
Reagentless protein-based electrochemical biosensors
Saimon M.
Silva
 *acde,
Miaosi
Li
a,
Alexandre Xavier
Mendes
bc and
Simon E.
Moulton
bcd
*acde,
Miaosi
Li
a,
Alexandre Xavier
Mendes
bc and
Simon E.
Moulton
bcd
aUniversal Biosensors, Rowville, Victoria 3178, Australia. E-mail: smoraessilva@swin.edu.au
bARC Centre of Excellence for Electromaterials Science, School of Science, Computing and Engineering Technologies, Swinburne University of Technology, Melbourne, Victoria 3122, Australia
cAikenhead Centre for Medical Discovery, St Vincent's Hospital Melbourne, Melbourne, Victoria 3065, Australia
dIverson Health Innovation Research Institute, Swinburne University of Technology, Melbourne, Victoria 3122, Australia
eDepartment of Chemistry and Physics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Victoria 3086, Australia
First published on 13th April 2023
Abstract
The creation of reagentless protein-based biosensors that are capable of monitoring molecular analytes directly in bodily fluids could revolutionize our understanding of biology and personalized health monitoring. The limited number of molecular sensors that are currently available in the market depends on the specific enzymatic or chemical reactivity of their target analytes and therefore are not applicable to many relevant biomarkers. Aiming to overcome this limited molecular sensing generality, a new class of reagentless protein-based electrochemical sensors has been introduced for the direct measurements of biomarkers in unprocessed biological fluids. This mini-review will discuss the most recent cutting-edge discoveries for the development of electroanalytical modular biosensors, where all the sensors’ components are integrated into a self-sufficient sensor allowing hence its autonomous functionality.
Introduction
For years the detection of clinically relevant proteins and other biomarkers has been attained in point-of-care settings mostly using fluorescence anisotropy (also often called fluorescence polarization).1–5 In fluorescence polarization, the occurrence of a specific protein–protein complex is measured through binding-induced variations in the tumbling of a surface-attached fluorophore.6 Such a technique is one of the most suitable methodologies for quantifying the levels of specific proteins in clinically relevant samples, as it does not necessitate washing steps to eliminate unbound reagents.7 However, the effectiveness of fluorescence polarization in the point-of-care is significantly hindered due to its numerous limitations. First, it requires considerably large sample volumes, demanding hence venous blood draws, which limits its direct application in the point-of-care. Second, when challenged with clinically complex fluids (e.g. unprocessed blood), it requires substantial signalling processing for background correction as it displays a modest signal gain.8,9 Typically, the intensity difference between the two polarizations (bound and unbound states) for an antibody–antigen complex is in the order of ∼15%, which needs to be measured against background polarizations of comparable magnitudes.10 Thirdly, fluorescence polarization cannot be straightforwardly applied for multiplex diagnostics.Therefore, this challenge of how to detect target biomolecules dynamically in unprocessed bodily fluids using self-generating signal biosensors remained unsolved until recently. Thus, having a new technology with the capability to monitor protein biomarkers in vivo or in point-of-care settings would create a powerful tool for disease diagnostics, monitoring of disease progression, and monitoring of treatment efficacies.11–13 A crucial prerequisite for this category of sensing applications is a reagentless assay format where all necessary components are integrated into a self-contained sensor to permit independent function. Recently, aiming to overcome these above-mentioned challenges, an innovative sensing concept has emerged in the electroanalytical field, the so-called reagentless, protein-based electrochemical biosensors. This new class of sensors is the electrochemical analogy of fluorescence polarization, and it can be applied to detect proteins, peptides, and other relevant biomarkers.14,15 The sensor surface chemistry comprises three main components: (1) a protein or antibody as the recognition element that specifically binds to the target analytes, (2) a flexible linker (i.e. short polypeptides, DNA strands, or even proteins), and (3) a redox reporter engineered at a specific location on the surface chemistry (e.g. within the recognition element or linker) (Fig. 1).15
 | ||
| Fig. 1 Scheme illustrating the three main components of protein-based electrochemical biosensors: the recognition elements engineered within a redox reporter and attached to an electrode surface using a flexible linker. | ||
In a general manner, all the reagentless protein-based electrochemical sensors that will be described here work based on the alteration of the efficiency with which the redox reporter approaches the electrode surface upon binding the recognition element with the target analyte (Fig. 2). This alteration leads to changes in redox current that can be directly measured using electrochemical voltammetric techniques. Despite the common working principle of these sensors relying on alterations in the efficiency of redox reporter communication with the electrode surface, the mechanisms causing these changes are particular to each sensor embodiment, and it will be discussed in detail in this review.
