
Viologen linker as a strong electron-transfer mediator in the covalent organic framework to enhance electrocatalytic CO2 reduction†
Xin
Zhang
ab,
Yin-Zong
Yuan
ab,
Hong-Fang
Li
*b,
Qiu-Jin
Wu
bc,
Hong-Jing
Zhu
bc,
Yu-Liang
Dong
ab,
Qiao
Wu
bc,
Yuan-Biao
Huang
 *bc and
Rong
Cao
*bcd
*bc and
Rong
Cao
*bcd
aCollege of Chemistry, Fuzhou University, Fuzhou, 350108, China
bState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, 350002, Fujian, China. E-mail: hongfangli@fjirsm.ac.cn; ybhuang@fjirsm.ac.cn; rcao@fjirsm.ac.cn
cUniversity of Chinese Academy of Science, Beijing 100049, China
dFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, 350108, Fujian, China
First published on 6th April 2023
Abstract
Covalent organic frameworks (COFs) are promising candidates for the CO2 electroreduction reaction (CO2RR) due to their advantages of tunable structures, abundant active sites, and strong CO2 adsorption enrichment. However, the poor conductivities of the traditional COFs usually result in a low current density in the CO2RR and limits their use in industrial applications. Herein, viologen units as strong electron-transfer mediators (ETMs) were inserted into the backbones of Co-porphyrin-based COF (Por(Co)-Vg-COF) nanosheets to enhance the electronic conductivity and improve the CO2RR activity. The obtained Por(Co)-Vg-COF displayed a good conductivity of 3.7 × 10−7 S m−1 and thus exhibited a very high selectivity towards CO production (>98%) in a wide range of applied potentials from −0.6 V to −0.9 V versus the reversible hydrogen electrode (RHE) in neutral aqueous solution, which surpassed all the conventional COF electrocatalysts. Moreover, Por(Co)-Vg-COF was employed as the first COF electrode in an acidic/alkaline system, and achieved a high FECO of up to 91% at −0.9 V versus RHE in acidic electrolyte and a current density of 251 mA cm−2 at −1.3 V versus RHE in 1 M KOH aqueous electrolyte. This work provides a facile strategy to enhance the CO2RR performance by improving the electronic conductivity of porous framework materials via the introduction of an ETM in their backbone.
Introduction
Clean and sustainable energy-conversion technologies are highly demanded due to the environmental and energy problems caused by large emissions of the greenhouse gas carbon dioxide (CO2) from conventional fossil fuel-based technologies.1 Among the clean technologies, the electrocatalytic reduction of CO2 for value-added products using clean electric energy stands out as a particularly attractive and promising solution to reduce carbon emissions.2–15 To date, many Au-, Ag-, and Cu-based nanostructured materials, metal–organic frameworks, and single-atom catalysts have been developed to apply in the CO2 electroreduction reaction (CO2RR).16–22 However, the CO2RR generally faces challenges in achieving high selectivity and a high current density due to the intrinsic thermodynamic stability of CO2 and the competitive hydrogen evolution reaction (HER), hindering its practical application. Therefore, the development of highly efficient electrode materials that can overcome these problems is desirable.Covalent organic frameworks (COFs) linked by organic building blocks with strong covalent bonds are regarded as promising candidates for the CO2RR due to their robust frameworks, tuneable functional groups and active sites, and strong CO2 adsorption enrichment ability.23–31 The enriched CO2 can easily diffuse to the exposed abundant active sites in porous COFs via channels. Thus, some metal porphyrin and metal phthalocyanine-based COFs have been synthesized and applied to the CO2RR to produce CO.1,27 However, the poor conductivity of the COFs is one of the most challenging issues still to overcome, limiting the current density and the efficiency of the energy conversion in the CO2RR. Therefore, in order to generate high current densities, it is highly desirable to create conductive 2D COFs with efficient electron-transfer abilities for the CO2RR. In addition, most of the COF electrocatalysts reported to date have been constructed by metastable imine bonds, which means they can be used only in H-type cells with neutral electrolytes and thus cannot afford large current densities (up to 100 mA cm−2) due to the limited CO2 concentration in neutral aqueous solutions (0.034 mol L−1).2,18,23,27 In contrast, when the CO2RR29 is operated in a flow cell with a gas-diffusion electrode in acidic/alkaline aqueous electrolyte, it can effectively tackle the above challenges through the faster CO2 diffusion to the catalyst surface.33–45 However, the majority of COFs with an imine bond connected usually show poor stability in acidic/alkaline systems. Thus, it is highly desirable to construct ultrastable COFs in acidic/alkaline aqueous solutions for the CO2RR with an appropriate industrial-current level density to meet the needs of commercial applications.
To address the above-mentioned problems, we turned our attention to the introduction of an electron-transport mediator (ETM) into chemically stable COFs to increase the electron-transfer ability and enhance the CO2RR in alkaline and acidic conditions. It is well known that viologens are a unique class of ETM molecules, which can undergo two- or one-electron reduction in steps.46–48 Dicationic 4,4′-bipyridinium core (Vg2+) is reversibly reducible to the corresponding radical cationic intermediate (Vg2+) or to neutral (Vg0).49 Such redox properties have been used to enhance electrocatalysis by improving the charge-transfer ability when combined with electron-deficient molecules.48,50–52 On the other hand, cobalt porphyrin is also considered to be an active site for CO2RR towards the production of CO.1,2,23,27 Moreover, cobalt porphyrin molecules are excellent electron acceptors or electron-transfer carriers because of their conjugated electron system. So, introducing viologens into the backbones of cobalt porphyrin-based COFs would be an effective strategy to improve the electronic conductivity and ultimately enhance the energy-conversion efficiency of the CO2RR.46,53–60 In addition, compared with most the COFs linked by imine bonds, the C–N bond is more stable in harsh conditions, which could make the COF materials constructed by C–N bonds applicable in acidic or alkaline electrolytes and thus improve the CO2RR activity.
Herein, we introduced viologen building blocks into a C–N bond-connected robust conductive Co-porphyrin-based COF (Por(Co)-Vg-COF) to enhance the CO2RR performance (Fig. 1(a)). The viologen groups served as ETMs to transfer the abundant electrons to the adjacent Co-porphyrin site in the Por(Co)-Vg-COF, and thus a highly efficient electrocatalytic porous material system was constructed for the CO2RR. The obtained Por(Co)-Vg-COF displayed a one order of magnitude higher electron-conduction value (3.7 × 10−7 S m−1) than that of the traditional COF-366-Co (2.08 × 10−8 S m−1). Thus, the Por(Co)-Vg-COF nanosheets exhibited high CO selectivity with 98.5% Faraday efficiency at −0.6 V versus the reversible hydrogen electrode (RHE, all the potentials quoted in this paper are with reference to the RHE) tested using the H-cell in KHCO3 electrolyte. Moreover, the robust Por(Co)-Vg-COF also showed excellent CO2RR performance in an acidic/alkaline system. It showed excellent CO selectivity above 90% in the wider potential range of −1.2 to −1.5 V and the CO partial current density reached −82 mA cm−2 at −1.5 V in 0.06 M H2SO4 with 0.5 M K2SO4 addictive. More impressively, the Por(Co)-Vg-COF achieved a nearly 100% FECO over a wide potential range from −0.3 V to −1.2 V and achieved an industrial CO current density of up to 251 mA cm−2 in a flow cell in 1 M KOH electrolyte.
