Improvements in the detection and characterization of engineered nanoparticles using spICP-MS with microsecond dwell times†
M. D.
Montaño
*a,
H. R.
Badiei
b,
S.
Bazargan
b and
J. F.
Ranville
a
aColorado School of Mines, Dept. of Chemistry and Geochemistry, Golden, CO 80401, USA. E-mail: mamontan@mines.edu
bPerkinElmer, Inc., Woodbridge, Ontario L4L 8H1, Canada
First published on 4th June 2014
Abstract
The imminent release of engineered nanomaterials (ENPs) into the environment has raised several questions regarding their fate, transport, and toxicity. However, their small size, expected low concentrations (ng L−1), and the high environmental background of naturally occurring nanomaterials make detection and characterization difficult. In recent years, single particle ICP-MS (spICP-MS) has been developed as a promising technique to detect and characterize engineered nanoparticles in biological and environmental matrices. Improvements in the spICP-MS technique were made in this study by employing 100 microsecond dwell times. Commercially available hardware and software were developed to fully capture multiple data points over the fast transient (~500 μs) nanoparticle events, which provides accurate particle sizing and counting. Reducing the background signal facilitated the characterization of Ag NPs even in the presence of ten-fold higher Ag+ concentration. By improving the time resolution between particle events, the upper limit of the dynamic range of Au NP concentration was increased to several μg L−1. These short dwell times also provide detection of two elements in the same nanoparticle, opening the door for possible environmental applications with the prospect of obtaining particle-by-particle elemental compositions. These improvements help further establish spICP-MS as a leading analytical technique for the detection and characterization of metal-containing ENPs, and introduces new possibilities for differentiating engineered nanomaterials from their naturally occurring analogues.
Nano impactDetection and characterization of engineered nanomaterials in the environment requires sophisticated analytical techniques capable of analysis across a wide working range of concentrations and amidst a high background of naturally occurring nanoparticles. Single particle ICP-MS has been a suitable technique for environmental ENP analysis, but has been ineffective at high particle number concentrations and amidst high dissolved analyte backgrounds. By utilizing microsecond dwell times, particle resolution is greatly improved, increasing the working range of this technique, while also significantly reducing signal generated from dissolved analyte. In addition, the utilization of these fast dwell times allows for the simultaneous detection of multiple elements within a single particle, opening the door for a possible means of differentiating engineered and naturally occurring nanomaterials. |
Introduction
The past two decades have witnessed an exponential growth in the use of engineered nanomaterials in commercial products.1 The novel properties that nanotechnology exhibits have proven to be worthwhile investments scientifically and economically.2 With the rapid pace of manufacturing and incorporation into everyday products, release of ENPs into the environment is likely inevitable.3–6 However, the environmental and ecological risk associated with these materials is still not well understood.7,8 Uncertainty regarding how many nanomaterial-containing products are in the commercial market and the minimal information on release rates from products, severely limits application of materials flow analysis in making accurate predictions of environmental concentrations.6,9–13 Despite these uncertainties, risk assessment of ENPs have relied heavily on these modeling approaches mainly because of the lack of analytical methods to quantify environmental concentrations of ENPs. In terms of analytical limitations, ENPs are expected to enter into systems containing naturally occurring nanomaterials at concentrations several orders of magnitude above the expected concentrations of the ENPs (low ng L−1) and most analytical methods are not selective to ENPs only.9,14 Developing techniques and methods to overcome these analytical challenges has become a priority.15,16Single-particle ICP-MS (spICP-MS) harnesses the specificity and sensitivity of ICP-MS to detect and characterize ENPs at low-ppt concentrations.17,18 Initially developed for aerosol particle analysis, spICP-MS has evolved for application to aqueous and complex matrices.19–24 Utilizing time-resolved analysis with short dwell times, a discrete pulse of intensity originating from particle vaporization and ionization can be detected and the signal generated by the ions can be correlated to particle mass. Assuming an appropriate particle geometry (i.e. spherical), particle diameter can be calculated utilizing transport efficiency determined from a standard Au NP and instrument calibration with dissolved analyte standards.25,26 This technique has recently been used for metallic (gold and silver) and metal oxide (TiO2, CeO2) nanoparticles, carbon nanotubes, and silver nanowires in matrices ranging from animal tissues, natural and processed waters, and macroinvertebrates.26–33 Despite its utility, specifically for environmentally relevant samples, spICP-MS has several analytical obstacles that limit its applicability.25,34
In previous literature reports, the most commonly used dwell time has been 10 milliseconds. A number of studies suggest that under most of the commonly applied ICP-MS conditions the ion cloud generated by the particle spans only a few hundred microseconds.35–38 Consequently, 10 millisecond dwell time windows are simply too large, allowing for the possibility of several nanoparticle events to occur within this time span and therefore requiring sample dilution, which may alter the representativeness of the sample and produce low counting statistics.36 The low counting statistics can be somewhat alleviated by increasing the analysis time, but this limits the use of spICPMS as a high throughput technique and also results in large data sets that must contain >90% background readings.
