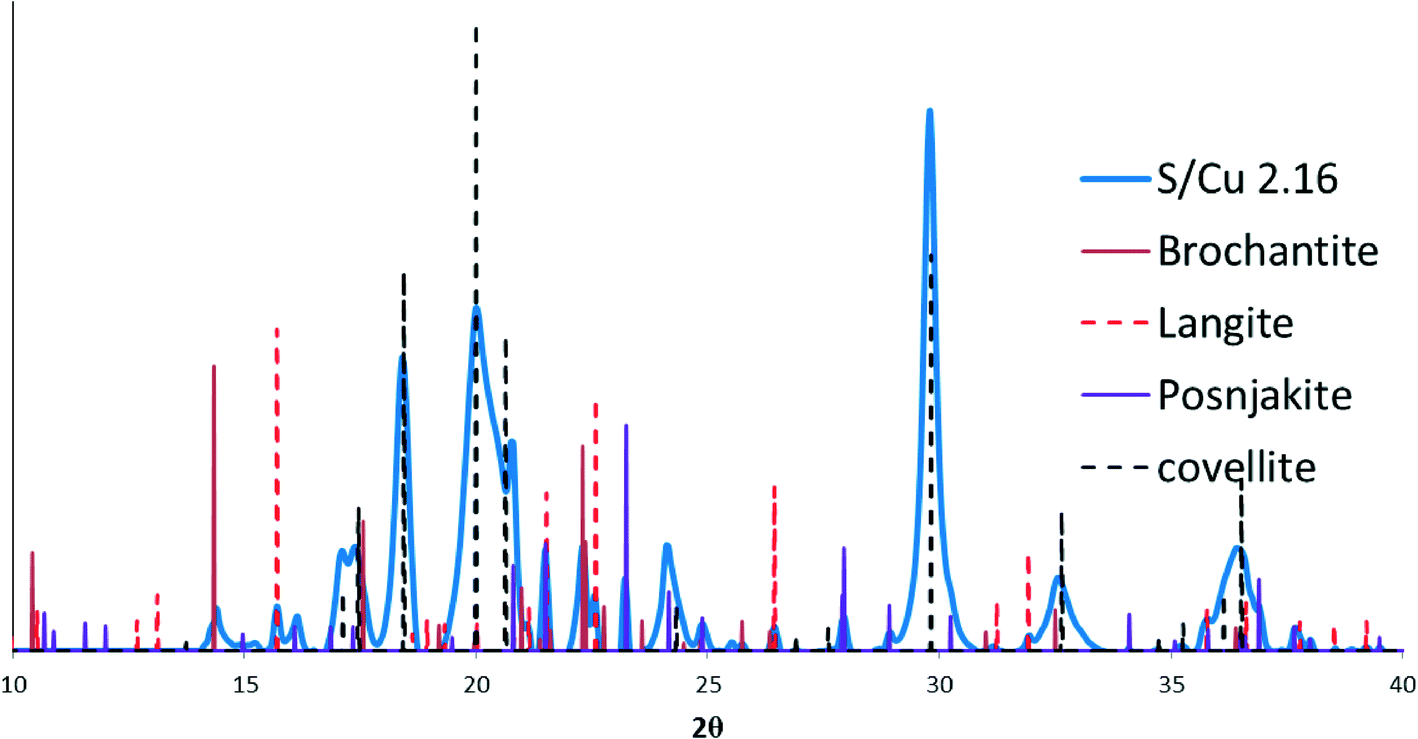 | ||
| Fig. 3 Peak matching results showing a mixture of CuS (covellite) and a copper sulfate hydroxides and two copper sulfate hydroxide hydrates: brochantite (64688): Cu4(SO4)(OH)6; posnjakite (100276): Cu4(SO4)(OH)6(H2O); and langite (030724): Cu4(SO4)(OH)6(H2O). | ||
Additional structural characterization of the sulfidized materials was carried out using XAS (Fig. 4). The extended X-ray absorption fine structure (EXAFS) spectra (Fig. 4a) clearly show a steady decrease in CuO-oscillations and the appearance of Cu–S oscillations with increasing sulfide concentration. Linear combination fitting (LCF) for k from 2 to 9 was conducted to identify the relevant phases for each sample (CuO, Cu2S or CuS). The best fits were obtained by including all three (CuO, Cu2S and CuS) of the model compounds used (Fig. S3 and Table S2†). This result suggests incomplete sulfidation and a mixture of covellite and chalcocite as reaction products. However, the linear combination fits were poor, especially for the NPs that had been sulfidized with a Cu/S ratio of less than ~1 (i.e. S/Cu ratios of 0.43, 0.63, and 0.86). The fits were somewhat better (R < 0.15) for the materials that were least sulfidized and those with S/Cu > 0.94. The relatively poor fits, especially for the intermediate Cu/S ratios, suggest that either CuxSy phases other than CuS and Cu2S had formed which were not among the model compounds used, or that the CuS and Cu2S formed was poorly ordered and therefore different than the crystalline CuS and Cu2S model compounds. The broadness of the Fourier transformed EXAFS spectra is consistent with poorly ordered materials. The most sulfidized material (S/Cu = 2.16) had only one peak in the FT corresponding to the expected Cu–S bond distance for copper sulfide (Fig. 4b). In contrast, the crystalline model compounds used all have FT features over an R + ΔR range of 6–8 Å indicating longer range order (Fig. 4b). This difference in long-range order may explain the relatively poor linear combination fits. In addition, the copper sulfate hydroxides identified by XRD were not represented in the model compound library used for LCF. To confirm the transition from CuO to CuS upon sulfidation, the first shell was fit using theoretical scattering paths generated by SixPack for Cu–S and Cu–O shells fixed at 1.95 and 2.25 Å, respectively. The Debye–Waller and the sigma parameters were set to 0.9 and 0.008, respectively. There is a clear decrease in the Cu–O coordination number while a simultaneous increase in the Cu–S coordination number as the particles become increasingly sulfidized (Fig. 4c). Even though a clear distinction between covellite and chalcocite was not possible, it is certain from XAS that the Cu–O character of the particles is readily replaced by Cu–S and it is relatively poorly ordered material.