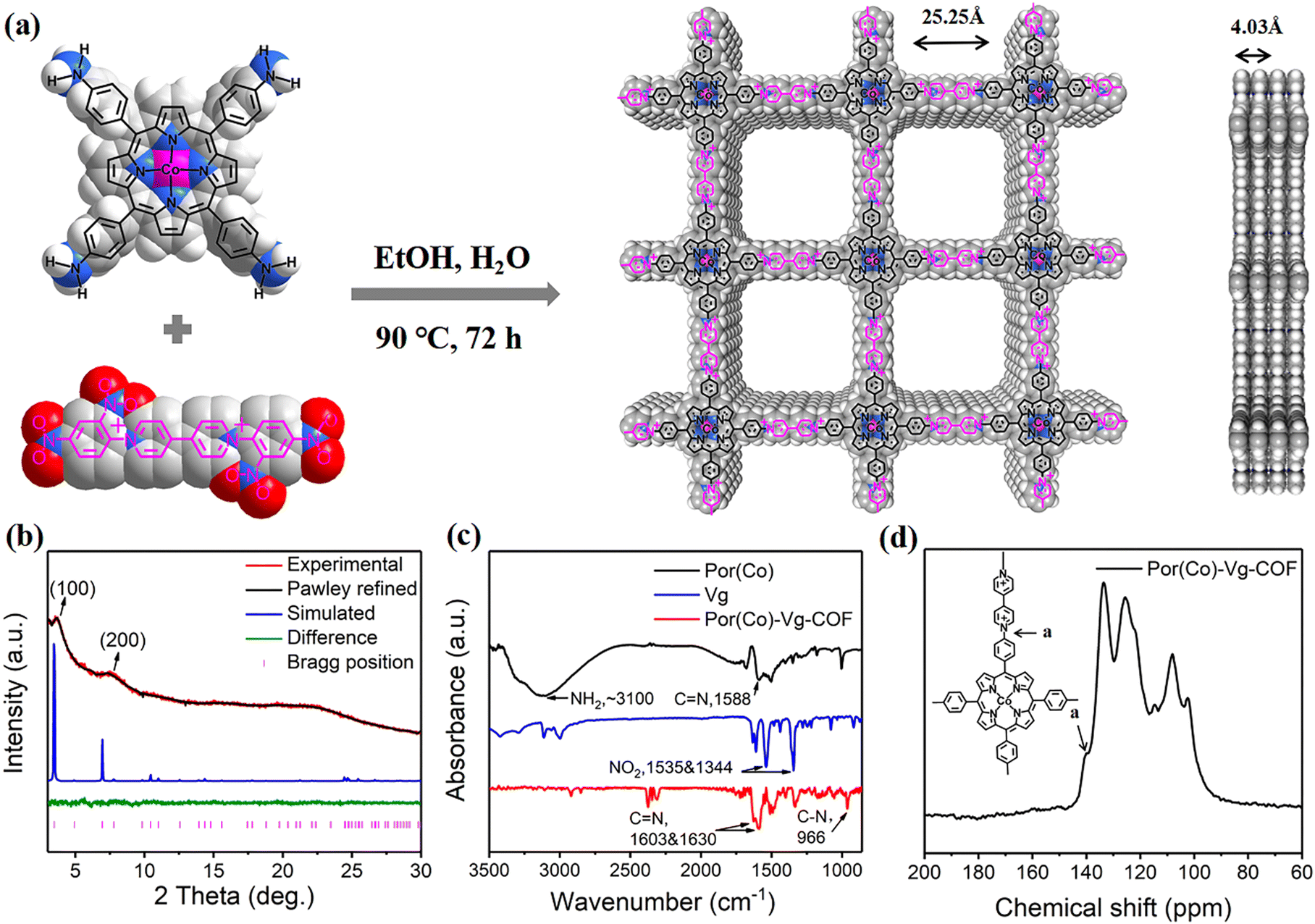 | ||
| Fig. 1 (a) Illustration of the preparation of Por(Co)-Vg-COF and top and side views of their structure with 2 × 2 square grids in an AA-stacking mode. (b) PXRD patterns of Por(Co)-Vg-COF. (c) FT-IR spectra of Por(Co) (black curve), Por(Co)-Vg-COF (red curve), and Vg (blue curve). (d) Solid-state 13C NMR spectrum of Por(Co)-Vg-COF. | ||
Results and discussion
The conjugated 2D COF Por(Co)-Vg-COF was prepared by the solvothermal Zincke reaction of 1,1′-bis(2,4-dinitrophenyl)-[4,4′-bipyridine]-1,1′-dium dichloride (TNV) and 5,10,15,20-tetrakis(4-aminophenyl)porphinatocobalt (Co-TAPP) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio in a mixed solvent of ethanol and water (4:1, v:v) for 72 h (Fig. 1(a)). In the process of the Zincke reaction, the Co-TAPP (Por(Co)) and 4-4-bipyridine cationic salt (Vg) building blocks of the Por(Co)-Vg-COF were connected by C–N bonds. Notably, the structure of viologen moieties in Por(Co)-Vg-COF was retained and was quite similar to the methyl viologen (MV) molecules, which can serve as ETMs to provide abundant electrons to the nearby Por(Co) sites. For comparison, the metal-free Por(H)-Vg-COF was also prepared using 5,10,15,20-tetrakis(4-cyanophenyl)porphyrin (TAPP) monomer instead of Por(Co) under similar conditions (Scheme S4, ESI†). To study the role of viologen in Por(Co)-Vg-COF, viologen-free COF-366-Co containing Por(Co) and 1,4-benzenedicarboxaldehyde (BDA) linked by typical imine bonds was also produced by the reported method. It should be noted that although the formation of Por(Co)-Vg-COF was driven by the irreversible Zincke reaction, only weak peaks were observed in the powder X-ray diffraction (PXRD) pattern. Thus, we simulated the Por(Co)-Vg-COF crystal structure using PXRD analysis and Pawley refinements in Materials Studio software. The peaks at the low 2θ angles of 3.70° and 7.46° were assigned to (100) and (200) facets, respectively (Fig. 1(b)), and were in good agreement with the simulated AA-stacking model.61 The Por(Co)-Vg-COF crystallized in the triclinic P1 space group with a unit cell parameter of a = 25.25 Å, b = 25.25 Å, c = 4.03 Å, and α = 90°, β = 90°, γ = 90°. The refinements showed a 1D channel along c-axis with theoretical pore sizes of 2.5 nm, and a distance between adjacent stacked 2D sheets of 4.03 Å. The Fourier-transform infrared (FT-IR) spectrum of Por(Co)-Vg-COF (Fig. 1(c)) showed that the –N–O vibrations bands at 1340 and 1530 cm−1 belonging to the TNV and the –NH2 vibrations bands at ∼3100 cm−1 attributed to the Por(Co) disappeared after the reaction. Moreover, –C–N– bonds that connected the Por(Co) and Vg stretching vibration band at ∼966 cm−1 appeared. In addition, a –C
:2 molar ratio in a mixed solvent of ethanol and water (4:1, v:v) for 72 h (Fig. 1(a)). In the process of the Zincke reaction, the Co-TAPP (Por(Co)) and 4-4-bipyridine cationic salt (Vg) building blocks of the Por(Co)-Vg-COF were connected by C–N bonds. Notably, the structure of viologen moieties in Por(Co)-Vg-COF was retained and was quite similar to the methyl viologen (MV) molecules, which can serve as ETMs to provide abundant electrons to the nearby Por(Co) sites. For comparison, the metal-free Por(H)-Vg-COF was also prepared using 5,10,15,20-tetrakis(4-cyanophenyl)porphyrin (TAPP) monomer instead of Por(Co) under similar conditions (Scheme S4, ESI†). To study the role of viologen in Por(Co)-Vg-COF, viologen-free COF-366-Co containing Por(Co) and 1,4-benzenedicarboxaldehyde (BDA) linked by typical imine bonds was also produced by the reported method. It should be noted that although the formation of Por(Co)-Vg-COF was driven by the irreversible Zincke reaction, only weak peaks were observed in the powder X-ray diffraction (PXRD) pattern. Thus, we simulated the Por(Co)-Vg-COF crystal structure using PXRD analysis and Pawley refinements in Materials Studio software. The peaks at the low 2θ angles of 3.70° and 7.46° were assigned to (100) and (200) facets, respectively (Fig. 1(b)), and were in good agreement with the simulated AA-stacking model.61 The Por(Co)-Vg-COF crystallized in the triclinic P1 space group with a unit cell parameter of a = 25.25 Å, b = 25.25 Å, c = 4.03 Å, and α = 90°, β = 90°, γ = 90°. The refinements showed a 1D channel along c-axis with theoretical pore sizes of 2.5 nm, and a distance between adjacent stacked 2D sheets of 4.03 Å. The Fourier-transform infrared (FT-IR) spectrum of Por(Co)-Vg-COF (Fig. 1(c)) showed that the –N–O vibrations bands at 1340 and 1530 cm−1 belonging to the TNV and the –NH2 vibrations bands at ∼3100 cm−1 attributed to the Por(Co) disappeared after the reaction. Moreover, –C–N– bonds that connected the Por(Co) and Vg stretching vibration band at ∼966 cm−1 appeared. In addition, a –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– stretching vibration band could be observed at ∼1603 and 1630 cm−1, belonging to the porphyrin and bipyridinium subunits, respectively.62 The 13C cross-polarization magic angle spinning (CP-MAS) solid-state NMR (Fig. 1(d)) spectrum exhibited a strong peak at 139.91 ppm, corresponding to the pyridyl carbon atoms connecting Por(Co) and Vg. Therefore, these results suggested that Por(Co)-Vg-COF had been successfully synthesized through the Zincke reaction. Por(Co)-Vg-COF was found to have a high Co content of 2.43 wt% by inductively coupled plasma (ICP) optical emission spectrometry (Table S1, ESI†). The CO2 sorption isotherms showed that the CO2 uptake value for Por(Co)-Vg-COF was 45 cm3 g−1 (Fig. S5, ESI†), which would be beneficial for promoting the CO2RR performance.
N– stretching vibration band could be observed at ∼1603 and 1630 cm−1, belonging to the porphyrin and bipyridinium subunits, respectively.62 The 13C cross-polarization magic angle spinning (CP-MAS) solid-state NMR (Fig. 1(d)) spectrum exhibited a strong peak at 139.91 ppm, corresponding to the pyridyl carbon atoms connecting Por(Co) and Vg. Therefore, these results suggested that Por(Co)-Vg-COF had been successfully synthesized through the Zincke reaction. Por(Co)-Vg-COF was found to have a high Co content of 2.43 wt% by inductively coupled plasma (ICP) optical emission spectrometry (Table S1, ESI†). The CO2 sorption isotherms showed that the CO2 uptake value for Por(Co)-Vg-COF was 45 cm3 g−1 (Fig. S5, ESI†), which would be beneficial for promoting the CO2RR performance.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to characterize the morphology of the as-prepared Por(Co)-Vg-COF.63 The SEM images showed that the Por(Co)-Vg-COF was composed of spherical particles (Fig. S6, ESI†). No aggregated Co or CoO particles were observed in the TEM (Fig. 2(a)) or high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 2(b)). In addition, elemental mapping by energy dispersive X-ray spectroscopy (EDS) showed that the elements C, N, O, Cl, and Co were uniformly distributed in Por(Co)-Vg-COF (Fig. 2(e)). To guarantee the accessibility of the cobalt porphyrin active sites for the electrolytes and CO2, ultrathin 2D Por(Co)-Vg-COF nanosheets were successfully exfoliated from its bulk via high-frequency sonication at room temperature. The atomic force microscopy (AFM) images in Fig. 2(c) and (d) clearly show nanosheets with a thickness of ca. 5.5 nm, which was close to thirteen layers of Por(Co)-Vg-COF. Such thin 2D Por(Co)-Vg-COF nanosheets would make the Co active sites highly exposed to the electrolytes and CO2 in the CO2RR.
