
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the polymerization potential of itaconic acid through methacrylate functionalization
Mary Hnatyshyn*a,
Matthew Halloran†
a,
Maxwell Laykisha,
Jim A. Nicellb,
Richard L. Leaska and
Milan Maric *a
*a
aDepartment of Chemical Engineering, McGill University, Montreal, Quebec, Canada. E-mail: milan.maric@mcgill.ca
bDepartment of Civil Engineering, McGill University, Montreal, Quebec, Canada
First published on 12th November 2025
Abstract
Itaconic acid (IA) is a bio-renewable molecule with increasing industrial availability. However, IA-based polymers have been limited by low molecular weights and conversions. In this work, we report the synthesis of two novel methacrylate-functionalized IA monomers. Using various reversible-deactivation radical polymerization methods, we achieved well-defined polymers with high conversions (≥98%) and moderate reaction times (e.g., 70 minutes by atom transfer radical polymerization at 80 °C). Homopolymers of these two monomers exhibited a range of properties, with glass transition temperatures (Tg) ranging from −40 °C for heptyl-functionalized moieties to 14 °C for benzyl-functionalized moieties. Controllable reaction kinetics enabled the synthesis of pre-designed AB-type diblock copolymers, demonstrating the potential of the heptyl-functionalized moiety as a soft block in phase-separated materials. The favorable reaction kinetics of these methacrylate-functionalized IA monomers make this approach one of the most promising pathways for incorporating renewably sourced IA into polymeric materials.
1. Introduction
The relevance of using renewable feedstocks has become overwhelmingly apparent in both emerging chemical synthesis pathways and polymer innovations alike, as evidenced by the growing number of research publications and patents referencing the concept.1–3 Moving away from fossil fuel and mining derived feedstocks is largely motivated by the goal of mitigating carbon emissions, but it also offers the benefit of revealing new chemical building blocks with unique features.4 The economic feasibility of these alternative chemical starting materials has also improved, as pushing for renewable options has grown the production of many to an industrial scale.5Itaconic acid (IA) is a naturally occurring chemical whose production via the citric acid cycle intermediate cis-aconitate using the fungus Aspergillus terreus in bioreactors has been commercialized.6 The potential for IA to contribute to a more sustainable chemical industry was highlighted previously, when it was listed as one of the “Top-12 Platform Chemicals” in a report commissioned by the US Department of Energy summarizing renewable feedstocks.7 However, IA was removed from this list in 2010 due to comparatively little progress in its research relative to other compounds such as bioethanol.8 Contributing to this fall are the difficulties faced when attempting to polymerize IA.9
The aspects of IA which make it an exciting chemical feedstock, namely its dicarboxylic acid structure with a naturally occurring unsaturated group, have also been the features that impede its use in the polymer industry. Early attempts of polymerization at the double bond using free radical polymerization (FRP) proceeded sluggishly to produce low molecular weight polymers, caused by the carboxylic acid stabilizing the propagating radical via resonance.9,10 While advancements in methods of reversible deactivation radical polymerization (RDRP) have brought greater success to the polymerization of IA and its derivatives than FRP, fine tuning of reaction conditions have still resulted in slow reactions yielding low molecular weights and suboptimal conversion.11 Recently, Fischer von Mollard et al. conducted a systematic investigation into the emulsion polymerization of itaconates where reactions successfully achieved residual monomer contents less than 2%.12 This, coupled with moderate molecular weights (e.g., 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), suggest emulsion as an improved avenue for itaconate polymerization, but dispersities (Đ) were still high (e.g., 4.7 up to 8 for residual monomer contents less than 2%).12
000 g mol−1), suggest emulsion as an improved avenue for itaconate polymerization, but dispersities (Đ) were still high (e.g., 4.7 up to 8 for residual monomer contents less than 2%).12
Looking towards polycondensation reactions of the carboxylic acids to produce polyesters is not more promising. In these polyesters, the unsaturated double bond can be cross-linked to yield unique materials such as hydrogel microspheres, UV cured coatings, or shape memory materials.13,14 However, attempting to reach high conversions tends to result in cross-linking via the double bonds, inducing gelling in the reaction mixture.15 A reduction in final molecular weight of these polymers is typical when attempts to increase IA content are made.16
Considering an alternate approach, various renewable and bio-sourced molecules have successfully been incorporated into novel polymeric materials via functionalization. For example, using plant oil triglycerides, Yuan et al. demonstrated a method of obtaining a series of methacrylate monomers, imino ether monomers, and cyclic norbornene monomers from which polymers with varying mechanical properties could be produced.17 Also, the methacrylate and acrylate functionalization of various terpene feedstocks by Atkinson et al. presented a pathway using waste streams to synthesize well-defined ABA triblock copolymers with promising qualities for application as pressure-sensitive adhesives and thermoplastic elastomers.18,19 Chin et al. demonstrated the functionalization of lignin-derived bio-molecules was suitable for thermoplastics manufactured via 3D vat polymerization.20 Vanillin, thymol, lactic acid, gum rosin, and abietic acid are several other examples of renewable feedstocks which have been functionalized with a methacrylate group to give polymeric materials with an increased green content.21–24 However, until now, this method of functionalization prior to polymerization has not been attempted with IA.
In the present study, we extend the strategy of methacrylate functionalization to derivatives of IA, to incorporate IA into polymeric materials without the difficulties previously experienced with direct IA polymerization approaches. A sequential hydroboration–oxidation and esterification reaction pathway transforms the unsaturated double bond on the itaconate backbone, that when applied to various itaconate derivative starting materials, presents the opportunity to establish a set of novel itaconate-derived methacrylate monomers. Here, we describe the synthesis, characterization, and polymerization of two novel, IA-based methacrylate monomers: diheptyl itaconyl methacrylate (DHIAMA) and dibenzyl itaconyl methacrylate (DBIAMA). Methods of reversible deactivation radical polymerization (RDRP) are used to obtain homopolymers of both, validating the facile incorporation of IA into polymers via this strategy. Initial screening of the homopolymers showed poly(DHIAMA) (herein referred to as “p(DHIAMA)”) to have a low glass transition (−40 °C) with poly(DBIAMA) (p(DBIAMA)) having a Tg of 14 °C. Adjusting the molecular weight of p(DHIAMA) demonstrated the potential to tune the polymer's rheological properties, and the suitability of p(DHIAMA) to serve in specialized materials was shown using p(DHIAMA) in targeted diblock architectures to yield hard–soft AB polymers.
