
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecyclable RAFT-3D printing
Xiaofeng
Pan
 ,
Xinggang
Luo
,
Xiangqiang
Pan
,
Jiajia
Li
* and
Jian
Zhu
*
,
Xinggang
Luo
,
Xiangqiang
Pan
,
Jiajia
Li
* and
Jian
Zhu
*
State Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, Department of Polymer Science and Engineering, College of Chemistry Chemical Engineering and Materials Science, Soochow University, China. E-mail: chemjji@suda.edu.cn; chemzhujian@suda.edu.cn
First published on 25th September 2025
Abstract
Recyclable 3D printing systems are urgently needed to address sustainability challenges in polymer manufacturing. Herein, we report a closed-loop 3D printing strategy based on reversible addition–fragmentation chain transfer (RAFT) step-growth polymerization, in which dynamic trithiocarbonate linkages are embedded into the polymer network. The resulting network is recyclable via RAFT interchange under 405 nm irradiation in the presence of excess RAFT agents, regenerating oligomers bearing reactive chain-end groups. These intermediates can be directly re-crosslinked by adding stoichiometric vinyl monomers, thereby reconstructing the original polymer structure. Moreover, upcycled materials can be easily obtained by varying the vinyl monomer concentration. Digital light processing (DLP)-based 3D printing of these resins yields robust objects with tunable mechanical properties (Young's modulus from 0.78 to 835 MPa). Post-printing functionalization and erasure via RAFT interchange further demonstrate the dynamic and “living” features of this system. This catalyst-free, modular approach eliminates the need for specialized dynamic covalent monomers, offering a practical and sustainable platform for recyclable RAFT-based 3D printing.
Introduction
3D printing, also known as additive manufacturing, has found widespread application owing to its flexibility, precision, and efficiency across diverse fields, including the aerospace industry, biomedical engineering, architecture, and education.1 Among various 3D printing technologies, digital light processing (DLP) stands out due to its high resolution and accuracy, rapid printing speed, and excellent surface quality.2,3 This technique relies on layer-by-layer photopolymerization, typically involving radical or cationic polymerization mechanisms. However, these conventional polymerization methods are generally non-living, offering limited control over molecular weight, molecular weight distribution (MWD), and other polymer structural parameters. In contrast, reversible deactivation radical polymerization (RDRP) has emerged as a powerful strategy that enables precise control over polymerization, allowing the synthesis of polymers with well-defined molecular weights, narrow MWDs, and programmable monomer sequences and topologies.4–6 In recent years, integrating RDRP with photoinduced 3D printing has opened new avenues for fabricating tailor-made polymeric materials, providing enhanced control over network formation and thus the final material properties.7–9 In particular, reversible addition–fragmentation chain transfer (RAFT) polymerization—one of the most versatile RDRP techniques—has been successfully incorporated into photoinduced 3D printing, allowing precise tuning of mechanical properties through modulation of the (macro) RAFT agent's concentration or structure.10–18 Furthermore, the thiocarbonylthio end group of RAFT polymers enables facile post-functionalization, which broadens the utility of printed materials in areas such as self-healing,19,20 polymerization-induced microphase separation (PIMS),21–23 biomedical applications,24–27 and 4D printing.13,28Like other 3D printing systems, RAFT-based 3D printed materials face challenges in recyclability, which is critical for reducing environmental impact and production costs.29 Moreover, the resins used for RAFT-based 3D printing typically require additional synthetic steps to prepare chain transfer agents (CTAs), initiators, or specialized monomers. Therefore, the development of recyclable resin systems for RAFT-mediated 3D printing is highly desirable. Currently, employing reversible chemical reactions triggered by external stimuli has demonstrated significant promise in enabling recyclable 3D printing. For example, Xie, Zheng, and coworkers reported a photopolymerization system based on the stepwise reaction of aldehydes and thiols to form dithioacetal linkages within the polymer network.30 These linkages can be reversed to the original aldehyde and thiol upon thermal treatment and neutralization. When a fresh photoacid generator (PAG) is introduced, the recycled liquid mixture can be reprinted into 3D objects with a network structure identical to the original, thereby achieving closed-loop 3D printing. In another example, Dove, Worch, and colleagues utilized lipoates bearing dynamic cyclic disulfides as DLP-printable resins.31 Upon light irradiation, these resins undergo ring-opening polymerization to form crosslinked networks, which can subsequently be depolymerized back to cyclic disulfides via base catalysis. Additionally, Boyer and Hawker developed a recyclable printing resin by combining acrylate monomers with a lipoic acid–based cross-linker containing a central disulfide bond, which imparts the printed materials with remarkable self-healing and degradability.32 In this paper, the authors also demonstrate reprinting at least 2 cycles was achieved with a good maintain of the mechanical properties. Other dynamic covalent chemistries have also been elegantly employed for recyclable 3D printing systems.33–41 However, such reversible chemistries have yet to be integrated into RAFT-based 3D printing. Moreover, some stimulus conditions used in these dynamic systems may be incompatible with RAFT agents (e.g. acid or base-catalyzed dynamic systems), potentially resulting in degradation of thiocarbonylthio groups. Some specialized monomers or catalysts required complex synthesis, restricting their practical applications. Notably, the thiocarbonylthio group in RAFT agents can itself serve as a dynamic linkage through radical addition–fragmentation chain transfer processes.42 This dynamic RAFT exchange, also known as RAFT interchange (Scheme 1A), has been applied to prepare self-healing materials;19 however, such systems typically involve irreversibly crosslinked acrylate networks that are not recyclable.