Some investigators have attempted to improve the spICP-MS method by using dwell times on the order of 3–5 milliseconds. While reducing the chance of coincidence, it introduces the problem of peak splitting, whereby the particle event is divided between two dwell times.35 This necessitates recombination of split peaks and to date this has only been achieved by a laborious manual process of scanning a dataset for these events, with some researchers describing in-house spreadsheet data processing. Consequently, most investigators have argued for analysis of dilute solutions utilizing dwell times >5 millisecond or so.35
A very different approach reduces the dwell times to time-scales shorter than the duration of the nanoparticle event. In contrast to millisecond spICP-MS, which relates a single point of intensity above the background to particle mass, microsecond spICP-MS produces a distribution of pulse intensities that correspond to partial sections of the ion cloud reaching the detector. In this case, multiple readings must be combined to reconstruct the particle pulse and thus obtain mass and particle number data. Previous reports have accomplished this feat through the use of sophisticated electronics and/or instrumentation that may not be widely available for most research institutions.36,39 Olesik et al. demonstrated the capability of microsecond data acquisitions by amplifying the analog output from a discrete dynode detector and converting the signal to a voltage which could then be measured by a digital oscilloscope at a rate of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz.36 Borovinskaya et al. developed a prototype time-of-flight ICP-MS for the analysis of short transient signals (~33 μs) which could also monitor multiple isotopes/elements.40 In this paper we present a microsecond spICP-MS methodology using a standard quadrapole ICP-MS and recently developed commercial software to facilitate microsecond spICP-MS. Furthermore we highlight its advantages for reducing particle coincidence, reducing background interference from dissolved ions, and the detection of multiple elements or isotopes in single particles. With this latter capability and some knowledge of naturally occurring elemental ratios in mineral nanoparticles, it may be possible to use this technique for detecting ENPs amidst a high background of naturally occurring particles.
000 Hz.36 Borovinskaya et al. developed a prototype time-of-flight ICP-MS for the analysis of short transient signals (~33 μs) which could also monitor multiple isotopes/elements.40 In this paper we present a microsecond spICP-MS methodology using a standard quadrapole ICP-MS and recently developed commercial software to facilitate microsecond spICP-MS. Furthermore we highlight its advantages for reducing particle coincidence, reducing background interference from dissolved ions, and the detection of multiple elements or isotopes in single particles. With this latter capability and some knowledge of naturally occurring elemental ratios in mineral nanoparticles, it may be possible to use this technique for detecting ENPs amidst a high background of naturally occurring particles.
Results and discussion
Improvements in particle resolution
With conventional millisecond dwell times, spICP-MS analyses have been relegated to using relatively low particle number concentrations (i.e. <5 × 106 particles L−1 for a 100 nm Au particle at 10 ms dwell times).These dilute concentrations are required to avoid coincidence, a phenomena that arises from two or more nanoparticles being detected within the same dwell time, effectively giving the appearance of a larger particle at a lower particle number concentration.18,41 It has generally been suggested that when using 10 millisecond dwell times, concentrations that result in a maximum of 5–10% of the total readings containing a particle will avoid coincidence.25,34To demonstrate the effect of dwell time on coincidence, a series of 100 nm gold nanoparticle solutions were created at five concentrations ranging from 50 ppt to 10 ppb by mass. Fig. 1 shows selected data from the analysis of a 2 ppb 100 nm gold nanoparticle solution. Fig. 1A, C, and E illustrate the effect of dwell time on the raw counts of the nanoparticle. In Fig. 1A, at 10 ms dwell times, one background and three particle events are observed. Analysis of the 50 ppt sample at 10 ms dwell time showed a single particle registers as about 400 counts (ESI†). Thus it is evident that the reading at 902 counts represents multiple particles being detected within the dwell time window. Decreasing the dwell time to 3 millisecond shows greater resolution between particle events (Fig. 1B), as evidenced by a greater abundance of background counts between the peaks. However, there are still some data points that represent a multiple particle detection event (643 counts). Upon reducing the dwell time to 0.1 ms (100 μs), particle resolution can be greatly enhanced to the point of defining the particle pulses (Fig. 1E). At 100 μs dwell times, the nanoparticle response is parsed into bins of signal intensity as determined by the dwell time window. This allows for small changes in the number of ions present across the ion cloud to be detected. Thus, even partial coincidence of particle events might be discernable if peak deconvolution methods are introduced into the signal processing. This is apparent in the first peak in Fig. 1E (inset in Fig. 1E), which shows two peaks occurring within 100 μs of each other.
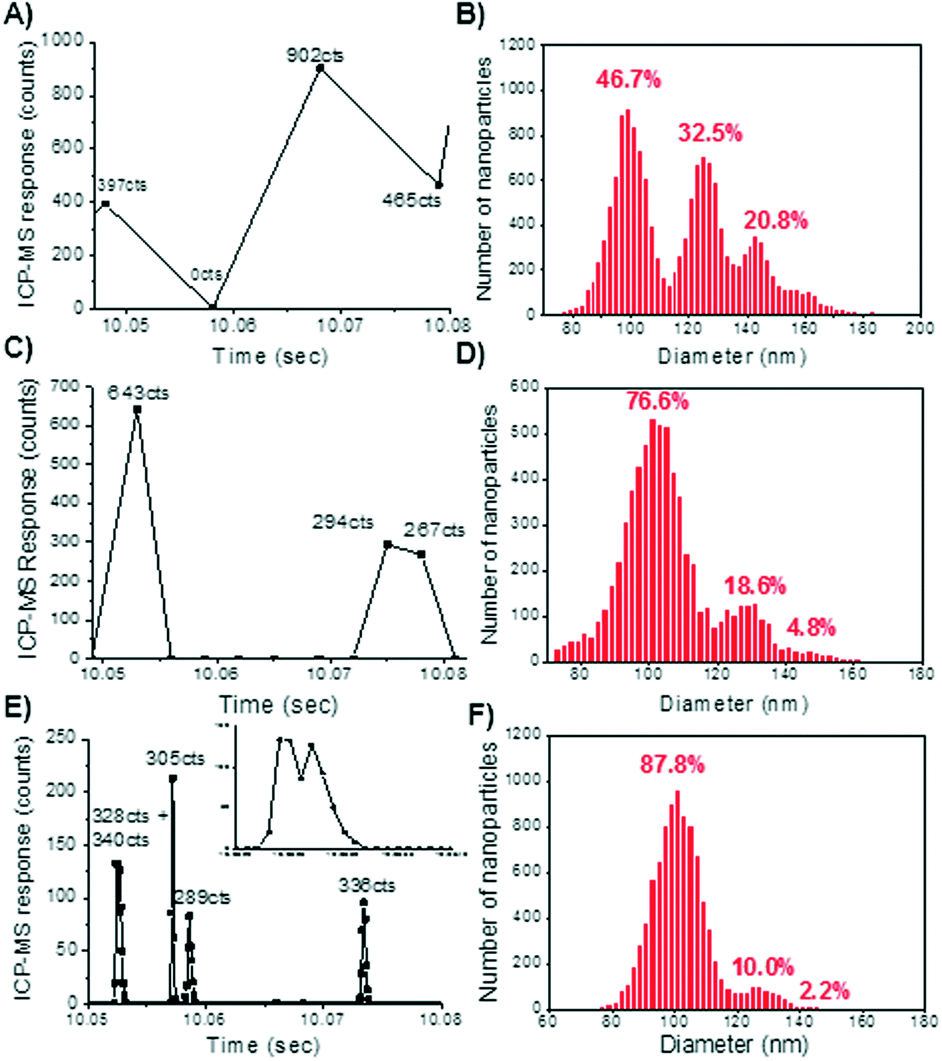 | ||