2. Results and discussion
2.1. Monomer synthesis
The presence of an unsaturated double bond in the backbone of IA was successfully exploited to functionalize this renewable feedstock with a methacrylate group. An overview of the synthesis scheme is shown in Fig. 1 where “R” functional groups are either heptanol or benzyl alcohol, and detailed methods for the synthesis are provided in the SI (Section S1.9). As a first step in the synthetic pathway, the carboxylic acid groups of IA are functionalized by an esterification reaction, allowing for a range of distinct monomers to be obtained by varying the choice of alcohol used in this step. The synthesis was completed independently using both heptanol and benzyl alcohol, serving as the “R” functional arms branching off from the IA core. These two groups were chosen to explore the potential of tuning the final polymeric properties of our IA-based monomer due to the flexible saturated carbon chains of heptanol contrasting the rigid nature of benzene rings. During the synthesis, the two-step hydroboration–oxidation saw the formation of a lactone side product. The formation could be minimized by ensuring anhydrous conditions in the hydroboration step, giving yields of 80%, with the lactone being removed during column chromatography after the oxidation step, but it highlights this synthesis pathway as a first route in obtaining methacrylate functionalized itaconates. For broader applications, it would be worthwhile to further refine the procedure as it is outlined here. Regardless, for both selected “R” groups, 1H NMR of the intermediates and final monomers confirmed the synthesis’ success in obtaining the desired products (SI, Section S1.9). | ||
| Fig. 1 Schematic overview for obtaining methacrylate monomers of IA derivatives using heptanol and benzyl alcohol. | ||
2.2. Polymerization by methods of reversible deactivation radical polymerization
To demonstrate polymerization ubiquity of the obtained methacrylate monomers, DHIAMA was subjected to reactions by three common methods of RDRP: namely, nitroxide mediated polymerization (NMP), atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer polymerization (RAFT). Kinetic plots displaying the polymerization results arising from the three employed methods are shown in Fig. 2. | ||
| Fig. 2 RDRP reaction kinetics for DHIAMA: number average molecular weight (Mn) by GPC, calculated theoretical Mn, and dispersity (Đ) throughout polymerization by: (A) NMP; (C) ATRP; and (E) RAFT. Linearized monomer conversion (semi-logarithmic) during polymerization is shown by: (B) NMP; (D) ATRP; and (F) RAFT. Reaction conditions are provided in section 2.1. | ||
600 g mol−1) and concluded reaction conversion and Đ were negatively influenced by IA content.25Here, the obtained diheptyl functionalized monomer was polymerized by NMP with BlocBuilder ([M]:[BB] = 66:1) at 90 °C as described in the SI (Section S1.10). Styrene was used as a controlling co-monomer in low concentrations (fST,0 = 0.1), due to methacrylate homopolymerizations displaying poor control from NMP using BlocBuilder (Đ = 1.5–4).27,28 Kinetic plots in Fig. 2, showing monomer conversion (X) as ln[1/1 − X] against time, reveal a strong linear dependency with reaction time, indicating that p(DHIAMA) formation proceeds via a controlled radical polymerization mechanism. Gel permeation chromatography (GPC) analysis of samples collected throughout the reaction enabled monitoring of both the Mn and Đ. A steady increase in Mn was observed up to a DHIAMA conversion of 83%, and when combined with relatively narrow molecular weight distributions for NMP (Đ < 1.4) suggest the polymerization mechanism was consistent with an active chain end.
:[CTA]:[I] = 50:1:1) at 80 °C (SI, Section S1.11). The direct polymerization of IA by ATRP has faced challenges including the deprotonation of IA during chain growth causing coordination complexes to form between conventional copper catalysts and the deprotonated IA, leading to catalyst deactivation and the cessation of polymerization.29 Previous studies showed using cyclic itaconimides prevents this unfavorable interaction, as does using itaconate derivatives, but homopolymers in literature made by these methods tend towards high dispersities (Đ > 1.5), and low conversions (<40%), respectively.28,30–32In the present study, these unfavorable interactions are avoided, due to the polymerization occurring at the methacrylate group. The pseudo first-order dependance of monomer conversion with reaction time is shown in Fig. 2 and displays a linear trend, after an induction period, indicating that the commonly used and commercially available catalyst, initiator and ligand Cu(I)Br, EbiB, and PMDETA, respectively, effectively controls polymerization. This, along with narrow molecular weight distributions, and an increasing, linear molecular weight plot with the conversion of 93% in a 70-minute reaction supports the application of ATRP for DHIAMA polymerization.
000 g mol−1, 70% conversion) relative to other RDRP methods, but, long reaction times are required (>150 hours) and the Đ are higher than typical for RAFT polymerization (>1.70).33,34 Here, 2-cyano-2-propyl dodecyl trithiocarbonate was selected as the RAFT chain transfer agent due to its commercial availability and known ability to effectively polymerize methacrylates.35 Detailed methods of the synthesis with [M]:[CTA]:[I] = 47:1:0.1 at 72 °C are provided in the SI (Section S1.12). As with other RDRP methods, kinetic plots show linear dependance of conversion expressed as ln[1/1 − X] with time (Fig. 2), and narrow molecular weight distributions are paired with a steady linear increase in molecular weight with conversion.It should be noted that the molecular weight determined by GPC calibrated using narrow molecular weight distribution p(MMA) standards deviates from the theoretical molecular weight determined by the monomer conversion at the reaction time, due to the unknown Mark–Houwink coefficients for this novel polymer. This deviation, however, is less pronounced for the reaction carried out by NMP. This may be due to the inclusion of styrene at fST,0 = 0.1, altering the reaction environment and influencing the extent of side reactions, such as chain transfer, that may occur. Regardless, in all three reversible deactivation radical methods, monomodal GPC traces and Đ < 1.3 indicate the polymerizations proceeded by controlled radical mechanisms, confirming the universality of DHIAMA to a range of polymerization methods.35
RAFT polymerization was selected as the preferred method for the preparation of a series of homopolymers for both DHIAMA and DBIAMA, due to its simplicity and ability to produce polymers exhibiting low Đ. The intent was to determine if the reaction could be controlled across a range of molecular weights, with target Mns ranging from 10000 g mol−1 to 400000 g mol−1. The polymerization of the series resulted in polymers with low Đ and Mn values near those targeted, as determined by GPC equipped with a light scattering (LS) detector. Tables 1 and 2 show the molecular weight characteristics of the DHIAMA and DBIAMA series of homopolymers, respectively. From the polymer series, the dn/dc value relating the sample concentration to the refractive index (RI) detector output of the GPC was determined as 0.066 ± 0.004 for p(DHIAMA) and 0.127 ± 0.006 for p(DBIAMA). Knowing the dn/dc allows for a more precise molecular weight estimate of the homopolymer samples to be known in future GPC samples, and the consistency between dn/dc values in the polymer series indicates reliable sample preparation and molecular weight estimates. Tables S1 and S2 of the SI provide the molecular weight and Đ data of the two series obtained by both RI, and LS detectors. Briefly, the RI indicates lower molecular weights and slightly increased Đ, giving, for example, an Mn of 57900 g mol−1 and Đ of 1.19 for H8. In all cases, the RAFT polymerization reaction was carried out as detailed in the experimental methods (SI, Section S1.12).
