Deposition of nanoparticles onto polysaccharide-coated surfaces: implications for nanoparticle–biofilm interactions†
Kaoru
Ikuma‡
a,
Andrew S.
Madden
b,
Alan W.
Decho
c and
Boris L. T.
Lau‡
*a
aDepartment of Geology, Baylor University, Waco, TX 76798, USA. E-mail: borislau@engin.umass.edu; Fax: (413) 545-2840; Tel: (413) 545-5423
bSchool of Geology and Geophysics, University of Oklahoma, Norman, OK 73019, USA
cDepartment of Environmental Health Sciences, University of South Carolina, Columbia, SC 29208, USA
First published on 28th January 2014
Abstract
While environmental biofilms have recently been implicated as a potential major sink for nanoparticles (NPs), the mechanisms of interactions remain largely unknown. Polysaccharides are a common component of biofilm extracellular polymeric substances (EPS) and an initial point of contact for NPs in early NP–biofilm interactions. In this study, the significance of polysaccharide coatings on the deposition of hematite and silica NPs was examined by quartz crystal microgravimetry (QCM) and in-depth characterization of surface properties. NP deposition was shown to be largely governed by electrostatic forces. However, bulk surface zeta potential values of the tested polysaccharide-coated surfaces were not sufficient in describing the varying extent of NP deposition. Surface charge density and distribution both appeared to contribute to different NP deposition behaviors. These results suggest that nanometer to micrometer spatial characterization of biofilm surface properties, including chemical composition and charge, is necessary to improve our understanding of NP–biofilm interactions.
Nano impactThe environmental partitioning of nanoparticles has been previously documented to be greatly influenced by the presence of biofilms. The physicochemical characteristics of biofilm surfaces, which are dependent on biofilm matrix components such as polysaccharides, are expected to impact the initial surface deposition of nanoparticles. This study demonstrates that nanoparticle deposition onto polysaccharide-coated surfaces is primarily governed by electrostatic interactions. Furthermore, we show that microscale and nanoscale differences in surface charge density, distribution, and heterogeneity could impact the deposition dynamics of nanoparticles onto surfaces coated with polysaccharide-rich biofilms. Therefore, our study highlights the importance of assessing both bulk and small-scale biofilm surface characteristics for future nanoparticle–biofilm interaction studies. |
Nano-sized particles are generated abundantly in nature through physicochemical and geological processes and are ubiquitously found in the environment. While such natural nanoparticles (NPs) influence important environmental processes,1 the fate and transport of NPs in the natural environment are still largely unknown. In particular, owing to their small size, NPs are likely to encounter many “bulk” surfaces with which they may interact; these NP–surface interactions remain largely understudied.
Microbial biofilms exist on virtually all environmental surfaces and are an essential component of natural systems.2 Due to their omnipresent nature, it is likely that NPs often interact with biofilm-coated surfaces. In fact, recent mesocosm studies have documented significant accumulations of gold and TiO2 NPs occurring in biofilms.3,4 These initial studies point to an important role of biofilms for influencing environmental partitioning of NPs within natural systems. Therefore, the mechanisms of deposition and accumulation of NPs onto biofilm matrices are fundamental steps in understanding their broader environmental fate. Similarly, macro-sized particles, such as latex beads,5 bacteria,6 and Cryptosporidium parvum oocysts,7 have been shown to readily partition to biofilms. In such particle–biofilm interactions, it appears that the physical structure of biofilms may be more important than their chemical features for particle retention and transient storage.7,8 However, due to the smaller sizes of NPs, micro- and nanoscale chemical differences at the biofilm–water interface will likely have greater impacts on NP–biofilm interactions. While several studies have examined NP–biofilm interactions in bulk systems using silver and other metallic NPs,9–14 a mechanistic understanding of such small-scale interactions is still lacking.
An initial step in the interactions between NPs and biofilms may be the deposition of NPs from the water column to the biofilm surface. A critical parameter for this step is how the complex chemistry of biofilms influences NP deposition. The biofilm matrix is mainly composed of extracellular polymeric substances (EPS), which include a range of molecules such as polysaccharides, proteins, and nucleic acids.15 While the composition of EPS varies between biofilms and even locally within a biofilm, polysaccharides are considered a major component in most biofilms, typically accounting for up to 80% of the total EPS (e.g.ref. 16 and 17). For this reason, our study focused on polysaccharides as an important component of NP–biofilm interactions. Polysaccharides are inherently complex molecules and are widely employed throughout biological systems. The compositional and steric conformational properties of the natural polysaccharides present in biofilms are also expected to vary widely across biofilms depending on microbial species present and environmental conditions.18–21 This variability is likely to result in significantly different physicochemical surface characteristics that may impact NP–biofilm interactions.