| Fig. 1 Comparison of a 2 ppb solution of 100 nm gold nanoparticles sizes at 10 ms, 3 ms, and 0.1 ms. (A) Raw counts at 10 ms dwell time. (B) Particle size histogram at 10 ms. (C) Raw counts of the gold nanoparticles at 3 ms dwell times. (D) Particle size histogram of 3 ms dwell time analysis. (E) Raw counts of the gold nanoparticles at 0.1 ms dwell times (inset: magnification of first peak showing particle resolution). (F) Particle size histogram at 0.1 ms dwell times. | ||
The effect of reducing dwell time on improving particle sizing is further demonstrated in Fig. 1B, D, and F. The percentages shown above each peak is a representation of the contribution of that peak toward the overall number of particles counted. The first peak represents the number of single 100 nm nanoparticles detected, where each subsequent peak is a result of 2 or more particles being detected simultaneously. Coincidence results in a large number of particle readings being over-sized, shifting the particle size distribution to larger diameters, which is most clearly seen at 10 millisecond dwell time (Fig. 1B). At 3 ms (Fig. 1D), the coincidence is greatly reduced, but diameters representing two particles still account for nearly 20 percent of the overall readings. At 100 μs dwell times (Fig. 1E), the coincidence peak has been reduced to only 10% of the total particle readings, and is most likely an artifact of current data processing methods being unable to quantitatively resolve the two peaks, rather than the actual readings of two nanoparticles reaching the detector simultaneously. As previously mentioned, further development of peak deconvolution methods may allow coincidence to be nearly completely eliminated over the concentration ranges for which the technique would be applied.
The need to reduce coincidence in spICP-MS is evidenced in Fig. 2, which demonstrates the single nanoparticle readings as a percentage of the overall particle readings (excluding background counts) as a function of mass concentration for 100 nm gold nanoparticles (error bars represent the standard deviation of triplicate measurements). At the conventional 10 ms dwell time, the ability to resolve single nanoparticle readings quickly diminishes at higher mass (particle number) concentrations. Despite being a monodisperse solution, the data collected at these millisecond dwell times would misrepresent the samples as a collection of aggregates or particles with diameters larger than 100 nm. Furthermore the measured particle number concentration will be underestimated at higher concentrations. The reduction in dwell time allows for better particle resolution, and a more accurate determination of the size distribution of the particle population even at concentrations as high as 10 μg Au L−1. This ability to improve the resolution of spICP-MS readings enhances the working range of the technique, expanding its applicability to a greater range of samples.
 | ||
| Fig. 2 Percentage of single nanoparticle readings with increasing mass (particle number) concentrations. | ||
Reduction of dissolved analyte signal
Many samples, particularly ones obtained from the environment, contain a variety of dissolved constituents of both inorganic and organic nature. In some cases these dissolved species may be of the same elemental composition as the nanoparticle. A common method of assessing elemental concentration in aqueous samples involves the acidification of the sample to ensure the metallic constituents are equally dispersed throughout the sample and performing an element-specific analytical technique. This technique however eliminates the particulate fraction of the sample, requiring additional complementary methods (filtration, field-flow fractionation, hydrodynamic chromatography) to determine the existence of nanoparticles in the sample. By quantifying the background signal and the signal from particle pulses, spICP-MS has the unique ability to differentiate between nanoparticulate and dissolved fractions of the sample without any additional sample preparation.30 In practice, the cut-off between background counts and counts arising from a nanoparticle event is done through an iterative calculation where an average of the entire dataset is calculated and added to three times its standard deviation. Events above this threshold are removed from the dataset and the calculation repeated until a convergent value is reached. This value is then determined to be the cut-off between nanomaterial readings and background counts. With millisecond spICP-MS, only low concentrations (ppt) of dissolved analyte can be overcome, as under these conditions the signal generated from a nanoparticle is sufficiently large when compared to the signal from the dissolved ions. In these cases, the quantification of the nanoparticle peak intensity is only a matter of subtracting the background intensity from the total intensity to generate the net intensity of the nanoparticle.25However, if dissolved concentrations reach a certain point, the signal of small nanoparticles will be lost in the noise of the background signal. This results from all ions of particular isotope being analyzed within a given dwell time, indiscriminate of particle or dissolved ion origin. An elevated background occurs where the average signal intensity of the baseline correlates to the concentration of dissolved background analyte. However, the signal generated by the particle remains the same and is a function of the mass of the analyte in the particle. As a result, once dissolved concentrations are high enough, they can mask the signal of the particle, preventing differentiation between particle and background intensities.