| Polymer | Target Mn [g mol−1] | Conversiona [%] | Mnb [g mol−1] | Đb | dn/dcb |
|---|---|---|---|---|---|
| a Calculated as the integral of monomer peaks in NMR.b Determined from GPC LS detector of final dried samples.c H1 was analyzed using the GPC RI detector due to inaccuracies of LS detector at low molecular weights. | |||||
| H1 | 10000 |
98 | 8600 |
1.07 | RI detectorc |
| H2 | 17000 |
89 | 15700 |
1.07 | 0.074 |
| H3 | 20000 |
99 | 28700 |
1.07 | 0.067 |
| H4 | 30000 |
97 | 37000 |
1.07 | 0.068 |
| H5 | 35000 |
95 | 39000 |
1.05 | 0.064 |
| H6 | 40000 |
95 | 64900 |
1.12 | 0.059 |
| H7 | 100000 |
95 | 93900 |
1.09 | 0.063 |
| H8 | 200000 |
97 | 148900 |
1.13 | 0.063 |
| H9 | 400000 |
94 | 198200 |
1.60 | 0.069 |
| Polymer | Target Mn [g mol−1] | Conversiona [%] | Mnb [g mol−1] | Đb | dn/dcb |
|---|---|---|---|---|---|
| a Calculated as the integral of monomer peaks in NMR.b Determined from GPC LS detector of final dried samples. | |||||
| B1 | 10000 |
97 | 15400 |
1.15 | 0.127 |
| B2 | 15000 |
94 | 22400 |
1.16 | 0.126 |
| B3 | 20000 |
99 | 30700 |
1.11 | 0.135 |
| B4 | 30000 |
99 | 45900 |
1.14 | 0.122 |
| B5 | 60000 |
99 | 88000 |
1.13 | 0.118 |
| B6 | 100000 |
91 | 105200 |
1.27 | 0.126 |
| B7 | 200000 |
87 | 136000 |
1.40 | 0.136 |
The RAFT polymerizations of DHIAMA consistently produced satisfactory results across a range of molecular weights. High conversions (>89%) were achieved in the standard polymerization time of 7 hours at 72 °C, and narrow monomodal GPC traces were obtained (i.e., Đ not higher than 1.13) from most of the polymers. The exception comes from H9, where the highest Mn was targeted. Although the GPC trace displayed a single peak, the obtained polymer had a higher Đ (1.60), indicating the reaction was not as controlled as the others. This result is not surprising as the RAFT polymerization of methacrylic monomers is known to be less successful when targeting high molecular weight polymers.36
The RAFT polymerization of DBIAMA, using the same conditions, gave defined polymers but with a higher Đ compared to the DHIAMA polymers. Monomer conversion expressed in terms of ln[1/1 − X] exhibited linear dependance with reaction time, and no significant plateauing of the molecular weight was observed with conversion. Kinetic plots for the polymerization of p(DBIAMA) are provided in the SI (Fig. S11 and S12). While the p(DBIAMA) series exhibited broader molecular weights than the p(DHIAMA) set, polymers with target Mn up to 100 kg mol−1 still produced Đ < 1.3 and monomodal GPC traces. Slight shouldering was observed in reactions targeting Mn > 100 kg mol−1, which coupled with the jump in Đ may indicate branching occurred. Both the faster polymerization and the possibility of branching are thought to be caused by the electron-dense benzyl side chains. This trend has been noted previously by other groups, where benzyl-containing monomers display higher propagation rate constants than saturated alternatives.37,38 Specifically, the most recent IUPAC benchmark values report the propagation rate constant at 25 °C of benzyl methacrylate as nearly double that of methyl methacrylate, at 643 and 325 L mol−1 s−1, respectively.39
2.3. Thermal properties
The influence of the side chain on the thermal properties of the resulting polymer was evaluated via thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). Both p(DHIAMA) and p(DBIAMA) were determined to have excellent thermal stability under standard processing temperatures, with the onset of 10 wt% decomposition occurring at 262 and 256 °C for p(DHIAMA) and p(DBIAMA), respectively, shown in Fig. 3. A clear two-step decomposition pattern is displayed by both p(DHIAMA) and p(DBIAMA), with 80% of the weight being decomposed in the first stage. This proportion of weight loss corresponds well to the pendant chain branching off the backbone, joined by the itaconate core. Decomposition of the methacrylate-resembling backbone follows, with the onset of 10 wt%, relative to the starting mass, of the second decomposition occurring at 398 and 387 °C for p(DHIAMA) and p(DBIAMA), respectively.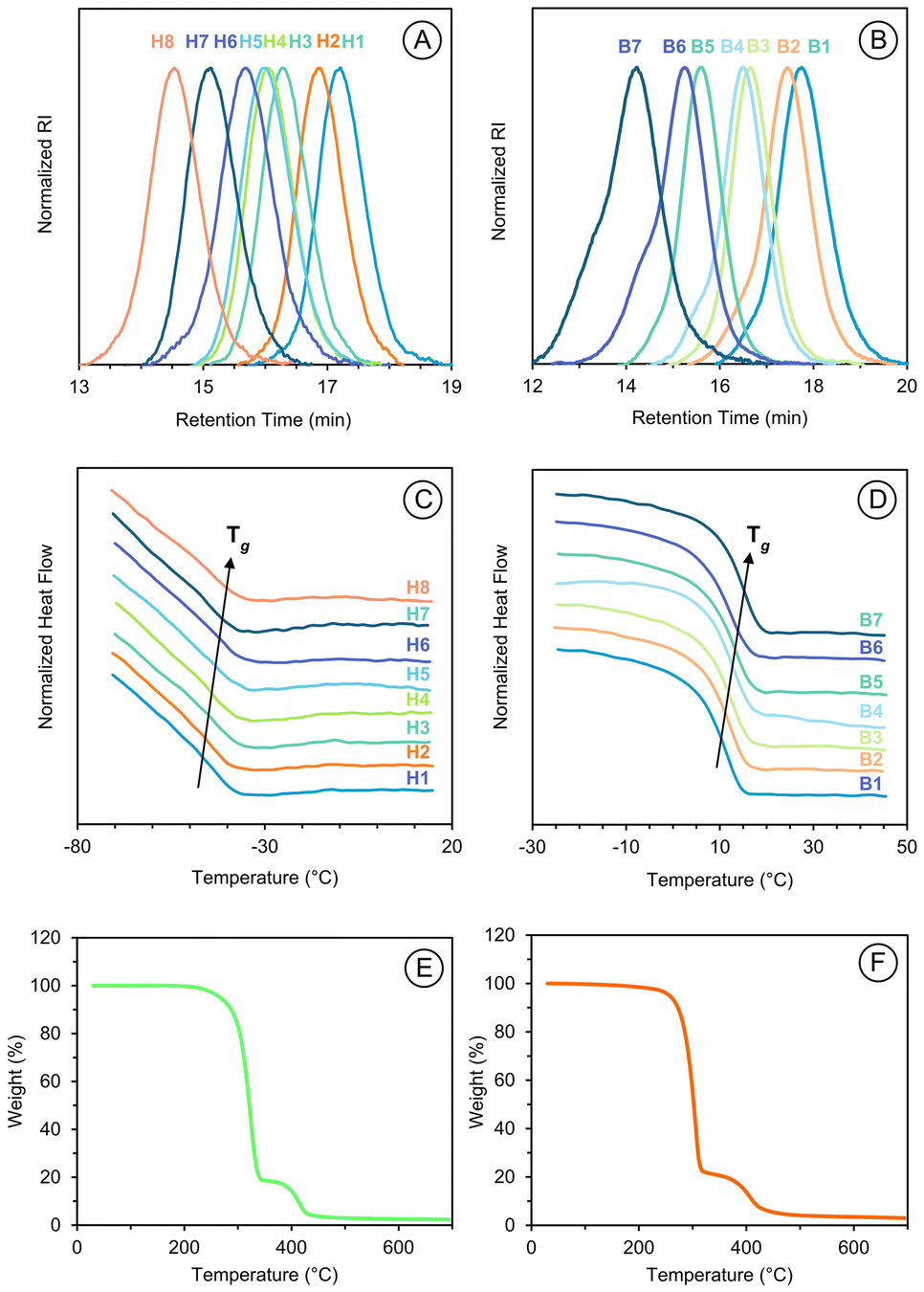 | ||
| Fig. 3 GPC traces of (A) p(DHIAMA) homopolymer series and (B) p(DBIAMA) homopolymer series. DSC traces of (C) p(DHIAMA) homopolymer series and (D) p(DBIAMA) homopolymer series. TGA trace for (E) p(DHIAMA) (H7) and (F) p(DBIAMA) (B4). | ||