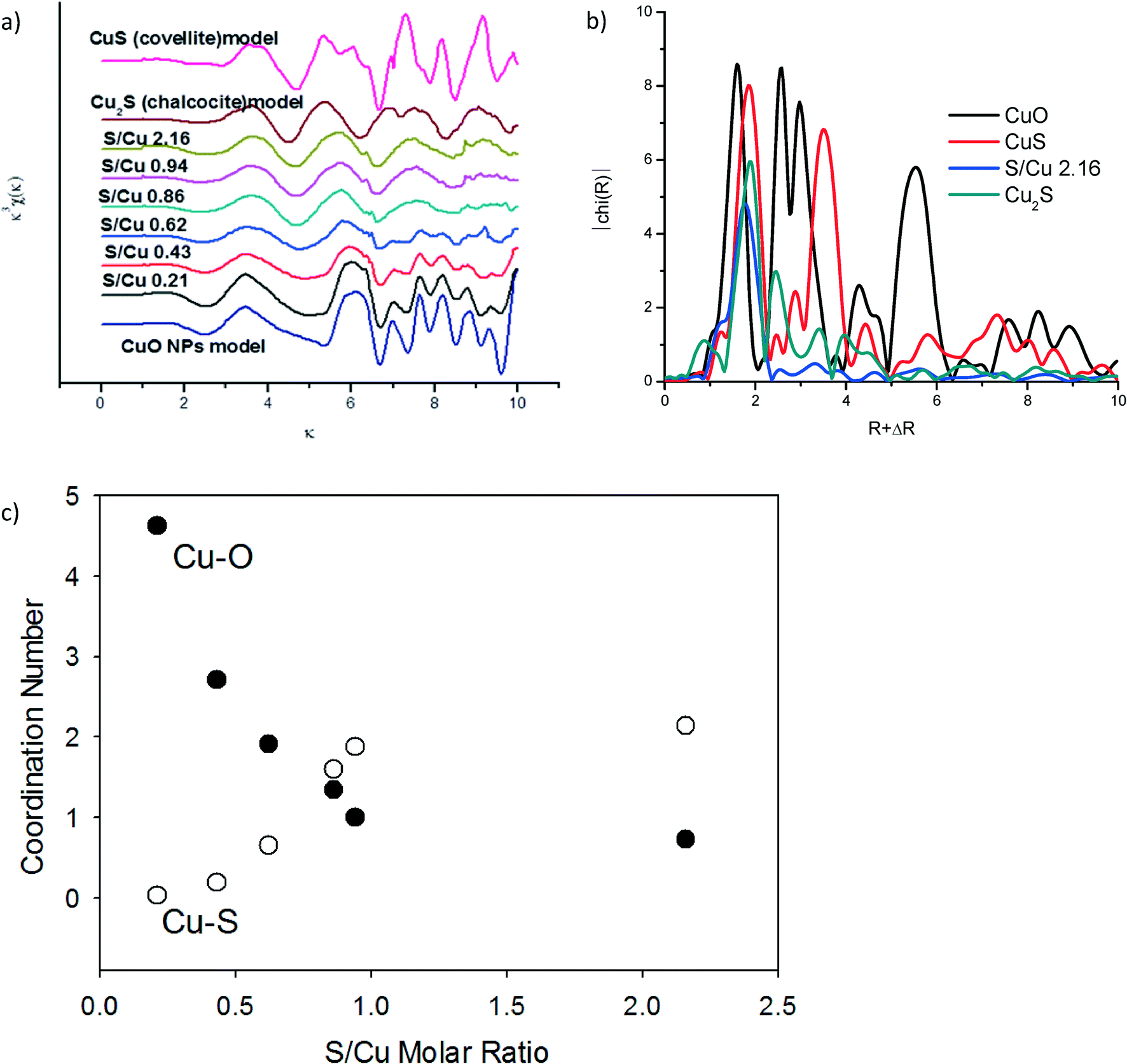 | ||
| Fig. 4 (a) EXAFS spectra of CuO, Cu2S (chalcocite), and CuS (covellite) model compound, and the partially sulfidized CuO particles for S/Cu from 0.22 to 2.16. (b) Fourier transformed EXAFS of the model compounds and the most sulfidized sample: S/Cu 2.16. (c) Fitted coordination numbers for theoretical Cu–S and Cu–O scattering paths for the partially sulfidized CuO particles for S/Cu from 0.22 to 2.16. | ||
The oxidation state of Cu in CuxSy species can vary from +I to +II. Both Cu2S (chalcocite) and CuS (covellite) have been described as containing predominantly Cu(I).26,27 The XANES region of our XAS data suggest that this is not the case for the CuxSy formed here. There was more than a 2 eV difference between the K edge of a Cu2S model compound and that of the CuxSy formed here at S/Cu = 2.16 (Fig. S3†). The oxidation state of Cu in the most sulfidized particles is therefore greater than one, so at least some, if not all, of the Cu in the sulfidized material is Cu(II).28
The EXAFS suggested that for higher S/Cu ratios Cu is primarily bound to S. This Cu–S phase was poorly ordered as evidenced by the lack of order after R + ΔR over 2.5 Å. XRD results showed formation of CuS, primarily. TEM analysis was performed to further assess the structure and morphology of the CuS phases formed.