The present study investigated the initial deposition characteristics of NPs onto surfaces coated with polysaccharides by quartz crystal microgravimetry (QCM). We hypothesized that the contribution of electrostatic forces in relation to other forces (e.g., hydrophobic interactions, van der Waals forces) is dominant in governing the deposition of bare NPs onto polysaccharide-coated surfaces. Furthermore, the distribution and heterogeneity of surface charges were expected to impact NP deposition.
Pseudo-hexagonal platelet hematite (α-Fe2O3; prepared as previously described22) and spherical silica (SiO2) NPs (nanoComposix, Inc., San Diego, CA) were used herein as positively and negatively-charged model NPs, respectively. NPs were characterized based on their sizes and zeta potentials as described in the ESI.† In 10 mM NaCl (pH 5.7), hematite NPs had an average hydrodynamic diameter of 73.6 ± 3.9 nm and a zeta potential of +25.7 ± 0.6 mV (electrophoretic mobility (EPM) of 2.02 ± 0.04 μm cm V−1 s−1). The average hydrodynamic diameter and zeta potential of silica NPs were 139.9 ± 3.1 nm and −14.8 ± 1.2 mV (EPM of −1.16 ± 0.03 μm cm V−1 s−1), respectively. These values show that hematite NPs were smaller and positively charged while silica NPs were approximately twice the size of hematite NPs and negatively charged. The NPs were verified to show minimal aggregation at the electrolyte concentration and pH used for all experiments (Fig. S2†).
We specifically utilized several model polysaccharides (sodium alginate, dextran sulfate, dextran, and chitosan) to represent a range of functional groups and surface charges that may be present in the polysaccharides of natural EPS.16,18,21 Their characteristics are provided in Table S1.† A Q-Sense (Stockholm, Sweden) E1 quartz crystal microbalance with dissipation monitoring (QCM-D) was used to first coat the silica sensors with polysaccharides to a similar thickness (~1 nm), followed by NP deposition following procedures described previously.23,24 These polysaccharide-coated surfaces were characterized for their surface zeta potential, surface wettability, surface topography and spatial distribution of the surface potential, and charge density (details of the procedures are provided in the ESI†). The surface zeta potential, contact angle, surface roughness (root mean square, RMS), and relative surface area are shown in Table 1. Silica surfaces coated with alginate and dextran sulfate had net negative charges, with dextran a net neutral charge and chitosan a net positive charge. Contact angle results indicate that while all surfaces were hydrophilic (<90°), dextran and chitosan were less hydrophilic compared to alginate or dextran sulfate (p < 0.05). On the other hand, surface topography examination by atomic force microscopy (AFM) revealed that alginate- and dextran-coated surfaces showed slightly higher surface roughness compared to dextran sulfate- and chitosan-coated surfaces (p < 0.05); however, the RMS roughness was less than 10 nm for all surfaces determined in 10 × 10 μm scans. AFM results indicate that most of the polysaccharide-coated surfaces had similar surface areas (p > 0.05). While alginate-coated surfaces had a significantly greater relative surface area than those coated with dextran sulfate (p = 0.022), all ratios showed that the surface areas only deviated from the flat projected area of an ideal sensor surface by up to 2%. These AFM analyses suggest that all four polysaccharide coatings had relatively smooth features.