Interestingly, the thermal stability of the methacrylic itaconates here is superior to comparable p(itaconates). The temperature of 5% weight decomposition occurs at 233 °C for p(dihexyl itaconate), and 211 °C for p(dibenzyl itaconate).40,41 With higher onsets of decomposition, p(DHIAMA) and p(DBIAMA) exhibit enhanced thermal tolerance.
Thermal transitions of polymers, as assessed by DSC, are influenced by several structural factors. The mobility of the polymer's backbone, intermolecular forces, the presence and architecture of pendant groups, and cross-linking between chains all influence the Tg, as well as crystalline and melting temperatures (Tc and Tm, respectively). DSC traces for both p(DHIAMA) and p(DBIAMA) lacked Tc and Tm transitions, indicating amorphous polymers (Fig. 3).
We then probed how the differing functional arms of p(DHIAMA) compared to p(DBIAMA), as well as the molecular weight within the two series, would influence the glassy transition of the polymers. According to the Flory–Fox equation for monodisperse polymers,42
 | (1) |
It is noteworthy that the Tgs for p(diheptyl itaconate) and p(dibenzyl itaconate) are −78 and 130 °C, respectively. Incorporation of the methacrylate group shifted the Tgs of the representative p(itaconates) towards that of p(MMA).43,44
The inclusion of heptyl side arms produced a soft material with a low Tg, and substituting these arms for rigid benzyl groups increased the Tg nearly 60 °C. Although the benzyl derivative has a transition temperature near ambient conditions, which may limit its range of applications, it provided a useful preliminary assessment of how the polymer's final properties can be influenced by employing functional groups of various structures in the first step of the monomer synthesis. Having a Tg well below ambient conditions, the heptyl derivative is deemed suitable for a range of soft-polymer applications including rubber toughening, flexible electronics, sensors, thermoplastic elastomers, or shape memory materials.45,46 It was therefore selected for further assessment.
2.4. Homopolymer rheological properties
Using the series of p(DHIAMA) homopolymers, frequency sweeps and viscosity measurements were completed to gauge their molecular weight dependence. As expected, higher molecular weights led to an increase in viscosity and required lower frequencies for the zero-shear viscous plateau to be observed. To estimate the molecular weight of entanglement, the zero-shear viscosities were plotted against the number average molecular weight of the polymer samples (SI, Fig. S14). However, the zero-shear viscosities exhibited a linear dependance with molecular weight, indicating the critical molecular weight (Mc) has likely not been obtained in our sample series. As DHIAMA is a highly bulky monomer, it is possible that the critical molecular weight is greater than H9 (198 kg mol−1), as p(MMA) alone has a Mc of 20 kg mol−1, and lauryl methacrylate, a monomer still half the molecular weight of DHIAMA, gives polymers with an Mc of 225 kg mol−1.47–49Across the molecular weights (i.e., 39 kg mol−1 to 198 kg mol−1) and the tested frequencies (0.01 to 100 rad s−1), the loss modulus was dominant (Fig. 4), indicating the polymer's tendency to flow. The strong dependence of the elastic modulus on the frequency further highlights this fluid behavior of the tested samples, with no rubbery plateau region appearing in the sweep range. Despite the common fluid-like characteristics of the DHIAMA-based polymers, adjusting the molecular weight does allow for the modulus to be tuned. As the molecular weight increased from 39 to 198 kg mol−1, an order of magnitude increase in the loss modulus was observed, with the storage modulus at low frequencies increasing by four orders of magnitude. Although the chains of the largest 198 kg mol−1 sample were not completely entangled, as was hypothesized from the viscosity measurements, more chain-to-chain interactions due to the larger size evidently imparted structure and elasticity. It should be noted that the rheological tests were completed at ambient conditions, as the storage modulus was below the detection threshold of the instrument when testing was done at conventional processing temperatures of 170 °C.