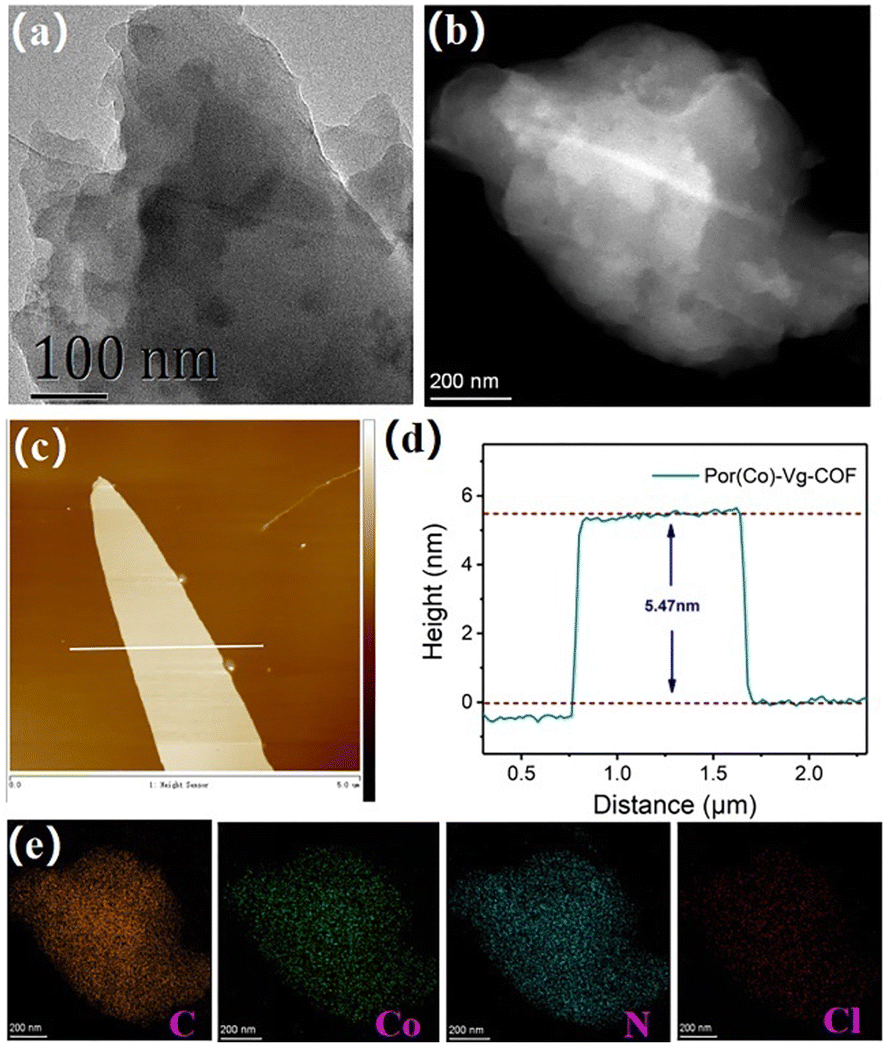 | ||
| Fig. 2 (a) TEM, (b) HAADF-STEM, and (c) AFM images of Por(Co)-Vg-COF. (d) The height profile along the white line in (c). (e) The elemental mapping of C, Co, N, and Cl for Por(Co)-Vg-COF. | ||
The X-ray photoelectron spectroscopy (XPS) survey spectrum of Por(Co)-Vg-COF (Fig. S7, ESI†) confirmed the presence of C, N, Cl, and Co elements in Por(Co)-Vg-COF, which well matched with the EDS result (Fig. 2(e)). The O resonance peak might have originated from the adsorbed water guest molecules or the residual unreacted nitro groups in the edges of Por(Co)-Vg-COF. The C 1s region (Fig. 3(a)) displayed three sets of peaks located at 284.82, 285.6, and 286.4 eV, assignable to CC/C–C, C–N+, and CN, respectively, which proved the viologen in the as-synthesized fresh Por(Co)-Vg-COF was in a dicationic state.32 The N 1s spectrum (Fig. S8(a), ESI†) of Por(Co)-Vg-COF was fitted with three sets of peaks: cationic pyridinic (398.51 eV), Co-porphyrin (399.13 eV), and N-oxide (401.31 eV), respectively, in which the N–Co bond further verified the Co species was coordinated by N atoms.64 To investigate the effects of the Vg changes during the electrocatalysis, XPS tests of the Por(Co)-Vg-COF after the CO2RR were conducted. As shown in Fig. S8(b), ESI,† the N 1s spectrum of Por(Co)-Vg-COF displayed one more peak at 400.9 eV after catalysis, which was ascribed to the neutral pyridinic species, and the Cl 2p spectrum of Por(Co)-Vg-COF disappeared after the catalysis, which proved Vg+ had changed to Vg0 during the electrocatalysis (Fig. S9, ESI†). The spectra of the Por(Co)-Vg-COF catalyst before and after the CO2RR displayed one pair of peaks arising from the spin–orbit doublet of Co 2p (Fig. S10, ESI†), which could be assigned to the Co 2p3/2 and Co 2p1/2 with the Co(II) state. The Co 2p3/2 and Co 2p1/2 peaks of after catalyst were located at 780.34 and 795.35 eV, which represented an apparent positive shift of ∼0.41 eV and ∼0.16 eV compared with the pristine Por(Co)-Vg-COF (Co 2p3/2, 780.75 eV and Co 2p1/2, 795.51 eV). These changes in binding energies provided direct evidence that the charge-carrier migration pathway might be from Vg to Por(Co).
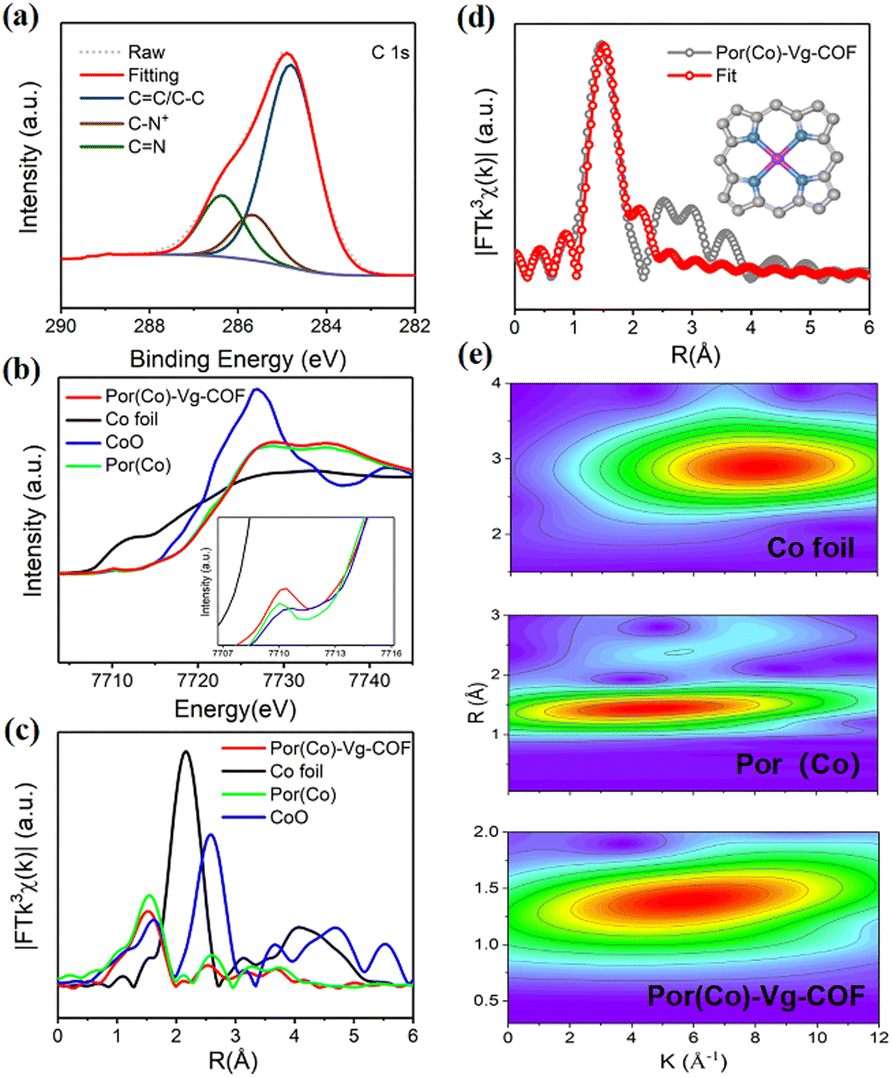 | ||
| Fig. 3 (a) High-resolution XPS spectra of C 1s for Por(Co)-Vg-COF; (b) Normalized Co K-edge XANES spectra of Por(Co)-Vg-COF, Co foil, CoO, and Por(Co); (c) Fourier-transform EXAFS spectra of Por(Co)-Vg-COF, Co foil, CoO, and Por(Co). (d) EXAFS fitting curves of Por(Co)-Vg-COF (inset: the Ni–N4 architecture). (e) Wavelet transforms for the k2-weighted EXAFS signals of different samples. | ||
X-Ray absorption spectroscopy was used to determine the electronic and coordination structures of cobalt in Por(Co)-Vg-COF. The Co K-edge XANES spectra of Por(Co)-Vg-COF showed obvious pre-edge peaks at 7710 eV, which were assigned to a dipole-forbidden 1s → 3d transition (Fig. 3(b)), and a Co K-edge located between those of CoO and Co foil, close to Por(Co), suggesting that the positively charged cobalt in Por(Co)-Vg-COF lay between Co(0) and Co(II).26 The Co-K edge of the extended X-ray absorption fine structure (EXAFS) appeared as the main signal at 1.50 Å, assigned to the Co–N scattering path (Fig. 3(c)). This was similar to that of Por(Co) (1.47 Å). No obvious signals ascribed to Co–Co (2.17 Å) and Co–O (2.57 Å) were observed, revealing that atomic Co–Nx species predominated and Co or CoO particles were not present in Por(Co)-Vg-COF. In order to determine the Co coordination environment, the EXAFS fitting results were obtained and showed that the coordination number of the Co species within the Por(Co)-Vg-COF was 4.5 (Fig. 3(d) and Table S2, ESI†), suggesting that the Co porphyrin structure was retained after the Zincke reaction. The EXAFS wavelet transform (WT) was further analyzed based on the radial distance resolution and k-space resolution to discriminate the backscattering atoms in R-space. An intensity maximum attributed to the Co–Co bond was observed in the EXAFS WT contour plots of the Co foil (Fig. 3(e)). While one intensity maximum was observed in the Por(Co) standard sample, which could be ascribed to the Co–N bond. Notably, only one intensity maximum at 5.7 Å−1 was observed, which was attributed to Co–N bonding, and no intensity maximum corresponding to Co–Co bonding was present in Por(Co)-Vg-COF, further demonstrating that atomic Co–Nx species predominated and Co particles were not rendered in Por(Co)-Vg-COF.65
To further prove the Vg0 state of the viologen moiety in Por(Co)-Vg-COF had been created during electrocatalysis, the electrochemical behaviour of the Por(Co)-Vg-COF electrode was evaluated by cyclic voltammetry (CV) measurements in 0.2 M Na2SO4 aqueous electrolyte.66 As shown by the CV curves in Fig. S11a, ESI,† two pairs of redox peaks were observed in Por(Co)-Vg-COF, which corresponded to the two-electron redox reaction of the viologen moieties (Fig. S11b, ESI†). Compared with the Vg, a more positive direction shift of the redox potential in Por(Co)-Vg-COF was observed, which may be because the electron-withdrawing group Por(Co) in the framework was more conducive to the redox reaction of Vg. Notably, the redox potential peaks were different from those of the Por(Co) (Fig. S11c, ESI†). So, it is believed that the viologen