
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced ruthenium-catalyzed alkene C–H-arylation at room temperature
Tanumoy
Mandal
a,
Stéphane
Golling
a,
Sven
Trienes
 ab and
Lutz
Ackermann
*ab
ab and
Lutz
Ackermann
*ab
aWöhler Research Institute for Sustainable Chemistry (WISCh), Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
bDZHK (German Centre for Cardiovascular Research), Potsdamer Straße 58, 10875 Berlin, Germany
First published on 30th September 2025
Abstract
C–H arylation has surfaced as a powerful tool for molecular sciences, with alkene C–H arylation thus far requiring either high reaction temperatures of 120 °C or stoichiometric amounts of RMgX. In sharp contrast, we herein report on room-temperature C–H arlyations of alkenes by means of ruthenium(II) catalysis with ample scope. This strategy also enabled late-stage diversification of structurally complex molecules and mechanistic studies provided strong evidence for photo-excitation of a ruthenacycle intermediate.
Transition metal catalysis has emerged as a transformative platform for the interconversion of functional groups, enabling the assembly of valuable organic molecules. Particularly, C–H activation represents arguably the most efficient strategy, avoiding lengthy and resource-demanding multi-step syntheses.1 The introduction of an aryl motif via C–H arylation provided access to distinct molecules by means of ruthenium catalysis,2 with various applications to crop protection, polymer chemistry, and drug discovery, among others.3,4 While most C(sp2)–H functionalizations predominantly focus on arene C–H bonds, the direct activation of vinylic C–H bonds continues to be underdeveloped.5 Thus, previous alkene C–H arylations relied as of yet on either high reaction temperatures of 120 °C,6–10 or very reactive Grignard reagents in stoichiometric amounts,11 thus severely compromising the viable functional group tolerance (Scheme 1A). In sharp contrast, we herein report on the unprecedented C–H-arylation of alkenes at ambient temperature. Key to success was represented by the unique features of light-enabled12–16 ruthena(II)17 photoredox catalysis, enabling efficient arylations of alkenes and 1,3-dienes with outstanding levels of chemo-, diastereo- and site-selectivities (Scheme 1B). Late-stage diversifications proved thereby viable at ambient temperature, while detailed mechanistic studies were suggestive of the photo-MLCT-excitation of the key ruthenacycle intermediate.
 | ||
| Scheme 1 (A) Previous work on directed, transition-metal-catalyzed arylation of alkenes. (B) This work: photo-induced ruthenium-catalyzed C–H-arylation of alkenes. | ||
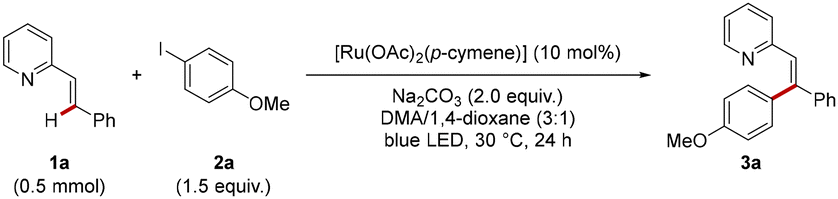
At the outset of our studies, vinylpyridine 1a and 4-iodoanisole 2a were probed for the photo-induced C(sp2)–H-arylation. Gratifyingly, with Na2CO3 as the base, 10 mol% of [Ru(OAc)2(p-cymene)] as the catalyst in a mixture of DMA/1,4-dioxane (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the corresponding tri-substituted olefin 3a was obtained in 81% yield (Table 1, entry 1). Lower catalytic performances were observed in 1,4-dioxane, DMA, or other solvent mixtures such as DMA/THF in a 3:1 ratio (entries 2 and 3). An inferior catalytic efficacy was observed with K2CO3 or K3PO4 as the base (entry 4). [Ru(MesCO2)2(p-cymene)] as the catalyst furnished the corresponding product 3a in a satisfying yield of 72% (entry 5). Interestingly, the use of the dimeric [RuCl2(p-cymene)]2 resulted in a lower yield of 32% which can be improved to 79% by adding a catalytic amount of NaOAc (entry 6), highlighting the importance of carboxylate assistance for efficient C–H activation.18 However, additional studies indicated a broader applicability and efficacy with [Ru(OAc)2(p-cymene)] as compared to [RuCl2(p-cymene)]2 in combination with acetate additives (see SI for detailed optimization). Other ruthenium sources, such as Ru3(CO)12 and RuCl3·nH2O completely failed to give the desired product 3a (Table 1, entry 7). Alternative electrophilic aryl donors proved viable, albeit with reduced yields of the desired product 3a (entry 8). Control experiments reflected the crucial importance of the base and light irradiation (entries 9–11). The essential role of the ruthenium catalyst was also confirmed (entry 12).