| Polysaccharide coating | Surface zeta potential (mV) | Contact angle, θ (°) | Surface roughness (root mean square, nm) | Surface area/projected area |
|---|---|---|---|---|
| Alginate | −56.8 (±2.7) | 32.3 (±5.4) | 7.11 (±3.02) | 1.015 (±0.014) |
| Dextran sulfate | −59.9 (±3.3) | 28.4 (±0.9) | 2.35 (±0.35) | 1.005 (±0.0004) |
| Dextran | −0.1 (±3.9) | 61.9 (±2.4) | 6.94 (±2.82) | 1.016 (±0.021) |
| Chitosan | 39.8 (±1.8) | 55.0 (±8.9) | 2.38 (±1.08) | 1.007 (±0.004) |
Surfaces coated with alginate and dextran sulfate showed similar negative zeta potentials (p > 0.05, Table 1). However, surface zeta potential values only present a bulk view on the average charge environment across the entire surface and may have limited sensitivity to small differences in the surface charge that may be significant at the nanoscale. Therefore, we further characterized the alginate- and dextran sulfate-coated surfaces for negative charge densities and surface potential heterogeneity. Results from charge density measurements by QCM indicate that alginate- and dextran sulfate-coated surfaces have average negative charge densities of 3.41 ± 0.15 and 2.21 ± 0.13 sites nm−2, respectively. These values suggest that even though they appear to have similar surface zeta potentials, alginate-coated surfaces have 1.54 ± 0.11 fold higher negative charge density compared to surfaces coated with dextran sulfate (p < 0.05). Kelvin probe force microscopy (KPFM) was used in this study to investigate spatial variations of the surface potential across each surface.25 Surfaces coated with alginate and dextran sulfate both appeared to have small patches (with average diameters of 186 ± 53 and 139 ± 44 nm, respectively) of more-negative potentials compared to the surrounding smoother areas (representative examples of KPFM images are shown in Fig. 1). These patches were more frequently observed and had lower potentials (average of 3.6 patches per 5 × 5 μm scan; 10.3–35.3 mV lower compared to the smooth areas) in alginate-coated samples compared to surfaces coated with dextran sulfate (1.3 patches per scan; 6.0–18.2 mV lower). These results together suggest that alginate-coated surfaces may have a larger average negative charge density and greater heterogeneity of charges on the surface compared to surfaces coated with dextran sulfate. These surface charge conditions could greatly affect interfacial interactions between the polysaccharide-coated surfaces and NPs.