 | ||
| Fig. 2 Scheme illustrating how binding of the recognition element to the target analyte leads to protein conformational changes and consequently alters the relative position of redox reporter to the electrode surface. This alteration in redox reporter position, in this case moving far away from the electrode surface, results in a measurable voltammetric current which is proportional to target analyte concentration. | ||
The reagentless molecular assays described here were inspired by the extraordinary success of the electrochemical aptamer-based (EAB) sensors. EAB sensors employ nucleic acid aptamer as recognition elements, which can reversibly and selectively bind to molecular targets even in complex biological fluids such as whole blood.16 In this sensing design, usually, nucleic acid aptamers are functionalized with redox reporters and covalently attached to electrode surfaces via an alkyl thiol self-assembled monolayer.17–19 EAB sensors presented many advantages, including modularity to allow the detection of any arbitrary target molecules for which aptamers are available. The successful use of EAB sensors for in vivo monitoring has already been reported for a dozen of molecular targets, and such technology is one step closer to translation to biomedical and clinical research, decentralized diagnostics, and other medical applications.20–22 The progress of the EAB sensors field has been discussed in many other excellent review articles,21,23–26 and we direct the reader for further information.
This review provides an overview of reagentless protein-based electrochemical sensors research. The first part of the review will concentrate on the sensors that are based on response signals arising directly from protein recognition elements that undergo conformational changes upon binding to target species. Then it will cover strategies where the recognition element does not change conformation, but the signal transduction mechanism is based on the incorporation of a flexible linker (i.e. DNA duplexes). The final part of the review sheds insight into future directions toward translation and clinical applications of such sensors.
Biomolecular sensing: mimicking tricks from nature
The versatileness, specificity, and high affinity of biomolecular recognition have inspired decades of research intended to adapt biomolecules into a general platform for molecular sensing. Lessons learnt from nature's remarkable capability of real-time molecular sensing in highly complex environments, for example, proteins or nucleic acids that specifically bind to their complementary target biomolecules, have consequently assisted in the development of enhanced sensing technologies. In particular, proteins present impressive engineering features such as three-dimensional shapes, specific binding affinities, and when they encounter their target biomolecules, they bind together, leading to precise binding-induced changes in conformation or oligomerization state.27 These conformational changes, sequentially, generate specific output signals, including the activation of an enzyme or the opening of an ion channel.28Thus, proteins or nucleic acids that reversibly adopt different conformations in response to the binding event of a specific target biomolecule can be called biomolecular switches.28 Numerous characteristics make such switches very appropriate for engineering new innovative biosensors. Firstly, binding-specific conformational changes create a reliable method of transducing binding events into output signals which are not effortlessly affected by non-specific binding of non-target biomolecules. Particularly, because proteins structure alterations are only instigated by the formation of various weak, non-covalent bonds (e.g. hydrogen bonding, hydrophobic effect, and van der Waals forces), and it is normally very specific to a given target biomolecule and therefore unsusceptible to the presence of other non-target species which are found in abundance in complex biological environments such as bodily fluids.28 Secondly, the protein and nucleic acid conformation changes are quick, and so is the signal transduction, they are reversible and reagentless, making these nanoscale switches suitable for application in continuous, real-time detection of target biomolecules even inside living bodies. Thirdly, due to the versatility of biomolecular switches, they can be engineered and used with electrochemical and optical methods. Lastly, the conformational equilibria of biomolecular switches are associated with both switch's underlying thermodynamics and target concentration. Therefore protein conformational based biosensors can be quantitative and offer the possibility of rationally optimizing the dynamic ranges without impacting proteins’ binding specificity.28 Here, we review the successful examples of reagentless and protein-based electrochemical biosensors, giving special focus on the sensor configuration, chosen recognition element, sensing mechanism, electrochemical performance, and we will discuss the possible pathways for translation of such technologies.