:1), the corresponding tri-substituted olefin 3a was obtained in 81% yield (Table 1, entry 1). Lower catalytic performances were observed in 1,4-dioxane, DMA, or other solvent mixtures such as DMA/THF in a 3:1 ratio (entries 2 and 3). An inferior catalytic efficacy was observed with K2CO3 or K3PO4 as the base (entry 4). [Ru(MesCO2)2(p-cymene)] as the catalyst furnished the corresponding product 3a in a satisfying yield of 72% (entry 5). Interestingly, the use of the dimeric [RuCl2(p-cymene)]2 resulted in a lower yield of 32% which can be improved to 79% by adding a catalytic amount of NaOAc (entry 6), highlighting the importance of carboxylate assistance for efficient C–H activation.18 However, additional studies indicated a broader applicability and efficacy with [Ru(OAc)2(p-cymene)] as compared to [RuCl2(p-cymene)]2 in combination with acetate additives (see SI for detailed optimization). Other ruthenium sources, such as Ru3(CO)12 and RuCl3·nH2O completely failed to give the desired product 3a (Table 1, entry 7). Alternative electrophilic aryl donors proved viable, albeit with reduced yields of the desired product 3a (entry 8). Control experiments reflected the crucial importance of the base and light irradiation (entries 9–11). The essential role of the ruthenium catalyst was also confirmed (entry 12).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield 3ab (%) |
|
a Reaction conditions: 1a (0.5 mmol), 2a (0.75 mmol), [Ru(OAc)2(p-cymene)] (10 mol%), Na2CO3 (2.0 equiv.), N2, 30 °C, 24 h, DMA/1,4-dioxane (3:1, 1.5 mL), 450 nm blue LED, 30 °C (fan cooling).
b Isolated yield. Yield in parentheses was determined by 1H-NMR using CH2Br2 as the internal standard.
c K2CO3 (2.0 equiv.) as base.
d 30 mol% NaOAc as additive.
|
||
| 1 | None | 81 |
| 2 | 1,4-Dioxane or DMA as sole solvent | 31/48 |
| 3c | DMA/THF (3:1) as solvent |
72 |
| 4 | K2CO3 or K3PO4 as base | 74/65 |
| 5 | [Ru(MesCO2)2(p-cymene)] as catalyst | 72 |
| 6 | [RuCl2(p-cymene)]2 as catalyst | (32), 79d |
| 7 | RuCl3·10H2O or Ru3(CO)12 as catalyst | 0d/0d |
| 8 | Ar–Br, Ar–Cl, or Ar–OTf as arylating reagent | 44/27/(19) |
| 9 | No light | (9) |
| 10 | Without Na2CO3 | (12) |
| 11 | Under air | 0 |
| 12 | Without [Ru(OAc)2(p-cymene)] | 0 |
With the optimized reaction conditions for the alkene C–H arylation at room temperature in hand, a viable substrate scope for the photo-induced alkene functionalization was investigated with different aryl iodides. The room-temperature direct arylation proved to be compatible with a large variety of functional groups on electron-rich as well as electron-deficient arenes, including ether (3a), ester (3f, 3h), ketone (3i), and cyano (3j). Halo derivatives were also well tolerated, furnishing the desired products 3l–3n, featuring valuable electrophilic handles. The synthesis potential of our photo-alkene arylation at room temperature was reflected by the efficient late-stage diversification of (−)-menthol (3w), (−)-myrtenol (3x), naproxen (3y), and indomethacin (3z) derivatives (Scheme 2A).
 | ||
| Scheme 2 Robustness of the photo-induced C–H activation at room temperature of (A) different aryl iodides and (B) vinyl-heteroarenes. Reaction conditions: 1 (0.5 mmol), aryl iodide 2 (0.75 mmol), [Ru(OAc)2(p-cymene)] (10 mol%), DMA/1,4-dioxane (1.5 mL), 30 °C (fan cooling). The product was obtained as single isomer. If the product was obtained as a mixture of (Z) and (E) isomers, the ratio is given in parenthesis. More details can be found in the SI. aPerformed on 0.25 mmol scale. | ||
Next, we explored the robustness of the photo-ruthenium-catalyzed C–H arylation at room temperature with a set of representative alkenes 1. Again, a variety of sensitive functional groups, such as cyano- and halo-substituted substrates, were well tolerated. Notably, a diene furnished product 3aj with excellent levels of chemo- and site-selectivities. It is noteworthy that a thiazole also enabled chelation-assisted C–H activation, thereby delivering product 3al in an efficient manner. Additionally, pyrazole was also identified as viable orienting group, furnishing the desired product 3am (Scheme 2B).