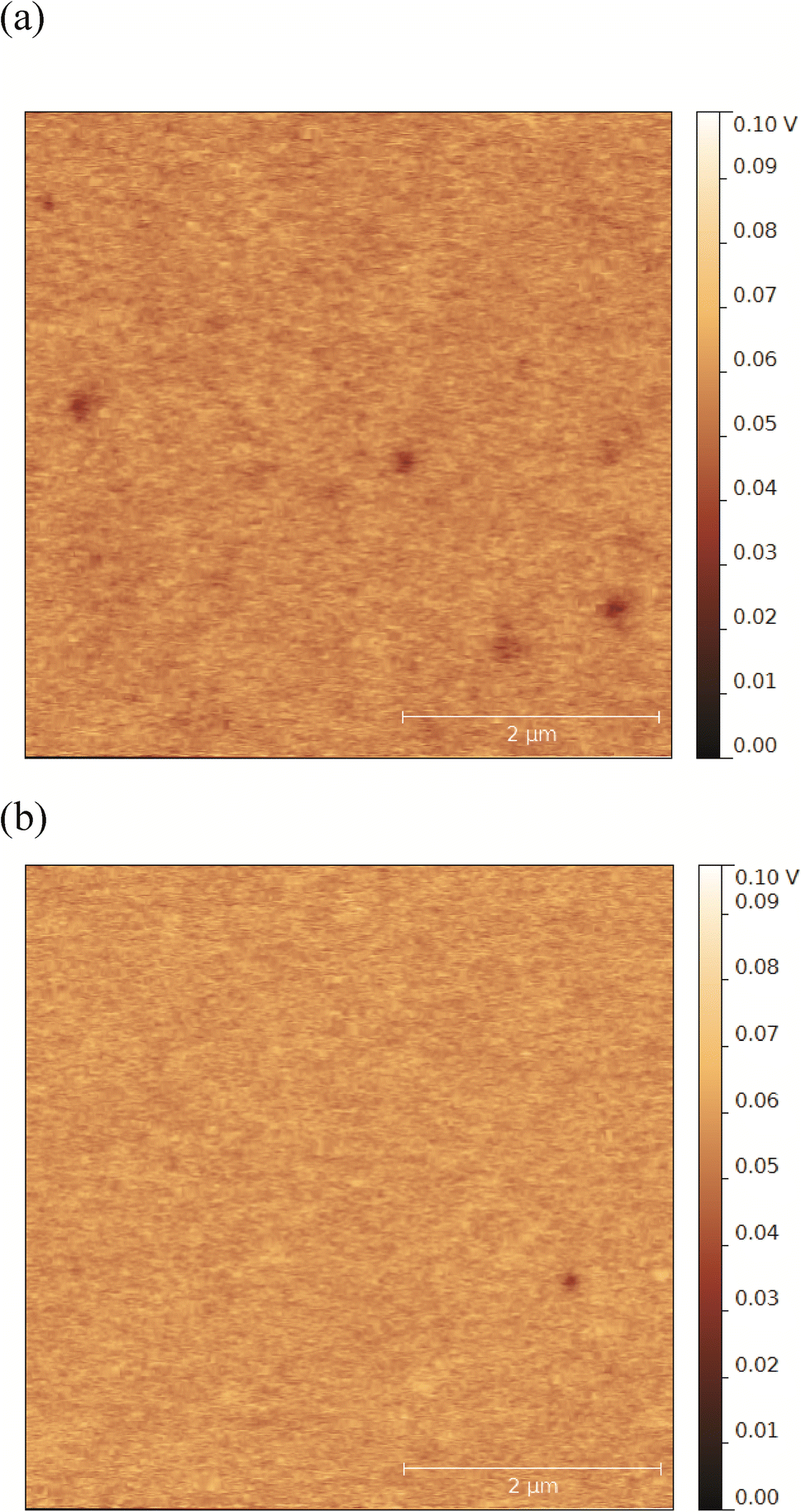 | ||
| Fig. 1 Representative KPFM surface potential images of silica QCM sensors coated with alginate (a) and dextran sulfate (b). Patches of lower surface potential are observed as areas of darker color. The absolute potential values are not directly comparable between KPFM images due to different values used for zeroing during analysis; therefore, the relative differences between the dark patches and the surrounding smooth areas are presented in the text. | ||
Following the polysaccharide coating on silica sensors, NP deposition was measured using the QCM-D by flowing through a 10 mg L−1 working suspension of NPs in 10 mM NaCl (pH 5.7). The detailed procedure is described in the ESI.† With the best signal-to-noise ratio, changes in resonance frequency (Δf) and resonance dissipation (ΔD) obtained from the third overtone are presented in this study (representative raw data shown in Fig. S1†). The mass deposited on the QCM sensor was calculated using the Sauerbrey equation.26 Real time dynamic light scattering (DLS) measurements were run simultaneously during QCM-D experiments to verify that there was a minimal size change of NPs over the experimental period (representative data shown in Fig. S2†). The deposition extents of hematite and silica NPs in 10 mM NaCl (pH 5.7) are shown in Fig. 2a and b, respectively. These extents were calculated as the total change in the deposited mass as a result of flowing through NP suspensions until a stable reading (within ±0.05 Hz s−1) was reached, followed by washing with clean 10 mM NaCl solution to remove unbound NPs. All values were normalized to relative surface areas of polysaccharide-coated sensor surfaces reported in Table 1. Both the total NP deposition extents (ng NP cm−2) and NP deposition extents normalized to the areal mass of the polysaccharide layer (ng NP per ng polysaccharide) are reported in the figures. Polysaccharide normalization was performed to examine the specific affinities of the different polysaccharides for NP deposition.
