Natural water chemistry (dissolved organic carbon, pH, and hardness) modulates colloidal stability, dissolution, and antimicrobial activity of citrate functionalized silver nanoparticles†
Lok R.
Pokhrel
ac,
Brajesh
Dubey
*b and
Phillip R.
Scheuerman
a
aDepartment of Environmental Health, College of Public Health, East Tennessee State University, Johnson City, TN 37614-1700, USA
bEnvironmental Engineering, School of Engineering, University of Guelph, 50 Stone Road East, Ontario, Guelph, Canada. E-mail: bdubey@uoguelph.ca; Tel: +1 519 824 4120 ext 52506
cUS Environmental Protection Agency, National Health and Environmental Effects Research Laboratory, 200 SW 35th St., Corvallis, OR 97333, USA
First published on 28th November 2013
Abstract
Knowledge about whether/how natural water chemistry influences the fate, dissolution, and toxicity of silver nanoparticles (AgNPs) should contribute to ecological risk assessment and informed decision making. The effects of three critical water chemistry parameters – dissolved organic carbon (DOC), pH, and hardness – were investigated on the colloidal stability, dissolution dynamics, and antimicrobial activity of citrate-functionalized AgNPs (citrate–AgNPs) against Escherichia coli. Toxicities of citrate–AgNPs and AgNO3 were also determined in the river water samples collected across three seasons (for seven months). Detectable changes in hydrodynamic diameter, surface charge, and plasmonic resonance revealed the modulating effects of the water chemistry parameters on the colloidal stability of citrate–AgNPs. Although, overall Ag release from citrate–AgNPs was low (0.33–3.62%), it increased with increasing DOC concentrations (0–20 mg L−1) but decreased with increasing pH (5–7.5) or hardness (150–280 mg L−1). Citrate–AgNP toxicity was 3–44 fold lower than of AgNO3 (Ag mass basis). Notably, higher DOC or pH conferred protection to E. coli against citrate–AgNPs or AgNO3; increasing solution hardness tended to enhance toxicity, however. Citrate–AgNPs or AgNO3 toxicity in the river water matrix revealed no seasonality. Generalized linear models developed, by parameterizing particle properties, could fairly predict empirically-derived nanotoxicity. Our results show that particle size, surface properties, ion release kinetics, and toxicity of citrate–AgNPs can be modified upon release into aquatic environments, suggesting potential implications to ecosystem health and functions.
Nano impactGrowing applications of silver nanoparticles in the myriad of consumer products warrant better understanding of their fate, dissolution and toxicity in the environment. In this paper we document significant modulating effects of natural water chemistry (dissolved organic carbon (DOC), pH, and hardness) on the colloidal stability, ion release kinetics, and antibacterial activity of silver nanoparticles against Escherichia coli. Notably, higher DOC or pH conferred protection to E. coli against citrate–AgNPs or AgNO3, while increasing solution hardness tended to enhance toxicity. The data are novel, timely and should contribute to risk assessment and informed decision making about engineered nanoparticles in aquatic systems. |
Introduction
As nanotoxicology is yet to mature as much as nanotechnology has,1 research efforts to better understand whether engineered nanoparticles (ENP) pose risks upon exposure are ongoing. Some recent studies employing in vitro or in vivo models, however, indicate potential environmental, health, and safety effects of ENPs.2–9 Because ENPs are becoming integral elements of the myriad of products (e.g., toothpaste, soap, sunscreen lotion, clothing, medical masks, plastic wares, electronics, cement, paint, etc.),10 the likelihood of environmental exposures to these nanoproducts vis-à-vis their leachable by-products (i.e., as nanoparticles, aggregates, or ions) is high.6,7,11,12Because the wet chemical methods are commonly used for ENP synthesis, precursor chemicals, coating materials, and reducing agents are likely present to some extent in the nanosuspension, depending on the method of purification employed.6–8,12 Whether the toxicity observed is due to the combined effects of impurities, released ions, and/or colloidal particles in the exposure medium, or if it is due to the change in particle size and/or surface characteristics, or if interactive effects of particle size, surface charge and the released ions occur during the experimental period are premises less explored or understood.7,8,13–15
Elucidating factors playing a significant role in ENP stability and potential toxicity has been a challenging opportunity for the nanoresearch community.9,13 Interactions of ENPs with the natural colloids, including with a multitude of environmental factors such as pH, monovalent and divalent cations, background electrolytes,17,18 and natural organic matters (e.g., humic and fulvic acids),18,19 could modify their colloidal stability upon entering aquatic systems by altering particle size, surface characteristics, and mobility,16,20–23 and thereby the toxicity.8,9,19,24 Only a few studies have assessed the fate and toxicity of silver nanoparticles (AgNP), one of the widely sought ENPs for broad spectrum antimicrobial and plasmonic properties,7,10 under multiple water chemistry conditions such as variable pH, monovalent and divalent cations, anions such as chloride and sulfide,25,26 other electrolyte types,16,20 or natural organic matters (e.g., humic and fulvic acids).18,19 Often results are confounded by multiple parameters such as the particle size, surface coating, or the toxicity of media used;7,27 perhaps due to less routine use of robust statistical methods (higher level designs),13,15,28 which could parameterize multiple variables to model their effects, enabling us to identify and explain the potential contribution of each variable on nanotoxicity.13,15,28
Recognized as a non-toxic coating/capping agent imparting negative surface charge to nanoparticles,6–8 citrate is also amongst the most widely sought reductants used in nanoparticle synthesis, which electrostatically stabilizes the particles in suspension.12 As a model AgNP of widespread use, we chose to use citrate-functionalized AgNPs (citrate–AgNPs) in this study. Here we investigated the potential effects of multiple water chemistry on the colloidal stability, dissolution rate, and antibacterial activity of citrate–AgNPs against Escherichia coli. Potential changes in particle stability are investigated by measuring hydrodynamic diameter (HDD), zeta (ζ) potential, and plasmonic resonance of citrate–AgNPs as a function of dissolved organic carbon (DOC), suspension pH, and hardness. Rate of Ag dissociated under variable DOC, pH, and hardness are evaluated to discern its contribution to the toxicity under the experimental conditions. Finally, the effects of variable DOC, pH, and hardness conditions on the stability and Ag dissociation rates are used to explain the toxicity of citrate–AgNPs using the previously reported high-throughput β-galactosidase (β-gal, hereafter) E. coli bioassay.7 The toxicity of AgNO3, as a source of free Ag+, is assessed concurrently under a range of DOC, pH, and hardness conditions, which allowed for mass-based toxicity comparison between citrate–AgNPs and free Ag+ (as AgNO3). Additionally, the toxicities of citrate–AgNPs and AgNO3 are assessed in the representative river water samples collected across three seasons (for seven months). Employing the generalized linear model (GLM), we also probe and quantify the effects of particle properties to explain nanotoxicity. We systematically show that dissolved organic carbon, pH, and hardness inherent to the natural waters can influence AgNP characteristics and alter its toxicity against E. coli.