TEM analysis of the pristine CuO NPs and the most sulfidized NP (S/Cu 2.16) (Fig. 5) show that the average size of the CuO NPs is 30–50 nm (Fig. 5a) and that they are crystalline (Fig. 5b). TEM images of the S/Cu 2.16 NPs indicated a range of NP sizes and the presence of both poorly ordered (Fig. 5c) and crystalline phases (Fig. 5d). Additional TEM images are provided in the ESI† (Fig. S4). In some cases, the sizes and range of sizes for the sulfidized NPs were found to be similar to those of the pristine CuO NPs, which suggests a direct solid–fluid sulfidation process in water at pH = 11.9 and the Cu/S ratios used here. The wide distribution of NP sizes (Fig. S4†) also suggests that a dissolution/precipitation mechanism is also occurring.
 | ||
| Fig. 5 TEM images of the pristine CuO NPs showing a) primary particle size of CuO is 30–50 nm, b) crystalline nature of the CuO material, c) poorly ordered CuxSy phases formed upon sulfidation, and d) crystalline CuxSy phases including CuS as determined from lattice fringe d-spacings of 3.05 Å. | ||
To confirm the identity of the crystalline phases in Fig. 5, the d-spacing of the lattice fringes was determined in selected regions using Gatan DigitalMicrograph. The d-spacings determined for the pristine CuO particles in Fig. 5a and b were 2.30 Å and 2.50 Å, corresponding with tenorite's two primary peaks. Analysis of Fig. 5d yielded a d-spacing of 3.05 Å, which is consistent with the (102) plane for covellite (CuS). The d-spacings for other crystalline materials in the TEM were 2.80 Å, which is also consistent with the CuS (covellite) (103) plane. The presence of some crystalline CuS is consistent with the presence of covellite peaks in the XRD pattern.
Dissolution
The rate and extent of dissolution of the pristine CuO NPs and the sulfidized NPs were determined at neutral pH in 10 mM NaNO3 (Fig. 6). In the presence of dissolved oxygen, after 2 weeks, dissolution of CuO and CuO/CuS NPs apparently reaches equilibrium. This time scale is longer than that observed for ZnO/ZnS NP dissolution but similar to that of Ag NP oxidative dissolution.29,30 CuO does not dissolve significantly at neutral pH without ligands present (e.g., amino acids). The highest measured dissolved copper released by CuO NPs was only 0.02 mg L−1. The partially and fully sulfidized particles have a higher apparent solubility than CuO. This result contrasts with expectations for metal sulfides and with previous studies which showed that sulfidation reduces dissolution and ion release from ZnO and Ag NPs.16,30 Wang et al. also found lower solubility of sulfidized CuO NPs compared to pristine CuO NPs, as well as reduced cytotoxicity of CuS compared to CuO NPs in RPMI 1640 medium.31 It also contradicts calculated solubility expected for the sulfide minerals Cu2S and CuS compared to CuO32 under the conditions used for dissolution (Table 1).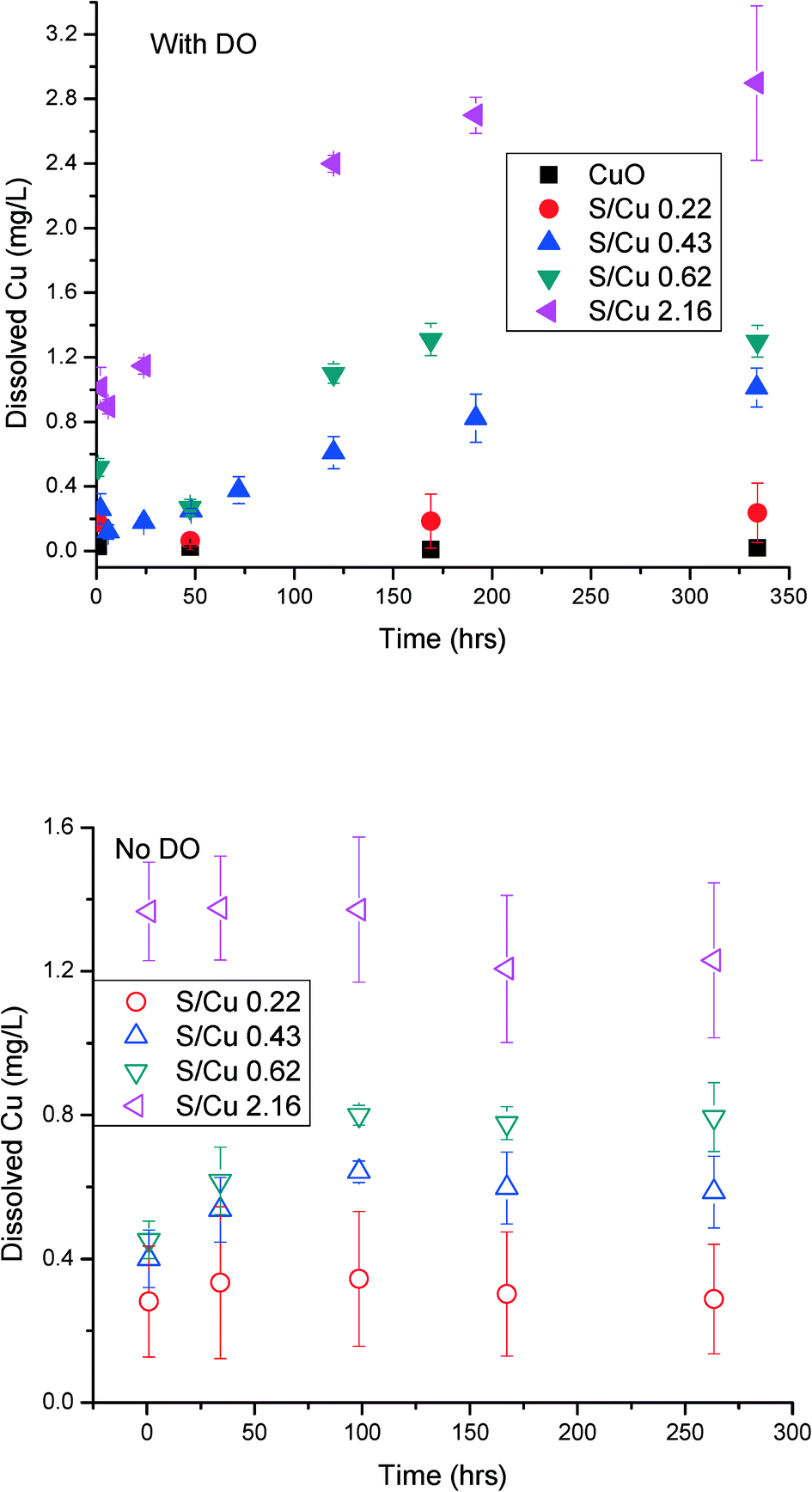 | ||