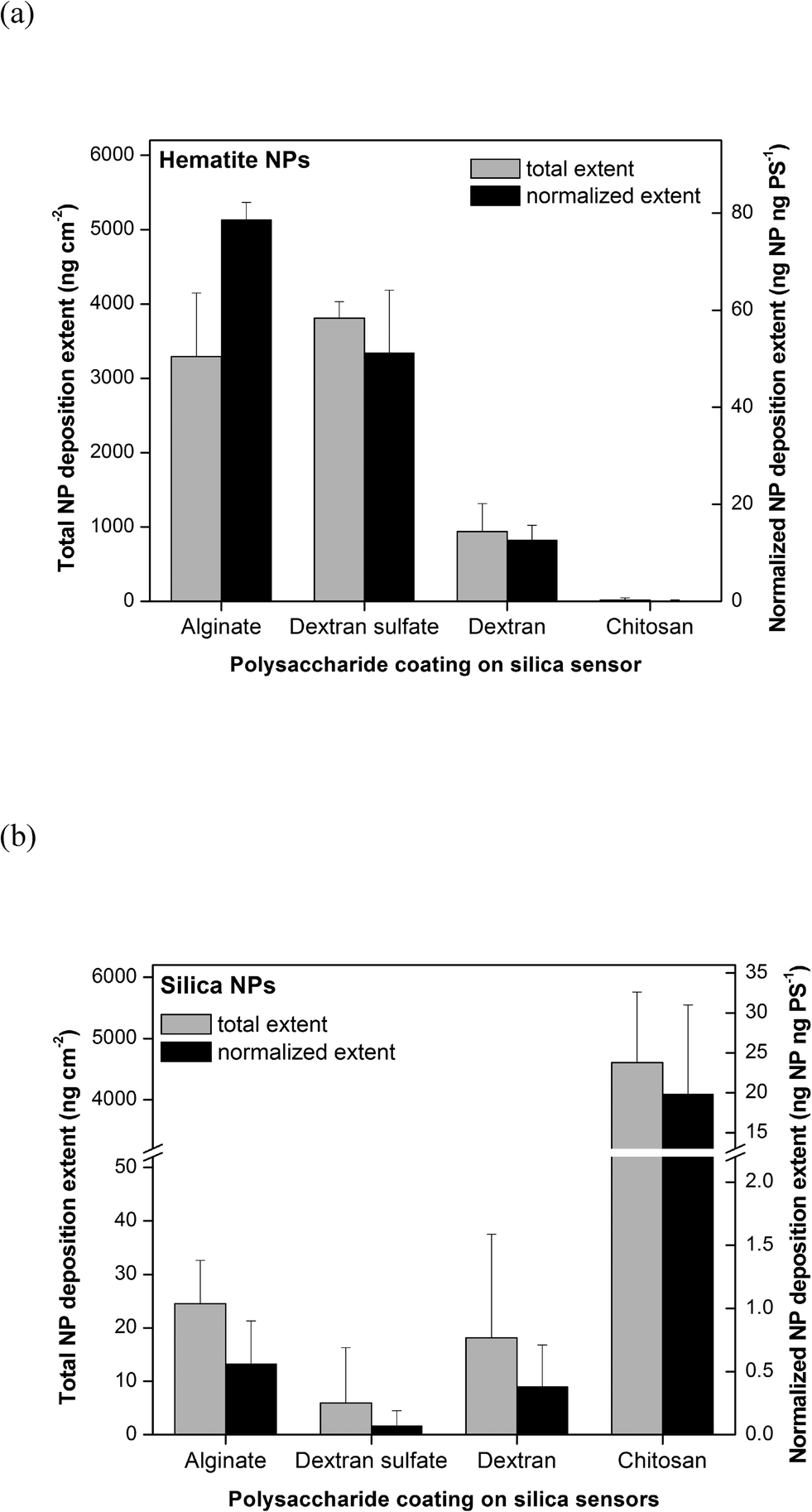 | ||
| Fig. 2 Total and polysaccharide-normalized deposition extents of (a) hematite NPs and (b) silica NPs onto silica sensors coated with various polysaccharides (PS) in 10 mM NaCl (pH 5.7) as determined by QCM. Total deposition extents were corrected for the specific surface area of each polysaccharide-coated surface. Error bars indicate the standard deviation of at least three replicate experiments. | ||
As shown in Fig. 2a, the total extents of hematite NP deposition were similar for surfaces coated with alginate and dextran sulfate (p > 0.05) and highest for the polysaccharide-coated surfaces tested. Significantly lower masses of hematite NPs were deposited onto dextran-coated surfaces, while almost none was deposited on chitosan-coated surfaces. These trends were in agreement with electrostatic attraction and repulsion occurring between the positively-charged hematite NPs and surfaces coated with negatively-charged alginate and dextran sulfate, neutrally-charged dextran, and positively-charged chitosan. The trends in the polysaccharide-normalized hematite NP deposition extent were similar to those in total extent for most of the polysaccharide-coated surfaces. However, the polysaccharide-normalized hematite NP deposition extents onto alginate-coated surfaces were approximately 1.8 fold higher compared to that onto dextran sulfate-coated surfaces. This difference may be due to alginate-coated surfaces having a larger average negative charge density and larger number of patches with more-negative charges than dextran sulfate-coated surfaces as reported above.
The trends observed for silica NP deposition were nearly opposite to those observed for hematite NPs. As shown in Fig. 2b, the total and normalized silica NP deposition extents were statistically similar (p > 0.05) and very low for surfaces coated with alginate, dextran sulfate, and dextran, but high for chitosan-coated surfaces. This trend was also in agreement with electrostatic attraction and repulsion occurring between negatively-charged silica NPs and polysaccharide-coated surfaces with varying charges. Electrosteric repulsion was evident for alginate- and dextran sulfate-coated surfaces, and it appeared that the differences in average negative charge densities had no effect on the degree of repulsion between the silica NPs and polysaccharide-coated surfaces. Furthermore, even though surfaces coated with chitosan and dextran were more hydrophobic, those slight increases in hydrophobicity did not appear to observably affect the deposition of NPs. There was no observable aggregation of NPs during all of the QCM experiments. These hematite NP and silica NP deposition characteristics together suggest that electrostatic interactions are a major force in governing the initial surface deposition of NPs onto polysaccharide-coated surfaces.
The deposition rates of hematite and silica NPs onto polysaccharide-coated surfaces are shown in Fig. 3. The differences in deposition rate for silica NPs follow the same trends as the deposition extents (Fig. 2b), in which surfaces coated with alginate, dextran sulfate, and dextran all have similar (p > 0.05) and very low silica NP deposition rates while chitosan-coated surfaces have higher rates. These observations suggest that the propensity of silica NP interactions with the polysaccharide-coated surfaces is governed primarily by electrostatic interactions and may be explained by the bulk surface zeta potential values. Deposition rates of hematite NPs were highest for surfaces coated with dextran sulfate. Hematite NPs were deposited onto surfaces coated with alginate at 0.7 fold the rate for dextran sulfate (p = 0.031), and onto dextran-coated surfaces at 0.7 fold the rate for alginate (p = 0.025). Chitosan-coated surfaces resulted in almost no hematite NP deposition and hence had very low deposition rates. As dextran-coated surfaces have a net neutral charge, it was expected that the hematite NP deposition rates onto those surfaces would be lower compared to those onto negatively-charged alginate or dextran sulfate. However, the hematite NP deposition rate onto dextran-coated surfaces was relatively high compared to the corresponding deposition extents. This high rate may suggest the interplay of other attractive forces such as van der Waals interactions between the NPs and the dextran-coated surface. The hematite NP deposition rate onto surfaces coated with dextran sulfate was higher than that onto alginate even though dextran sulfate-coated surfaces had lower average negative charge densities compared to those coated with alginate. This may be due to the difference in surface potential heterogeneity as observed by KPFM (Fig. S1†). The localized patches of more-negative charges observed on alginate-coated surfaces may hinder the rapid accessibility of some less favorably-charged sites for NP deposition. A similar effect called the “hydrodynamic bump” illustrates that when particles come into contact with a heterogeneous bulk surface, the probability of particle deposition onto a less favorably-charged surface site is reduced when such sites are close to more favorably-charged areas.27 Such a localized distribution of charges may not result in observable differences in total NP deposition extents but may have greater effects on NP deposition rates. It is important to note that the NP deposition rates may be controlled not only by bulk surface charges or average charge densities on the surface but also by the distribution and heterogeneity of charges across the surface.