Materials and methods
Nanoparticle synthesis and purification
Citrate-functionalized silver nanoparticles (citrate–AgNP) were synthesized following the previously established procedure (detailed in ESI†).16 Using 10 kD hollow fiber membranes (P/N: X31S-300-02P, surface area = 145 cm2) connected to KrosFlo Research IIi tangential flow filtration (TFF) system (Spectrum Laboratories, CA, USA), citrate–AgNPs were purified. The details of TFF were reported previously (ESI† Fig. S1, Table S1).6,7 Purified samples were digested using the USEPA method 3050B, and Ag concentration in the stock nanosuspension was determined (as total Ag) using flame-atomic absorption spectroscopy (AAS).Nanoparticle characterization
Citrate–AgNPs were tested for volume-weighted hydrodynamic diameter (HDD) and zeta (ζ) potential using the dynamic light scattering (DLS) and phase analysis light scattering (PALS), respectively, prior to- and post-purification. The measurement accuracy of the DLS unit was assessed at 23 °C using Duke 500 (491 nm) NIST 3490A standard (PSS NICOMP Particle Sizing Systems, CA), which preceded sample measurements for particle size and surface charge. The localized SPR (surface plasmon resonance) spectra were recorded for citrate–AgNPs using an UV–vis spectrophotometer (Shimadzu PharmaSpec UV-1700). A transmission electron microscope (Philips TEM 420) operated at 120 kV in the bright-field mode allowed visualization of particle morphology. Using an ImageJ 1.44,29 particle circularity and size distributions (PSD) were estimated from the TEM images.Toxicity bioassay
The toxicity of biolabile citrate–AgNPs or free Ag+ (as AgNO3) was measured as β-gal (β-galactosidase) activity inhibition in Escherichia coli.7,30 β-gal bioassay involves an hydrolysis of glycosidic bond in chlorophenol-red β-galactopyranoside (CPRG), a chromogenic substrate, leading to the formation of galactopyranose and chlorophenol red as the reaction byproducts.7 Colorimetric quantification of chlorophenol red at 570 nm in a 96 well-plate using a Multiskan microplate reader offered an estimate proportional to the activity of intracellular β-gal.7,30 1 mg Cu2+ L−1 (as CuSO4) and moderately hard water (MHW, as Evian water) represented the positive and negative controls, respectively. The detailed physico-chemical characteristics of Evian water (used as MHW) can be found in ref. 31. At least triplicate samples were analyzed for each concentration. The incubation temperature and time required for β-gal assay are 35 °C and 3–4 h, respectively; the details of the β-gal assay are previously described (schematic in ESI† Fig. S2).7 The bacterial reagent and CPRG were procured through M2B Research & Innovative Technologies, LLC, Gainesville, FL.Preparation of test solutions and exposure conditions
Leonardite humic acid (LHA, International Humic Substances Society (IHSS); cat# 1S104H-5) was prepared in MHW stirring overnight, and filtered through a 0.45 μm pore size Millipore filter. Following SM-5310C method coupled with persulfate-UV oxidation, the filtrate was analyzed for dissolved organic carbon (DOC) concentration. Concurrently, multiple concentrations of DOC were verified by developing a calibration curve at 280 nm (ESI† Fig. S3), which tends to offer meaningful information on the aromaticity and molecular weight of the humic acid molecules.32,33 The solution was then stored at 4 °C in the dark until use. Detailed chemical characteristics of this LHA are documented by Thorn et al.34Potential intracellular inhibition of β-gal activity in E. coli upon exposure to citrate–AgNPs or free Ag+ (as AgNO3) was investigated under multiple water chemistry conditions, i.e., under different solution pH, hardness, and DOC concentrations. Because a narrow pH range of 5–7.5 is recommended for the bioassay, we chose four different pH values (i.e., 5, 6, 7, and 7.5) to test for pH effects on citrate–AgNP stability, ion release rate and, subsequently, on toxicity. The test solution hardness (280 mg L−1 as CaCO3) and DOC concentrations (2 mg L−1) were held constant while testing for the effects of pH. At 25 °C, the pH of MHW was 7.0, requiring no pH adjustment. Using nanopure water, Evian water hardness (280 mg L−1 as CaCO3) was adjusted to obtain the desired hardness of 280, 250, 200, and 150 mg L−1, which was analytically confirmed by EDTA titration method (Hach method 10247). Potential changes in the colloidal stability, ion release rate, and toxicity of citrate–AgNPs were evaluated using the hard water while maintaining pH (neutral) and DOC (2 mg L−1) constant. Eight different concentrations in the range 0.05–10 mg L−1 for AgNO3 (as total Ag), and seven comparable concentrations in the range 0.1–10 mg L−1 for citrate–AgNPs, were assessed for toxicity under various water chemistry conditions as stated above, including in the Watauga River samples. Toxicity was also evaluated using the Watauga River water matrix sampled four times over the period of seven months (three seasons), and used for comparison. A 2 mg L−1 DOC is representative of the natural water samples collected from the Watauga River, near Elizabethton, TN, USA (36.3339° N, −82.2704° W). To assess the impacts of DOC on citrate–AgNP stability and antimicrobial activity, five different DOC concentrations (0, 2, 5, 10, and 20 mg L−1) were applied while maintaining pH 7 and a hardness 280 mg L−1 (as CaCO3). DOC suspension prepared in MHW (pH 7.0) only required minor pH adjustment which was accomplished, prior to citrate–AgNPs addition into DOC suspension, using a dilute solution of HNO3 or NaOH. A minimum of triplicate samples were run for particle stability and enzyme activity tests. Potential changes in HDD and ζ potential measured under the multiple water chemistry conditions using the DLS method were used to characterize particle stability in the test media,17,20,35 which was further compared with the likely changes observed in the corresponding SPR spectra.32 For this, citrate–AgNPs were incubated at 35 °C for 4 h (conditions required for β-gal bioassay) under variable DOC, hardness, and pH levels before the DLS or UV–vis measurements were recorded. The DLS data obtained from the triplicate samples are reported as an average ± 1 standard deviation.
Measuring ion release under variable water chemistry conditions
Separate experiments were conducted in duplicates to determine the rates of dissolved Ag released from the colloidal citrate–AgNP suspension under variable water chemistry conditions stated above. For this, 25 mL of citrate–AgNPs (10 mg L−1) were incubated at 35 °C for 4 h under a range of DOC concentrations, hardness, and pH levels, similar to those used for evaluating toxicity. Additionally, the kinetics of dissolved Ag released from pure citrate–AgNP suspension at 1, 3, 6 and 10 mg L−1 levels were also evaluated. Each sample was then ultracentrifuged (Thermo Scientific Sorvall WX Ultra Series Centrifuge SN# N13V-427288-NV) at 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (205835g at the bottom, 146347g at the middle, and 86858g at the top-part of the bottle) in a polycarbonate bottle (Thermo Scientific cat# 314348) for an hour; this was followed by pipetting off 3 mL clear supernatant, which was soon digested with the same volume of conc. HNO3 (trace metal grade) before total Ag concentration was determined using graphite furnace-AAS.
000 rpm (205835g at the bottom, 146347g at the middle, and 86858g at the top-part of the bottle) in a polycarbonate bottle (Thermo Scientific cat# 314348) for an hour; this was followed by pipetting off 3 mL clear supernatant, which was soon digested with the same volume of conc. HNO3 (trace metal grade) before total Ag concentration was determined using graphite furnace-AAS.