moieties in Por(Co)-Vg-COF could be reduced to Vg0 through a two-electron redox reaction under the applied potentials (Scheme S5, ESI†), which was well in accordance with the XPS results. To further reveal the valence change of viologen moieties in Por(Co)-Vg-COF under the negative potentials applied, the UV–vis spectra were recorded in real time during the charging/discharging procedure (Fig. S12, ESI†).49 During the reduction reaction process of the Por(Co)-Vg-COF electrode, upon the two-electron reduction of the viologen moiety, the absorption band at 411 nm increased proportionally, and in which the position change of the UV absorption peak was consistent with Vg changing to Vg0. These further proved Vg0 of the viologen moiety in Por(Co)-Vg-COF had been produced. Such a change of the viologen moiety from a cationic state to neutral form could improve the electronic conductivity of Por(Co)-Vg-COF, as verified by electrochemical impedance spectroscopy (EIS). As shown in Fig. S13a, ESI,† the semicircle values of Por(Co)-Vg-COF decreased with more negative potentials applied. This indicated that the electron-transfer ability had been significantly enhanced after the Vg2+ has changed to Vg0 in Por(Co)-Vg-COF, which might be ascribed to the formation of the large π conjugation plane including the Vg and Por(Co) parts via the CC bond of Vg0. In addition, the Por(Co)-Vg-COF displayed a smaller semicircle than the COF-366-Co (Fig. S13b, ESI†). This suggests that the viologen mediator played an important role in the transfer of electrons.
To confirm the charge transfer from the viologen moieties to the electron-acceptor Co-porphyrin in Por(Co)-Vg-COF, differential pulse voltammetry (DPV) tests and solid-state UV spectra measurements of its monomers were conducted.67 As shown in Fig. S14, ESI,† the first oxidation wave for the Por(Co) was at EOX = 0.91 V and the oxidation wave of the neutral form of Vg was at EOX = −0.37 V. Based on the Tauc plots of the solid-state UV spectra (Fig. S15, ESI†), the band gap (Eg) for Por(Co) was calculated to be 1.84 eV and for Vg0 was 3.67 eV.44 The relative positions of the highest occupied molecular orbital and the lowest unoccupied molecular orbital (HOMO–LUMO) were obtained according to the formula: HOMO = [(eEOX − eE(Fc/Fc+) + 4.8 V)] eV, and LUMO = HOMO − Eg. The LUMO of Vg with the Vg0 form possessed a higher potential of −0.31 eV than that of Por(Co) −3.2 eV, which meant it could more easily realize electron transfer from the viologen moieties to Por(Co) (Fig. S16, ESI†). The electrical conductivity in air was measured by the powder particle double electrode method to investigate the superiority of Por(Co)-Vg-COF over COF-366-Co. (Fig. S17 and S18, ESI†).68 Interestingly, the Por(Co)-Vg-COF had a one order of magnitude higher electron-conduction value (3.7 × 10−7 S m−1) than that of COF-366-Co (2.08 × 10−8 S m−1), which further revealed that the viologen building blocks as ETMs played an important role in promoting the electron transfer and electronic conductivity.
The CO2RR performances of the Por(Co)-Vg-COF nanosheets were first evaluated in a typical three-electrode system using a gas-tight H-type electrolysis cell with two compartments separated by a proton exchange membrane in 0.5 M KHCO3 aqueous solution.7 As shown in Fig. 4(a), a more positive onset potential (ca. −0.50 V) and significantly higher current densities were obtained in CO2-saturated electrolyte compared to in the N2-saturated electrolyte, suggesting a higher CO2 reactivity than for the HER for the Por(Co)-Vg-COF. To further confirm that the activity was derived from the CO2RR, the gas and liquid products at different applied potentials from −0.6 to −1.1 V were analyzed by gas chromatography and off-line 1H NMR spectroscopy, respectively. In particular, only CO and H2 were detected as gaseous products over the entire range of potentials applied, and no liquid product was found in the electrolyte solution (Fig. S19, ESI†). As shown in Fig. 4(b), the Por(Co)-Vg-COF exhibited a very high selectivity towards CO production (>98%) in a wide range of the applied potentials of −0.6 V to −1.1 V, which was found to be higher than that of COF-366-Co at the same potential and of other COFs that have been reported (Fig. 4(c), (d) and Table S3, ESI†). In addition, the CO partial current density continuously increased with the increase in potential, reaching −11.2 mA cm−2 at −0.9 V, which was higher than for COF-366-Co (Fig. 4(c)). The superior CO2RR performance for Por(Co)-Vg-COF highlighted the effectiveness of the viologen moiety with the neutral state could as an ETM to deliver electrons fast to Co centres. Moreover, the Por(Co)-Vg-COF displayed long-term stability and the FECO could retain over 90% after 10 h chronoamperometric test at a fixed potential of −0.6 V in CO2-saturated 0.5 M KHCO3 solution (Fig. 4(e)). The TEM images (Fig. S20, ESI†) of the Por(Co)-Vg-COF after the CO2RR revealed that its morphology was unchanged. Furthermore, no significant Co NPs were observed in the TEM images, indicating that the Por(Co) active sites were not reduced to Co(0). XPS analysis (Fig. S21, ESI†) showed no change in the valence state of Co(II) after electrocatalysis. The above results indicated that the Por(Co)-Vg-COF linked by viologen units was a robust framework.