| Fig. 6 Dissolution of pristine CuO NPs and sulfidized CuO NPs (S/Cu 0.22, 0.43, 0.62, and 2.16) over two weeks in the presence of dissolved oxygen (top) and with deoxygenated water (bottom). The initial concentration of particles was 100 mg L−1, in 10 mM NaNO3 and 2 mM HEPES buffer. The solution pH for all dissolution studies was 7.4. The error bar indicates ±standard deviation of duplicate reactors. Note that the y-axis scales are not the same in both figures to better indicate the slope of the dissolution curve. | ||
| Solid | LogKsp |
∑[Cu]diss (M) | Dominant aq. species (% total) |
|---|---|---|---|
|
a CuO + 2H+ = Cu2+ + 2H2O.
b CuS + H+ = Cu2+ + HS−.
c H+ + 0.15S(s) + 0.85Cu1.18S = Cu2+ + HS−.
d LogKsp taken from Shea and Helz.33
e Cu2S + H+ = 2Cu+ + HS−.
f Cu4SO4(OH)6 + 6H+ = 4Cu2+ + SO42− + 6H2O.
g Cu4SO4(OH)6·H2O + 6H+ = 4Cu2+ + SO42− + 7H2O.
h Cu4SO4(OH)6·H2O + 6H+ = 4Cu2+ + SO42− + 7H2O.
i LogKsp taken from Zittlau et al.34
|
|||
| Tenorite | 7.644a | 2.45 × 10−7 | Cu2+ (67), CuOH+ (30) |
| Covellite | −22.300b | 1.91 × 10−15 | Cu2+ (68), CuOH+ (31) |
| am-CuS | −18.90c,d | 8.49 × 10−14 | N/A |
| Chalcocite | −34.920e | 8.39 × 10−15 | Cu+ (100) |
| Brochantite | 15.220f | 5.41 × 10−6 | Cu2+ (65), CuOH+ (30) |
| Langite | 17.489g | 1.52 × 10−5 | Cu2+ (62), CuOH+ (29) |
| Posnjakite | 17.6h,i | 1.25 × 10−5 | Cu2+ (63), CuOH+ (29) |
Both the solubility and the rate of dissolution increased with increased sulfidation. The most sulfidized particles (S/Cu 2.16) (~85% sulfidized Cu) have the highest solubility of 2.9 mg L−1 (4.6 × 10−5 M). This solubility is much higher than the modeled values for either crystalline CuS or crystalline Cu2S (Table 1). It is also higher than expected for poorly crystalline Cu1.18S.33 This solubility is more consistent with that predicted for the high solubility Cu sulfate hydroxide species identified by XRD and may explain the higher than expected solubility. However, it appears from XRD that these sulfate hydroxide species are not a large fraction of the solids formed. Moreover, the washing processes prior to the measurement of solubility should have partially or completely removed these soluble species. Assuming that the DI water rinse solutions come to equilibrium with small fraction of the total solid mass as Cu–SO4–OH minerals, complete dissolution could be achieved during the first or second rinse. If equilibrium is not obtained due to kinetic limitations, the most likely solid to remain is the less soluble brochantite. Copper solubility observed for the S/Cu = 0.22 dissolution experiment, approximately 0.3 mg L−1, could be explained by a small fraction of brochantite in the solid phase. However, the solubility measured in the more sulfidized systems cannot be reconciled by thermodynamics of observed crystalline phases. Therefore, dissociation of Cu2+ from crystalline CuS or copper sulfate hydroxide alone is not likely controlling the solubility for the sulfidized NPs in our experiments. The high dissolved copper concentration could result from either a higher apparent solubility due to formation of poorly ordered CuxSy phases, the presence of very small CuS nanoclusters, or oxidative dissolution of the formed CuS NPs according to eqn (1) and (2).