Statistical analysis
The EC50 (i.e., effective concentrations for 50% enzyme activity inhibition) values were estimated using linear regression analysis.7 Potential toxicity of citrate–AgNPs or free Ag+ are reported for different water chemistry conditions as an average EC50 obtained from at least triplicate test runs. Data satisfied normal probability distribution (Kolmogorov–Smirnov [K–S] test, p > 0.1) and were used untransformed,36 except for hardness (K–S test, p = 0.04) for which the non-parametric Kruskal–Wallis test was used to adjust for unequal variance. One-way analysis of variance (ANOVA) was used to test for a significant difference between the sample EC50s and the controls, followed by the Dunnett t test (post-hoc) for multiple comparisons. Generalized linear (regression) models (GLM) were developed to probe and quantify the main and interactive effects of particle properties under the variable DOC, pH, and hardness conditions to explain the observed toxicity.28 The model deviance value was compared with the other models to test the goodness-of-fit of the final model based on the information criteria that ‘small-is-better’. Significant difference was established at the p ≤ 0.05 using IBM SPSS ver. 20.37Results and discussion
Characteristics of citrate–AgNPs
The synthesized citrate–AgNPs (prior to- and post-purification) had an average HDD of ~11 nm (SD = 0.7 nm; ESI† Fig. S1, Table S1) and a TEM diameter of 56.5 nm (SD = 19.2 nm; n = 208 particles; ESI† Fig. S4A, B, D). The discrepancy in particle diameter as observed using different tools (e.g., DLS vs. TEM) can be attributed to the underlying differences in the methods used for sizing particles and data reporting.15 Fairly similar ζ potentials before (−21.43 mV) and after (−25.13 mV) purification revealed that the particles were unchanged during the TFF and were stably dispersed in aqueous suspension, which is in agreement with the particles observed on the TEM imagery (ESI† Fig. S4A).7 As predicted by Mie theory,38 its characteristic SPR peak was observed at 445 nm (ESI† Fig. S4C). Following purification, citrate–AgNPs were found to be of high circularity of 0.88 and purity; the latter verified by the change in electrical conductivity (from initial 1095 μS cm−1 to final 5 μS cm−1; ESI† Table S2),7 significantly lower amount of dissolved Ag (~65 μg L−1 as total Ag) quantified in the permeate/wastewater,7 and the X-ray photoelectron spectroscopy and nuclear magnetic resonance analyses as performed previously.8Modulation of AgNP stability by DOC, pH, and hardness
Interparticle bridging of AgNPs with humic acid molecules,39 particularly at higher DOC concentrations, resulted in significantly larger particles as shown by the HDDs (Fig. 1A) vis-à-vis a gradual decrease in ζ potentials with increasing DOC concentrations as confirmed by the DLS (Fig. 1B). This corresponded with increasing absorbance and broadening, with slight blue-shift, of citrate–AgNP SPR spectra with increasing DOC concentrations (Fig. 2A), suggesting potential agglomeration or sedimentation of the particles upon interactions with humic acid molecules.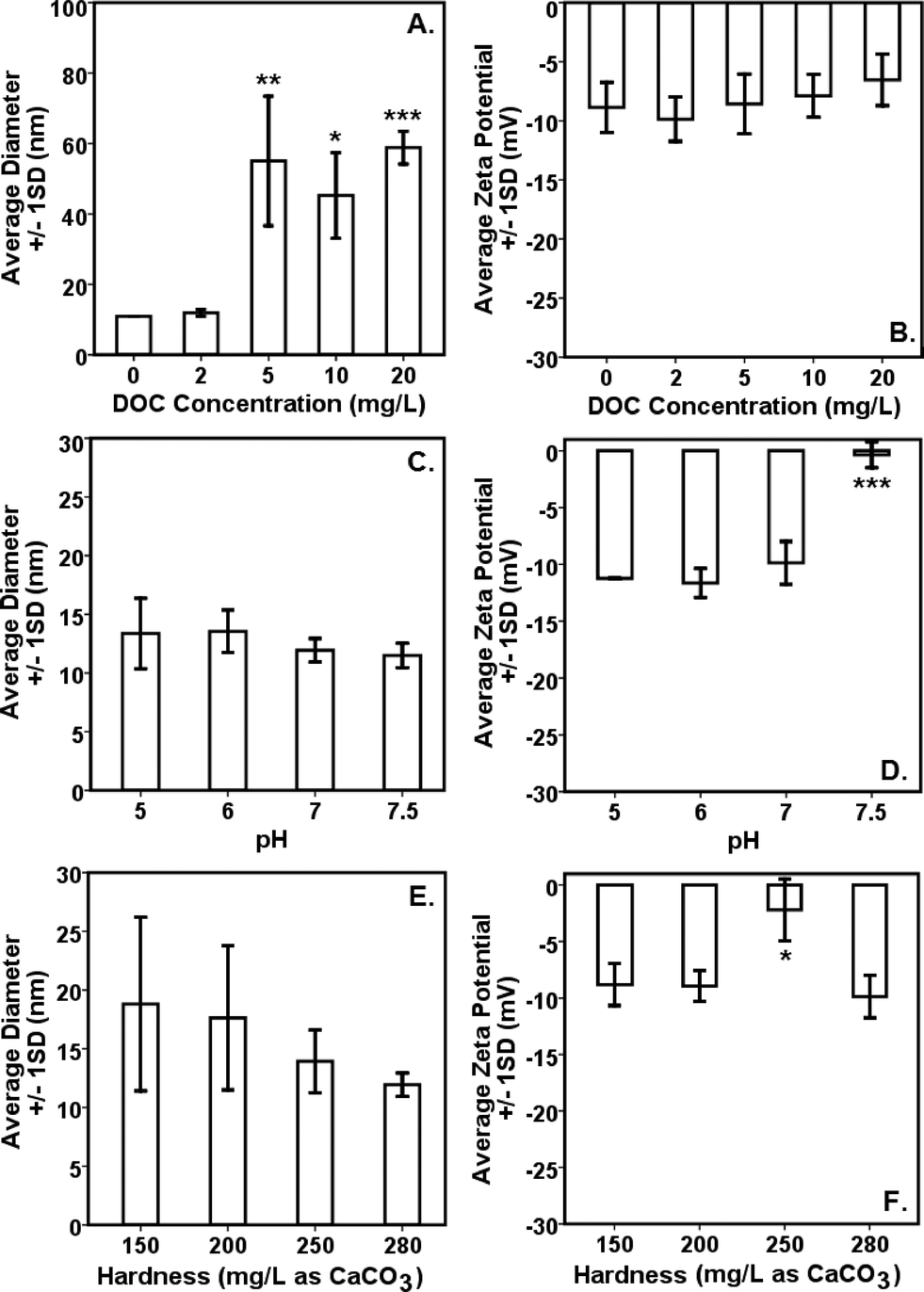 | ||
| Fig. 1 Effects of variable water chemistry conditions on the (A, C, E) hydrodynamic diameter (HDD) and (B, D, F) surface charge (as ζ potential) of citrate–AgNPs. To obtain reliable DLS measurements for HDD and ζ potential, 9.1 mg L−1 citrate–AgNPs were used under a range of water chemistry conditions. DOC, dissolved organic carbon. ‘*’ denotes p ≤ 0.01; ‘**’ denotes p ≤ 0.001; and ‘***’ denotes p ≤ 0.0001 as compared to the baseline of 2 mg DOC L−1, pH 7, and 280 mg hardness L−1 (as CaCO3); significant difference was analyzed by ANOVA followed by Dunnett t test for multiple comparisons. Two (of the three) parameters were held constant while evaluating the effects of the third. | ||
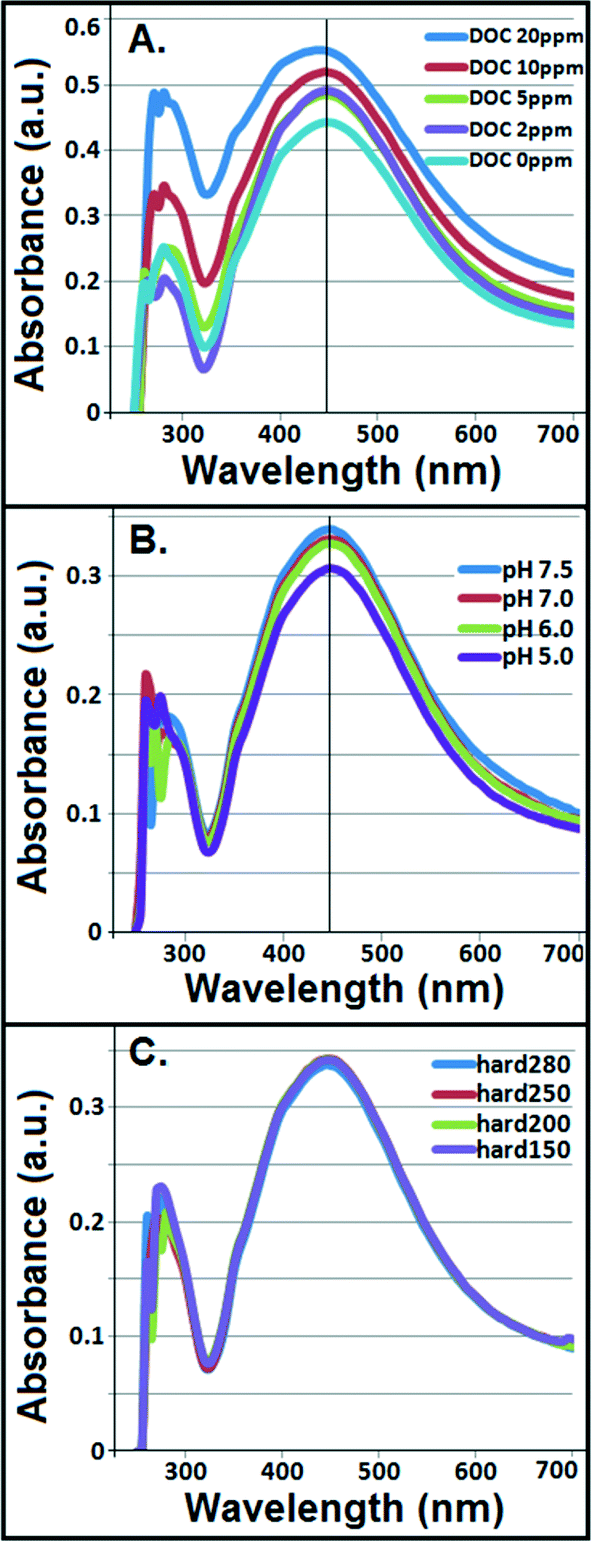 | ||
| Fig. 2 Potential changes in the localized surface plasmon resonance (SPR) spectra of citrate–AgNPs in the presence of variable (A) concentrations of dissolved organic carbon (DOC), (B) pH, and (C) hardness (mg L−1 as CaCO3). Two (of the three) parameters were held constant while evaluating the effects of the third. ‘hard’ denotes hardness. | ||
Although not statistically significant, increasing pH (5–7.5) tended to slightly decrease particle size (Fig. 1C), which corresponded to no significant change in ζ potential in the same pH range (Fig. 1D).8 However, at pH 7.5 surface charge of citrate–AgNPs significantly declined and reached to circumneutral, on average (ζ = −0.35 mV; Fig. 1D). Overall, the DLS data are consistent with the UV–vis absorbance data, revealing no change in SPR peak and an absence of blue/red shift of the plasmonic spectra (Fig. 2B) together indicating no agglomeration or sedimentation of citrate–AgNPs under the measured pH range.20,35
With increasing solution hardness (150–280 mg L−1 as CaCO3), average particle diameter (HDD) showed a decreasing, but not significantly different (p > 0.1), trend (Fig. 1E). In agreement were the ζ potentials that remained unchanged on average, but, to our surprise, at hardness 250 mg L−1 (as CaCO3) it approached an isoelectric point (mean ζ potential = −3.07 mV; Fig. 1F) indicating an approximately equal number of oppositely charged species in the diffuse layer. Increasing ionic strength or hardness can compete with counter-ions causing the electrical double layer to contract, thereby resulting in smaller HDDs.16,39 No change in SPR peak or UV–vis absorbance in the measured hardness range (Fig. 2C) indicated stable particle persistence in the test conditions.