Next, we explored the robustness of the photo-ruthenium-catalyzed C–H arylation at room temperature with a set of representative alkenes 1. Again, a variety of sensitive functional groups, such as cyano- and halo-substituted substrates, were well tolerated. Notably, a diene furnished product 3aj with excellent levels of chemo- and site-selectivities. It is noteworthy that a thiazole also enabled chelation-assisted C–H activation, thereby delivering product 3al in an efficient manner. Additionally, pyrazole was also identified as viable orienting group, furnishing the desired product 3am (Scheme 2B).
To gain insights into the catalyst's mode of action, we first probed the performance of independently synthesized cyclometallated complex Ru-I. Interestingly, under photoexcitation, the desired product 3a was obtained in 77% yield, indicating that a cyclometallated intermediate is likely involved. In contrast, a significantly diminished efficacy was observed in the absence of light at ambient temperature or at 30 °C, highlighting the crucial role of light beyond catalyst activation through p-cymene decoordination, being suggestive of photo-excitation of a cyclometallated ruthenium species (Scheme 3A). Second, radical scavenger experiments were performed. While BHT and TEMPO led to strongly diminished yields, galvinoxyl and DPPH completely inhibited the reaction, and the corresponding radical adducts were observed by HRMS, supporting the formation of an aryl radical (Scheme 3B). Third, we analyzed the role of the blue LED irradiation by an on/off experiment, showing a significant inhibition of the reaction in the dark. These findings again suggest that continuous light irradiation is required, hence rendering a sole arene decoordination unlikely to be operative (Scheme 3C). Instead, photo-excitation of a ruthenacycle is more likely. Moreover, a quantum yield of 2% renders a radical chain mechanism unlikely to be operative (see SI for details). Additionally, detailed UV/Vis spectroscopy studies (see SI, Fig. S2) support the formation of a new ruthenium species upon light irradiation. Thus, the generation of a cyclometalated intermediate, most likely, plays a key role in the catalytic cycle.
 | ||
| Scheme 3 Key mechanistic findings including (A) reactions with a ruthenacycle, (B) radical scavenger experiments, and (C) on/off experiment. | ||
Based on these mechanistic studies and literature precedents,17 a plausible mechanism is proposed in Scheme 4. A carboxylate-assisted C–H activation first forms complex A, followed by coordination of the aryl iodide to give intermediate B,17c which is excited by light to form singlet species B*via MLCT. Intersystem crossing (ISC) then generates the triplet state B**, enabling SET to generate an aryl radical and complex C. Radical recombination results in complex D, followed by reductive elimination to afford the desired product 3a and complex E after ligand exchange. Afterwards, further C–H activation leads to the regeneration of the active species A.
 | ||
| Scheme 4 Mechanistic proposal for the ruthenium-catalyzed photoinduced C–H-arylation. | ||
In conclusion, we reported on a photo-induced C–H-arylation of alkenes. Contrary to previous state of the art, the ruthenium catalysis bypasses high temperature or the use of Grignard reagents through photoexcitation and is thereby compatible with a large variety of otherwise sensitive functional groups. Thereby, 39 multi-substituted heterocycle-incorporated olefins were assembled, including several biorelevant derivatives. Mechanistic studies highlighted the crucial role of blue light in this transformation.
The authors gratefully acknowledge generous support by the DZHK, the DFG (Gottfried Wilhelm Leibniz award to L. A.), the ERC Advanced Grant no. 101021358 (L. A.), and DAAD (NAMASTE+) for the financial support (T. M.).
Conflicts of interest
There are no conflicts to declare.Data availability
All data associated with this study are available in the article and supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc04527d.Notes and references
- (a) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS; (b) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
- (a) H. Simon, A. Zangarelli, T. Bauch and L. Ackermann, Angew. Chem., Int. Ed., 2024, 63, e202402060 CrossRef CAS; (b) G. McArthur, J. H. Docherty, M. D. Hareram, M. Simonetti, I. J. Vitorica-Yrezabal, J. J. Douglas and I. Larrosa, Nat. Chem., 2024, 16, 1141–1150 CrossRef CAS PubMed; (c) T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 15640–15645 CrossRef CAS PubMed.
- (a) T. Michiyuki, I. Maksso and L. Ackermann, Angew. Chem., Int. Ed., 2024, 63, e202400845 CrossRef CAS PubMed; (b) L. Ackermann, Org. Proc. Res. Dev., 2015, 19, 260–269 CrossRef CAS; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed; (d) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed.