 | ||
| Fig. 4 Frequency sweeps using a fixed strain of 0.1% for: (A) H5; (B) H7; and (C) H9. | ||
2.5. Block copolymer synthesis
Many of the cited value-added applications rely on well-defined and complex polymer architectures, which can be synthesized using RDRP methods. This approach enables precise tuning of physical properties through the selection and pairing of suitable copolymers, with block architectures emerging as an increasingly popular strategy for enhancing the performance and applicability of bio-based polymers.50 To test the potential of DHIAMA in obtaining targeted architectures, a series of diblock copolymers were synthesized via RAFT polymerization. Methyl methacrylate (MMA) was chosen as the comonomer block segment due to its commercial availability and high glass transition temperature, which is expected to provide a strong contrast against the soft p(DHIAMA) segment. To ensure copolymers exhibit microphase separation between the blocks, the Flory–Huggins enthalpic interaction parameter χ was determined (SI, Section S3.1), and used to estimate the segregation strength of the system. χ can be calculated using eqn (2),51 as follows:
 | (2) |
![[V with combining overline]](https://www.rsc.org/images/entities/i_char_0056_0305.gif) is the average molar volume of the monomer unit approximated by
is the average molar volume of the monomer unit approximated by  ; δ is the solubility parameter for the monomer unit and is determined for DHIAMA by group contribution theory while MMA's is found from experimental data;52 R is the universal gas constant; and T is the temperature of the system. Then, χ and the total degree of polymerization, N, are used as measures of segregation strength. For a symmetric and monodisperse diblock polymer, the strong segregation limit (SSL) indicates microphase separation will occur when (χN) exceeds a value of 10.5.53 The segregation strength of the system was calculated (SI, Sections S3.1 and S3.2) and used to design a series of diblock polymers with varying DHIAMA content displaying (χN) > 10.5. Detailed methods of the diblock synthesis are provided in the SI (Section S1.13) with kinetic results in Section S3.3. In short, a macro-RAFT agent was first synthesized by polymerizing MMA and then re-initiating it with DHIAMA in extensions targeting DHIAMA molar compositions of 0.20 to 0.75. The molecular weight results of the macro-RAFT agent and diblock polymer series are found in Table 3.
; δ is the solubility parameter for the monomer unit and is determined for DHIAMA by group contribution theory while MMA's is found from experimental data;52 R is the universal gas constant; and T is the temperature of the system. Then, χ and the total degree of polymerization, N, are used as measures of segregation strength. For a symmetric and monodisperse diblock polymer, the strong segregation limit (SSL) indicates microphase separation will occur when (χN) exceeds a value of 10.5.53 The segregation strength of the system was calculated (SI, Sections S3.1 and S3.2) and used to design a series of diblock polymers with varying DHIAMA content displaying (χN) > 10.5. Detailed methods of the diblock synthesis are provided in the SI (Section S1.13) with kinetic results in Section S3.3. In short, a macro-RAFT agent was first synthesized by polymerizing MMA and then re-initiating it with DHIAMA in extensions targeting DHIAMA molar compositions of 0.20 to 0.75. The molecular weight results of the macro-RAFT agent and diblock polymer series are found in Table 3.
| Polymer | Macro-RAFT Mna [g mol−1] | Macro-RAFT Đa | Block two target Mn [g mol−1] | Block two conversionb [%] | Mn, GPCa [g mol−1] | Mn, NMRc [g mol−1] | Đa |
|---|---|---|---|---|---|---|---|
| a Measured by GPC using p(MMA) standards.b Calculated as the integral of areas under monomer peaks in 1H NMR.c Calculated from the 1H NMR of the dried sample knowing p(MMA) polymer peak represented 10000 g mol−1. |
|||||||
| MbH1 | 10000 |
1.12 | 10500 |
91 | 19600 |
21500 |
1.14 |
| MbH2 | 10000 |
1.12 | 24000 |
91 | 26800 |
36700 |
1.15 |
| MbH3 | 10000 |
1.12 | 85000 |
90 | 52200 |
115800 |
1.29 |
| MbH4 | 10000 |
1.12 | 118000 |
98 | 65900 |
162800 |
1.38 |
In the chain extensions, DHIAMA added onto the macro-RAFT agent in a controlled manner, achieving molecular weights close to those targeted for the extension. The absence of shoulders in the GPC traces (Fig. 5) indicates the successful re-initiation of the macro-RAFT agent across the series. In the case of MbH1 and MbH2, low Đ < 1.15 indicates there was a controlled addition of the DHIAMA, resulting in a well-defined block copolymer. Targeting higher molecular weights for MbH3 and MbH4 resulted in increased Đ, which is again likely due to the reported observations of RAFT polymerization being less controlled in methacrylate polymerizations targeting high degrees of polymerization.36 As was the case in the homopolymerizations, DHIAMA exhibited high conversions in the diblock syntheses. Its ease of polymerization under moderate conditions, and ability to maintain end group fidelity (see SI Sections S1.14 and S3.8 for details on chain extending MbH3 to obtain triblocks) shows its potential for use as a soft block in applications like thermoplastic elastomers.