Modulation of ion release by DOC, pH, and hardness
Fig. 3A–C illustrate Ag ion released/dissociated from citrate–AgNPs under different water chemistry conditions during the 4 h incubation period. The dissociation rate increased as a function of DOC concentrations; it increased to nearly 5× at 20 mg L−1 DOC concentration compared to the one without added DOC (Fig. 3A). Generally, smaller sized AgNPs are known for their lower redox potentials, which facilitate higher oxidative dissolution compared to the larger size AgNPs or other forms of bulk Ag.40,41 Greater dissolution that occurred under higher DOC concentrations can be attributed to higher aromaticity (fa = 0.58; the value determined from quantitative C-13 nuclear magnetic resonance spectra by Thorn et al.,34 using a ratio of spectrum area from 110–65 ppm to total spectrum area) of the LHA molecule (compared to several other humic/fulvic acids standards and reference chemicals, including the Suwanee River humic acid [for SRHA fa = 0.37], commercially sold by IHSS and well-characterized by Thorn et al.34), thereby offering more reactive sites and promoting oxidative dissolution of citrate–AgNPs,18 despite some aggregation that might have occurred as shown by the HDDs and ζ potentials (Fig. 1A, B), including the SPR peaks that blue-shifted (Fig. 2A). Consistent with the LHA's higher aromaticity and reactivity observed in this study, Fukushima et al.42 demonstrated enhanced reactivity of the highly aromatic humic acid molecules. The wide differences in the molecular characteristics of the humic substances are primarily due to the nature of precursor compounds and the environmental conditions which supported their formation. A previous study, in contrast to our results, reported a linear decrease in the rates of Ag released from their citrate-coated AgNPs (synthesized by a different method than ours) upon exposure to increasing concentrations of added natural organic matters (i.e., SRHA and SR fulvic acid) in a 1 day experiment.18 These contrasting differences in results can be attributed to the distinct differences in the molecular structures, offering variability in aromatic and/or aliphatic characteristics, of the LHA (used in this study) versus the SRHA or SRFA (for SRFA fa = 0.24 as reported in ref. 32) used in the previous study.18 There was no detectable amount of Ag in 20 mg L−1 DOC when tested alone as the absorbance value (using GF-AAS) for Ag was an order of magnitude lower (0.012 au) than that of the method blank (0.10 au), confirming that the source of released Ag under various DOC concentrations was citrate–AgNPs, not the DOC (LHA) itself. | ||
| Fig. 3 Dynamics of Ag ion release (measured as total Ag) from citrate–AgNPs in a four hour period under variable water chemistry conditions: (A) different concentrations of dissolved organic carbon (DOC), (B) hardness (mg L−1 as CaCO3), and (C) pH levels. Two (of the three) parameters were held constant while evaluating the effects of the third. Concentration-dependent inhibition of β-galactosidase activity in Escherichia coli upon exposure to (D) citrate–AgNPs or (E) free Ag+ (as AgNO3); error bars represent ±1 standard deviation (SD) of at least triplicate test runs. (F) Toxicity of citrate–AgNPs or free Ag+ (as AgNO3) against E. coli in Watauga River water (36.3339° N, −82.2704° W) matrix measured over a period of seven months covering three seasons; EC50s over the course of seven months were not significantly different from each other (ANOVA: p > 0.5) or with the baseline control using moderately hard water (as Evian water with DOC 2 mg L−1, pH 7, and hardness 280 mg L−1 as CaCO3; ANOVA: p > 0.5) for both the citrate–AgNPs and Ag+ ions. The sample collection dates (mm/dd/yy) are shown on X-axis. | ||
Increasing hardness or pH, however, caused a decrease in Ag ion release (Fig. 3B, C). While decreasing protonation (lower H+ concentration) of the citrate–AgNP surface with increasing pH may explain lower Ag ion release,16 competing Ca+2 and Mg+2 ions including other carbonate (CO2, HCO3−1, CO3−2) and noncarbonate (NCH) species that increased as a function of hardness in MHW can be attributed to lower Ag dissociation from citrate–AgNPs. Ag ion release was about 3.5× greater at hardness 150 mg L−1 (as CaCO3) than at 280 mg L−1 (as CaCO3). At pH 5, the release rate was 1.5× greater than at pH 7.5 (Fig. 3C). Under the tested water chemistry conditions relevant to the freshwater environments, overall Ag ion release was in the narrow range 0.33–3.62% (Fig. 3A–C). Only 0.75% (75.1 μg L−1) of Ag emanated in dissolved form from the stock citrate–AgNP (10 mg L−1) suspensions. Dissolution kinetics of citrate–AgNPs in the range 1–10 mg L−1 (as total Ag) in nanopure water in a 4 h period revealed a unique pattern: while total dissolved Ag released from citrate–AgNPs increased as a function of AgNP concentrations, the rate of dissolution (%) decreased with increasing AgNPs concentrations (Fig. 4). These results are consistent with the previous studies utilizing similarly coated AgNPs. For example, Stebounova et al.43 observed 0.03–0.07% dissolution of two commercial AgNPs; Ma et al.44 measured 0.99% dissolution for comparable TEM size and organic-coated AgNPs; Navarro et al.14 reported 1% dissolution of carbonate-coated AgNPs; Huynh and Chen17 found 1.3% dissolution of citrate–AgNPs; Laban et al.45 reported 0.45–3.7% dissolution for two commercial AgNPs (NanoAmor- and Sigma-AgNPs); while Liu and Hurt18 documented 5.5% dissolution for citrate–AgNPs at pH 7.4. Total Ag recovery was in the range 104.8–112.4% in this study.