| Cu2S + 2.5O2 + 2H+ = 2Cu2+ + SO42− + H2O | (1) |
| CuS + 2O2 = Cu2+ + SO42− | (2) |
In order to evaluate the potential for oxidative dissolution, the dissolution of the NPs was also measured after purging the dissolved oxygen from the water (Fig. 6 bottom). The dissolution rate and extent was lower for S/Cu ratios of 0.43, 0.62, and 2.16 compared to dissolution in the presence of DO. Thus, it appears that an oxidative dissolution mechanism occurs in the dissolution of sulfidized CuO NPs when dissolved oxygen is present. This is consistent with a the finding of Wang et al.,31 who showed that addition of H2O2 to the sulfidized CuO NPs rapidly solubilized those particles.
Non-zero dissolved Cu concentrations without dissolved oxygen was also noted. In the absence of oxygen, solubility equilibrium was reached rapidly: within 1 h for S/Cu 2.16 and within 100 h for other ratios. Because oxidative dissolution was excluded under anoxic conditions, the apparent rapid dissolution is most likely due to the combination of 1) presence of poorly ordered, higher solubility phases, 2) presence of copper sulfate hydroxide species, and 3) formation of very small clusters of CuS (e.g. tetrameric Cu–S structures noted by Luther et al.26) that can pass through the filter with a MWCO of 3 kDa. The latter suggestion is most consistent with the non-zero initial concentration in the dissolution experiments. Small (passing a 3 kDa MWCO filter) metal sulfide clusters were previously found to account for more than 40% of the total metal in river water in Connecticut, USA.35
Mechanistic insights on the sulfidation process
Information from the combined XAS, XRD, TEM, and dissolution measurements provide some mechanistic insights on sulfidation of CuO NPs. EXAFS indicates the presence of poorly ordered CuxSy. XRD shows the presence of some crystalline CuS (covellite) as well as copper sulfate hydroxide phases. TEM confirms a mixture of poorly ordered and crystalline CuS phases. TEM also indicates that some of the sulfidized NPs are similar in size to that of the pristine CuO NPs, while others are not, suggesting that sulfidation occurs through dissolution and reaction with sulfide as well as a direct solid–fluid sulfidation. This kind of Kirkendall effect was demonstrated previously for copper sulfidation in organic solvents under an argon atmosphere36 and in water under mild conditions.24,25 This study suggests that this mechanism is also occurring for sulfidation in water under the conditions used here. Dissolution measurements show the possibility of formation of small clusters (<1 nm).A potential set of reactions for the sulfidation of CuO consistent with the species identified are given in eqn (3) through (6).
| 8CuO + 5S2− + 8H+ = 4Cu2S + SO42− + 4H2O | (3) |
| CuO + S2− + 2H+ = CuS + H2O | (4) |
| 8Cu2+ + 5S2− + 4H2O = 4Cu2S + SO42− + 8H+ | (5) |
| Cu2+ + S2− = CuS | (6) |
Eqn (3) and (5) are redox reactions in which Cu(II) is reduced to Cu(I) while S(−II) in sulfide is oxidized to S(VI) in sulfate. Eqn (4) and (6) are not redox reactions. The standard Gibbs free energy of reaction of eqn (3) and (4) are −1432 and −246.8 kJ mol−1, respectively. For eqn (4) under our reaction conditions, ΔGr = ΔGr0 + RT log(1/([S2−][H+]2)) = −92.63 kJ mol−1 indicating a spontaneous reaction. Thus, the formation of CuS and Cu2S are thermodynamically favorable. At pH 11.9 and for all S/Cu ratios, the free Cu2+ ion concentration is extremely low (as calculated by MINEQL+). Hence eqn (3) and (4) may be the major pathways for sulfidation under the experimental conditions used here. However, the formation of larger CuS particles than the initial CuO and small CuS clusters suggests that the dissolution and precipitation reaction pathways in eqn (5) and (6) are also active to some degree. The proposed sulfidation process is summarized in Fig. 7.