By reducing the dwell time window, the numbers of background ions that generate a response are reduced relative to the number of nanoparticle ions that generate a signal. This effect is seen in Fig. 3, where the raw signal is shown for a solution of 50 ppt 60 nm silver nanoparticles with a dissolved background of 500 ppt dissolved silver. At 10 ms, the readings generated by background and nanoparticle ions are indistinguishable. As the dwell time is decreased from 10 to 0.1 millisecond the background is reduced by a factor of 100 whereas the signal from the nanoparticle is only split between 2–3 readings. Thus the relative proportion of nanoparticle ions versus dissolved ions generating a signal increases such that the particle signal can be observed. The inset in Fig. 3 demonstrates that a 60 nm nanoparticle can be resolved from a background of 500 ppt Ag+ at 100 μs dwell time observations.
 | ||
| Fig. 3 Raw counts of 50 ppt 60 nm silver nanoparticles with a background of 500 ppt dissolved silver (inset: of 0.1 ms data showing particle readings above background). | ||
The ability of microsecond spICP-MS to improve the signal-to-noise ratio is further demonstrated in Fig. 4. Here a range of dissolved silver concentrations were added to a constant concentration of 50 ppt 60 nm citrate-capped silver nanoparticles. In a typical distribution of count intensities, a large initial peak of background/dissolved counts are present followed by a significantly smaller peak representing the counts generated from nanoparticles. As shown with conventional 10 ms data, the resolution between these two peaks decreases rapidly at increasing concentrations of dissolved analyte to a point where dissolved background and nanoparticle counts are indistinguishable. At 100 μs dwell times however, the resolution between the nanoparticle signal and the dissolved background signals are preserved even at dissolved analyte concentrations 10× higher than that of the nanoparticle mass concentration. This improved ability to detect and characterize nanoparticles amidst high backgrounds of dissolved analyte (i.e. where dissolved ion concentrations are ten times higher than particle mass concentrations) may prove essential for the analysis of ENPs in situ, where environmental concentrations of dissolved species are variable.
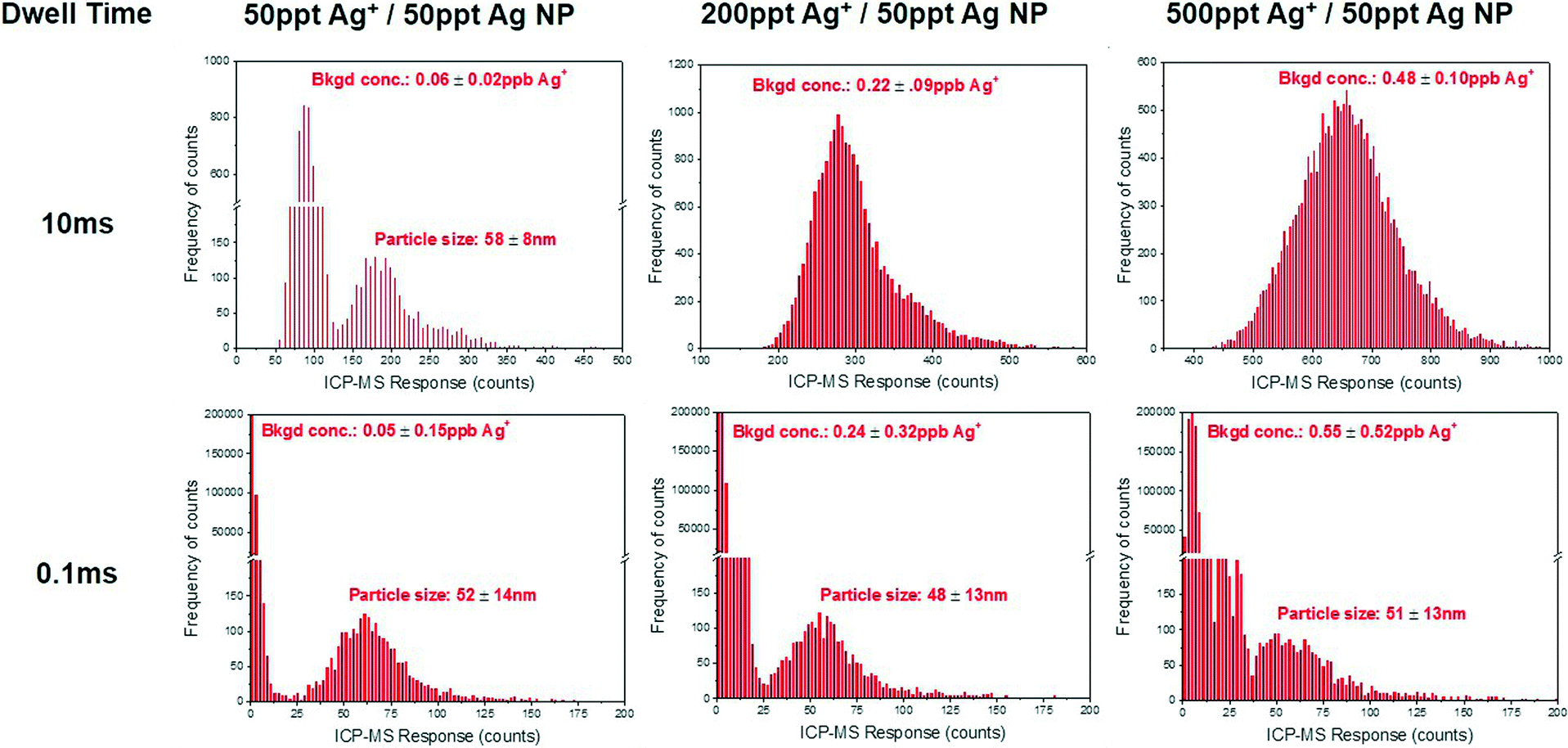 | ||
| Fig. 4 Frequency distribution of raw counts for 50 ppt 60 nm silver nanoparticles with increasing concentrations of dissolved silver. Dissolved background concentrations and particle size are included as an average and standard deviation of triplicate measurements. | ||
Dual-element spICP-MS
In conventional millisecond spICP-MS analyses, the analysis of a single element would encompass the entirety of the nanoparticle reading. Yet, as the nanoparticle is parsed into separate intensities in microsecond analysis, the intensities of two separate elements within a single particle can be compared and analyzed. This ability allows for isotopic and elemental ratios to be determined within a single particle if sufficient mass of the respective isotopes are present. This analysis require that both the read and settling times are on the order of 100 microseconds or less.This ability to perform detection of multiple isotopes at microsecond dwell times had been previously reported using a prototype time-of-flight ICP-MS.42 The software reported in this study allows for dual-element detection using an unmodified quadrupole ICP-MS, without the need for any additional ion separation apparatus. Though the ability to detect the full concentration of ions for a particular isotope may be lost, the ability to achieve elemental ratios could be achieved on a particle-by-particle basis.
Fig. 5 shows the analysis of a 1 ppb solution of 60 nm silver nanoparticles. As the two isotopes of silver (107Ag and 109Ag) occur in nearly equal ratios, it was expected that the counts generated from both isotopes would not only be detected, but in approximately equal intensities. For the data shown in Fig. 5, the percent abundances in the single particle shown are 46% (109Ag) and 54% (107Ag). An averaging of the intensities over the entire data set (n = 125000) gives a ratio of 107Ag: 80609 counts (50.7%) and 109Ag: 78245 counts (49.2%) respectively. This is very similar to the natural abundance of silver 107Ag: 51.35% and 109Ag: 48.65%.43 The ability to determine the isotopic ratio within a given nanoparticle sample adds another dimension to spICP-MS analysis that can be exploited for stable isotope analysis.