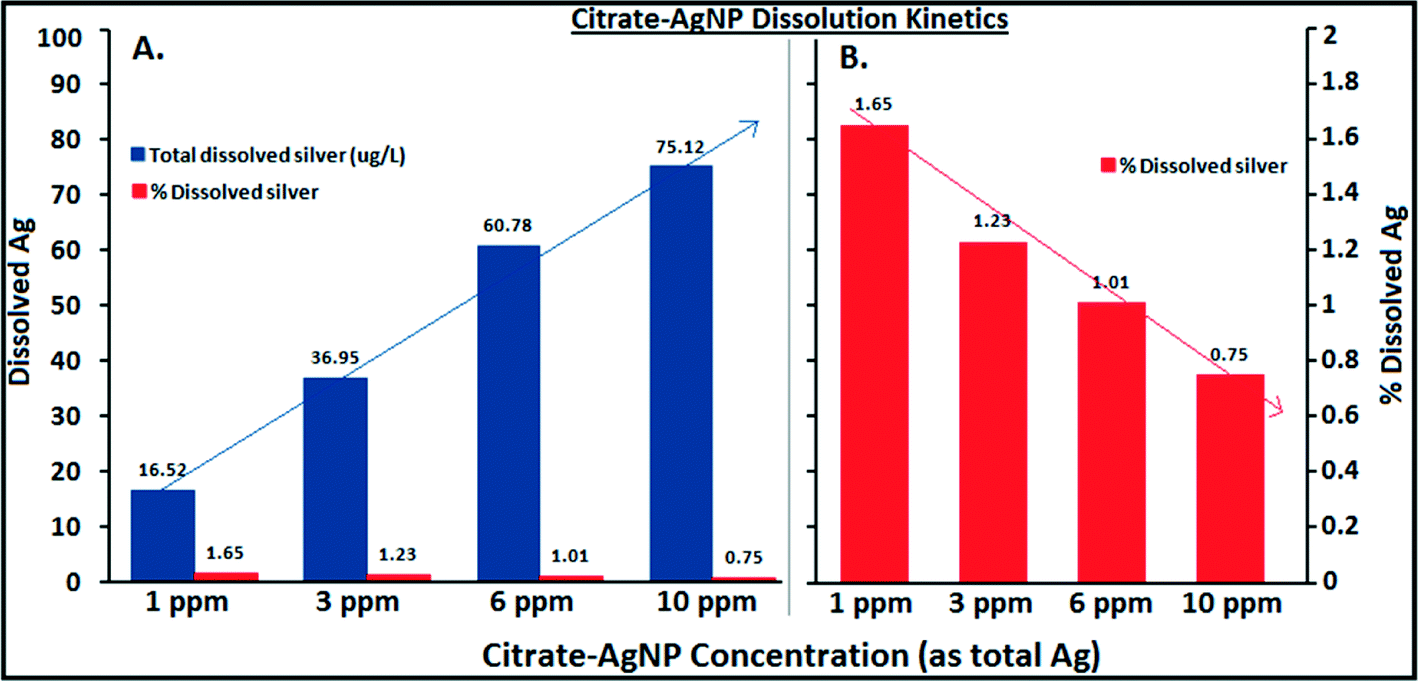 | ||
| Fig. 4 Dissolution kinetics of citrate–AgNPs in a four hour period in the concentration range 1–10 mg L−1 in nanopure water showing that total dissolved Ag increased as a function of citrate–AgNPs concentrations (A), while a steep decrease in the rate of dissolution (%) occurred with increasing citrate–AgNPs concentrations (B). The lines are only meant as a guide to eyes. | ||
Modulation of AgNP toxicity by DOC, pH, and hardness
A concentration-dependent antibacterial activity (β-gal inhibition in E. coli) was observed for both the citrate–AgNPs and free Ag+ (as AgNO3; Fig. 3D, E). Under the condition: pH 7, hardness 280 mg L−1 (as CaCO3) and without added DOC, the EC50 for citrate–AgNPs was 5.79 mg L−1 (as total Ag) versus 0.36 mg L−1 (as total Ag) for free Ag+ (as AgNO3) – an evidence showing lower (by 16 times) toxicity of citrate–AgNPs compared to that of free Ag+.A significant decrease in antibacterial activity was also observed with increasing pH (5–7.5) for both the citrate–AgNPs and free Ag+ (as AgNO3; Fig. 5C, D). At pH 5, the toxicity of citrate–AgNP was 3.2× and 13.8× higher than at pH 7 and 7.5, respectively; while for AgNO3, the toxicity at pH 5 was 6.8× and 6.4× higher than at pH 7 and 7.5, respectively. Comparison of EC50 values in the measured pH range showed about 10–44× lower antimicrobial activity of citrate–AgNPs than of AgNO3 (Fig. 5C, D). Significant attenuation of citrate–AgNPs or free Ag+ toxicity with increasing pH (5–7.5) can be attributed to increasing OH− concentrations, which could compete for the prevalent binding sites on the cell-surface,8 likely lowering potential Ag bioavailability and reducing the toxicity. Although a linear decline in Ag release rate was evident with an increasing pH, the amount released in this study was not enough to cause 50% β-gal activity inhibition.
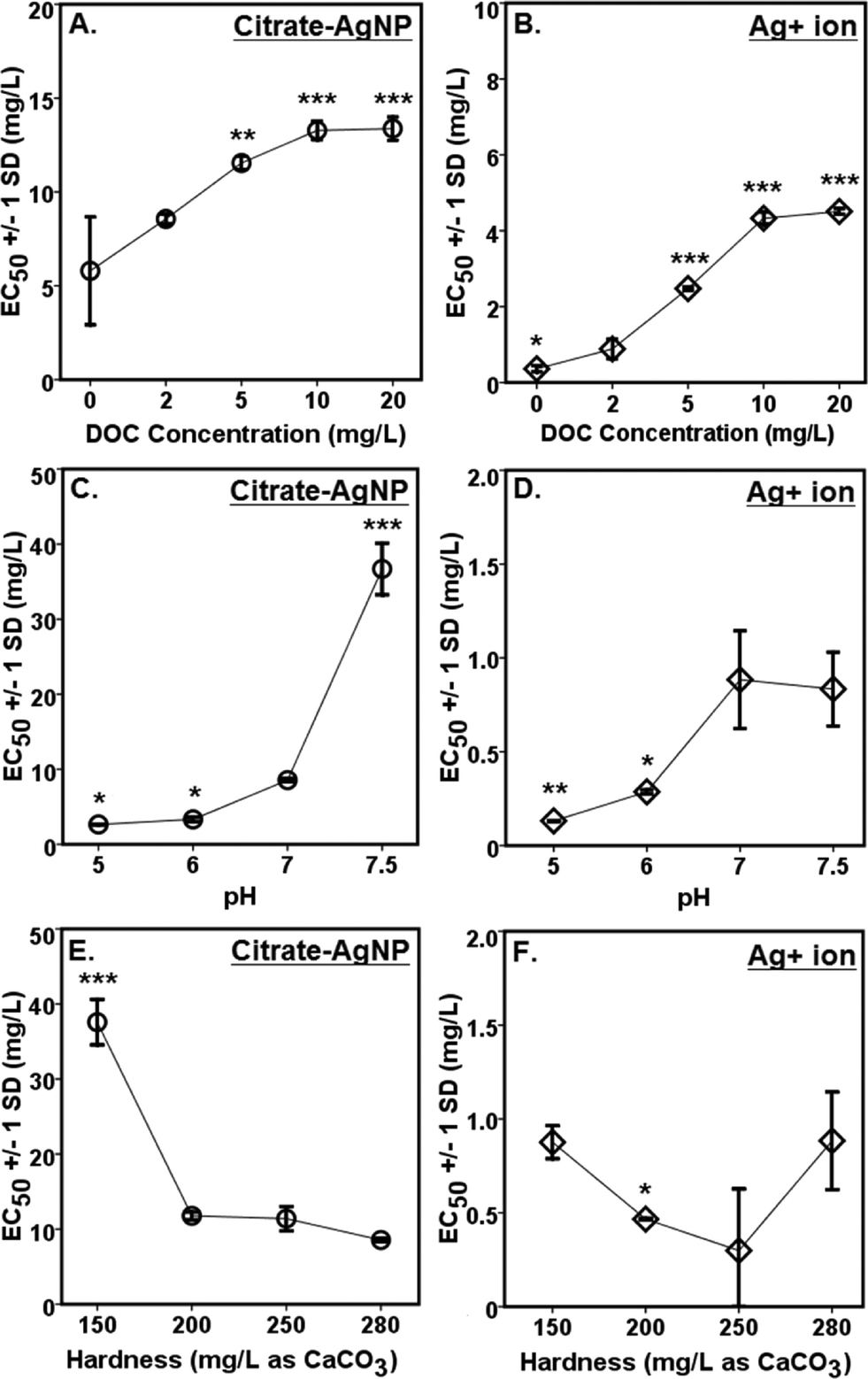 | ||
| Fig. 5 Effects of water chemistry on the toxicity of (A, C, E) citrate–AgNPs or (B, D, F) free Ag+ (used as AgNO3) measured as inhibition of β-galactosidase activity in Escherichia coli. Error bars represent ± 1 standard deviation (SD) of at least triplicate test runs. Note, higher EC50 (mg L−1 as total Ag) values denote lower toxicity and vice versa. DOC, dissolved organic carbon. ‘*’ denotes p ≤ 0.01; ‘**’ denotes p ≤ 0.001; and ‘***’ denotes p ≤ 0.0001 as compared to the baseline of 2 mg DOC L−1, pH 7, and 280 mg hardness L−1 (as CaCO3); significant difference was analyzed by ANOVA followed by Dunnett t (post-hoc) test. Two (of the three) parameters were held constant while evaluating the effects of the third. | ||
Although hardness has been known to confer protective effects against the toxic metals in the natural waters,46–49 a study by Ryan et al.50 contradicts this showing greater toxicity of copper with increasing hardness. With increasing solution hardness, the toxicity tended to increase (lower EC50s) for citrate–AgNPs and AgNO3 in this study (Fig. 5E, F). At lower hardness (150 mg L−1 as CaCO3), citrate–AgNP toxicity was 4.4× lower than at higher hardness (280 mg L−1 as CaCO3). For Ag+, the toxicity was not significantly different between 150 mg hardness L−1 and 280 mg hardness L−1 (as CaCO3; p > 0.05). For citrate–AgNPs, an assessment of EC50 values did not support the premise that the competing Ca+2 and Mg+2 ions and different carbonate (CO2, HCO3−1, CO3−2) and noncarbonate (NCH) species present in the hard water attenuate its toxicity. Instead, the toxicity increased significantly when hardness increased from 150 mg L−1 to 200 mg L−1 (Fig. 5E), suggesting that an increased hardness might have promoted Ag biouptake, thereby leading to greater intracellular β-gal inhibition upon citrate–AgNP treatment. At higher hardness, however, the toxicity of citrate–AgNPs leveled off (Dunnett t test, p > 0.05; Fig. 5E); using the Kruskal–Wallis test to adjust for unequal variance did not change the statistical significance for both the citrate–AgNPs and free Ag+. It is possible that the small sample size might explain some of the differences observed. Mass-based (total Ag) comparison of EC50 values under the tested hardness range showed about 10–43× lower toxicity of citrate–AgNPs than of AgNO3.