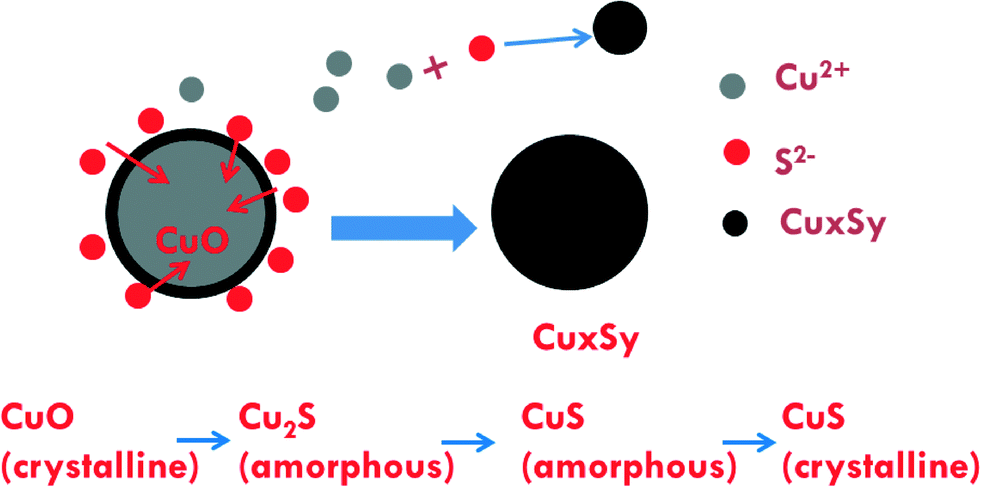 | ||
| Fig. 7 Proposed sulfidation process of CuO NPs with the dominant pathway being direct solid–fluid sulfidation accompanied by a lower amount of dissolution–precipitation formation of small CuxSy clusters. | ||
Environmental implications
Sulfidation and its impacts on the properties of the sulfidized NPs have important environmental implications. Transformations and the environmental risks of metal and metal oxide NPs are often controlled by sulfidation reactions.17,37–39 The facile sulfidation of commercial 40 nm CuO NPs at ambient temperature in water to form a variety of CuxSy NPs, soluble copper-hydroxide-sulfates, and likely the formation of CuxSy nanoclusters suggests that these different copper sulfide species may be those likely found in the environment, rather than the pristine CuO NPs. This finding is consistent with expectations based on previous studies showing that copper sulfide is the predominant form of copper in sediment under sulfate-reducing conditions, and in sewer pipes and wastewater treatment plants.40–42Sulfidation increased the dissolved fraction of copper compared to the pristine CuO NPs under environmentally relevant neutral pH. This finding is opposite to that found for Ag and ZnO NPs after sulfidation, where sulfidation decreases solubility and metal availability. The increased release of Cu2+ and/or CuS nanoclusters from sulfidized NPs compared to CuO NPs suggests that toxicity studies with pristine CuO may be misleading in environments where sulfidation is expected, considering the complex mixture of sulfidized products that may be formed at pH and redox potentials expected in the environment (Fig. 8) and their potentially different toxicity to organisms.
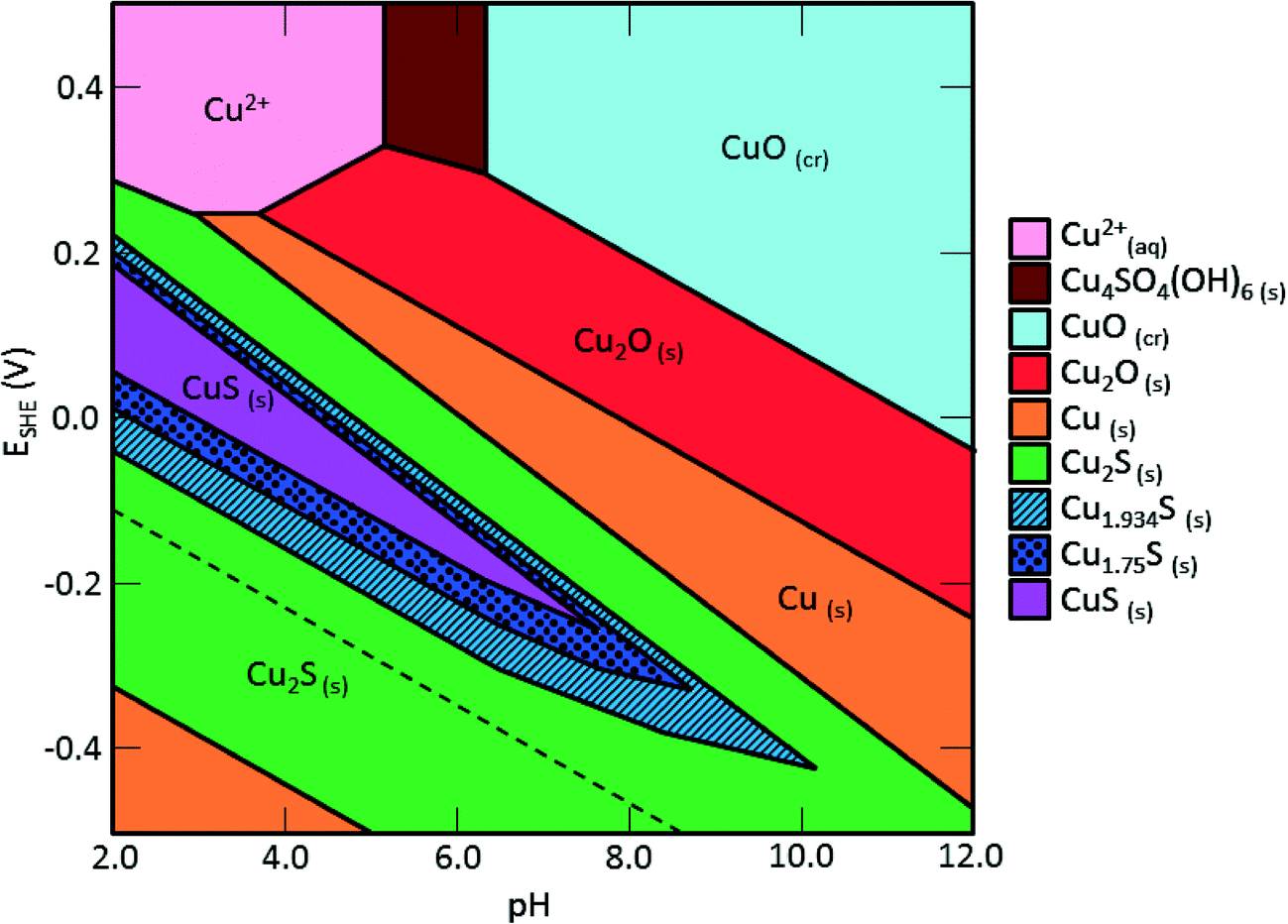 | ||
| Fig. 8 Pourbaix diagram of the Cu–O–S system generated by Hydromedusa. [HS−]tot = 2 mM, [Cu]tot = 1 mM, [Na+] = [NO3−] = 10 mM. Under the conditions of the sulfidation used here and at environmentally relevant pH and Eh, there are a number of non-stoichiometric CuxSy species that may form. | ||
The formation of some small CuxSy structures (5–10 nm) and nanoclusters (<1 nm) were observed by TEM or implied in the dissolution experiments. If these structures are formed during sulfidation of CuO in the environment, they may have different transport properties. Both the pristine and sulfidized NPs aggregated (intensity average size of 587 and 487 nm, respectively in 10 mM NaNO3) and rapidly settled from solution (within 30 min). However, the smaller and poorly crystalline CuxSy structures formed may have increased mobility and higher bioavailability compared with the larger particles of CuO or CuS formed.
Sulfidation has proved to be an important transformation for some metal and metal oxide nanoparticles. This study suggests that CuO may become sulfidized in the environment, and that the resulting properties relevant to toxicity, e.g. solubility, will be affected. Thus it is prudent to use environmentally transformed nanoparticles in fate, transport, and toxicity studies rather than focusing solely on the pristine materials. However the implications of sulfidation differ for Ag, ZnO, and CuO NPs, with sulfidation decreasing the solubility of Ag and ZnO NPs, but increasing apparent solubility for CuO. Studies are also still needed to (1) identify the nature of the CuxSy nanoclusters, (2) assess the toxicity of sulfidized CuO NPs and CuxSy nanoclusters; (3) assess the stability of very small metal sulfide clusters (Ag, Zn, and Cu) against oxidation under environmental and biological conditions; and (4) assess how sulfidation of CuO NPs occurs in situ at relevant CuO/S concentration ratios, and how this affects their bioavailability (e.g., plant uptake) under realistic exposure scenarios.
Acknowledgements
This material is based on work supported by the US EPA Science to Achieve Results program (R834574), Transatlantic Initiative for Nanotechnology and the Environment (TINE), and the National Science Foundation (NSF and the Environmental Protection Agency (EPA) under NSF Cooperative Agreement EF-0830093, Center for the Environmental Implications of Nanotechnology (CEINT). Any opinions, findings, conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the NSF or the EPA. This work has not been subjected to EPA review and no official endorsement should be inferred. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL) beamline 4-3 and 11-2, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. We also thank the beamline scientists at SSRL (BL 4-3 and 11-2) for their support. The authors also acknowledge use of the facilities and the assistance of Christopher Winkler at the Nanoscale Characterization and Fabrication Laboratory at Virginia Polytechnic Institute.References
- E. Ebrahimnia-Bajestan, H. Niazmand, W. Duangthongsuk and S. Wongwises, Int. J. Heat Mass Transfer, 2011, 54, 4376–4388 CrossRef CAS PubMed.
- K. Guo, Q. Pan, L. Wang and S. Fang, J. Appl. Electrochem., 2002, 32, 679–685 CrossRef CAS.
- S. Magdassi, M. Grouchko and A. Kamyshny, Materials, 2010, 3, 4626–4638 CrossRef CAS PubMed.
- A. Saha, D. Saha and B. C. Ranu, Org. Biomol. Chem., 2009, 7, 1652–1657 CAS.
- K. Sahithi, M. Swetha, M. Prabaharan, A. Moorthi, N. Saranya, K. Ramasamy, N. Srinivasan, N. C. Partridge and N. Selvamurugan, J. Biomed. Nanotechnol., 2010, 6, 333–339 CrossRef CAS PubMed.
- Y. Wu, C. Wadia, W. L. Ma, B. Sadtler and A. P. Alivisatos, Nano Lett., 2008, 8, 2551–2555 CrossRef CAS PubMed.
- M. E. Leitch, E. Casman and G. V. Lowry, J. Nanopart. Res., 2012, 14 Search PubMed.
- I. A. Mudunkotuwa, J. M. Pettibone and V. H. Grassian, Environ. Sci. Technol., 2012, 46, 7001–7010 CrossRef CAS PubMed.
- O. Bondarenko, K. Juganson, A. Ivask, K. Kasemets, M. Mortimer and A. Kahru, Arch. Toxicol., 2013, 87, 1181–1200 CrossRef CAS PubMed.
- A. J. Bone, B. P. Colman, A. P. Gondikas, K. M. Newton, K. H. Harrold, R. M. Cory, J. M. Unrine, S. J. Klaine, C. W. Matson and R. T. Di Giulio, Environ. Sci. Technol., 2012, 46, 6925–6933 CrossRef CAS PubMed.
- C. Gunawan, W. Y. Teoh, C. P. Marquis and R. Amal, ACS Nano, 2011, 5, 7214–7225 CrossRef CAS PubMed.
- H. Y. Zhang, Z. X. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. J. Lin, X. Wang, Y. P. Liao, M. Y. Wang, L. J. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Madler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS PubMed.
- S. M. Webb, Phys. Scr., T, 2005, 115, 1011–1014 CrossRef.
- H. L. Karlsson, P. Cronholm, J. Gustafsson and L. Moller, Chem. Res. Toxicol., 2008, 21, 1726–1732 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, B. P. Colman, A. L. Dale, L. Truong, X. Y. Yang, A. J. Bone, G. E. Brown, R. L. Tanguay, R. T. Di Giulio, E. S. Bernhardt, J. N. Meyer, M. R. Wiesner and G. V. Lowry, Environ. Sci. Technol., 2013, 47, 13440–13448 CrossRef CAS PubMed.
- R. Ma, C. Levard, F. M. Michel, G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2013, 47, 2527–2534 CrossRef CAS PubMed.
- J. Y. Liu, K. G. Pennell and R. H. Hurt, Environ. Sci. Technol., 2011, 45, 7345–7353 CrossRef CAS PubMed.
- B. C. Reinsch, C. Levard, Z. Li, R. Ma, A. Wise, K. B. Gregory, G. E. Brown and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 6992–7000 CrossRef CAS PubMed.
- R. G. Pearson, J. Chem. Educ., 1968, 45, 643–648 CrossRef CAS.
- E. Donner, D. L. Howard, M. D. de Jonge, D. Paterson, M. H. Cheah, R. Naidu and E. Lombi, Environ. Sci. Technol., 2011, 45, 7249–7257 CrossRef CAS PubMed.
- C. O. Dimkpa, D. E. Latta, J. E. McLean, D. W. Britt, M. I. Boyanov and A. J. Anderson, Environ. Sci. Technol., 2013, 47, 4734–4742 CrossRef CAS PubMed.
- P. Leidinger, R. Popescu, D. Gerthsen, H. Lunsdorf and C. Feldmann, Nanoscale, 2011, 3, 2544–2551 RSC.
- S. H. Jiao, L. F. Xu, K. Jiang and D. S. Xu, Adv. Mater., 2006, 18, 1174 CrossRef CAS.
- H. T. Zhu, J. X. Wang and D. X. Wu, Inorg. Chem., 2009, 48, 7099–7104 CrossRef CAS PubMed.
- Z. H. Yang, D. P. Zhang, W. X. Zhang and M. Chen, J. Phys. Chem. Solids, 2009, 70, 840–846 CrossRef CAS PubMed.
- G. W. Luther, S. M. Theberge, T. F. Rozan, D. Rickard, C. C. Rowlands and A. Oldroyd, Environ. Sci. Technol., 2002, 36, 394–402 CrossRef CAS.
- S. W. Goh, A. N. Buckley, R. N. Lamb, R. A. Rosenberg and D. Moran, Geochim. Cosmochim. Acta, 2006, 70, 2210–2228 CrossRef CAS PubMed.
- P. Kumar, R. Nagarajan and R. Sarangi, J. Mater. Chem. C, 2013, 1, 2448–2454 RSC.
- I. A. Mudunkotuwa, T. Rupasinghe, C. M. Wu and V. H. Grassian, Langmuir, 2012, 28, 396–403 CrossRef CAS PubMed.
- C. Levard, B. C. Reinsch, F. M. Michel, C. Oumahi, G. V. Lowry and G. E. Brown, Environ. Sci. Technol., 2011, 45, 5260–5266 CrossRef CAS PubMed.
- Z. Wang, A. von dem Bussche, P. K. Kabadi, A. B. Kane and R. H. Hurt, ACS Nano, 2013, 7, 8715–8727 CrossRef CAS PubMed.
- G. E. Lagos, C. A. Cuadrado and M. V. Letelier, J. – Am. Water Works Assoc., 2001, 93, 94–103 Search PubMed.
- D. Shea and G. R. Helz, Geochim. Cosmochim. Acta, 1989, 53, 229–236 CrossRef CAS.
- A. H. Zittlau, Q. Shi, J. Boerio-Goates, B. F. Woodfield and J. Majzlan, Chem. Erde, 2013, 73, 39–50 CrossRef CAS PubMed.
- T. F. Rozan, G. Benoit and G. W. Luther, Environ. Sci. Technol., 1999, 33, 3021–3026 CrossRef CAS.
- G. Cheng and A. R. H. Walker, Anal. Bioanal. Chem., 2010, 396, 1057–1069 CrossRef CAS PubMed.
- R. Kaegi, A. Voegelin, C. Ort, B. Sinnet, B. Thalmann, J. Krismer, H. Hagendorfer, M. Elumelu and E. Mueller, Water Res., 2013, 47, 3866–3877 CrossRef CAS PubMed.
- E. Lombi, E. Donner, E. Tavakkoli, T. W. Turney, R. Naidu, B. W. Miller and K. G. Scheckel, Environ. Sci. Technol., 2012, 46, 9089–9096 CrossRef CAS PubMed.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- A. F. Hofacker, A. Voegelin, R. Kaegi, F. A. Weber and R. Kretzschmar, Geochim. Cosmochim. Acta, 2013, 103, 316–332 CrossRef CAS PubMed.
- A. E. Lewis, Hydrometallurgy, 2010, 104, 222–234 CrossRef CAS PubMed.
- F. A. Weber, A. Voegelin, R. Kaegi and R. Kretzschmar, Nat. Geosci., 2009, 2, 267–271 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Molar ratios of Cu to S used in sulfidation, TGA weight loss over temperature of CuO NPs, peak matching of XRD standard peak from mineral database for S/Cu = 0.22 and S/Cu = 2.16 sulfidized particles, linear combination fitting results and XANES spectra from XAS, additional TEM images of the sulfidized CuO particles. See DOI: 10.1039/c4en00018h |
| This journal is © The Royal Society of Chemistry 2014 |