- (a) S. G. Ouellet, A. Roy, C. Molinaro, R. Angelaud, J.-F. Marcoux, P. D. O’Shea and I. W. Davies, J. Org. Chem., 2011, 76, 1436–1439 CrossRef CAS PubMed; (b) A. Schischko, H. Ren, N. Kaplaneris and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1576–1580 CrossRef CAS.
- (a) M. Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed; (b) J. Zhang, X. Lu, C. Shen, L. Xu, L. Ding and G. Zhong, Chem. Soc. Rev., 2021, 50, 3263–3314 RSC; (c) S. Tang, K. Liu, C. Liu and A. Lei, Chem. Rev., 2015, 44, 1070–1082 CAS.
- S. Oi, K. Sakai and Y. Inoue, Org. Lett., 2005, 7(18), 4009–4011 CrossRef CAS PubMed.
- L. Ackermann, R. Born and P. lvarez-Bercedo, Angew. Chem., Int. Ed., 2007, 46, 6364–6367 CrossRef CAS PubMed.
- L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed.
- D. Zell, S. Warratz, D. Gelman, S. J. Garden and L. Ackermann, Chem. – Eur. J., 2016, 22, 1248–1252 CrossRef CAS.
- M. Kim, J. Kwak and S. Chang, Angew. Chem., Int. Ed., 2009, 48, 8935–8939 CrossRef CAS PubMed.
- L. Ilies, S. Asako and E. Nakamura, J. Am. Chem. Soc., 2011, 133(20), 7672–7675 CrossRef CAS PubMed.
- For representative reviews, see: (a) P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed; (b) J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed; (c) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed.
- For rhodium catalysis, see: (a) J. Tanaka, Y. Nagashima, A. J. Araujo Dias and K. Tanaka, J. Am. Chem. Soc., 2021, 143, 11325–11331 CrossRef CAS PubMed; (b) J. Thongpaen, R. Manguin, V. Dorcet, T. Vives, C. Duhayon, M. Mauduit and O. Baslé, Angew. Chem., Int. Ed., 2019, 58, 15244–15248 CrossRef CAS PubMed.
- For copper catalysis, see: (a) S. Trienes, J. Xu and L. Ackermann, Chem. Sci., 2024, 15, 7293–7299 RSC; (b) C. Li, B. Chen, X. Ma, X. Mo and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2130–2134 CrossRef CAS PubMed; (c) F. Yang, J. Koeller and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 4759–4762 CrossRef CAS PubMed.
- For iron catalysis, see: A. M. Messinis, T. von Münchow, M. Surke and L. Ackermann, Nat. Catal., 2024, 7, 273–284 CrossRef CAS.
- For recent ruthenium catalysis, see: (a) Y. Wang, S. Chen, X. Chen, A. Zangarelli and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202205562 CrossRef CAS PubMed; (b) J. Struwe, K. Korvorapun, A. Zangarelli and L. Ackermann, Chem. Eur. J., 2021, 27, 16237–16241 CrossRef CAS PubMed; (c) K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 18103–18109 CrossRef CAS PubMed; (d) A. Sagadevan, A. Charitou, F. Wang, M. Ivanova, M. Vuagnat and M. F. Greaney, Chem. Sci., 2020, 11, 4439–4443 RSC; (e) P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS; (f) T. Michiyuki, I. Maksso and L. Ackermann, Angew. Chem., Int. Ed., 2024, 63, e202400845 CrossRef CAS PubMed; (g) S. Trienes, S. Golling, M. H. Gieuw, M. Di Matteo and L. Ackermann, Chem. Sci., 2024, 15, 19037–19043 RSC; (h) J. Pöhlmann, B. Yuan, J. Wu and L. Ackermann, ACS Catal., 2025, 15, 10542–10549 CrossRef PubMed; (i) S. Chen, Z. Xu, B. Yuan, X.-Y. Gou and L. Ackermann, Nat. Synth., 2025, 4, 655–663 CrossRef CAS PubMed; (j) Y. Wang, B. Yuan, X. Chang and L. Ackermann, Chem, 2025, 11, 102387 CrossRef CAS; (k) A. Dey, R. Kancherla, K. Pal, N. Kloszewski and M. Rueping, Commun. Chem., 2024, 7, 295 CrossRef CAS PubMed.
- (a) T. Rogge, J. C. A. Oliveira, R. Kuniyil, L. Hu and L. Ackermann, ACS Catal., 2020, 10, 10551–10558 CrossRef CAS; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed; (c) L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed; (d) L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032–5035 CrossRef CAS PubMed; (e) L. Ackermann, R. Vicente and A. Althammer, Org. Lett., 2008, 10, 2299–2302 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |