A bimetallic iron(III) catalyst for CO2/epoxide coupling†‡
Antoine
Buchard
a,
Michael R.
Kember
a,
Karl G.
Sandeman
b and
Charlotte K.
Williams
*a
aDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: c.k.williams@imperial.ac.uk; Tel: +44 (0)20 7594 5790
bDepartment of Physics, Imperial College London, London, SW7 2AZ, UK
First published on 27th September 2010
Abstract
A novel di-iron(III) catalyst for the copolymerisation of cyclohexene oxide and CO2 to yield poly(cyclohexene carbonate), under mild conditions, is reported. The catalyst selectivity was completely changed on addition of an ammonium co-catalyst to yield only the cis-isomer of the cyclic carbonate, also under mild conditions. Additionally, the catalyst was active for propylene carbonate and styrene carbonate production at 1 atm pressure.
Carbon dioxide is an attractive, renewable C-1 source: it is non-toxic, highly abundant, relatively inexpensive and a waste product of many processes.1–4 The metal catalysed coupling of CO2 and epoxides (Scheme 1) is one of the few processes that actually consume CO2 and it provides a sustainable alternative synthesis of carbonates. The catalyst, and reaction conditions, control formation of either a cyclic carbonate5 or polycarbonate;6–8 the trans-cyclic carbonate is the thermodynamic product. Aliphatic polycarbonates are applied as binders, adhesives and coatings, whilst cyclic carbonates are used as high boiling solvents, electrolytes, fuel additives and as sustainable reagents.
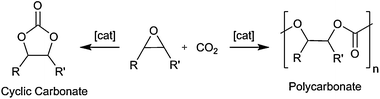 | ||
| Scheme 1 Coupling of CO2/epoxides. | ||
Various metal complexes are successful copolymerisation catalysts, usually having Zn(II), Co(II/III), Cr(III) or Al(III) active sites.6,7 There is much scope for new metal catalysts, including those that operate at low CO2 pressure, thereby improving the energy balance and net CO2 consumption. Recently, we reported various bimetallic Zn(II) and Co(II/III) copolymerisation catalysts which displayed impressive activity, at only 1 atm CO2 pressure.9–11 Here, we report a novel di-iron(III) catalyst which is selective for the production of either poly(cyclohexene carbonate) (PCHC) or cyclohexene carbonate (CHC). Iron is a particularly attractive metal due to its ready availability and low-cost, yet it has received scant attention in either catalysis. Double metal cyanides, e.g. [Zn3[Fe(CN)6]2], are viable heterogeneous copolymerisation catalysts (although 50–60 atm CO2 are required),12 however, the homogeneous analogues were less active, under the same conditions.13,14 Some heterobimetallic tert-butoxide iron complexes were also reported.15 Importantly, these species all rely on Zn(II) or La(III) centres as the active site. Furthermore, although Co(III)/Cr(III) salen complexes are excellent catalysts, the Fe(III) analogues were inactive.16,17
The macrocyclic pro-ligand, H2L, was prepared following a two-step procedure, in 84% overall yield.9 It was deprotonated, using potassium hydride, and reacted with two equivalents of [FeCl3·DME] (DME = dimethoxyethane) to yield an air stable blue di-iron tetrachloride complex 1 (85%, unoptimised) (Scheme 2).
![Synthesis of 1. Reagents and conditions: (i) 2 KH, THF, −30 to 25 °C, 2 h; (ii) 2 [FeCl3(DME)], THF, 25 °C, 20 h.](/image/article/2011/CC/c0cc02205e/c0cc02205e-s2.gif) | ||
| Scheme 2 Synthesis of 1. Reagents and conditions: (i) 2 KH, THF, −30 to 25 °C, 2 h; (ii) 2 [FeCl3(DME)], THF, 25 °C, 20 h. | ||
The complex's paramagnetism rendered NMR spectroscopy ineffective, but the stoichiometry was confirmed by elemental analysis and mass spectrometry. The UV-Vis spectrum (Fig. S1, ESI‡) showed a strong ligand-to-metal charge transfer absorption at 590 nm.18 No other d–d transitions were detected, in agreement with the complex having two high-spin (HS) iron(III) centres in octahedral coordination environments. The effective magnetic moment, determined by Evans' NMR method,19 at 293 K was 8.2 μB (8.37 μB is calculated for two isolated HS Fe(III)). Further investigation, using a Oxford Instruments Vibrating Sample Magnetometer in a field of 8 Tesla, shows the saturation moment (at 4.2 K) is approximately 9 μB (Fig. S2, ESI‡). These findings are consistent with a bimetallic complex in which both Fe(III) centres are in the HS (S = 5/2) state,20 possibly with some ferromagnetic coupling.