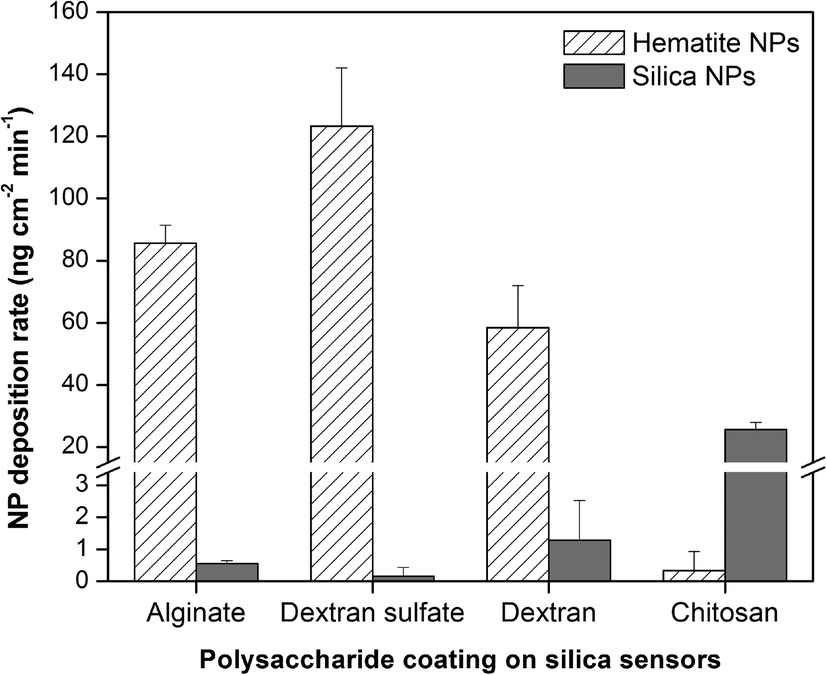 | ||
| Fig. 3 Deposition rates of hematite and silica NPs onto QCM silica sensors coated with various polysaccharides (PS) in 10 mM NaCl (pH 5.7) as determined by QCM. Error bars indicate the standard deviation of at least three replicate experiments. | ||
Previous studies showed that NP attachment onto surfaces was increased in the presence of biofilms,12,28,29 resulting in deviations from the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory on NP deposition behavior. Lerner et al. (2012) indicated that such NP deposition onto biofilms follows a polymer-mediated steric model that takes both DLVO and steric interactions into account.12 However, as Tong et al. (2010) suggested, the physicochemical characteristics of biofilms and EPS are likely to impact NP–biofilm interactions29 in a way that may not be well predicted by existing models. In fact, we showed herein that while the interactions between NPs and surfaces coated with pure polysaccharides may be largely governed by electrostatic forces, even microscale and nanoscale differences in the surface charge could impact such interactions. In environmental biofilms, these differences could not only be due to differences in the composition and identities of polysaccharides and other organic molecules but also due to the conformations and interactions of molecules and moieties on the surface. As the NP deposition extent and kinetics at the biofilm surface can greatly change the overall NP–biofilm interactions, our results suggest that both bulk and small-scale biofilm surface characteristics should be taken into account for future NP–biofilm studies.
Polysaccharides are ubiquitous in the environment as a major component of biofilms15 and natural organic matter30 and occur in pure forms as well as in complexes with proteins, peptides, and lipids.18 While typical chemical characterization of biofilms often treats all polysaccharides as one entity, our results suggest that the small-scale chemical and electrochemical identities of the polysaccharides presented may play important roles in the initial surface attachment of NPs. As the physicochemical characteristics of bacteria and biofilms are likely to be extremely heterogeneous in both composition and distribution,15,31,32 closer identification and characterization of surface molecules and properties are necessary for better prediction of NP attachment onto environmental surfaces.
Acknowledgements
This work was supported in part by the C. Gus Glasscock, Jr. Endowed Fund for Excellence in Environmental Science from Baylor University. We thank Tue Hassenkam for the assistance in KPFM measurements. ASM gratefully acknowledges support from NASA contract NNX11AH11G.References
- M. F. Hochella, S. K. Lower, P. A. Maurice, R. L. Penn, N. Sahai, D. L. Sparks and B. S. Twining, Science, 2008, 319, 1631–1635 CrossRef CAS PubMed.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed.
- J. L. Ferry, P. Craig, C. Hexel, P. Sisco, R. Frey, P. L. Pennington, M. H. Fulton, I. G. Scott, A. W. Decho, S. Kashiwada, C. J. Murphy and T. J. Shaw, Nat. Nanotechnol., 2009, 4, 441–444 CrossRef CAS PubMed.
- T. J. Battin, F. von der Kammer, A. Weilhartner, S. Ottofuelling and T. Hofmann, Environ. Sci. Technol., 2009, 43, 8098–8104 CrossRef CAS PubMed.
- W. J. Drury, W. G. Characklis and P. S. Stewart, Water Res., 1993, 27, 1119–1126 CrossRef CAS.
- C. M. Buswell, Y. M. Herlihy, L. M. Lawrence, J. T. M. McGuiggan, P. D. Marsh, C. W. Keevil and S. A. Leach, Appl. Environ. Microbiol., 1998, 64, 733–741 CAS.