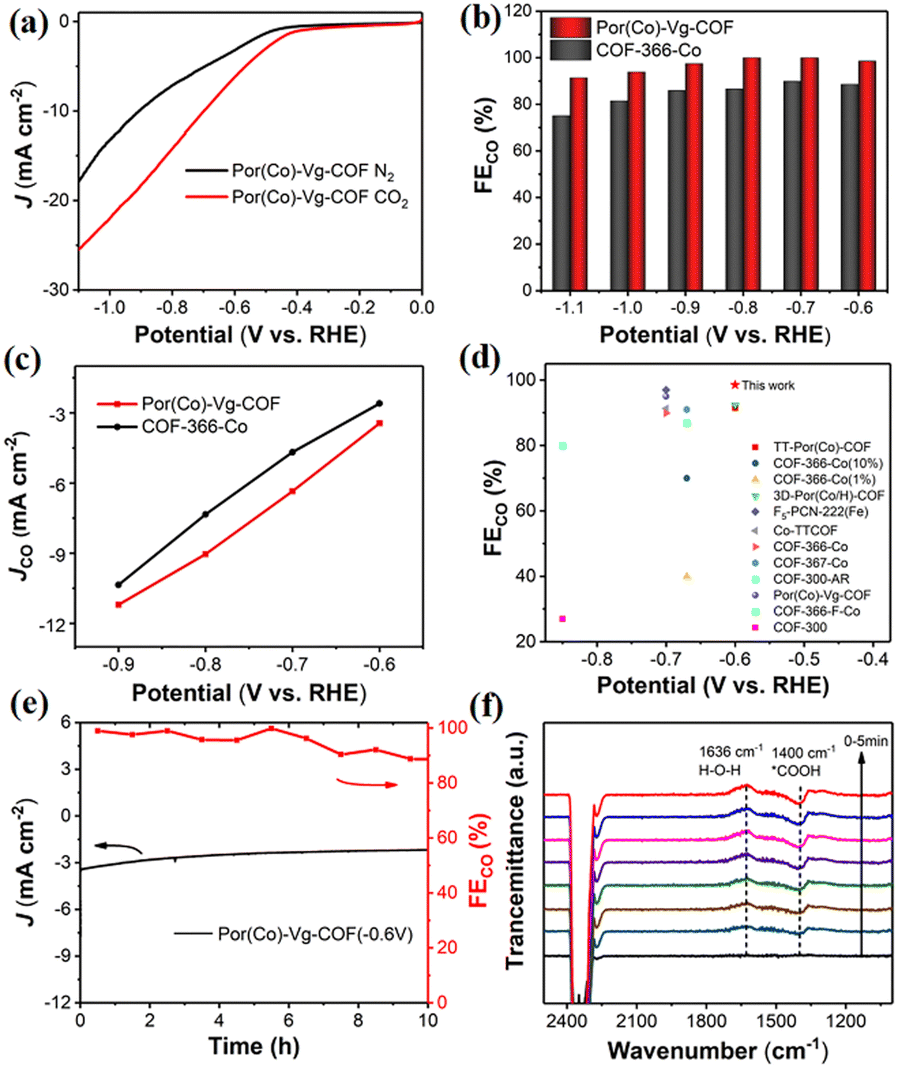 | ||
| Fig. 4 (a) LSV curves of Por(Co)-Vg-COF in CO2-saturated and N2-saturated 0.5 M KHCO3 electrolyte at a scan rate of 10 mV S−1. (b) FECO from −0.6 to −0.9 V versus RHE of Por(Co)-Vg-COF and COF-366-Co. (c) JCO from −0.6 to −0.9 V versus RHE of Por(Co)-Vg-COF and COF-366-Co. (d) Comparison of the optimal FECO among the 2D conductive Por(Co)-Vg-COF and the reported COF electrocatalysts evaluated in a H-type electrochemical cell. (e) Stability test (total current density and FE for CO) at −0.6 V for 10 h. (f) In situ ATR-IR of Por(Co)-Vg-COF at −0.9 V. | ||
To confirm the active sites of Por(Co)-Vg-COF, we tested the catalytic CO2RR of Por(H)-Vg-COF without cobalt species under the same conditions. As shown in Fig. S22, ESI,† Por(H)-Vg-COF showed negligible catalytic activity for the CO2RR and only the HER happened at various applied potentials. This result indicated that the cobalt centre in Por(Co)-Vg-COF was the active centre for CO2-to-CO conversion. In addition, the Por(Co)-Vg-COF exhibited a higher FECO of 98.5% at −0.6 V compared with COF-366-Co without viologen groups. So the efficient electron transfer from viologen to the porphyrin cobalt centre in Por(Co)-Vg-COF promoted the conversion from CO2 to CO. To confirm that the CO produced was indeed from the reduction of CO2, 13C isotope labelling experiments were carried out. When 13CO2 was used as a substrate, 13CO (m/z = 29) was finally detected by means of mass spectrometry, which confirmed that the CO produced came from the reactant CO2 and not from the decomposition of the catalysts (Fig. S23, ESI†).
Operando electrochemical attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) measurements were performed at −0.6 V in CO2-saturated 0.5 M KHCO3 to verify the possible reaction intermediates in the CO2RR. The band at ∼1400 cm−1 in the Por(Co)-Vg-COF spectrum was assigned to the C–O stretching of *COOH, as seen in the ATR-IR spectra (Fig. 4(f)). The intensity of this *COOH signal gradually increased with the time taken to respond, reaching a maximum after 120 s and then reaching dynamic equilibrium. Therefore, for the CO2RR catalyzed by Por(Co)-Vg-COF, we propose that the formation of the *COOH intermediate was the rate-determining step (RDS).23 This speculation was consistent with the Tafel slope results (Fig. S24, ESI†), where the values of Por(Co)-Vg-COF were greater than the theoretical value of 118 mV dec−1. This suggests that the formation of *COOH from the adsorbed CO2via a proton-coupled electron-transfer (PCET) process was the RDS.26
To meet the requirements for commercial applications, the excellent CO2RR performance of Por(Co)-Vg-COF in neutral electrolyte encouraged us to further improve its electrocatalytic performance with a large current density using a gas-diffusion electrode (GDE) in acidic media, which could offer an avenue to reduce carbonate formation and eliminate CO2 crossover, and this was the first COF catalyst reported for CO2RR in acidic/alkaline conditions.37,63–78 The CO2RR of Por(Co)-Vg-COF in 0.06 M H2SO4 with a 0.5 M K2SO4 additive was first tested. As shown in Fig. 5(a), a more positive onset potential (−0.75 V) and significantly higher current densities were obtained in CO2-saturated electrolyte compared to COF-366-Co, suggesting a higher CO2 reactivity than COF-366-Co for the Por(Co)-Vg-COF in acidic electrolyte. As shown in Fig. 5(b), with the increase in potential, the CO partial current density continuously increased and reached −82 mA cm−2 at −1.5 V, which was much higher than that of COF-366-Co. Furthermore, the Por(Co)-Vg-COF exhibited a high selectivity towards CO production (>90%) in the range of the applied potentials of −1.3 V to −1.5 V, and a high FECO of 91% at −1.3 V (Fig. 5(c)). However, the COF-366-Co only had 20% selectivity towards CO production at the same potential. These results indicated the superior CO2RR performance for Por(Co)-Vg-COF than COF-366-Co in strong acidic electrolyte and proved that the Por(Co)-Vg-COF was stable in acidic conditions. Besides, we also tested the CO2RR performance of Por(Co)-Vg-COF in 1 M KOH aqueous electrolyte. As shown in Fig. 5(d), the total current density reached −251 mA cm−2 at −1.3 V and the CO partial current density reached −172 mA cm−2 at −1.3 V (Fig. 5(e)). The FECO was as high as 95% at the wide potential at −0.3 V to −1.2 V (Fig. 5(f)). Compared with the H-cell, the current density of the CO2RR was significantly improved in the GDE by the faster CO2 diffusion to the catalyst surface. ICP optical emission spectrometry demonstrated that it had a low Co content of 0.032 mg L−1 and 0.0106 mg L−1 in acidic and alkaline aqueous electrolytes, respectively (Table S4, ESI†). Moreover, the Por(Co)-Vg-COF displayed long-term stability in 6 h chronoamperometric tests at a fixed potential of −1.3 V in 0.06 M H2SO4 with a 0.5 M K2SO4 solution and in 9 h chronoamperometric test at a fixed potential of −0.9 V in 1 M KOH solution (Fig. S25 and S26, ESI†). As shown in Fig. S27, ESI,† we obtained the FT-IR spectra of Por(Co)-Vg-COF after soaking in acid and alkali for 4 h respectively, and found that the spectrum did not change. These results suggest that the C–N was more stable in acidic/alkaline aqueous electrolyte and proved the Por(Co)-Vg-COF had excellent CO2RR performance under harsh conditions.
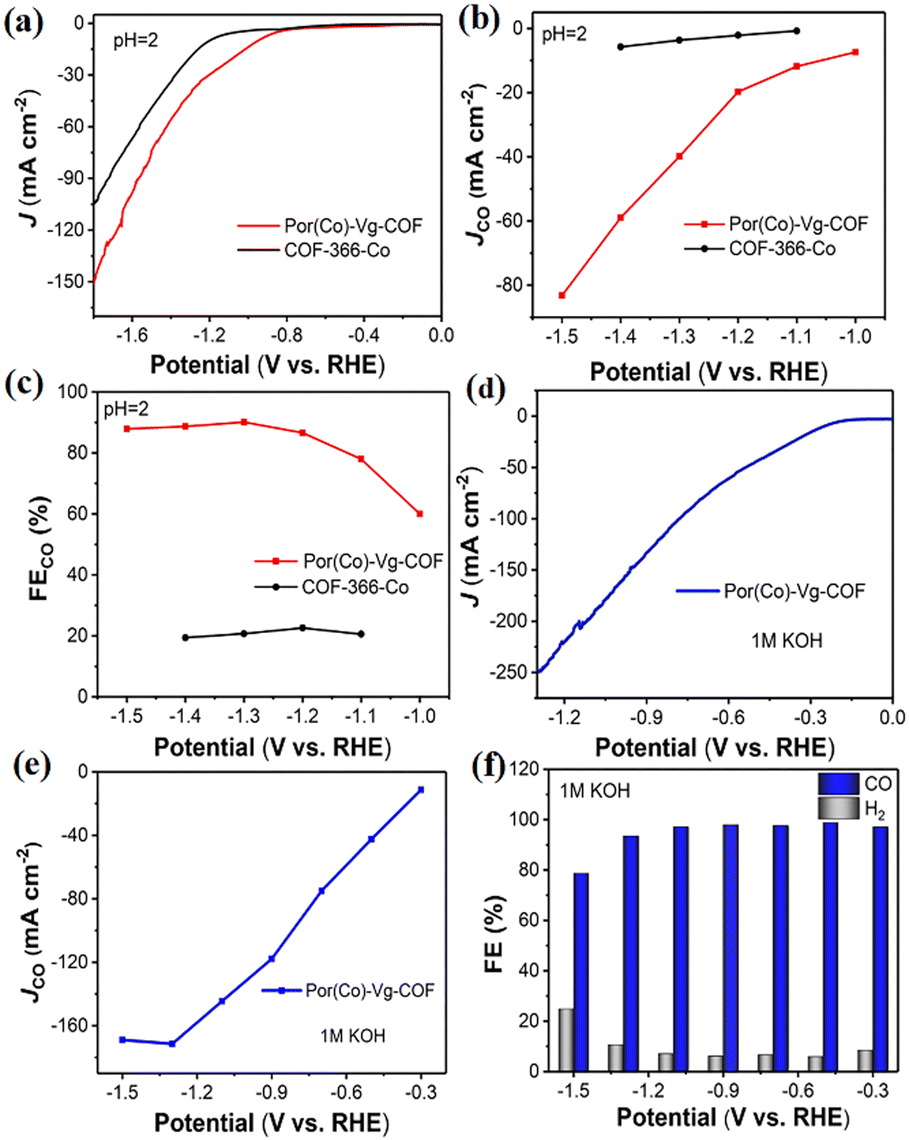 | ||
| Fig. 5 (a) LSV curves for Por(Co)-Vg-COF and COF-366-Co in 0.06 M H2SO4 with a 0.5 M K2SO4 additive electrolyte at a scan rate of 10 mV S−1. (b) JCO and (c) FECO from −1.0 to −1.5 V versus RHE of Por(Co)-Vg-COF and COF-366-Co in 0.06 M H2SO4 with a 0.5 M K2SO4. (d) LSV curves for Por(Co)-Vg-COF in 1 M KOH under CO2 flow. (e) JCO and (f) FECO and FEH2 from −0.3 to −1.5 V of Por(Co)-Vg-COF in 1 M KOH under CO2 flow. | ||
Conclusions