Complex 1 was evaluated as a cyclohexene oxide (CHO) copolymerisation catalyst, reactions were conducted in neat CHO, at 80 °C and under 1–10 atm of carbon dioxide (Table 1). It was active at just 1 atm of CO2 (entry 1), producing a copolymer in 93% yield, with 7% trans-cyclohexene carbonate (CHC). However, the quality of the copolymer was sub-optimal, with only 66% carbonate linkages. The TON and TOF values were slightly lower than those of analogous Zn(II)/Co(II/III) species, but still showed good values at such low CO2 pressure.9–11
| Entry | Catalyst (mol%) | p(CO2)/atm | Time/h | % CHO conversionb | TONc |
TOF/h−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d d |
% CHC | % PCHC [% carbonate]b,e | M n [Mw/Mn]f |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: neat CHO, 80 °C. b From the normalised integrals, in the 1H NMR spectra, of the methylene resonances, including PCHC carbonate (δ: 4.65 ppm), PCHC ether (δ: 3.45 ppm), and CHC (δ: 3.9 (trans) or 4.63 ppm (cis)). c TON = molCHO used/mol11. d TOF = TON/h. e % Carbonate = (copolymer carbonate linkages)/(copolymer carbonate + ether linkages). f Determined by GPC, in THF, using narrow Mw polystyrene standards as calibrants. | |||||||||
| 1 | 0.1% 1 | 1 | 48 | 29 | 290 | 6 | 7 (trans) | 93 [66] | 2000 [1.55] |
| 2 | 0.1% 1 | 10 | 5 | 24 | 235 | 47 | 1 (trans) | 99 [99] | 3100 [1.18] |
| 3 | 0.1% 1 | 10 | 24 | 70 | 694 | 29 | 1 (trans) | 99 [99] | 11700 [1.13] |
| 4 | 0.01% 1 | 10 | 24 | 25 | 2570 | 107 | 1 (trans) | 99 [99] | 17200 [1.03] |
| 8100 [1.06] | |||||||||
| 5 | 0.1% 1 + 0.4% [PPN]Cl | 1 | 48 | 41 | 410 | 9 | 100 (99% cis) | — | — |
| 6 | 0.1% 1 + 0.2% [PPN]Cl | 1 | 120 | 33 | 330 | 3 | 100 (97% cis) | — | — |
| 7 | 0.1% 1 + 0.1% [PPN]Cl | 1 | 24 | 20 | 200 | 8 | 89 (96% cis) | 11 [27] | — |
| 8 | 1% 1 + 2% [PPN]Cl | 1 | 24 | 90 | 90 | 4 | 100 (99% cis) | — | — |
| 9 | 0.1% 1 + 0.2% [PPN]Cl | 10 | 24 | 70 | 700 | 29 | 76 (96% cis) | 24 [99] | 2300 [1.26] |
On increasing the pressure to 10 atm (entry 2), the rate of copolymerisation was significantly increased with the activity being ∼8 times greater than at 1 atm. Also, the quality of PCHC was excellent: only trace quantities of the trans-CHC by-product and >99% carbonate linkages in the copolymer were observed. The copolymerisation was run until solidification prevented further conversion (70%, entry 3), yielding PCHC with good productivity (TON: 694), indeed the new di-iron catalyst was as active under these conditions as the recently reported dizinc catalyst.913C{1H}NMR spectroscopy showed that the PCHC was atactic (Fig. S6, ESI‡).21Gel permeation chromatography (GPC) indicated a molecular weight of 11700 and narrow polydispersity index (1.13) (Fig. S7, ESI‡). At low conversion (entry 2, 24%), the MALDI-ToF mass spectrum showed a major series of peaks corresponding to PCHC with chloride end-groups (Fig. S10–S12, ESI‡).
A copolymerisation mechanism is proposed (Scheme 3) where the Fe–Cl bond(s) initiate the ring-opening of CHO to generate an Fe–OR species, which undergo CO2 insertion to produce an iron–carbonate intermediate. Propagation and copolymerisation occurs by sequential repetition of the ring-opening and insertion reactions. It is likely that the unprecedented activity of 1 is due to the bimetallic active site and the macrocyclic ligand coordination environment.