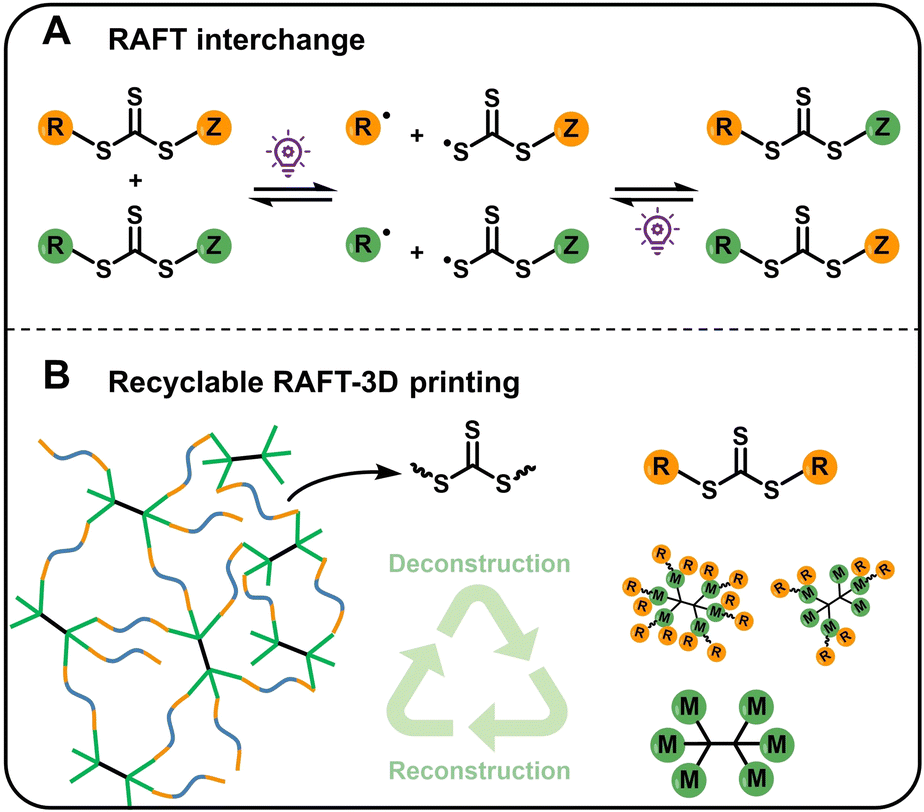 | ||
| Scheme 1 (A) Simplified RAFT interchange process; (B) the recyclable RAFT-3D printing. | ||
The RAFT single unit monomer insertion (RAFT-SUMI), first proposed by Moad et al. in 2011,43 has since enabled significant advances in the synthesis of sequence-defined polymers.44–46 Recently, Tanaka, You and our group developed an A2 + B2-type RAFT step-growth polymerization system by employing a RAFT SUMI reaction of a bifunctional vinyl monomer and a symmetric trithiocarbonate (TTC), yielding polymers with TTC linkages embedded in each repeat unit.46–51 This poly(trithiocarbonates) can be deconstructed into ABA-type RAFT oligomers in the presence of excess RAFT agents (the A2 monomer) under light irradiation. Upon adding a stoichiometric amount of vinyl monomer (the B2 component), the system can be repolymerized into the original polymer, achieving a closed-loop cycle. Building on this strategy, we propose using multifunctional monomers (functionality > 2) to generate polymer networks via RAFT step-growth polymerization, incorporating TTC linkages in each repeat unit of the network backbone (Scheme 1B). These networks can be deconstructed under visible light irradiation in the presence of excess RAFT agents, generating multifunctional RAFT intermediates that can be re-crosslinked by simply adding multifunctional vinyl monomers. This approach obviates the need for custom-synthesized monomers containing dynamic covalent bonds, while enabling recyclable RAFT-based 3D printing networks without requiring additional catalysts.