In summary, strong ETM viologen units were introduced into cobalt porphyrin-based COF (Por(Co)-Vg-COF) through a Zincke reaction to enhance the electrocatalytic performance of the CO2RR. As the viologens could undergo a two-electron reduction to achieve an excellent electron-transfer ability, the obtained Por(Co)-Vg-COF nanosheets showed a very high selectivity of 98.5% towards CO production and a large CO partial current density of 9.04 mA cm−2 at −0.8 V, which was superior to the insulating COF electrocatalysts. This Por(Co)-Vg-COF material constructed by C–N bond was very stable and is the first COF catalyst reported for the CO2RR in acidic/alkaline conditions, which could achieve a high FECO of up to 91% at −1.3 V, and a large CO partial current density up to −82 mA cm−2 at −1.5 V in 0.06 M H2SO4 with a 0.5 M K2SO4 electrolyte. At the same time, the Por(Co)-Vg-COF achieved a nearly 100% FECO over a wide potential range of −0.3 V to −1.2 V and reached an industrial level CO current density as high as 251 mA cm−2 in a flow cell in 1 M KOH electrolyte. This study provides a theoretical basis for the rational design of high-conductivity porous skeleton electrocatalytic materials, as well as an effective strategy for improving the current density and product selectivity of the CO2RR.Author contributions
X. Z., H. L., Y. H. and R. C. conceived the project and wrote the manuscript. X. Z., Y. Y., Q. W., H. Z., Y. D. and Q. W. performed the experiments and collected the data. H. L. and Y. H. polished the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2018YFA0208600, 2018YFA0704502), NSFC (U22A20436, 22071245, 22033008, 22220102005), Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (2021ZZ103). We thank the beamline BL14W1 station for XAS measurements at the Shanghai Synchrotron Radiation Facility, China.Notes and references
- Q. Wu, M.-J. Mao, Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Construction of Donor–Acceptor Heterojunctions in Covalent Organic Framework for Enhanced CO2 Electroreduction, Small, 2021, 17, 2004933 CrossRef CAS PubMed.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Efficient Electron Transmission in Covalent Organic Framework Nanosheets for Highly Active Electrocatalytic Carbon Dioxide Reduction, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- Z. B. Zhang, T. Vasiliu, F. F. Li, A. Laaksonen, F. Mocci and X. Y. Ji, Electrochemically Driven Efficient Enzymatic Conversion of CO2 to Formic Acid with Artificial Cofactors, J. CO2 Util., 2021, 52, 8 Search PubMed.
- Y. H. Zhang, L. Ma, Y. Q. Lv and T. W. Tan, Facile Manufacture of Cof-Based Mixed Matrix Membranes for Efficient CO2 Separation, Chem. Eng. J., 2022, 430, 9 Search PubMed.
- X.-D. Zhang, T.-Y. Liu, C. Liu, D.-S. Zheng, J.-M. Huang, Q.-W. Liu, W.-W. Yuan, Y. Yin, L.-R. Huang, M. Xu, Y. Li and Z.-Y. Gu, Asymmetric Low-Frequency Pulsed Strategy Enables Ultralong CO2 Reduction Stability and Controllable Product Selectivity, J. Am. Chem. Soc., 2023, 145, 2195–2206 CrossRef CAS PubMed.
- P. H. Zhang, Z. F. Wang, P. Cheng, Y. Chen and Z. J. Zhang, Design and Application of Ionic Covalent Organic Frameworks, Coord. Chem. Rev., 2021, 438, 15 Search PubMed.
- M.-D. Zhang, D.-H. Si, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, Conductive Phthalocyanine-Based Covalent Organic Framework for Highly Efficient Electroreduction of Carbon Dioxide, Small, 2020, 16, 2005254 CrossRef CAS PubMed.
- Y. Yang, Y. Lu, H.-Y. Zhang, Y. Wang, H.-L. Tang, X.-J. Sun, G. Zhang and F.-M. Zhang, Decoration of Active Sites in Covalent–Organic Framework: An Effective Strategy of Building Efficient Photocatalysis for CO2 Reduction, ACS Sustainable Chem. Eng., 2021, 9, 13376–13384 CrossRef CAS.
- W.-F. Xiong, D.-H. Si, J.-D. Yi, Y.-B. Huang, H.-F. Li and R. Cao, Morphology and Composition Dependence of Multicomponent Cu-Based Nanoreactor for Tandem Electrocatalysis CO2 Reduction, Appl. Catal., B, 2022, 314, 11 CrossRef.
- X. Yang, Q.-X. Li, S.-Y. Chi, H.-F. Li, Y.-B. Huang and R. Cao, Hydrophobic Perfluoroalkane Modified Metal-Organic Frameworks for the Enhanced Electrocatalytic Reduction of CO2, SmartMat., 2022, 3, 163–172 CrossRef CAS.
- M.-S. Yang, J.-Q. Sun, Y.-J. Qin, H. Yang, S.-S. Zhang, X.-J. Liu and J. Luo, Hollow Cofe-Layered Double Hydroxide Polyhedrons for Highly Efficient CO2 Electrolysis, Sci. China Mater., 2022, 65, 536–542 CrossRef CAS.
- Q.-Y. Wang, K. Chen, S.-H. Wang, D.-H. Jiang, C.-Q. Ma, L.-X. Zhu and X.-L. Xu, An in situ-fabricated p-Co3O4@N–ZnO surface heterojunction photocatalyst for solar-to-fuel conversion of CO2, Mater. Chem. Front., 2023, 7, 523–534 RSC.
- X.-Q. Wei, Z.-J. Li, H. Jang, M.-G. Kim, Q. Qin and X. Liu, Lattice Strain and Interfacial Engineering of a Bi-Based Electrocatalyst for Highly Selective CO2 Electroreduction to Formate, Sci. China Mater., 2023, 66, 1398–1406 CrossRef CAS.
- D. Wang, Z.-N. Chen, Q.-R. Ding, C.-C. Feng, S.-T. Wang, W. Zhuang and L. Zhang, Rational Preparation of Atomically Precise Non-Alkyl Tin-Oxo Clusters with Theoretical to Experimental Insights into Electrocatalytic CO2 Reduction Applications, CCS Chem., 2021, 3, 250–259 Search PubMed.
- F.-L. Ren, W.-J. Hu, C. Wang, P. Wang, W. Li, C.-P. Wu, Y.-F. Yao, W.-J. Luo and Z.-G. Zou, An Extrinsic faradaic Layer on CuSn for High-Performance Electrocatalytic CO2 Reduction, CCS Chem., 2022, 4, 1610–1618 CrossRef CAS.
- J. Liang, Y.-Q. Xie, X.-S. Wang, Q. Wang, T.-T. Liu, Y.-B. Huang and R. Cao, An Imidazolium-Functionalized Mesoporous Cationic Metal-Organic Framework for Cooperative CO2 Fixation into Cyclic Carbonate, Chem. Commun., 2018, 54, 342–345 RSC.
- X. Liang, Y. Tian, Y. Yuan and Y. Kim, Ionic Covalent Organic Frameworks for Energy Devices, Adv. Mater., 2021, 33, e2105647 CrossRef PubMed.
- L.-L. Ling, L. Jiao, X.-S. Liu, Y. Dong, W.-J. Yang, H.-J. Zhang, B.-J. Ye, J. Chen and H.-L. Jiang, Potassium-Assisted Fabrication of Intrinsic Defects in Porous Carbons for Electrocatalytic CO2 Reduction, Adv. Mater., 2022, 34, 2205933 CrossRef CAS PubMed.
- Z. Chen, K. Wang, Y.-M. Tang, L. Li, X. Hu, M.-X. Han, Z.-Y. Guo, H.-B. Zhan and B.-L. Chen, Reticular Synthesis of One-Dimensional Covalent Organic Frameworks with 4-C Sql Topology for Enhanced Fluorescence Emission, Angew. Chem., Int. Ed., 2023, 62, e202213268 CAS.
- Y.-R. Wang, H.-M. Ding, S.-N. Sun, J.-W. Shi, Y.-L. Yang, Q. Li, Y. Chen, S.-L. Li and Y.-Q. Lan, Light, Heat and Electricity Integrated Energy Conversion System: Photothermal-Assisted Co-Electrolysis of CO2 and Methanol, Angew. Chem., Int. Ed., 2022, 61, e202212162 CAS.
- X. Yang, J.-Q. Yi, T. Wang, Y.-N. Feng, J.-W. Wang, J. Yu, F.-L. Zhang, Z. Jiang, Z.-S. Lv, H.-C. Li, T. Huang, D.-H. Si, X. Wang, R. Cao and X.-D. Chen, Wet-Adhesive on-Skin Sensors Based on Metal–Organic Frameworks for Wireless Monitoring of Metabolites in Sweat, Adv. Mater., 2022, 34, 2201768 CrossRef CAS PubMed.
- Y.-Z. Wang, T. Sun, H. B. Mostaghimi Amir, J. Goncalves Tiago, Z.-Z. Liang, Y.-Y. Zhou, W. Zhang, Z.-H. Huang, Y.-H. Ma and R. Cao, Two-Dimensional Metal-Organic Frameworks with Unique Oriented Layers for Oxygen Reduction Reaction: Tailoring the Activity through Exposed Crystal Facets, CCS Chem., 2022, 4, 1633–1642 CrossRef CAS.
- S.-Y. Chi, Q. Chen, S.-S. Zhao, D.-H. Si, Q.-J. Wu, Y.-B. Huang and R. Cao, Three-Dimensional Porphyrinic Covalent Organic Frameworks for Highly Efficient Electroreduction of Carbon Dioxide, J. Mater. Chem. A, 2022, 10, 4653–4659 RSC.
- S. Lin, C.-S. Diercks, Y.-B. Zhang, N. Kornienko, E.-M. Nichols, Y.-B. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C.-J. Chang, Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- A. Mal, S. Vijayakumar, R. K. Mishra, J. Jacob, R. S. Pillai, B. S. Dileep Kumar and A. Ajayaghosh., Supramolecular Surface Charge Regulation in Ionic Covalent Organic Nanosheets: Reversible Exfoliation and Controlled Bacterial Growth, Angew. Chem., Int. Ed., 2019, 59, 8713–8719 CrossRef PubMed.
- Q.-J. Wu, D.-H. Si, Q. Wu, Y.-L. Dong, R. Cao and Y.-B. Huang, Boosting Electroreduction of CO2 over Cationic Covalent Organic Frameworks: Hydrogen Bonding Effects of Halogen Ions, Angew. Chem., Int. Ed., 2023, 62, e202215687 CAS.
- Q. Wu, R.-K. Xie, M.-J. Mao, G.-L. Chai, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, Integration of Strong Electron Transporter Tetrathiafulvalene into Metalloporphyrin-Based Covalent Organic Framework for Highly Efficient Electroreduction of CO2, ACS Energy Lett., 2020, 5, 1005–1012 CrossRef CAS.
- Y. Li, J.-F. Sui, L.-S. Cui and H.-L. Jiang, Hydrogen Bonding Regulated Flexibility and Disorder in Hydrazone-Linked Covalent Organic Frameworks, J. Am. Chem. Soc., 2023, 145, 1359–1366 CrossRef CAS PubMed.
- S. Dou, J. Song, S. Xi, Y. Du, J. Wang, Z.-F. Huang, Z. J. Xu and X. Wang, Boosting Electrochemical CO2 Reduction on Metal–Organic Frameworks via Ligand Doping, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 CrossRef CAS PubMed.
- H. Xiao, H. Zhu, W. Weng, K. Li, W. Li and W. Xiao, Electrochemical fixation of CO2 over a Mo plate to prepare a Mo2C film for electrocatalytic hydrogen evolution, Mater. Chem. Front., 2021, 5, 4963–4969 RSC.
- F. Lü, H. Bao, F. He, G. Qi, J. Sun, S. Zhang, L. Zhuo, H. Yang, G. Hu, J. Luo and X. Liu, Nitrogen dopant induced highly selective CO2 reduction over lotus-leaf shaped ZnO nanorods, Mater. Chem. Front., 2021, 5, 4225–4230 RSC.
- W.-Q. Li, Z.-Y. Zhao, W.-B. Hu, Q.-Q. Cheng, L.-J. Yang, Z. Hu, Y. A. Liu, K. Wen and H. Yang, Design of Thiazolo[5,4-D]Thiazole-Bridged Ionic Covalent Organic Polymer for Highly Selective Oxygen Reduction to H2O2, Chem. Mater., 2020, 32, 8553–8560 CrossRef CAS.
- J. Szczesny, A. Ruff, A. R. Oliveira, M. Pita, I. A. C. Pereira, A. L. De Lacey and W. Schuhmann., Electroenzymatic CO2 Fixation Using Redox Polymer/Enzyme-Modified Gas Diffusion Electrodes, ACS Energy Lett., 2020, 5, 321–327 CrossRef CAS.
- Y. Qiao, W.-C. Lai, K. Huang, T.-T. Yu, Q.-Y. Wang, L. Gao, Z.-L. Yang, Z.-S. Ma, T.-L. Sun, M. Liu, C. Lian and H.-W. Huang, Engineering the Local Microenvironment over Bi Nanosheets for Highly Selective Electrocatalytic Conversion of CO2 to HCOOH in Strong Acid, ACS Catal., 2022, 12, 2357–2364 CrossRef CAS.
- J. E. Huang, F.-W. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S.-J. Liu, S.-Z. Zhang, M.-C. Luo, X. Wang, Y.-W. Lum, Y. Xu, K. Bertens, R.-K. Miao, C.-T. Dinh, D. Sinton and E. H. Sargent, CO2 Electrolysis to Multicarbon Products in Strong Acid, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, Competition between CO2 Reduction and Hydrogen Evolution on a Gold Electrode under Well-Defined Mass Transport Conditions, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- J. Lee, J. Lim, C.-W. Roh, H. S. Whang and H. Lee, Electrochemical CO2 Reduction Using Alkaline Membrane Electrode Assembly on Various Metal Electrodes, J. CO2 Util., 2019, 31, 244–250 CrossRef CAS.
- H.-F. Li, T.-F. Liu, P.-F. Wei, L. Lin, D.-F. Gao, G.-X. Wang and X. Bao, High-Rate CO2 Electroreduction to C2+ Products over a Copper-Copper Iodide Catalyst, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- J. Li, Z.-Y. Wang, C. McCallum, Y. Xu, F.-W. Li, Y.-H. Wang, C. M. Gabardo, D. Cao-Thang, T.-T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Constraining Co Coverage on Copper Promotes High-Efficiency Ethylene Electroproduction, Nat. Catal., 2019, 2, 1124–1131 CrossRef CAS.
- R.-R. Li, F. Liu, Y.-H. Zhang, M.-M. Guo and D. Liu, Nitrogen, Sulfur Co-Doped Hierarchically Porous Carbon as a Metal-Free Electrocatalyst for Oxygen Reduction and Carbon Dioxide Reduction Reaction, ACS Appl. Mater. Interfaces, 2020, 12, 44578–44587 CrossRef CAS PubMed.
- T. Moeller, W. Ju, A. Bagger, X.-L. Wang, F. Luo, T. Trung Ngo, A. S. Varela, J. Rossmeisl and P. Strasser, Efficient CO2 to CO Electrolysis on Solid Ni-N-C Catalysts at Industrial Current Densities, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- M. C. O. Monteiro, F. Dattila, N. Lopez and M. T. M. Koper, The Role of Cation Acidity on the Competition between Hydrogen Evolution and CO2 Reduction on Gold Electrodes, J. Am. Chem. Soc., 2022, 144, 1589–1602 CrossRef CAS PubMed.
- C. P. O'Brien, R.-K. Miao, S.-J. Liu, Y. Xu, G. Lee, A. Robb, J. E. Huang, K. Xie, K. Bertens, C. M. Gabardo, J. P. Edwards, C.-T. Dinh, E. H. Sargent and D. Sinton, Single Pass CO2 Conversion Exceeding 85% in the Electrosynthesis of Multicarbon Products Via Local CO2 Regeneration, ACS Energy Lett., 2021, 6, 2952–2959 CrossRef.
- Y. Wang, M.-Y. Wang, Z.-S. Zhang, Q. Wang, Z. Jiang, M. Lucero, X. Zhang, X.-X. Li, M. Gu, Z.-X. Feng and Y.-Y. Liang, Phthalocyanine Precursors to Construct Atomically Dispersed Iron Electrocatalysts, ACS Catal., 2019, 9, 6252–6261 CrossRef CAS.
- Z.-L. Yin, H.-Q. Peng, X. Wei, H. Zhou, J. Gong, M.-M. Huai, L. Xiao, G.-W. Wang, J.-T. Lu and L. Zhuang, An Alkaline Polymer Electrolyte CO2 Electrolyzer Operated with Pure Water, Energy Environ. Sci., 2019, 12, 8 Search PubMed.
- W. Shin, S. Lee, J. Shin, S. Lee and Y. Kim, Highly Selective Electrocatalytic Conversion of CO2 to CO at −0.57 V (NHE) by Carbon Monoxide Dehydrogenase from Moorella Thermoacetica, J. Am. Chem. Soc., 2003, 125, 14688–14689 CrossRef CAS PubMed.
- B. C. De Simone, T. Marino and N. Russo, TDDFT Investigation on Methylviologen, 3,7-Diazabenzophosphole, and Helical Helquat Electrochromic Systems, Theor. Chem. Acc., 2016, 135, 9 Search PubMed.
- J.-J. Ding, C.-N. Zheng, L.-X. Wang, C.-B. Lu, B. Zhang, Y. Chen, M.-Q. Li, G.-Q. Zhai and X.-D. Zhuang, Viologen-Inspired Functional Materials: Synthetic Strategies and Applications, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- Z. Wang, X. Jia, P. Zhang, Y. Liu, H. Qi, P. Zhang, U. Kaiser, S. Reineke, R. Dong and X. Feng, Viologen-Immobilized 2D Polymer Film Enabling Highly Efficient Electrochromic Device for Solar-Powered Smart Window, Adv. Mater., 2021, 34, 9 Search PubMed.
- V. Fourmond and C. Leger, Dinitrogen Reduction: Interfacing the Enzyme Nitrogenase with Electrodes, Angew. Chem., Int. Ed., 2017, 56, 4388–4390 CrossRef CAS PubMed.