 | ||
| Fig. 5 Diblock copolymer characterization: (A) GPC traces of macro-RAFT agent and final p(MMA-b-DHIAMA) diblock polymers; (B) SAXS spectra of the p(MMA-b-DHIAMA) series indicating the presence of microdomains; (C) schematic of p(MMA-b-DHIAMA) polymer; and (D) differential scanning calorimetry trace of MbH1 presenting two Tgs corresponding to the Tgs of the homopolymers. | ||
2.6. Block copolymer phase separation
After obtaining the series of diblock copolymers, it was desired to know if phase separation was achieved across the compositions. While the χN values were estimated to be above the SSL by over an order of magnitude (SI, Section S3.2), the applicability of exceeding this value to achieve phase separation in the asymmetric diblock polymers of this study is unknown, as the minimum χN value was developed specifically for symmetric diblock polymers.53 Thus, more tangible evidence was sought to verify the existence of microdomains. DSC was performed, with all the traces shown in the SI (Fig. S22). For polymers MbH1 and MbH2, two Tgs can be observed (Fig. 5), supporting distinct p(DHIAMA) phases from the p(MMA) phases. Due to the low p(MMA) content in MhB3 and MhB4, a second glass transition temperature (Tg) is not detected. However, the absence of p(MMA)'s Tg does not conclusively indicate miscibility, as a miscible system would exhibit a single Tg corresponding to the weight-average of the individual Tgs. In both MbH3 and MbH4, the observed Tg remains close to that of DHIAMA, suggesting limited or no miscibility.To substantiate the findings from DSC, small angle X-ray scattering (SAXS) experiments were completed on solvent cast films of the copolymers. Shown in Fig. 5, the SAXS patterns indicate microphase separation occurs for all the polymers in the series. From the scattering profiles, the interdomain spacing, d, can be calculated using eqn (3),54 as follows:
 | (3) |
The interdomain spacing was smallest for MbH1 at 17.1 nm and increased with both molecular weight and DHIAMA content (Table 4). MbH4, with the highest molecular weight, exhibited the largest interdomain spacing of 28.3 nm and showed scattering peaks at √3q* and √12q*, consistent with spherical domain structures. Although MbH4's elevated molecular weight strongly promotes microphase separation, the SAXS results indicating the system will readily phase separate even when one domain is substantially smaller are satisfying, with ϕMMA being 5% of the system. This behavior is particularly advantageous for applications such as thermoplastic elastomers, where small, well-dispersed hard domains are needed within a soft, elastic matrix.
| Polymer | FDHIAMAa | ΦDHIAMA | wt% DHIAMAa | Interdomain spacingb [nm] | Microstructure |
|---|---|---|---|---|---|
| a Calculated from the dried sample's NMR using the integrals of the polymer peaks.b Calculated from the principial peak q* of SAXS results. | |||||
| MbH1 | 0.22 | 0.57 | 0.54 | 17.1 | Lamella |
| MbH2 | 0.39 | 0.75 | 0.73 | 19.9 | Hexagonal |
| MbH3 | 0.72 | 0.92 | 0.91 | 25.1 | Spherical |
| MbH4 | 0.79 | 0.95 | 0.94 | 28.3 | Spherical |
The SAXS indicates microphase separation, but the microstructure of the regions is less apparent. The absence of every characteristic peak, and distinct long-range order may be attributed to fast evaporation of the solvent during casting, as well as a lack of chain uniformity as observed with the Đ exceeding 1.3 for the largest block copolymer.55 At best, tentative microstructures can be assigned using the broad SAXS peaks compounded with knowledge of the composition, shown in Table 4. MbH1 displays a peak at 2q* and may correspond to a lamellar morphology given its volume composition near parity. MbH2, with ϕDHIAMA being 0.75, could be expected to follow a bicontinuous or cylindrical morphology, with p(MMA) cylinders surrounded by a p(DHIAMA) matrix. It exhibits SAXS peaks at √4q* and √12q*, aligning with the characteristic peaks of hexagonally packed cylinders. Finally, MbH3 shows peaks at √3q* and √10q* supporting the presence of unordered spherical patterns.54
2.7. Block copolymer rheological properties
To allow for comparison of rheological properties between block copolymers of varying p(DHIAMA) content and homopolymer p(DHIAMA), the molecular weights of the polymers must be similar. An additional block copolymer (MbH5) with decreased p(DHIAMA) content and the same molecular weight as MbH4 was therefore synthesized. Fig. S23 (SI) shows the SAXS pattern of MbH5 as well as of H9. Scattering peaks in MbH5 at √6q* and √14q* indicate that a bicontinuous microphase exists, whereas the absence of peaks in H9 confirms the homopolymer exists as one homogenous phase.56,57 Table 5 shows the composition of block copolymers used in rheological analyses.| Polymer | Macro-RAFT Mna [g mol−1] | Macro-RAFT Đa | Mn, GPC RIa [g mol−1] | Mn, NMRb [g mol−1] | Đa | FDHIAMAb | ΦDHIAMA | wt% DHIAMAb |
|---|---|---|---|---|---|---|---|---|
| a Determined by GPC RI detector calibrated using p(MMA) standards.b Calculated from the dried sample's NMR using the integrals of the polymer peaks and known p(MMA) block weight.c Determined by GPC LS detector. | ||||||||
| H9 | — | — | 81700 |
198200c |
1.39 | 1.00 | 1.00 | 1.00 |
| MbH4 | 10000 |
1.12 | 65900 |
162800 |
1.38 | 0.79 | 0.95 | 0.94 |
| MbH5 | 32200 |
1.15 | 61000 |
160000 |
1.46 | 0.49 | 0.82 | 0.80 |
To begin the comparison of viscoelastic behaviour among the diblocks, frequency sweeps were conducted and are shown in Fig. 6. p(DHIAMA) was shown above to have tunable rheological properties by adjusting the molecular weight, however, the suspected high Mc caused the homopolymers to produce soft, flowing semi-solid materials. This inherent softness, when combined with hard segments in a diblock copolymer architecture, enables the possibility of thermoplastic elastomer-like behavior.
 | ||
| Fig. 6 Frequency sweep results using a fixed 0.1% strain for diblock p(MMA-b-DHIAMA) contrasted with p(DHIAMA) homopolymer at 25 °C: (A) elastic modulus; (B) complex viscosity; and (C) tanδ. | ||
Pure p(DHIAMA) exhibits no intersection of the loss (G″) and elastic (G′) moduli within the tested frequency range (0.1–100 s−1); the loss modulus dominates throughout, indicating a structurally relaxed material. When a p(MMA) block comprising 5% by volume of the final polymer is introduced (MbH4), the behavior changes dramatically. Hard inclusions act as anchors within the soft p(DHIAMA) matrix, resisting shear deformation, and yielding a 300-fold increase in elastic modulus for the block copolymer MbH4 compared to pure p(DHIAMA) at 25 °C and 0.1 s−1. This effect is even more pronounced in MbH5, where the volume fraction of p(MMA) is 18%, resulting in an elastic modulus over 6 500 times greater than that of H9 under the same conditions.