Results and discussion
A commercially available acrylate monomer was selected for constructing the printable resin due to its abundant availability from both bio-based and petroleum-derived sources. We demonstrated the successful RAFT-SUMI model reaction by using a monofunctional trithiocarbonate (ethyl 2-(((butylthio)carbonothioyl)thio)-2-methylpropanoate, BTMPE) and butyl acrylate (Fig. S1 and S2). To enable efficient RAFT step-growth polymerization, a symmetrical bis-trithiocarbonate (diethyl 2,2′-(thiocarbonylbis(sulfanediyl))bis(2-methylpropanoate), DMET) bearing two tertiary ester R groups was synthesized. We initially explored the A2 + B2-type step-growth polymerization of DMET with a bifunctional acrylate monomer, 1,6-hexanediol diacrylate (HDDA). The polymerization progress was monitored by 1H NMR spectroscopy and size exclusion chromatography (SEC). As shown in Fig. S3 and Table S1, DMET and HDDA consumed equally with the polymerization progressed. In the meantime, the RAFT-SUMI adduct generated quantitatively. The SEC traces shifted towards higher molecular side, however, no significant shift toward higher molecular weight in the later stages of polymerization, likely due to dynamic TTC exchange among polymer chains (Fig. S4). All these data supported a step-growth polymerization mechanism (Fig. S5).The RAFT interchange process was further investigated by introducing exogenous DMET ([TTC group]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[DMET]0 = 1:5) to deconstruct the polymer (Mw = 6300, Đ = 1.32) under 405 nm LED irradiation. After 8 hours, the Mw decreased to approximately 1100, matching well with the theoretical molecular weight (919.3 g mol−1) of a RAFT-SUMI adduct comprising two DMET and one HDDA units,45 indicating the establishment of equilibrium (Fig. 1 and S6). The minor peak in the higher molecular weight side in the SEC curve after deconstruction indicated the existence of some oligomers, however, these oligomers still have DMET on each chain end, which can be utilized for repolymerization. Next, HDDA was added for repolymerization in an equivalent amount to the DMET used for deconstruction. The SEC trace shifted toward higher molecular weight, confirming successful reformation of the polymer (Fig. 1). The 1H NMR spectrum of the repolymerized material was consistent with that of the original polymer (Fig. S7).
:[DMET]0 = 1:5) to deconstruct the polymer (Mw = 6300, Đ = 1.32) under 405 nm LED irradiation. After 8 hours, the Mw decreased to approximately 1100, matching well with the theoretical molecular weight (919.3 g mol−1) of a RAFT-SUMI adduct comprising two DMET and one HDDA units,45 indicating the establishment of equilibrium (Fig. 1 and S6). The minor peak in the higher molecular weight side in the SEC curve after deconstruction indicated the existence of some oligomers, however, these oligomers still have DMET on each chain end, which can be utilized for repolymerization. Next, HDDA was added for repolymerization in an equivalent amount to the DMET used for deconstruction. The SEC trace shifted toward higher molecular weight, confirming successful reformation of the polymer (Fig. 1). The 1H NMR spectrum of the repolymerized material was consistent with that of the original polymer (Fig. S7).