A significant change in ζ potential (toward positive value) at pH 7.5 or at hardness 250 mg L−1 (as CaCO3) might be explained by free Ag+ adsorption on the surface of AgNPs. Dissolution profiles of AgNPs as shown in Fig. 3 support the earlier supposition as measurably less dissolved Ag was detected in the supernatant at pH 7.5 or at hardness 250 mg L−1. The forms (species) in which dissolved Ag might be present in the test suspension remained unclear and, therefore, if more free Ag+ were available at pH 7.5 or at hardness 250 mg L−1 (as CaCO3) can be known following on-going speciation studies under similar test conditions as applied in this study.
Increasing DOC concentrations caused a significant decline in antibacterial activity (evidenced by linearly increasing EC50 values) for both the citrate–AgNPs and free Ag+ (as AgNO3; Fig. 5A, B). On a total Ag mass basis, comparison of EC50 values under various DOC concentrations revealed citrate–AgNPs to be 3–16× less toxic than AgNO3 (Fig. 5A, B). The greatest amount of dissolved Ag released from citrate–AgNPs was under the highest DOC (20 mg L−1) concentration, the former being equal to the EC50 value for Ag+ ion (as AgNO3 without DOC).7 Ag+ toxicity was significantly attenuated upon DOC (2–20 mg L−1) addition; it was 12.5× lower at 20 mg L−1 DOC than without added DOC (Fig. 5B). This strongly suggests that the released Ag+ under the experimental conditions did not fully account for the observed citrate–AgNPs toxicity;7,14 16× less toxic citrate–AgNPs were 37× less toxic than Ag+ upon 20 mg L−1 DOC addition into nanosuspension (Fig. 5A, B). Clearly, DOC conferred strong protective effects not only against citrate–AgNP exposure, but also against AgNO3. The solution containing (i) 20 mg L−1 DOC (with pH 7 and hardness 280 mg L−1 as CaCO3), (ii) pH 7.5 (with 2 mg L−1 DOC and hardness 280 mg L−1 as CaCO3), and (iii) hardness 150 mg L−1 as CaCO3 (with 2 mg L−1 DOC and pH 7) appeared to be the media that conferred highest mitigating effects against citrate–AgNPs (Fig. 5A). A previous study has reported released Ag+ effects to be more prominent than of AgNPs in an anaerobic environment.10 Under an aerobic environment with variable water quality characteristics investigated herein, the effect of released Ag+ from citrate–AgNP suspension was rather less prominent and, interestingly, our evidence of lower toxic potency of citrate–AgNPs compared to Ag+ (either released from AgNPs or added as free Ag+, the latter source being AgNO3) clearly shows protective effects of citrate–AgNP suspension (citrate–AgNPs combined with released Ag ions) to that of its ionic counterpart alone. DOC molecules that chemisorb onto the citrate–AgNP surface, modified particle size and surface properties, which may explain its lower antibacterial activity. It is likely that the released Ag+ and/or citrate–AgNPs were prevented from internalization into E. coli cells due to potential DOC–AgNP–Ag+ complexation, likely resulting in lower biouptake and evidently lower toxicity.
Comparison of citrate–AgNP surface charge at pH 7.5 (ζ potential = −0.35 ± 1.14 mV) to that of E. coli cells (in Evian water, ζ potential = −11 mV) suggests an occurrence of dominant attraction force between citrate–AgNPs and the bacterial cell-surface as compared to under other (lower) pH values tested. According to a previous study,51 the greater cell-surface interactions that might occur at nano-bio interface at pH 7.5 should have led to higher toxicity due to potential physical contact; which, to our surprise, the results do not support as lowest toxicity was observed at pH 7.5 than at other pH environments (Fig. 5C). Alternately, increased OH− ions at pH 7.5 might have favored competitive binding of, and subsequently low internalization of, citrate–AgNPs and/or released Ag ions and therefore lower toxicity. At hardness 250 mg L−1 (as CaCO3), unlike other degrees of hardness investigated, circumneutral surface charge (ζ potential = −3.07 ± 2.58 mV) of citrate–AgNPs also corresponded to slightly lower toxicity compared to the hard water with 200 or 280 mg L−1 as CaCO3 (Fig. 5E).
Toxicity of citrate–AgNPs and free Ag+ (as AgNO3) in Watauga River water matrix was also measured over the period of seven months (at four different times) and was not significantly different among themselves (ANOVA: p > 0.5; Fig. 3F) or with the baseline control using MHW (as Evian water with DOC = 2 mg L−1, pH = 7, and hardness = 280 mg L−1 as CaCO3; ANOVA: p > 0.5) for citrate–AgNPs or free Ag+ (Fig. 5). Evaluation of these natural water samples over the course of seven months showed comparable physicochemical properties (ESI† Table S7), which explains the observed similarity in EC50 values for both the citrate–AgNPs and free Ag+ assessed at different time periods with the toxicity measured in MHW (DOC = 2 mg L−1, pH = 7, and hardness = 280 mg L−1 as CaCO3). It is important to note the limitation of using a natural water matrix owing to the fact that its composition and other physicochemical characteristics generally vary spatially and temporally within and between the aquatic systems.52 Here, the purpose of using a natural water matrix was to investigate how the toxicity of colloidal Ag and its free ions would vary in the matrix representative of natural surface water system and how that would compare with the toxicity in the laboratory water. No seasonal variation in antibacterial activity of citrate–AgNPs or free Ag+ was observed and that the synthetic laboratory water (MHW) could adequately capture the toxicity of AgNPs or free Ag+.
Using the generalized linear model (GLM), we probed and quantified the main and interactive effects of the particle properties under variable test conditions to explain the observed toxicity of citrate–AgNPs. Modeling the changeable particle properties data synthesized under a range of DOC concentrations, as presented in eqn (1), our GLM predicted significant main and interactive effects of HDD, ζ potential, and Ag release rate % (Agdiss) explaining the nanotoxicity (as EC50(DOC)). Under variable pH environments, however, only the HDD showed a significant main effect, while the two-way interactive effects of HDD and ζ potential, and HDD and Agdiss, including the three-way interactions among HDD, ζ potential and Agdiss could significantly explain the observed nanotoxicity (as EC50(pH); eqn (2)). Interestingly, for the variable hardness conditions both HDD and Agdiss showed significant main effects, including the significant interactive effects of HDD, ζ potential, and Agdiss, to predict citrate–AgNP toxicity (as EC50(Hardness); eqn (3)). εi,εii, and εiii are the respective error terms of the models representing any variance unaccounted for by each model. The detailed model effects and their parameter estimates are presented in ESI† Tables S3–S5.
| EC50(DOC) = 0.157 × HDD − 0.25 × ζ potential + 2.815 × Agdiss + 0.005(HDD × ζ potential × Agdiss) + εi | (1) |
| EC50(pH) = 10.995 × HDD + 0.819(HDD × ζ potential) − 8.992(HDD × Agdiss) − 0.691(HDD × ζ potential × Agdiss) + εii | (2) |
| EC50(Hardness) = − 0.621 × HDD + 32.049 × Agdiss − 0.057(HDD × ζ potential × Agdiss) + εiii | (3) |
Generally, the interaction term's (HDD × ζ potential × Agdiss) contribution to the models (eqn (1) and (2)) was relatively smaller, although statistically significant, as depicted by their smaller coefficient values (ESI† Tables S3–S5). The precision of the models to correctly predict citrate–AgNP toxicity under variable test conditions was calculated using the equation:13 % precision = 100 (GLM-predicted EC50/experimental EC50), and the results are presented in ESI† Table S6. Under variable DOC concentrations, our model precision was in the range 88.3–105.4%, suggesting the model's ability to effectively predict AgNP toxicity. Under different pH levels, however, the model precision ranged from 75.8–112.4%, and for variable hardness the model was less precise to predict the observed toxicity of citrate–AgNPs (ESI† Table S6), which could possibly be due to (i) larger standard error of the means associated to Ag dissociation rate % (ESI† Table S5), and (ii) the inherent complexity and heterogeneity regarding the water quality encountered in natural systems, as evaluated in this study particularly under the range of pH and hardness conditions, which cannot be adequately captured even by the complex algorithm such as the GLM.
Studies suggest that AgNPs can act as a Ag ions reserve with potential for continual environmental release.19,20,22 Environmental exposures to AgNPs include effects such as: (i) reactive oxygen species (ROS) generation leading to oxidative stress in nitrifying bacteria,53 (ii) structural damage to the phospholipid bilayer, causing pits on the cell wall and altering membrane permeability,54 (iii) inhibition of β-galactosidase activity in E. coli thereby leading to cell death,7 (iv) potential internalization of AgNPs into the cell could cause DNA damage, and potential Ag ion release from internalized AgNPs could affect ion-exchange and cellular respiration,55 (v) direct physical interactions of AgNPs due to charge difference leading to cell death,9 and (vi) potential disruption of Na+ regulation by citrate–AgNPs in the gills of young rainbow trout (Oncorhynchus mykiss).56 This study provides an indirect evidence of cell internalization of Ag, though unclear if internalization occurred as citrate–AgNPs or dissolved Ag ions or both, leading to inhibition of intracellular β-gal activity in E. coli under various conditions that prevail in the aquatic environments and reaffirms the usefulness of the β-galactosidase bioassay as a high-throughput screening (HTS) tool for metal nanoparticle toxicity evaluation in aqueous media.7
Environmental implications
Understanding the potential toxic effects of AgNPs under prevailing environmental conditions using the previously described high throughput β-gal E. coli bioassay is limited to a few studies.2,9 In this study, we systematically evaluated citrate–AgNP toxicity under three significant natural water chemistry conditions (DOC, pH, and hardness), and concurrently evaluated the potential changes in particle surface properties and size, including the rates of dissolved Ag released under those conditions. Our results show water chemistry conditions such as variable pH, DOC, and hardness could significantly modulate citrate–AgNP or AgNO3 (as source of ionic Ag+) toxicity toward Gram negative E. coli. Clearly, a lower release rate of dissolved Ag+ emanating from citrate–AgNP surface layer(s) under different water quality conditions was inadequate to fully account for citrate–AgNP toxicity. Overall, citrate–AgNPs were ca. 3–44 fold less toxic than AgNO3 (total Ag mass basis) under the test environments. While added Leonardite humic acid (source of DOC) and increasing pH could confer protective effects, increasing hardness however tended to promote antibacterial effects in E. coli, which was counterintuitive though. Detectable changes in the hydrodynamic diameter, ζ potential, and plasmon resonance are indicative of modulating effects of the water chemistry conditions on the colloidal stability of citrate–AgNPs in aquatic environments. The data presented are of ecological significance as they suggest that engineered or incidental AgNPs upon release into aquatic systems can modify particle size, surface characteristics, and ion release kinetics, and subsequently alter the toxicity. This study underscores the need to evaluate aquatic toxicity of AgNPs, owing to its continual use as a broad-spectrum antimicrobial agent in the myriad of consumer and biomedical products,10 under additional water chemistry conditions (e.g., other natural organic matter such as fulvic acid, polysaccharides, variable monovalent/polyvalent cations) to better understand the dynamics underlying their fate, mobility, and persistence (as discrete particles, agglomerates, and/or source of ion release) in aquatic environments as they may impact ecosystem health and functions.5–9,11,13–20,24,27,54,57–61Acknowledgements
This research was supported, in part, by the East Tennessee State University (ETSU) RDC grant# 82064 and the research grant# 83003 from the Office of Research and Sponsored Program (ETSU). Special thanks to Kurt Maier (ETSU), Tricia Metts (ETSU), Thabet Tolaymat (USEPA) and Richard Guinn (Eastman Chemicals) for their insights and thoughtful suggestions. We thank Dr. Reza Mohseni (ETSU) for providing access to the UV–vis spectrophotometer, and TEM Analysis Services Lab, TX for support with NP characterization. This study has not been subjected to the US EPA internal review, and the opinions expressed are those of the authors and do not reflect that of the associated institutions. Any mention of the trade names does not imply endorsement or recommendation for use.References
- J. A. Shatkin, L. C. Abbott, A. E. Bradley, R. A. Canady, T. Guidotti, K. M. Kulinowski, R. E. Lofstedt, G. Louis, M. MacDonell, A. D. Maynard, G. Paoli, L. Sheremeta, N. Walker, R. White and R. Williams, Risk Anal., 2010, 30, 1680–1687 CrossRef PubMed.
- J. Gao, S. Youn, A. Hovsepyan, V. L. Llaneza, Y. Wang, G. Bitto and J. J. Bonzongo, Environ. Sci. Technol., 2009, 43, 3322–3328 CrossRef CAS.
- C. A. Poland, R. Duffin, I. Kinloch, A. Maynard, W. A. H. Wallace, A. Seaton, V. Stone, S. Brown, W. MacNee and K. Donaldson, Nat. Nanotechnol., 2008, 3, 423–428 CrossRef CAS PubMed.
- O. V. Tsyusko, J. M. Unrine, D. Spurgeon, E. Blalock, D. Starnes, M. Tseng, G. Joice and P. M. Bertsch, Environ. Sci. Technol., 2012, 46, 4115–4124 CrossRef CAS PubMed.
- S. George, S. Lin, Z. Ji, C. R. Thomas, L. J. Li, M. Mecklenburg, H. Meng, X. Wang, H. Zhang, T. Xia, J. N. Hohman, S. Lin, J. I. Zink, P. S. Weiss and A. E. Nel, ACS Nano, 2012, 6, 3745–3759 CrossRef CAS PubMed.
- L. R. Pokhrel and B. Dubey, Environ. Sci. Technol., 2012, 46, 7755–7762 CrossRef CAS PubMed.
- L. R. Pokhrel, B. Dubey, T. Silva, A. M. El Badawy, T. M. Tolaymat and P. R. Scheuerman, Sci. Total Environ., 2012, 426, 414–422 CrossRef CAS PubMed.
- A. M. El Badawy, R. G. Silva, B. Morris, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2011, 45, 283–287 CrossRef CAS PubMed.
- Z. Xiu, Q. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef CAS PubMed.
- Project on Emerging Nanotechnologies Inventory. http://www.nanotechproject.org/inventories/consumer/.
- T. Benn, B. Cavanagh, K. Hristovski, J. D. Posner and P. Westerhoff, J. Environ. Qual., 2010, 39, 1875–1882 CrossRef CAS.
- T. M. Tolaymat, A. M. El Badawy, A. Genaidy, K. G. Schekel, T. P. Luxton and M. T. Suidan, Sci. Total Environ., 2010, 408, 999–1006 CrossRef CAS PubMed.
- T. U. Silva, L. R. Pokhrel, B. Dubey, K. J. Maier, T. M. Tolaymat and X. Liu, Sci. Total Environ., 2014, 468–469, 968–976 CrossRef PubMed.