Comparing the results of complex viscosity tests corroborates the elasticity that microphase separation has imparted. With the plateau modulus indicating a readiness to deform and rearrange under applied shear, H9 is demonstrating a highly fluid-like and thermoplastic behavior at low frequency shear rates. Conversely, MbH5 and MbH4 lack a plateau modulus indicating microphase separation has imparted cross-linked like behaviour to the block copolymers as they are resisting flow across the entire range of tested frequencies. This trend is again confirmed by the tanδ traces of the three polymers. The tanδ of H9 is not only greatly dominated by G″ across the frequencies, but is highly time dependent, revealing its lack of structural stability at long time scales. Including hard regions of microphase separation has been shown previously to give superior structural stability to block copolymer systems, and the phenomenon is confirmed here by the weaker frequency dependent tanδ values of MbH4 and MbH5.58–60
With the ability to produce block copolymers of pre-designed architectures, a range of microphase structures can be synthesized. In turn, this allows for the flow behavior of p(DHIAMA) to be controlled by imparting microphase separation with high Tg inclusions, giving way to elastic behavior. Thus, in this study, varying the molecular weight of p(DHIAMA) altered the magnitude of the elastic and viscous moduli, while tuning of their ratio (tanδ) was achieved by incorporating a phase-separated hard block.
3. Conclusion
Hydroboration–oxidation of itaconic acid derivatives followed by esterification allowed for the synthesis of a new set of itaconate-based methacrylates, allowing IA to readily be incorporated into polymeric materials. Two novel monomers, heptyl functionalized DHIAMA and benzyl functionalized DBIAMA, were synthesized. Various methods of RDRP were used in DHIAMA polymerizations with high monomer conversion (>98%), to produce polymers with pre-designed molecular weights and low Đ. A range of p(DHIAMA) and p(DBIAMA) polymers were obtained by RAFT polymerization in a controlled manner; however, targeting higher molecular weights led to slight broadening of Đ. Changing the functional group used in the first step of the synthesis offers the ability to alter the final polymer's behavior, with the glass transition temperatures of p(DHIAMA) and p(DBIAMA) being −40 and −14 °C, respectively.Adjusting the molecular weight of p(DHIAMA) polymers gave the ability to tune the rheological properties, which were characterized as loss-dominant due to the low glass transition temperature and hypothesized high Mc of the polymer. Designing phase-separated diblock copolymers, where p(MMA) acted as a macro-RAFT agent in chain extensions, demonstrated the consistent polymerization of DHIAMA could be utilized to reliably obtain well-defined block copolymers. Phase separation of these diblocks was confirmed by SAXS to give various microphase morphologies, which in turn allowed for elastic-dominant rheological properties to be attained.
Author contributions
Mary Hnatyshyn: Conceptualization, methodology, validation, investigation, writing – original draft, review and editing, visualization. Matthew Halloran: Conceptualization, methodology. Maxwell Laykish: Investigation. Jim A. Nicell: Supervision, funding acquisition, writing – review and editing. Richard L. Leask: Supervision, funding acquisition, writing – review and editing. Milan Maric: Supervision, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including all raw data tabulated in Microsoft Excel or CSV format for DSC, TGA, GPC, Rheology, FTIR and 1H NMR measurements are available at McGill University Dataverse [URL- https://borealisdata.ca/dataverse/mcgill)] at (URL- https://doi.org/10.5683/SP3/IXFMHA).Supplementary information (SI): full experimental methods and characterization, along with additional data. See DOI: https://doi.org/10.1039/d5py00911a.
Acknowledgements
This research was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC, Discovery Grant RGPIN-2024-04619 (MM), RGPIN-2023-03900 (RL) and RGPIN-2016-03792 (JN)). The authors would also like to thank the Eugene Ulmer-Lamothe Award (Mary Hnatyshyn and Matt Halloran) for the financial support. The authors also thank the McGill Institute for Advanced Materials (MIAM) and Centre Québécois sur les Matériaux Fonctionnels (CQMF) for providing access to SAXS, DSC, and TGA facilities.References
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- D. J. C. Constable, ACS Sustainable Chem. Eng., 2020, 8, 14657–14667 CrossRef CAS.
- C. Valderrama, H. Huamán, A. Valencia-Arias, M. Vasquez-Coronado, S. Cardona-Acevedo and J. Delgado-Caramutti, Sustainability, 2023, 15, 13946 CrossRef.
- T. Robert and S. Friebel, Green Chem., 2016, 18, 2922–2934 RSC.
- J. De Carvalho, A. Magalhaes and C. Soccol, Chim. Oggi, 2018, 36, 56 CAS.
- K. Kirimura, Y. Honda and T. Hattori, in Comprehensive Biotechnology, ed. M. Moo-Young, Elsevier, Amsterdam, 2nd edn, 2011, vol. 3, pp. 143–147 Search PubMed.
- T. Werpy and G. Peterson, Top Value Added Chemicals from Biomass, U.S. Department of Energy, United States, 2004, Report No.: DOE/GO-102004-1992 Search PubMed.
- S. Jain and S. Kumar, Energy, 2024, 296, 131130 CrossRef CAS.
- T. U. S. Nagai and K. Yoshida, Kobunshi Kagaku, 1958, 15, 550–554 CrossRef.
- C. S. Marvel and T. H. Shepherd, J. Org. Chem., 1959, 24, 599–605 CrossRef CAS.
- L. Sollka and K. Lienhamp, Macromol. Rapid Commun., 2021, 42, 1022–1336 CrossRef PubMed.
- S. C. Fischer von Mollard, N. Ueberschaar, M. Schreiber, F. Kamphuis, S. Zechel and M. D. Hager, J. Polym. Sci., 2025, 63, 1284–1296 CrossRef CAS.
- B. Guo, Y. Chen, Y. Lei, L. Zhang, W. Y. Zhou, A. B. M. Rabie and J. Zhao, Biomacromolecules, 2011, 12, 1312–1321 CrossRef CAS PubMed.
- J. Dai, S. Ma, X. Liu, L. Han, Y. Wu, X. Dai and J. Zhu, Prog. Org. Coat., 2015, 78, 49–54 CrossRef CAS.
- M. Y.-P. J. Retuert, F. Martínez and M. Jeria, Bull. Chem. Soc. Jpn., 1993, 66, 1707–1708 CrossRef.
- Q. Liu and X. M. Zhou, J. Macromol. Sci., 2015, 52, 745–751 CrossRef CAS.