- K. E. Searcy, A. I. Packman, E. R. Atwill and T. Harter, Appl. Environ. Microbiol., 2006, 72, 6242–6247 CrossRef CAS PubMed.
- T. J. Battin, L. A. Kaplan, J. D. Newbold and C. M. E. Hansen, Nature, 2003, 426, 439–442 CrossRef CAS PubMed.
- O. Choi, C.-P. Yu, G. E. Fernandez and Z. Hu, Water Res., 2010, 44, 6095–6103 CrossRef CAS PubMed.
- J. Fabrega, J. C. Renshaw and J. R. Lead, Environ. Sci. Technol., 2009, 43, 9004–9009 CrossRef CAS PubMed.
- J. Fabrega, R. Zhang, J. C. Renshaw, W.-T. Liu and J. R. Lead, Chemosphere, 2011, 85, 961–966 CrossRef CAS PubMed.
- R. N. Lerner, Q. Lu, H. Zeng and Y. Liu, Water Res., 2012, 46, 975–985 CrossRef CAS PubMed.
- E. Sahle-Demessie and H. Tadesse, Surf. Sci., 2011, 605, 1177–1184 CrossRef CAS PubMed.
- S. Tripathi, D. Champagne and N. Tufenkji, Environ. Sci. Technol., 2012, 46, 6942–6949 CrossRef CAS PubMed.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CAS.
- K. E. Eboigbodin and C. A. Biggs, Biomacromolecules, 2008, 9, 686–695 CrossRef CAS PubMed.
- A. R. Badireddy, B. R. Korpol, S. Chellam, P. L. Gassman, M. H. Engelhard, A. S. Lea and K. M. Rosso, Biomacromolecules, 2008, 9, 3079–3089 CrossRef CAS PubMed.
- I. W. Sutherland, Microbiology, 2001, 147, 3–9 CAS.
- B. Vu, M. Chen, R. J. Crawford and E. P. Ivanova, Molecules, 2009, 14, 2535–2554 CrossRef CAS PubMed.
- C. Mayer, R. Moritz, C. Kirschner, W. Borchard, R. Maibaum, J. Wingender and H.-C. Flemming, Int. J. Biol. Macromol., 1999, 26, 3–16 CrossRef CAS.
- Y. Jiao, G. D. Cody, A. K. Harding, P. Wilmes, M. Schrenk, K. E. Wheeler, J. F. Banfield and M. P. Thelen, Appl. Environ. Microbiol., 2010, 76, 2916–2922 CrossRef CAS PubMed.
- U. Schwertmann and R. M. Cornell, Iron Oxide in the Laboratory: Preparation and Characterization, 2nd edn, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS.
- K. L. Chen and M. Elimelech, Langmuir, 2006, 22, 10994–11001 CrossRef CAS PubMed.
- B. Moores, F. Hane, L. Eng and Z. Leonenko, Ultramicroscopy, 2010, 110, 708–711 CrossRef CAS PubMed.
- I. Reviakine, D. Johannsmann and R. P. Richter, Anal. Chem., 2011, 83, 8838–8848 CrossRef CAS PubMed.
- M. Elimelech, J. Y. Chen and Z. A. Kuznar, Langmuir, 2003, 19, 6594–6597 CrossRef CAS.
- X. Jiang, X. Wang, M. Tong and H. Kim, Environ. Pollut., 2013, 174, 38–49 CrossRef CAS PubMed.
- M. Tong, J. Ding, Y. Shen and P. Zhu, Water Res., 2010, 44, 1094–1103 CrossRef CAS PubMed.
- G. E. Schaumann, J. Plant Nutr. Soil Sci., 2006, 169, 145–156 CrossRef CAS.
- S. S. Branda, A. Vik, L. Friedman and R. Kolter, Trends Microbiol., 2005, 13, 20–26 CrossRef CAS PubMed.
- I. Sokolov, D. S. Smith, G. S. Henderson, Y. A. Gorby and F. G. Ferris, Environ. Sci. Technol., 2001, 35, 341–347 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3en00075c |
| ‡ Present Address: Department of Civil & Environmental Engineering, University of Massachusetts Amherst, Marston Hall, 130 Natural Resources Road, Amherst, MA 01003-9293, USA. |
| This journal is © The Royal Society of Chemistry 2014 |