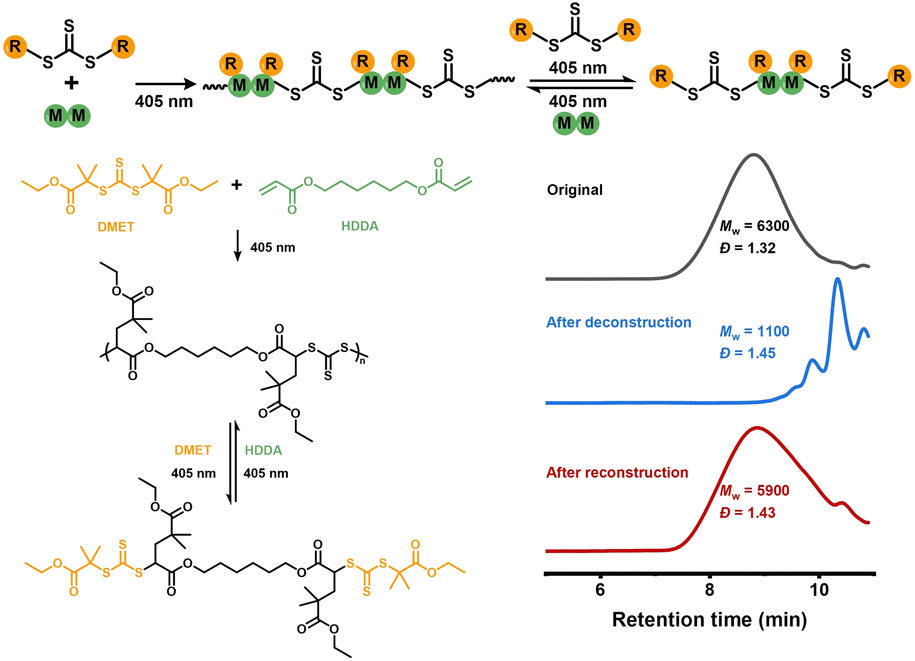 | ||
| Fig. 1 Deconstruction and repolymerization of RAFT step-growth polymer prepared with [DMET]0:[HDDA]0 = 1:1 for 12 hours under irradiation of a 405 nm LED. | ||
To extend this system to 3D printing, a commercial hexafunctional acrylate (Dipentaerythritol hexaacrylate, DPHA) was employed to build a crosslinked network via RAFT step-growth polymerization (Fig. 2A). A Type I photoinitiator (Diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide, TPO), was added to accelerate photocuring during DLP printing.52 Resin formulations with varying TPO concentrations were screened using UV-differential scanning calorimetry (UV-DSC, Fig. S8). The optimal TPO concentration was determined to be 0.2 molar equivalents, at which the curing rate plateaued (Fig. 2B). Accordingly, a formulation with [DMET]0:[DPHA]0:[TPO]0 = 3:1:0.2 was selected for DLP-3D printing. A dumbbell-shaped sample was printed and tested for mechanical performance, yielding a Young's modulus of 49.5 MPa (Fig. 2C).
 | ||
| Fig. 2 (A) Schematic diagram of DLP-3D printing; (B) polymerization rates measured by UV-DSC with varying TPO contents in the resin formulations; (C) tensile test of the printed sample. | ||
To evaluate recyclability, a printed Chinese knot object was cut into pieces and treated with a THF solution containing DMET ([TTC]0:[DMET]0 = 1:5) under 405 nm LED irradiation (Fig. 3A–C and Scheme S1). After 12 hours, the object fully dissolved, and the slightly turbid solution—attributed to oligomeric species—was collected for reprinting. After THF removal, DPHA and TPO were added (DPHA equal to DMET used for deconstruction) and the mixture was subjected to reprinting. The reprinted object has become less transparent, while the design features have also become blurred (Fig. 3E). Moreover, we conducted a resolution test before and after recycling. Pleasingly, the reprinted object maintained high resolution with slightly decline after one cycle (Fig. S9). However, the second batch of printed objects exhibited significantly defects, likely due to RAFT agent degradation.
 | ||
| Fig. 3 Recyclable 3D printing. (A) The initial resin; (B) the printed Chinese knot; (C) the crushed Chinese knot; (D) the recycled resin; (E) the reprinted Chinese knot. | ||
However, tensile testing of dumbbell samples revealed that the mechanical performance remained comparable to the original prints (Fig. 4B and Table S2), confirming successful recyclability and closed-loop 3D printing. Moreover, we also examined the amount of DMET for achieving the deconstruction. According to Carothers' gel point theory, a system will not undergo crosslinking when the average functionality is less than 2. Based on this, we calculated that polymer network completely deconstruction should occur when the ratio of [TTC]0:[DMET]0 > 3:2. Considering potential photodecomposition of DMET and other side reactions, we selected three low DMET ratios ([TTC]0:[DMET]0 = 1:0.8, 1, 1.2) for deconstruction. As shown in Fig. S10, the solid disappeared after 12 hours.