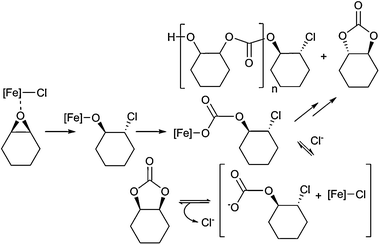 | ||
| Scheme 3 Proposed mechanism for CO2/CHO coupling (for clarity, only (R,S)-CHO is represented here although the same reactions occur for the enantiomer, yielding a racemic mixture of cis-CHC and atactic PCHC). | ||
At lower catalyst loading (0.01%), the TON (2570) and TOF (107 h−1) increased markedly as did the molecular weight (entry 4). However, GPC analysis revealed a bimodal distribution and the MALDI-ToF mass spectrum of the lower molecular weight series (Mn: 8100) revealed chains end-capped with cyclohexenyl groups and hydroxyl groups (Fig. S14, ESI‡).10 The cyclohexenyl end-group is proposed to arise by chain transfer reactions with cyclohex-1-enol, produced by elimination of HCl from the expected chloro-cyclohexanolate iron species. Indeed, traces of cyclohex-1-enol have indeed been identified in the control reaction between 1 (0.1%) and CHO, in the absence of CO2.
Researchers working with salen metal halide catalysts (or closely related derivatives) have observed increased productivity in the presence of ionic co-catalysts, such as ammonium salts.7,22,23 Indeed, catalyst 1 is less active than the best [(salen)M(III)] catalysts (M = Co/Cr) in the presence of -onium salts.8 However, under mild conditions (1 atm, 80 °C), 1 produced exclusively cis-cyclohexene carbonate on activation with bis(triphenylphosphino)iminium chloride ([PPN]Cl) (entries 5–7). It is notable that [PPN]Cl was completely inactive when used on its own. On increasing the loading (entry 8), 90% conversion to cis-CHC was achieved in 24 h. The cis-CHC product was characterised by a carbonyl stretching frequency in the FTIR spectrum at 1804 cm−1 (vs. 1825 cm−1 for the trans-isomer) and by the chemical shift of the methyne protons, at 4.63 ppm (vs. 3.90 ppm for the trans-isomer) in the 1H NMR spectrum (Fig. S3 and S4, ESI‡).24
To our knowledge, this is the first example of an Fe active site for either the copolymerisation or cyclisation of epoxides/CO2.5–7 Furthermore, the formation of cis-CHC is unusual: the trans-isomer is more commonly formed, usually by back-biting reactions under thermodynamic control (Scheme 3).7,25 However, the low pressure and moderate temperature used here are insufficient to yield significant quantities of trans-CHC (cf. entries 1–4). The formation of the cis-isomer requires a mechanism involving a double-inversion of CHO stereochemistry (Scheme 3). It is proposed that [PPN]Cl facilitates exchange reactions between the initiating iron carbonate species and chloride ions. The putative anionic carbonate species would be significantly more nucleophilic than the metal bound carbonate and so undergo intramolecular nucleophilic substitution, chloride elimination and formation of the cis-CHC. Such a mechanism is consistent with the finding that as the number of equivalents of [PPN]Cl is reduced so the percentage of cis-isomer reduces (entries 5–7). If a single equivalent of [PPN]Cl is used, some copolymerisation also occurs, highlighting the importance of excess chloride (vs.1) to prevent competitive binding of epoxide at the initiating Fe-carbonate species and competing copolymerisation. Increasing the pressure of CO2 to 10 atm, keeping all other conditions the same, led to an increase in production of PCHC (with 99% carbonate linkages), with low amounts of the trans-CHC also being observed (entry 9). There are only a few reports of cis-CHC production from CO2,13,26–28 indeed, many common cyclic carbonate catalysts are inactive for CHO.5,29,30 Interestingly, one of the homogeneous DMC model compounds was cis-CHC selective, however, relatively harsh conditions (52 atm CO2) were necessary.13
In order to establish the generality of the catalyses, 1 was tested with propylene (PO) and styrene oxide (SO) (Table 2).
| Entry |
Catalyst mol% 1∶mol% [PPN]Cl |
Epoxide | t/h | T/°C | % Conv.b | TONc |
TOF/h−1d |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: neat epoxide, 1 atm CO2. b From the normalised integrals, in the 1H NMR spectra, of the methylene resonances. c TON = molCHO/mol11. d TOF = TON/h. | |||||||
| 1 | 0.1∶0.2 |
PO | 24 | 25 | 18 | 180 | 8 |
| 2 | 0.1∶0.2 |
PO | 24 | 34 | 50 | 500 | 21 |
| 3 | 0.5∶1 |
PO | 48 | 25 | 91 | 182 | 4 |
| 4 | 0.1∶0.2 |
SO | 24 | 25 | 3 | 30 | 1 |
| 5 | 0.1∶0.2 |
SO | 24 | 80 | 83 | 830 | 35 |
| 6 | 0.5∶1 |
SO | 24 | 25 | 17 | 34 | 1 |
| 7 | 0.5∶1 |
SO | 20 | 80 | 98 | 196 | 10 |
Catalyst 1 did not yield any copolymer, even under 10 atm CO2 and at higher temperature (40 °C for PO, 80 °C for SO). The use of 1 and two equivalents of [PPN]Cl gave a good catalyst for production of propylene carbonate (PC) and styrene carbonate (SC). It was even active under mild conditions: 25 °C and 1 atm CO2 (entries 1, 4). Although, the activity was much improved by increasing the temperature (entries 2, 5, 7) and/or the catalyst loading (entries 3, 6, 7). Thus, using 0.5% of 1 and 1% of [PPN]Cl, 91% conversion of PO into cyclic propylene carbonate was achieved within 48 h at 25 °C (entry 3). At 80 °C, the conversion of SO was >80% after 24 h (entry 5), or within 6 h at higher catalyst loadings. There are only a few cyclic carbonate catalysts active under such mild conditions, the best of which are di-aluminium salen complexes.30–33Catalyst 1 is slightly less active than these catalysts (cf. under related conditions to entry 6, di-Al has TON = 86 and TOF 4 h−1).30 Interestingly, the di-Al catalysts also required two equivalents of co-catalyst; a mechanism was proposed whereby one Al centre binds and ring-opens the epoxide and the other delivers the CO2, which was pre-coordinated as a carbamate salt.33
In conclusion, a novel di-iron catalyst for CO2/epoxide coupling is reported. The catalyst shows good productivity and activity for the sequential copolymerisation of CHO and CO2, under mild conditions. The combination of the new catalyst and more than two equivalents of [PPN]Cl leads to the selective formation of cis-cyclohexene carbonate. The catalyst can be generally applied, yielding propylene carbonate and styrene carbonate, under only 1 atm pressure CO2. This is the first example of an iron catalyst for either the production of cyclohexene cyclic carbonate or polycarbonate. Furthermore it is a rare example of catalysis at 1 atm for these processes and is a cis-selective carbonate catalyst.
Notes and references
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975–2992 RSC.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010 10.1039/c004106h.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2010 10.1039/c0cc02207a.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- W. J. Kruper Jr. and D. J. Swart, The Dow Chemical Company, 1985, US 4,500,704 Search PubMed.
- D. J. Darensbourg, M. J. Adams and J. C. Yarbrough, Inorg. Chem., 2001, 40, 6543–6544 CrossRef CAS.
- D. J. Darensbourg, M. J. Adams, J. C. Yarbrough and A. L. Phelps, Inorg. Chem., 2003, 42, 7809–7818 CrossRef CAS.
- A. V. Nikitinskii, L. N. Bochkarev, S. Y. Khorshev and M. N. Bochkarev, Russ. J. Gen. Chem., 2004, 74, 1197–1200 CrossRef CAS.
- D. J. Darensbourg, C. G. Ortiz and D. R. Billodeaux, Inorg. Chim. Acta, 2004, 357, 2143–2149 CrossRef CAS.
- G. A. Luinstra, G. R. Haas, F. Molnar, V. Bernhart, R. Eberhardt and B. Rieger, Chem.–Eur. J., 2005, 11, 6298–6314 CrossRef CAS.
- K. K. Nanda, S. K. Dutta, S. Baitalik, K. Venkatsubramanian and K. Nag, J. Chem. Soc., Dalton Trans., 1995, 1239–1244 RSC.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- K. S. Murray, Coord. Chem. Rev., 1974, 12, 1–35 CrossRef CAS.
- K. Nozaki, K. Nakano and T. Hiyama, J. Am. Chem. Soc., 1999, 121, 11008–11009 CrossRef CAS.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869–10878 CrossRef CAS.
- K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC.
- D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS.
- D. J. Darensbourg, P. Bottarelli and J. R. Andreatta, Macromolecules, 2007, 40, 7727–7729 CrossRef CAS.
- C. Qi, H. Jiang, Z. Wang, B. Zou and S. Yang, Synlett, 2007, 255–258 CAS.
- W. J. Kruper and D. D. Dellar, J. Org. Chem., 1995, 60, 725–727 CrossRef CAS.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
- A. Decortes, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580–4582 RSC.
- J. Melendez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323–3326 CrossRef CAS.
- X.-B. Lu, B. Liang, Y.-J. Zhang, Y.-Z. Tian, Y.-M. Wang, C.-X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CrossRef CAS.
- A. Berkessel and M. Brandenburg, Org. Lett., 2006, 8, 4401–4404 CrossRef CAS.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS.
Footnotes |
| † This article is part of the ‘Emerging Investigators’ themed issue for ChemComm. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures; spectral data for 1 and PCHC. See DOI: 10.1039/c0cc02205e |
| This journal is © The Royal Society of Chemistry 2011 |