- E. Navarro, F. Piccapietra, B. Wagner, F. Marconi, R. Kaegi, N. Odzak, L. Sigg and R. Behra, Environ. Sci. Technol., 2008, 42, 8959–8964 CrossRef CAS.
- L. R. Pokhrel, B. Dubey and P. R. Scheuerman, Expert. Opin. Environ. Biol., 2013, 2, 1–6 Search PubMed.
- A. M. El Badawy, T. P. Luxton, R. G. Silva, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Environ. Sci. Technol., 2010, 44, 1260–1266 CrossRef CAS PubMed.
- K. A. Huynh and K. L. Chen, Environ. Sci. Technol., 2011, 45, 5564–5571 CrossRef CAS PubMed.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS PubMed.
- S. M. Wirth, G. V. Lowry and R. D. Tilton, Environ. Sci. Technol., 2012, 46, 12687–12696 CrossRef CAS PubMed.
- A. M. El Badawy, K. G. Scheckel, M. T. Suidan and T. M. Tolaymat, Sci. Total Environ., 2012, 429, 325–231 CrossRef CAS PubMed.
- X. Li, J. J. Lenhart and H. W. Walker, Langmuir, 2010, 26, 16690–16698 CrossRef CAS PubMed.
- S. R. Kanel and S. R. Al-Abed, J. Nanopart. Res., 2011, 13, 4035–4047 CrossRef CAS.
- G. V. Lowry, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- Y. Yang, H. Zhu, V. L. Colvin and P. J. J. Alvarez, Environ. Sci. Technol., 2011, 45, 4988–4994 CrossRef CAS PubMed.
- C. Levard, S. Mitra, T. Yang, A. D. Jew, A. R. Badireddy, G. V. Lowry and G. E. Brown Jr., Environ. Sci. Technol., 2013, 47, 5738–5745 CrossRef CAS PubMed.
- R. Ma, C. Levard, F. M. Michel, G. E. Brown Jr. and G. V. Lowry, Environ. Sci. Technol., 2013, 47, 2527–2534 CrossRef CAS PubMed.
- X. Jin, M. Li, J. Wang, C. Marambio-Jones, F. Peng, X. Huang, R. Damoiseaux and E. M. V. Hoek, Environ. Sci. Technol., 2010, 44, 7321–7328 CrossRef CAS PubMed.
- L. R. Pokhrel and B. Dubey, Expert. Opin. Environ. Biol., 2013, 2, 1–2 Search PubMed.
- W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, 1997–2008 Search PubMed.
- G. Bitton, K. Jung and B. Koopman, Arch. Environ. Contam. Toxicol., 1994, 27, 25–28 CrossRef CAS.
- NSF International, 2011, http://www.evian.com/files/Evian%20AWQR%202012%20ENG%20California.pdf.
- Y. P. Chin, G. R. Aiken and E. O'Loughlin, Environ. Sci. Technol., 1994, 28, 1853–1858 CrossRef CAS PubMed.
- L. Wang, F. Wu, R. Zhang, W. Li and H. Liao, J. Environ. Sci., 2009, 21, 581–588 CrossRef CAS.
- K. A. Thorn, D. W. Folan and P. MacCarthy, U.S. Geological Survey, Water-Resources Investigations Report 89-4196, Denver, CO, 1989 Search PubMed.
- F. Piccapietra, L. Sigg and R. Behra, Environ. Sci. Technol., 2012, 46, 818–825 CrossRef CAS PubMed.
- L. R. Pokhrel, I. Karsai, M. K. Hamed and T. F. Laughlin, Ethology Ecology & Evolution, 2013, 25, 214–226 Search PubMed.
- IBM SPSS 20.0 User's guide, SPSS Inc., Chicago, IL Search PubMed.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- N. Akaighe, S. W. Depner, S. Banerjee, V. K. Sharma and M. Sohn, Sci. Total Environ., 2012, 441, 277–289 CrossRef CAS PubMed.
- M. Mostafavi, J. L. Marignier, J. Amblard and J. Belloni, Radiat. Phys. Chem., 1989, 34, 605–617 CrossRef.
- O. S. Ivanova and F. P. Zamborini, J. Am. Chem. Soc., 2010, 132, 70–72 CrossRef CAS PubMed.
- M. Fukushima, Y. Tanabe, K. Morimoto and K. Tatsumi, Biomacromolecules, 2007, 8, 386–391 CrossRef CAS PubMed.
- L. V. Stebounova, E. Guio and V. J. Grassian, J. Nanopart. Res., 2011, 13, 233–244 CrossRef CAS.
- R. Ma, C. Levard, S. Marinakos, Y. Cheng, J. Liu, F. M. Michel, G. E. Brown Jr. and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 752–759 CrossRef CAS PubMed.
- G. Laban, L. F. Nies, R. F. Turco, J. W. Bickham and M. S. Sepulveda, Ecotoxicology, 2010, 19, 185–195 CrossRef CAS PubMed.
- H. Nelson, D. A. Benoit, R. J. Erickson, V. R. Mattson and J. Lindberg, EPA/600/S3-86/023, U.S. Environmental Protection Agency, Duluth, MN, 1986 Search PubMed.
- D. M. Di Toro, H. E. Allen, H. L. Bergman, J. S. Meyer, P. R. Paquin and R. C. Santore, Environ. Toxicol. Chem., 2001, 20, 2383–2396 CrossRef CAS.
- K. L. Sciera, J. J. Isely, J. R. Tomasso Jr. and S. J. Klaine, Environ. Toxicol. Chem., 2004, 23, 2900–2905 CrossRef.
- A. Bianchini and C. M. Wood, Ecotoxicol. Environ. Saf., 2008, 71, 32–40 CrossRef CAS PubMed.
- A. C. Ryan, J. R. Tomasso and S. J. Klaine, Environ. Toxicol. Chem., 2009, 28, 1663–1670 CrossRef CAS PubMed.
- G. A. Sotiriou, A. Meyer, J. T. N. Knijnenburg, S. Panke and S. E. Pratsinis, Langmuir, 2012, 28, 15929–15936 CrossRef CAS PubMed.
- United Nations Environment Program and the World Health Organization. ISBN 0 419 22320 7 (Hbk) 0 419 21730 4 (Pbk); 1996. http://www.who.int/water_sanitation_health/resourcesquality/wqmchap12.pdf.
- O. Choi and Z. Q. Hu, Environ. Sci. Technol., 2008, 42, 4583–4588 CrossRef CAS.
- J. Fabrega, J. C. Renshaw and J. R. Lead, Environ. Sci. Technol., 2009, 43, 9004–9009 CrossRef CAS PubMed.
- H. T. Ratte, Environ. Toxicol. Chem., 1999, 18, 89–108 CrossRef CAS.
- A. G. Schultz, K. J. Ong, T. MacCormack, G. Ma, J. G. C. Veinot and G. G. Goss, Environ. Sci. Technol., 2012, 46, 10295–10301 CAS.
- L. Truong, K. S. Saili, J. M. Miller, J. E. Hutchison and R. L. Tanguay, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2012, 155, 269–2674 CAS.
- M. R. Smith, M. G. Boenzli, V. Hindagolla, J. Ding, J. M. Miller, J. E. Hutchison, J. A. Greenwood, H. Abeliovich and A. T. Bakalinsky, Appl. Environ. Microbiol., 2013, 79, 728–733 CrossRef CAS PubMed.
- L. R. Pokhrel and B. Dubey, Sci. Total Environ., 2013, 452–453, 321–332 CrossRef CAS PubMed.
- A. J. Kennedy, M. A. Chappell, A. J. Bednar, A. C. Ryan, J. G. Laird, J. K. Stanley and J. A. Steevens, Environ. Sci. Technol., 2012, 46, 10772–10780 CrossRef CAS PubMed.
- L. R. Pokhrel, B. Dubey and P. R. Scheuerman, Environ. Sci. Technol., 2013, 47, 12877–12885 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Citrate–AgNP synthesis protocol; purification of citrate–AgNPs using TFF system; β-gal protocol; DOC calibration curve using UV–vis absorbance; TEM/UV–vis characterization of AgNPs; PSD and effect of dilution in moderately hard water of citrate–AgNPs; mechanism of β-galactosidase activity in E. coli; QA/QC; GLMs model effects and parameter estimates; GLM precision to predict toxicity; and physicochemical characteristics of Watauga River water samples. See DOI: 10.1039/c3en00017f |
| This journal is © The Royal Society of Chemistry 2014 |