 | ||
| Fig. 4 (A) Schematic diagram of upcycling process; (B) tensile test of recycled samples using high DPHA ratios; (C) tensile test of recycled samples using low DPHA ratios. | ||
Furthermore, a comprehensive investigation was subsequently undertaken to evaluate the effects of the amount of added DPHA during the reprinting. Beyond a certain threshold, polymerization behavior would transition from step-growth to chain-growth, resulting in brittle plastic-like properties. As shown in Fig. 4B and Table S2, as DPHA content increased, Young's modulus increased while elongation at break decreased, attributed to higher crosslinking density. At a DPHA ratio of 1.025, the reprinted samples exhibited properties nearly identical to the originals. On the other hand, decreasing the amount of DPHA could decrease the Young's modulus gradually as the crosslinking density decreased (Fig. 4C and Table S2). Overall, the Young's modulus of reprinted materials could be modulated across a wide range (0.78–835 MPa) simply by altering the DPHA added during reprinting, highlighting the upcyclability of the system.
Finally, to verify the “living” feature of the RAFT-3D printing system, surface post-functionalization of a printed film made by using a formulation of [DMET]0:[DPHA]0:[TPO]0 = 3:1:0.2 was carried out via grafting a fluorescent acrylate monomer (PyMMA).11 As shown in Fig. 5, a “Soochow University” logo was clearly visible under 365 nm UV light on the modified film, confirming successful surface modification. Furthermore, the grafted fluorescent pattern could be erased via the RAFT interchange process using excess RAFT agent. The film was immersed in a DMSO solution containing DMET and irradiated with 405 nm light for 2 minutes, after which the pattern disappeared under UV light, demonstrating the potential of this approach for renewable anti-counterfeiting applications.
 | ||
| Fig. 5 (A) Schematic diagram of post-modification with PyMMA and erasure with DMET; (B, D and F) samples before and after modification and after erasure under natural light; (C, E and G) under UV light. | ||
Conclusion
We have developed a dynamic RAFT-based step-growth polymerization system that enables closed-loop 3D printing via photoreversible trithiocarbonate chemistry. The polymer networks formed through this strategy are fully recyclable under mild LED irradiation without the need for catalysts. This is achieved by embedding RAFT agents directly into each repeat unit of the polymer backbone, allowing both deconstruction and reconstruction through successive RAFT interchange and step-growth polymerization. Our approach allows for the facile modulation of mechanical properties, as well as surface post-functionalization and erasure, demonstrating the “living” and reprogrammable nature of the printed materials. This study establishes a new paradigm for sustainable additive manufacturing, bridging the gap between controlled polymerization chemistry and practical 3D printing technologies.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. Supplementary information: detailed experimental procedures, characterization methods, supplementary figures (Fig. S1−S14), supplementary tables (Tables S1−S2) and supplementary scheme (Scheme 1). See DOI: https://doi.org/10.1039/d5sc06472d.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22371199 and No. 22571219), Suzhou Cutting-edge Technology Research Project (SYG202350), National Outstanding Youth Foundation of China (21925107), the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the Program of Innovative Research Team of Soochow University.Notes and references
- C. M. González-Henríquez, M. A. Sarabia-Vallejos and J. Rodriguez-Hernandez, Prog. Polym. Sci., 2019, 94, 57–116, DOI:10.1016/j.progpolymsci.2019.03.001.
- R. Levato, O. Dudaryeva, C. E. Garciamendez-Mijares, B. E. Kirkpatrick, R. Rizzo, J. Schimelman, K. S. Anseth, S. C. Chen, M. Zenobi-Wong and Y. S. Zhang, Nat. Rev. Methods Primers, 2023, 3, 19, DOI:10.1038/s43586-023-00231-0.
- J. X. Cheng, S. Y. Yu, R. Wang and Q. Ge, Int. J. Extreme Manuf., 2024, 6, 24, DOI:10.1088/2631-7990/ad4a2c.
- Y.-N. Zhou, J.-J. Li, T.-T. Wang, Y.-Y. Wu and Z.-H. Luo, Prog. Polym. Sci., 2022, 130, 101555, DOI:10.1016/j.progpolymsci.2022.101555.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588, DOI:10.1016/j.chempr.2020.06.014.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311, DOI:10.1016/j.progpolymsci.2020.101311.
- V. A. Bobrin, J. Zhang, N. Corrigan and C. Boyer, Adv. Mater. Technol., 2022, 8, 2201054, DOI:10.1002/admt.202201054.
- A. Bagheri, C. M. Fellows and C. Boyer, Adv. Sci., 2021, 8, 2003701, DOI:10.1002/advs.202003701.
- N. Corrigan and C. Boyer, Trends Chem., 2020, 2, 689–706, DOI:10.1016/j.trechm.2020.05.001.
- A. Bagheri, Macromolecules, 2023, 56, 1778–1797, DOI:10.1021/acs.macromol.2c02585.
- A. Bagheri, K. E. Engel, C. W. A. Bainbridge, J. Xu, C. Boyer and J. Jin, Polym. Chem., 2020, 11, 641–647, 10.1039/c9py01419e.
- Y. Lee, C. Boyer and M. S. Kwon, Chem. Soc. Rev., 2023, 52, 3035–3097, 10.1039/d1cs00069a.
- N. C. C. B. Zhiheng Zhang, Angew. Chem., Int. Ed., 2019, 58, 17954–17963 CrossRef PubMed.
- C. W. A. B. Ali Bagheri, K. E. Engel, G. G. Qiao, J. Xu, C. Boyer and J. Jin, ACS Appl. Polym. Mater., 2020, 2, 782–790, DOI:10.1021/acsapm.9b01076.
- X. Shi, V. A. Bobrin, Y. Yao, J. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2022, e202206272, DOI:10.1002/anie.202206272.
- Z. Yuan, G. Li, C. Yang, W. Zhu, J. Li and J. Zhu, Chem.–Asian J., 2024, e202400648, DOI:10.1002/asia.202400648.
- Z. Zhang, N. Corrigan and C. Boyer, Macromolecules, 2021, 54, 1170–1182, DOI:10.1021/acs.macromol.0c02691.
- Z. Zhang, N. Corrigan, A. Bagheri, J. Jin and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 17954–17963, DOI:10.1002/anie.201912608.
- Z. Zhang, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2022, 61, e202114111, DOI:10.1002/anie.202114111.
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 1620–1628, DOI:10.1021/acs.macromol.1c02521.
- V. A. Bobrin, Y. Yao, X. Shi, Y. Xiu, J. Zhang, N. Corrigan and C. Boyer, Nat. Commun., 2022, 13, 3577, DOI:10.1038/s41467-022-31095-9.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2023, 62, e202307329, DOI:10.1002/anie.202307329.
- K. Lee, Y. Shang, V. A. Bobrin, R. Kuchel, D. Kundu, N. Corrigan and C. Boyer, Adv. Mater., 2022, 34, e2204816, DOI:10.1002/adma.202204816.
- M. Asadi-Eydivand, T. C. Brown and A. Bagheri, ACS Appl. Polym. Mater., 2022, 4, 4940–4948, DOI:10.1021/acsapm.2c00500.
- A. Bagheri, M. Asadi-Eydivand, A. A. Rosser, C. M. Fellows and T. C. Brown, Adv. Eng. Mater., 2023, 25, 2201785, DOI:10.1002/adem.202201785.
- H. J. B. Chua, A. A. Rosser, C. M. Fellows, T. C. Brown and A. Bagheri, ACS Appl. Polym. Mater., 2024, 7, 547–566, DOI:10.1021/acsapm.4c02501.
- S. P. Wankhede, A. Bagheri and X. Du, ACS Appl. Polym. Mater., 2025, 7, 5846–5854, DOI:10.1021/acsapm.4c04021.
- A. Bagheri, H. Ling, C. W. A. Bainbridge and J. Jin, ACS Appl. Polym. Mater., 2021, 3, 2921–2930, DOI:10.1021/acsapm.1c00048.
- E. M. Maines, M. K. Porwal, C. J. Ellison and T. M. Reineke, Green Chem., 2021, 23, 6863–6897, 10.1039/d1gc01489g.
- B. Yang, T. T. Ni, J. J. Wu, Z. Z. Fang, K. X. Yang, B. He, X. Q. Pu, G. C. Chen, C. J. Ni, D. Chen, Q. Zhao, W. Li, S. J. Li, H. Li, N. Zheng and T. Xie, Science, 2025, 388, 170–175, DOI:10.1126/science.ads3880.
- T. O. Machado, C. J. Stubbs, V. Chiaradia, M. A. Alraddadi, A. Brandolese, J. C. Worch and A. P. Dove, Nature, 2024, 629, 1069–1074, DOI:10.1038/s41586-024-07399-9.