- L. Yuan, Z. Wang, N. M. Trenor and C. Tang, Macromolecules, 2015, 48, 1320–1328 CrossRef CAS.
- M. F. Sainz, J. A. Souto, D. Regentova, M. K. G. Johansson, S. T. Timhagen, D. J. Irvine, P. Buijsen, C. E. Koning, R. A. Stockman and S. M. Howdle, Polym. Chem., 2016, 7, 2882–2887 RSC.
- R. L. Atkinson, O. R. Monaghan, M. T. Elsmore, P. D. Topham, D. T. W. Toolan, M. J. Derry, V. Taresco, R. A. Stockman, D. S. A. De Focatiis, D. J. Irvine and S. M. Howdle, Polym. Chem., 2021, 12, 3177–3189 RSC.
- K. C. H. Chin, J. Cui, R. M. O'Dea, T. H. Epps III and A. J. Boydston, ACS Sustainable Chem. Eng., 2023, 11, 1867–1874 CrossRef CAS.
- K. Parkatzidis, S. Boner, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2022, 11, 841–846 CrossRef CAS PubMed.
- Y. Zheng, K. Yao, J. Lee, D. Chandler, J. Wang, C. Wang, F. Chu and C. Tang, Macromolecules, 2010, 43, 5922–5924 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 68, 488–502 CrossRef CAS.
- M. Purushothaman, P. S. G. Krishnan and S. K. Nayak, J. Appl. Polym. Sci., 2014, 131, 0021–8995 Search PubMed.
- S. Kardan, O. Garcia Valdez, A. Métafiot and M. Maric, Processes, 2019, 7, 254 CrossRef CAS.
- T. Hirano, R. Takeyoshi, M. Seno and T. Sato, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2415–2426 CrossRef CAS.
- G. Moad, A. G. Anderson, F. Ercole, C. H. J. Johnson, J. Krstina, C. L. Moad, E. Rizzardo, T. H. Spurling and S. H. Thang, ACS Symp. Ser., 1998, 685, 332–360 CrossRef CAS.
- M. Y. Zaremski, A. V. Plutalova, M. B. Lachinov and V. B. Golubev, Macromolecules, 2000, 33, 4365–4372 CrossRef CAS.
- A. Y. Sankhe, S. M. Husson and S. M. Kilbey, Macromolecules, 2006, 39, 1376–1383 CrossRef CAS.
- S. Okada and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 822–827 CrossRef CAS.
- M. Fernández-García, M. Fernández-Sanz, J. L. de la Fuente and E. L. Madruga, Macromol. Chem. Phys., 2001, 202, 1213–1218 CrossRef.
- Z. Szablan, A. A. Toy, A. Terrenoire, T. P. Davis, M. H. Stenzel, A. H. E. Müller and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3692–3710 CrossRef CAS.
- Z. Szablan, A. A. Toy, T. P. Davis, X. Hao, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2432–2443 CrossRef CAS.
- K. Satoh, D.-H. Lee, K. Nagai and M. Kamigaito, Macromol. Rapid Commun., 2014, 35, 161–167 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. E. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed.
- S. Beuermann, M. Buback, T. P. Davis, N. García, R. G. Gilbert, R. A. Hutchinson, A. Kajiwara, M. Kamachi, I. Lacík and G. T. Russell, Macromol. Chem. Phys., 2003, 204, 1338–1350 CrossRef CAS.
- K. Yokota and A. Kondo, Makromol. Chem., 1973, 171, 113–122 CrossRef CAS.
- S. Beuermann, S. Harrisson, R. A. Hutchinson, T. Junkers and G. T. Russell, Polym. Chem., 2022, 13, 1891–1900 RSC.
- I. G. Popović, Thermochim. Acta, 1988, 134, 127–132 CrossRef.
- I. G. Popović, V. Galogaža, L. Katsikas and J. Veličkovic, Polym. Bull., 1991, 25, 107–114 CrossRef.
- T. G. Fox and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef CAS.
- J. M. G. Cowie, Z. Haq, I. J. McEwen and J. Velićkovič, Polymer, 1981, 22, 327–332 CrossRef CAS.
- J. Veličković, J. Filipovic, M. Plavsic, D. Petrović-Djakov, Z. Petrović and J. Budinski-Simendic, Polym. Bull., 1991, 27, 331–336 CrossRef.
- L. Li, L. Han, H. Hu and R. Zhang, Mater. Adv., 2023, 4, 726–746 RSC.
- J. Wang, X. Zhang, L. Jiang and J. Qiao, Prog. Polym. Sci., 2019, 98, 101160 CrossRef CAS.
- G. Huang, N. Wu, X. Wang, G. Zhang and L. Qiu, Macromol. Rapid Commun., 2022, 43, 2200149 CrossRef CAS PubMed.
- D. P. Chatterjee and B. M. Mandal, Macromolecules, 2006, 39, 9192–9200 CrossRef CAS.
- J. Choi, W. Kim, D. Kim, S. Kim, J. Chae, S. Q. Choi, F. S. Kim, T.-S. Kim and B. J. Kim, Chem. Mater., 2019, 31, 3163–3173 CrossRef CAS.
- F. L. Hatton, Polym. Chem., 2020, 11, 220–229 RSC.
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- J. C. Arnold, in Comprehensive Structural Integrity, ed. I. Milne, R. O. Ritchie and B. Karihaloo, Elsevier, Oxford, 3rd edn, 2003, vol. 6, pp. 281–319 Search PubMed.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- I. W. Hamley and V. Castelletto, Prog. Polym. Sci., 2004, 29, 909–948 CAS.
- G. Kim and M. Libera, Macromolecules, 1998, 31, 2569–2577 CrossRef CAS.
- I. Shibata, A. Sugawara-Narutaki and R. Takahashi, Chem. Sci., 2025, 16, 7921–7928 RSC.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 7641–7644 CrossRef CAS.
- N. Migliore, F. Picchioni and P. Raffa, Soft Matter, 2020, 16, 2836–2846 RSC.
- F. Asempour, E. Laurent, T. Bride and M. Maric, Eur. Polym. J., 2024, 219, 113402 CrossRef CAS.
- W. Jakubowski, A. Juhari, A. Best, K. Koynov, T. Pakula and K. Matyjaszewski, Polymer, 2008, 49, 1567–1578 CrossRef CAS.
Footnote |
| † Current affiliation: Department of Chemistry and Biochemistry, University of California, San Diego, CA, USA. |
| This journal is © The Royal Society of Chemistry 2025 |