- H. Noh, C.-W. Kung, K.-I. Otake, A. W. Peters, Z.-Y. Li, Y.-J. Liao, X.-Y. Gong, O. K. Farha and J. T. Hupp., Redox-Mediator-Assisted Electrocatalytic Hydrogen Evolution from Water by a Molybdenum Sulfide-Functionalized Metal-Organic Framework, ACS Catal., 2018, 8, 9848–9858 CrossRef CAS.
- L. Chen, C. Lin and R. G. Compton, Single Entity Electrocatalysis: Oxygen Reduction Mediated Via Methyl Viologen Doped Nafion Nanoparticles, Phys. Chem. Chem. Phys., 2018, 20, 15795–15806 RSC.
- T. Skorjanc, D. Shetty, F. Gandara, L. Ali, J. Raya, G. Das, M. A. Olson and A. Trabolsi, Remarkably Efficient Removal of Toxic Bromate from Drinking Water with a Porphyrin–Viologen Covalent Organic Framework, Chem. Sci., 2020, 11, 845–850 RSC.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gandara, M. Lusi, D. S. Shankar Rao, S. Vimala, S. Krishna Prasad, J. Raya, D. S. Han, R. Jagannathan, J. C. Olsen and A. Trabolsi, Viologen-Based Conjugated Covalent Organic Networks Via Zincke Reaction, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- L. Liu, Q. Liu, R. Li, M.-S. Wang and G.-C. Guo, Controlled Photoinduced Generation of “Visual” Partially and Fully Charge Separated States in Viologen Analogues, J. Am. Chem. Soc., 2021, 143, 2232–2238 CrossRef CAS PubMed.
- G. A. Leith, C. R. Martin, A. Mathur, P. Kittikhunnatham, K. C. Park and N. B. Shustova, Dynamically Controlled Electronic Behavior of Stimuli-Responsive Materials: Exploring Dimensionality and Connectivity, Adv. Energy Mater., 2021, 12, 23 Search PubMed.
- T. Komura, T. Yamaguchi, K. Furuta and K. Sirono, Irreversible Transformation of Polypyrrole-Bound Viologen with Two-Electron Reduction in Acidic Aqueous Solutions, J. Electroanal. Chem., 2002, 534, 123–130 CrossRef CAS.
- X. Li, S. Xi, L. Sun, S. Dou, Z. Huang, T. Su and X. Wang, Isolated FeN4 Sites for Efficient Electrocatalytic CO2 Reduction, Adv. Sci., 2020, 7, 2001545 CrossRef CAS PubMed.
- L. Sun, Z. Huang, V. Reddu, T. Su, A. C. Fisher and X. Wang, A Planar, Conjugated N4-Macrocyclic Cobalt Complex for Heterogeneous Electrocatalytic CO2 Reduction with High Activity, Angew. Chem., Int. Ed., 2020, 59, 17104–17109 CrossRef CAS PubMed.
- J. Wang, X. Huang, S. Xi, J.-M. Lee, C. Wang, Y. Du and X. Wang, Linkage Effect in the Heterogenization of Cobalt Complexes by Doped Graphene for Electrocatalytic CO2 Reduction, Angew. Chem., Int. Ed., 2019, 58, 13532–13539 CrossRef CAS PubMed.
- N. Li, D.-H. Si, Q.-J. Wu, Q. Wu, Y.-B. Huang and R. Cao, Boosting Electrocatalytic CO2 Reduction with Conjugated Bimetallic Co/Zn Polyphthalocyanine Frameworks, CCS Chem., 2022, 1–14 Search PubMed.
- T. Skorjanc, D. Shetty, F. Gándara, L. Ali, J. Raya, G. Das, M. A. Olson and A. Trabolsi, Remarkably Efficient Removal of Toxic Bromate from Drinking Water with a Porphyrin-Viologen Covalent Organic Framework, Chem. Sci., 2020, 11, 845–850 RSC.
- C. Liu, X.-D. Zhang, J.-M. Huang, M.-X. Guan, M. Xu and Z.-Y. Gu, In Situ Reconstruction of Cu–N Coordinated MOFs to Generate Dispersive Cu/Cu2O Nanoclusters for Selective Electroreduction of CO2 to C2H4, ACS Catal., 2022, 12, 15230–15240 CrossRef CAS.
- Q.-X. Li, D.-H. Si, W. Lin, Y.-B. Wang, H.-J. Zhu, Y.-B. Huang and R. Cao, Highly Efficient Electroreduction of CO2 by Defect Single-Atomic Ni–N-3 Sites Anchored on Ordered Micro-Macroporous Carbons, Sci. China: Chem., 2022, 65, 1584–1593 CrossRef CAS.
- J.-D. Yi, D.-H. Si, R. Xie, Q. Yin, M.-D. Zhang, Q. Wu, G.-L. Chai, Y.-B. Huang and R. Cao, Conductive Two-Dimensional Phthalocyanine-Based Metal–Organic Framework Nanosheets for Efficient Electroreduction of CO2, Angew. Chem., Int. Ed., 2021, 60, 17108–17114 CrossRef CAS PubMed.
- L. Chen, H. Willcock, C.-J. Wedge, F. Hartl, H.-M. Colquhoun and B.-W. Greenland, Efficient Access to Conjugated 4,4′-Bipyridinium Oligomers Using the Zincke Reaction: Synthesis, Spectroscopic and Electrochemical Properties, Org. Biomol. Chem., 2016, 14, 980–988 RSC.
- K. Qian, X. Guan, N. Sun and H.-L. Jiang, Precise Fabrication of Ternary Ordered Covalent Organic Frameworks for Photocatalysis, Sci. China: Chem., 2023, 66, 436–442 CrossRef CAS.
- N. Keller, M. Calik, D. Sharapa, H. R. Soni, P. M. Zehetmaier, S. Rager, F. Auras, A. C. Jakowetz, A. Görling, T. Clark and T. Bein, Enforcing Extended Porphyrin J-Aggregate Stacking in Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140, 16544–16552 CrossRef CAS PubMed.
- L. Sun, S. S. Park, D. Sheberla and M. Dincă, Measuring and Reporting Electrical Conductivity in Metal–Organic Frameworks: Cd2(TTFTB) as a Case Study, J. Am. Chem. Soc., 2016, 138, 14772–14782 CrossRef CAS PubMed.
- H. Zhao, H. Cao, Z. Zhang and Y.-G. Wang, Modeling the Potential-Dependent Kinetics of CO2 Electroreduction on Single-Nickel Atom Catalysts with Explicit Solvation, ACS Catal., 2022, 12, 11380–11390 CrossRef CAS.
- Y. Wu, Q. Chen, J. Zhu, K. Zheng, M. Wu, M. Fan, W. Yan, J. Hu, J. Zhu, Y. Pan, X. Jiao, Y. Sun and Y. Xie, Selective CO2-to-C2H4 Photoconversion Enabled by Oxygen-Mediated Triatomic Sites in Partially Oxidized Bimetallic Sulfide, Angew. Chem., Int. Ed., 2023, 62, e202301075 CrossRef CAS PubMed.
- Y. Li, J. Sui, L.-S. Cui and H.-L. Jiang, Hydrogen Bonding Regulated Flexibility and Disorder in Hydrazone-Linked Covalent Organic Frameworks, J. Am. Chem. Soc., 2023, 145, 1359–1366 CrossRef CAS PubMed.
- B. Sheng, D. Cao, H. Shou, W. Xu, C. Wu, P. Zhang, C. Liu, Y. Xia, X. Wu, S. Chu, J. Zhang, L. Song and S. Chen, Anomalous Ru Dissolution Enabling Efficient Integrated CO2 Electroreduction in Strong Acid, Chem. Eng. J., 2023, 454, 9 Search PubMed.
- Z. Jiang, Z. S. Zhang, H. Li, Y. R. Tang, Y. B. Yuan, J. Zao, H. Z. Zheng and Y. Y. Liang, Molecular Catalyst with near 100% Selectivity for CO2 Reduction in Acidic Electrolytes, Adv. Energy Mater., 2023, 13, 2203603 CrossRef CAS.
- J.-C. Hu, J. Wu, B.-Y. Deng and F. Wang, An assembled ternary photocatalyst CoPh/CdSe@TiO2 for simultaneous photocatalytic CO2 and proton reduction, Mater. Chem. Front., 2023, 7, 514–522 RSC.
- Y. Li, M. Karimi, Y. N. Gong, N. Dai, V. Safarifard and H. L. Jiang, Integration of Metal-Organic Frameworks and Covalent Organic Frameworks: Design, Synthesis, and Applications, Matter, 2021, 4, 2230–2265 CrossRef CAS.
- Y. Duan, X.-L. Zhang, F.-Y. Gao, Y. Kong, Y. Duan, X.-T. Yang, X.-X. Yu, Y.-R. Wang, S. Qin, Z. Chen, R. Wu, P.-P. Yang, X.-S. Zheng, J.-F. Zhu, M.-R. Gao, T.-B. Lu, Z.-Y. Yu and S.-H. Yu, Interfacial Engineering of Ni/V2O3 Heterostructure Catalyst for Boosting Hydrogen Oxidation Reaction in Alkaline Electrolytes, Angew. Chem., Int. Ed., 2023, 62, e202217275 CAS.
- M.-D. Zhang, J.-D. Yi, Y.-B. Huang and R. Cao, Covalent Triazine Frameworks-Derived N,P Dual-Doped Porous Carbons for Highly Efficient Electrochemical Reduction of CO2, Chin. J. Struct. Chem., 2021, 40, 1213–1222 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qm00218g |
| This journal is © the Partner Organisations 2023 |