 | ||
| Fig. 5 Multi-element spICP-MS analysis of silver nanoparticles for 107Ag and 109Ag isotopes. (A) Raw counts of silver isotope data. (B) A single peak enhanced to show temporal proximity of one isotope to the other. | ||
Perhaps more importantly than the ability to detect different isotopes of the same element, Fig. 6 demonstrates the capability of microsecond spICP-MS to simultaneously detect two different elements within the same particle. Here a 30 nm gold core with a 15 nm silver shell (60 nm nanometer diameter in total) was analyzed using microsecond spICP-MS. Using this technique, the two elements (197Au and 107Ag) were detected within each individual particle. As expected, the silver signal was much larger as it comprises the outer shell, requiring a greater mass of silver for each subsequent layer of particle diameter. The ratio of silver to gold count intensities was consistent in dual element analysis such as in Fig. 5 (107Ag: 255 (77.7%) and 197Au: 73 counts (22.3%); n = 62) and by single particle analysis performed individually on both elements (107Ag: 880763 (71.2%) and 197Au: 356047 counts (28.8%); n = 60000). The structure and composition of these Au/Ag core–shell nanoparticle was further confirmed with FFF-ICP-MS and transmission electron microscopy with energy dispersive X-ray analysis (ESI† Fig. S2–S4).
 | ||
| Fig. 6 Multi-element spICP-MS analysis of 30 nm gold core, 30 nm silver shell nanoparticle. | ||
The implications of detecting isotopic and elemental ratios in a single particle are significant. Of the several obstacles present in the detection and characterization of nanoparticles in the environment, the most challenging is quantification of engineered nanomaterials in the presence of significantly more ubiquitous, naturally-occurring nanomaterials. In the past, different fractionation techniques have been utilized (centrifugation, filtration, FFF) in combination with ICP-MS in an attempt to examine only the size fraction of nanomaterials under consideration. However this approach can introduce artifacts that may significantly alter the original state of the material.
This promising development in spICP-MS could allow for the simultaneous detection of multiple elements, which can then be used to discriminate between naturally occurring nanomaterials and ENPs. Nanomaterials found in the environment generally consist of a chemically complex elemental make-up, when contrasted to the elementally pure ENPs.14 One such example is cerium and lanthanum which occurs in the environment in a ratio of 1.70 ± 0.54 as reported for 807 samples found in the Geochemical Atlas of Europe.44 By contrast, an engineered cerium oxide nanoparticle would likely be enriched in cerium. Utilizing the dual-element capability of micro-second dwell time spICP-MS, the detection of engineered ceria nanoparticles would be realized by particle containing only cerium, as opposed to naturally occurring analogues containing a fixed ratio of cerium and lanthanum as shown in Fig. 7. Here two samples were analysed using dual-element spICP-MS with 100 μs dwell time and a 100 μs settling time. A sample of stream water collected from Clear Creek in Golden, Colorado, U.S.A was monitored for both 139La and 140Ce (Fig. 7a and c). In Fig. 7b and d, cerium dioxide nanoparticles (80–100 nm) were spiked into the stream water, shifting the ratio of cerium to lanthanum counts towards higher cerium content. This is further demonstrated by the presence of cerium only peaks shown in Fig. 7d. A limiting factor will be the difference in masses between the two elements being analysed, as this will determine the shortest settling time achievable by the instrument. In addition, both elements will need to be present in appreciable amounts to be detected, as the absence of the “natural” element may result in false positives of ENP detection. Despite these limitations, this proposed method may be able to extend to different naturally occurring nanoparticles and emerging ENPs. Further development of microsecond spICP-MS may allow researchers to exploit this approach for environmental fate studies of ENPs.
 | ||
| Fig. 7 Analysis of Clear Creek stream water using dual element microsecond spICP-MS. a) Raw data of Clear Creak stream water. B) Raw data of Clear Creek stream water with 80–100 nm spiked CeO2 ENPs. C) Magnified section of Fig. 7a showing presence of particles containing both cerium and lanthanum. D) Magnified section of Fig. 7b showing presence of particles containing cerium and lanthanum as well as only cerium, which may indicate the presence of engineered CeO2. | ||
Materials and methods
Reagents
To generate Au and Ag calibration curves of known mass concentrations, a set of dissolved analyte standards was used. For the dissolved silver standards, a stock solution of silver nitrate (Perkin Elmer Pure, atomic spectroscopy standard) was diluted using 2% optima grade nitric acid (Fisher Scientific) to concentrations from 0 to 500 ppb. Dissolved gold standards (Spex CertiPrep, spectroscopy standard) were made through the dilution of a stock solution of gold chloride using 2% optima grade hydrochloric acid (Fisher Scientific) to concentrations of 0 to 10 ppb. Gold nanoparticles (100 nm) capped with a citrate stabilizer to prevent aggregation were purchased from BBI Solutions and used to determine transport efficiency. Citrate-capped 60 and 100 nm silver nanoparticles were purchased from NanoComposix, Inc. Au/Ag particles were custom synthesized with a 30 nm gold core and 30 nm silver shell for a total diameter of 60 nm as purchased from NanoComposix, Inc. Cerium oxide (CeO2) nanoparticles with diameters between 50–80 nm were purchased from Inframat® Advanced Materials™ as a solid power and were suspended in water via sonication. All nanoparticle solutions were prepared by dilution of the stock solution using ultrapure deionized water, 18.2 mΩ resistivity, from a Barnstead International Nanopure Diamond™ purification system. Clear Creek stream water was collected in 250 mL polypropylene containers and diluted prior to analysis by spICP-MS.Instrumentation
A quadrapole ICP-MS (Nexion 300Q, Perkin Elmer), equipped with a Type-C Miramist nebulizer and baffled cyclonic spray chamber, was operated using instrumental conditions described in ESI.† Software developed for this project provided for both data collection at microsecond dwell times and, for single element measurements, the elimination of the quadrapole settling time between readings. The analysis of gold core–silver shell nanoparticles (Au/Ag NPs) was performed using a 100 microsecond dwell time and 100 microsecond settling time for each mass. The settling time was used to account for ion flight time through the quadrupole mass analyzer and to the detector. This was initially calculated based on the most probable ion kinetic energies for gold and silver ions and later verified in-lab using gold and silver standard aqueous solutions. Settling times shorter than 100 microseconds caused a discernable drop in Au and Ag signal intensities, indicating that the mass analyzer electronics are potentially switching faster than the ions are able to clear the mass analyzer. As a result, 100 microseconds was chosen as a suitable settling time. To further validate the composition of the Au/Ag NPs sedimentation field-flow fractionation (CF2000 PostNova Analytics) and asymmetrical flow-field flow fractionation-ICP-MS (AF4-2000 PostNova Analytics) were coupled to the NexION 300Q. (ESI† Fig. S5).Nebulizer transport efficiency
An important aspect of single particle ICP-MS analysis is the determination of the nebulizer transport efficiency. This term represents the fraction of aqueous sample that reaches the plasma. A mass-based nebulization efficiency was determined for the work presented here. For this method, a standard particle (i.e. gold) of known size is used and its count intensity is compared to that of a series of dissolved gold standards to determine the transport efficiency.25,45 This transport efficiency is then applied to the remaining analytes of interest, Ag in this study, to develop an accurate mass-flux calibration curve which is used in converting the counts generated by a nanoparticle into a mass.A more straightforward method is to compare the measured pulse number frequency of a known particle standard to the expected particle number concentration, as the ratio of these two values being the transport efficiency. The absence of a particle number standard in the commercial market, and potential NP losses during sample storage, diminishes the feasibility of this method. The software reported here allows for the determination of nebulization efficiency by either a mass-based method or a number-based method. In addition, multiple standard particle sizes can be used to create a particle size calibration curve.
Tuoriniemi et al. discuss different alternative methods to determine transport efficiency by taking into account the sample flow rate, waste flow rate and the respective analyte signals in both the sample and waste fractions.45 Another means of improving nebulization efficiency is to utilize micro-droplet generators which can produce micro-droplets of a fixed diameter containing the analyte.37 By accelerating the desolvation of the micro-droplets through the use a lighter carrier gas such as helium, nebulization efficiencies can improve to as high as 100%.38 This technique however requires extra instrumentation beyond the typical nebulizer setups commonly found for most ICP-MS configurations.
Data collection and processing
A commercially available software package, Syngistix™ Nano Application Module, designed for NexION ICP-MS by Perkin Elmer was used throughout the experiments for data acquisition and further processing of the raw intensity data to particle size distribution information, as shown ESI† Fig. S6 and S7. The software package enables the instrument to acquire transient data continuously with short dwell times (e.g. 0.1 ms used for this work). Background count threshold in the raw data is determined real-time using a method which employs an average plus 3σ described previously (Pace et al.) and nanoparticle events are detected based on the calculated background threshold.34 Peak area for each nanoparticle event is determined by summing the data points that construct the event. The peak area information is then converted to particle size assuming spherical particle shape and based on calibration curves constructed from solution or nanoparticle standards.Conclusions
At a time where there is a great need for sophisticated instrumentation and more sensitive analytical techniques, spICP-MS has proven to be a powerful tool for the detection and characterization of ENPs in environmental samples. By utilizing microsecond dwell times, the resolution and working range of this technique has been improved to analyze an even greater breadth of environmental samples and increasingly complex matrices. Obstacles arising from relatively high particle number concentrations and high backgrounds of dissolved analyte can be overcome to provide a more accurate representation of the number and size of engineered nanoparticles in solution. Moreover, the addition of dual-element detection capability could prove instrumental in distinguishing naturally occurring particle from their engineered counter-parts without significant sample preparation. The simultaneous development of the Nano software not only makes microsecond spICP-MS possible, it puts this tool into the hands of analysts who which to add spICPMS capabilities to their laboratory.Though the microsecond technique helps overcome a number of obstacles of conventional millisecond spICP-MS, it does have a number of obstacles. Some data at the edges of the pulse may be lost to the background signal, and thus be excluded from the overall intensity of the particle. Additionally, since the dwell time is reduced, the count intensity generated by the nanoparticle is parsed into 2–4 reading according to the dwell time window. Consequently, the spacing between sampling events decreases, the intensity within the sampling event will also decrease. As a result, even though resolution between particle events increases, the overall detection limit with respect to size increases.
Work remains to be done to further develop this technique and improve its sensitivity, but in its current state, it is a premiere technique for engineered nanomaterial detection and characterization.
Acknowledgements
This body of work was supported by the Semiconductor Research Corporation (CSM Task Order 425.040).The authors would like to thank Pritesh Patel and Dylan MacNair (Perkin Elmer, Inc.) for their development of the software necessary for the analysis of nanoparticles at microsecond dwell times.
Soheyl Tadjiki (PostNova Analytics) for his instruction and assistance in acquiring sedimentation-FFF fractograms of the gold–silver nanocomposites.
In addition, the authors would thank Gary Zito and John Chandler (Colorado School of Mines) for their assistance in obtaining images of the gold–silver nanocomposites by scanning electron microscopy. Chris Higgins provided valuable discussions on the spICPMS method development.
Notes and references
- M. E. Leitch, E. Casman and G. V. Lowry, J. Nanopart. Res., 2012, 14, 1–23 CrossRef.
- G. C. Delgado, Technol. Soc., 2010, 32, 137–144 CrossRef PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- S. Gavankar, S. Suh and A. Keller, Int. J. Life Cycle Assess., 2012, 17, 295–303 CrossRef.
- S. J. Klaine, P. J. J. Alvarez, G. E. Batley, T. F. Fernandes, R. D. Handy, D. Y. Lyon, S. Mahendra, M. J. McLaughlin and J. R. Lead, Environ. Toxicol. Chem., 2008, 27, 1825–1851 CrossRef CAS.
- B. Nowack, J. F. Ranville, S. Diamond, J. A. Gallego-Urrea, C. Metcalfe, J. Rose, N. Horne, A. A. Koelmans and S. J. Klaine, Environ. Toxicol. Chem., 2012, 31, 50–59 CrossRef CAS PubMed.
- P. Westerhoff and B. Nowack, Acc. Chem. Res., 2012, 46, 844–853 CrossRef PubMed.
- A. Nel, T. Xia, H. Meng, X. Wang, S. Lin, Z. Ji and H. Zhang, Acc. Chem. Res., 2012, 46, 607–621 CrossRef PubMed.
- F. Gottschalk, T. Sonderer, R. W. Scholz and B. Nowack, Environ. Sci. Technol., 2009, 43, 9216–9222 CrossRef CAS PubMed.
- N. C. Mueller and B. Nowack, Environ. Sci. Technol., 2008, 42, 4447–4453 CrossRef CAS.
- B. Nowack, H. F. Krug and M. Height, Environ. Sci. Technol., 2011, 45, 1177–1183 CrossRef CAS PubMed.
- C. Beaudrie and M. Kandlikar, J. Nanopart. Res., 2011, 13, 1477–1488 CrossRef.
- C. E. H. Beaudrie, M. Kandlikar and T. Satterfield, Environ. Sci. Technol., 2013, 47, 5524–5534 CrossRef CAS PubMed.
- F. von der Kammer, P. L. Ferguson, P. A. Holden, A. Masion, K. R. Rogers, S. J. Klaine, A. A. Koelmans, N. Horne and J. M. Unrine, Environ. Toxicol. Chem., 2012, 31, 32–49 CrossRef CAS PubMed.
- P. J. J. Alvarez, V. Colvin, J. Lead and V. Stone, ACS Nano, 2009, 3, 1616–1619 CrossRef CAS PubMed.
- M. Hassellov, J. Readman, J. Ranville and K. Tiede, Ecotoxicology, 2008, 17, 344–361 CrossRef PubMed.
- M. S. Jiménez, M. T. Gómez, E. Bolea, F. Laborda and J. Castillo, Int. J. Mass Spectrom., 2011, 307, 99–104 CrossRef PubMed.
- F. Laborda, J. Jimenez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2011, 26, 1362–1371 RSC.
- H. Kawaguchi, N. Fukasawa and A. Mizuike, Spectrochim. Acta, Part B, 1986, 41, 1277–1286 CrossRef.
- C. Degueldre and P.-Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef CAS.
- C. Degueldre and P.-Y. Favarger, Talanta, 2004, 62, 1051–1054 CrossRef CAS PubMed.
- C. Degueldre, P. Y. Favarger and C. Bitea, Anal. Chim. Acta, 2004, 518, 137–142 CrossRef CAS PubMed.
- C. Degueldre, P.-Y. Favarger, R. Rosse and S. Wold, Talanta, 2006, 68, 623–628 CrossRef CAS PubMed.
- C. Degueldre, P.-Y. Favarger and S. Wold, Anal. Chim. Acta, 2006, 555, 263–268 CrossRef CAS PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS PubMed.
- J. Tuoriniemi, G. Cornelis and M. Hassellöv, Anal. Chem., 2012, 84, 3965–3972 CrossRef CAS PubMed.
- J. G. Coleman, A. J. Kennedy, A. J. Bednar, J. F. Ranville, J. G. Laird, A. R. Harmon, C. A. Hayes, E. P. Gray, C. P. Higgins and G. Lotufo, Environ. Toxicol. Chem., 2013, 32, 2069–2077 CrossRef CAS PubMed.
- L. D. Scanlan, R. B. Reed, A. V. Loguinov, P. Antczak, A. Tagmount, S. Aloni, D. T. Nowinski, P. Luong, C. Tran, N. Karunaratne, D. Pham, X. X. Lin, F. Falciani, C. P. Higgins, J. F. Ranville, C. D. Vulpe and B. Gilbert, ACS Nano, 2013, 7, 10681–10694 CrossRef CAS PubMed.
- R. B. Reed, D. G. Goodwin, K. L. Marsh, S. S. Capracotta, C. P. Higgins, D. H. Fairbrother and J. F. Ranville, Environ. Sci.: Processes Impacts, 2013, 15, 204–213 CAS.
- D. M. Mitrano, E. K. Lesher, A. Bednar, J. Monserud, C. P. Higgins and J. F. Ranville, Environ. Toxicol. Chem., 2012, 31, 115–121 CrossRef CAS PubMed.
- E. P. Gray, J. G. Coleman, A. J. Bednar, A. J. Kennedy, J. F. Ranville and C. P. Higgins, Environ. Sci. Technol., 2013, 47, 14315–14323 CrossRef CAS PubMed.
- K. Loeschner, J. Navratilova, C. Købler, K. Mølhave, S. Wagner, F. Kammer and E. Larsen, Anal. Bioanal. Chem., 2013, 405, 8185–8195 CrossRef CAS PubMed.
- R. B. Peters, Z. Rivera, G. Bemmel, H. P. Marvin, S. Weigel and H. Bouwmeester, Anal. Bioanal. Chem., 2014, 1–11 Search PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, E. P. Gray, C. P. Higgins and J. F. Ranville, Environ. Sci. Technol., 2012, 46, 12272–12280 CrossRef CAS PubMed.
- J. Liu, K. E. Murphy, R. I. MacCuspie and M. R. Winchester, Anal. Chem., 2014, 86, 3405–3414 CrossRef CAS PubMed.
- J. W. Olesik and P. J. Gray, J. Anal. At. Spectrom., 2012, 27, 1143–1155 RSC.
- S. Gschwind, L. Flamigni, J. Koch, O. Borovinskaya, S. Groh, K. Niemax and D. Gunther, J. Anal. At. Spectrom., 2011, 26, 1166–1174 RSC.
- J. Koch, L. Flamigni, S. Gschwind, S. Allner, H. Longerich and D. Gunther, J. Anal. At. Spectrom., 2013, 28, 1707–1717 RSC.
- G. Cornelis and M. Hassellov, J. Anal. At. Spectrom., 2014, 29, 134–144 RSC.
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Günther, J. Anal. At. Spectrom., 2013, 28, 226–233 RSC.
- F. Laborda, J. Jimenez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2013, 28, 1220–1232 RSC.
- O. Borovinskaya, B. Hattendorf, M. Tanner, S. Gschwind and D. Gunther, J. Anal. At. Spectrom., 2013, 28, 226–233 RSC.
- J. R. White and A. E. Cameron, Phys. Rev., 1948, 74, 991–1000 CrossRef CAS.
- R. Salminen , M. J. Batista , M. Bidovec , A. Demetriades , B. De Vivo , W. De Vos , M. Duris , A. Gilucis , V. Gregorauskiene , J. Halamić , P. Heitzmann , A. Lima , G. Jordan , G. Klaver , P. Klein , J. Lis , J. Locutura , K. Marsina , A. Mazreku , P. J. O’Connor , S. A. Olsson , R.-T. Ottesen , V. Petersell , J. A. Plant , S. Reeder , I. Salpeteur , H. Sandstrom , U. Siewers , A. Steenfelt and T. Tarvainen, Geochemical Atlas of Europe, Part 1, Background Information, Methodology and Maps, Geological Survey of Finland , 2005. Search PubMed ISBN: 951-690-921-3.
- J. Tuoriniemi, G. Cornelis and M. Hasselloev, J. Anal. At. Spectrom., 2014, 29, 743–752 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Table of typical NexION 300Q operating conditions, TEM images of particles used in this study with accompanying EDX spectra, additional plots of improvements in coincidence and reduction in dissolved background signals, screen captures of commercial software in use for the analysis of nanoparticles. See DOI: 10.1039/c4en00058g |
| This journal is © The Royal Society of Chemistry 2014 |