- S. Han, V. A. Bobrin, M. Michelas, C. J. Hawker and C. Boyer, ACS Macro Lett., 2024, 13, 1495–1502, DOI:10.1021/acsmacrolett.4c00600.
- M. Caliari, F. Vidal, D. Mantione, G. Seychal, M. Campoy-Quiles, L. Irusta, M. Fernandez, X. L. de Pariza, T. Habets, N. Aramburu, J. M. Raquez, B. Grignard, A. J. Müller, C. Detrembleur and H. Sardon, Adv. Mater., 2025, 37, 2417355, DOI:10.1002/adma.202417355.
- X. Lopez de Pariza, O. Varela, S. O. Catt, T. E. Long, E. Blasco and H. Sardon, Nat. Commun., 2023, 14, 5504, DOI:10.1038/s41467-023-41267-w.
- L. Yue, Y. L. Su, M. Li, L. Yu, X. Sun, J. Cho, B. Brettmann, W. R. Gutekunst, R. Ramprasad and H. J. Qi, Adv. Mater., 2024, 36, 2310040, DOI:10.1002/adma.202310040.
- G. Zhu, J. Zhang, J. Huang, Y. Qiu, M. Liu, J. Yu, C. Liu, Q. Shang, Y. Hu, L. Hu and Y. Zhou, Chem. Eng. J., 2023, 452, 139401, DOI:10.1016/j.cej.2022.139401.
- J. Cui, F. Liu, Z. Lu, S. Feng, C. Liang, Y. Sun, J. Cui and B. Zhang, Adv. Mater., 2023, 35, 2211417, DOI:10.1002/adma.202211417.
- Y. Guo, S. Chen, L. Sun, L. Yang, L. Zhang, J. Lou and Z. You, Adv. Funct. Mater., 2020, 31, 2009799, DOI:10.1002/adfm.202009799.
- Y. Jia, F. Ling, J. Qian, Q. Chen, Z. Zhao and Y. Li, ACS Macro Lett., 2023, 12, 719–724, DOI:10.1021/acsmacrolett.3c00116.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831, DOI:10.1038/s41467-018-04292-8.
- S. Wei, J. Smith-Jones, R. F. Lalisse, J. C. Hestenes, D. Chen, S. P. O. Danielsen, R. C. Bell, E. M. Churchill, N. A. Munich, L. E. Marbella, O. Gutierrez, M. Rubinstein, A. Nelson and L. M. Campos, Adv. Mater., 2024, 36, 2313961, DOI:10.1002/adma.202313961.
- R. Nicolaÿ, J. Kamada, A. Van Wassen and K. Matyjaszewski, Macromolecules, 2010, 43, 4355–4361, DOI:10.1021/ma100378r.
- M. Chen, M. Häussler, G. Moad and E. Rizzardo, Org. Biomol. Chem., 2011, 9, 6111–6119 RSC.
- J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2016, 56, 8376–8383, DOI:10.1002/anie.201610223.
- J. Xu, Macromolecules, 2019, 52, 9068–9093, DOI:10.1021/acs.macromol.9b01365.
- C. Boyer, M. Kamigaito, K. Satoh and G. Moad, Prog. Polym. Sci., 2023, 138, 101648, DOI:10.1016/j.progpolymsci.2023.101648.
- J. Li, J. Tanaka, Q. Li, C. J. Jing Wang, S. Sheiko, S. M. Clouthier, J. Zhu and W. You, Chem. Sci., 2024, 15, 4910–4919, 10.1039/d3sc06736j.
- Z. Li, J. Li, X. Pan, Z. Zhang and J. Zhu, ACS Macro Lett., 2022, 11, 230–235, DOI:10.1021/acsmacrolett.1c00762.
- J. Tanaka, N. E. Archer, M. J. Grant and W. You, J. Am. Chem. Soc., 2021, 143, 15918–15923, DOI:10.1021/jacs.1c07553.
- J. Tanaka, J. Li, S. M. Clouthier and W. You, Chem. Commun., 2023, 59, 8168–8189, 10.1039/d3cc01087b.
- M. Kamigaito, Bull. Chem. Soc. Jpn., 2024, 97, uoae069, DOI:10.1093/bulcsj/uoae069.
- K. Lee, N. Corrigan and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 8839–8850, DOI:10.1002/anie.202016523.
| This journal is © The Royal Society of Chemistry 2025 |