Protein sensors that rely directly on protein's conformational changes
The first example of an electrochemical sensor that used a protein as a recognition element as well as its folding switching to generate electrochemical output signals, was reported by Kevin Plaxco and co-workers.15 In this first sensor design, the authors used a bacterial chemotaxis protein (CheY) as the recognition element. CheY presents two binding target analytes, the P2 domain of the chemotaxis kinase (CheA-P) and the 16-residue target region of the flagellar switch protein (FliM).15 The reason why the authors chose CheY as a first proof-of-concept for the reagentless protein-based sensors is that the structure of CheY and its folding behaviors upon binding to the targets FliM and CheA have been explored extensively in the literature, and hence it could be used a good model system.15 To create the functional protein capable of generating electrochemical signals, the authors produced a pool of CheY variants containing a carboxy-terminal hexa-His-tag, where each variant presented a single cysteine amino acid. The cysteine group was then used for conjugation with methylene blue maleimide derivative (the redox reporter) through thiol chemistry. Copper complexation with the His-tag was employed to tether the CheY recognition element to the gold electrode surface. The gold electrode also contained an alkanethiol self-assembled monolayer (Fig. 3a).15 When the CheY's binding targets are absent, and the sensor is interrogated using voltammetry, the methylene blue redox reporter is fairly free to collide with the electrode surface, generating hence a large faradaic current at the redox potential expected for methylene blue redox reaction. The faradaic current is then reduced when the sensor is exposed to a sample solution containing either of its binding targets (Fig. 3b and c). With this new sensing design, it was possible to detect the target analytes in concentrations as low as micromolar range, the sensor also responded well when challenged in 20% human blood serum.15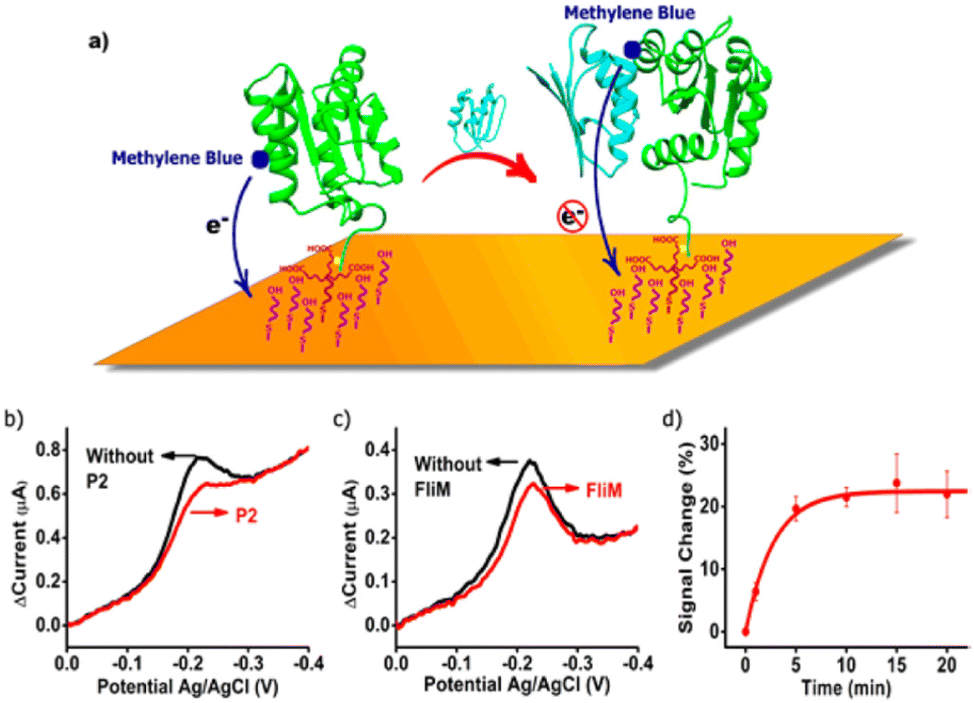 | ||
| Fig. 3 (a) Figure showing the first reagentless protein-based electrochemical biosensor. Electrochemical response occurs when a target protein binds to the surface tethered protein recognition element, reducing the efficacy with which the attached reporter transfers electrons to the electrode surface when interrogated using a voltammetric technique. (b and c) Alteration in voltammetric response when the sensor was measured in a solution containing its binding partners (CheA-P2 or FliM). Figure reproduced from ref. 15, with permission from American Chemical Society, Copyright © 2017. | ||
The same research group further investigated this category of biosensors for the detection of SH3 domain-binding peptide (VSL12) in whole blood.29 Similarly to the example cited above, the authors coupled a methylene blue redox reporter to a SH3 domain from human Fyn kinase (FynSH3), the recognition element. The methylene blue-FynSH3 was anchored to a gold electrode surface using a 7-carbon alkyl-thiol linker on its amino-terminus, which was further bound to an electrode surface passivated with a mercapto-hexanol self-assembled monolayer.29 The constructed sensor presented a signal gain of approximately 30%, and a limit of the detection of 2.5 μM for VSL12 in a clean buffer solution. The sensor presented good selectivity for VSL12, where it showed practically no binding to p85α-2, a peptide that can also bind to FynSH3 but with low binding affinity. This same sensor was successfully challenged in unprocessed whole blood for continuous, real-time measurement of VSL12. However, in order to function directly in whole blood, the protein recognition element necessitated being further engineered.29 This was due to the FynSH3 protein stabilization in high ionic strength solutions and causing the sensor to be insensitive to the addition of target VSL12. To overcome this, the FynSH3 was engineered by introducing a I50L substitution in the hydrophobic core of the protein, which caused the folding equilibrium to be shifted back to the unfolded state.29
This state-of-art sensing approach described here creates many opportunities for diagnostic and clinical applications as it does not depend on the specific chemical reactivity of the target analyte. Instead, reagentless protein-based electrochemical biosensors accomplish detection by monitoring the binding-induced changes in electron transfer between a surface tethered, redox reporter modified protein recognition element and the electrode surface. This sensing mechanism is inspired by the behavior of naturally occurring chemoreceptors in the body. Beyond creating many opportunities for the analytical detection of important biomarkers, it is also a new methodology for studying the thermodynamic consequences of protein–surface interactions, which so far have been mostly attained using theoretical or computational studies.30 It is well known that proteins behave differently when interacting with artificial surfaces, where they often fold, adopting unexpected configurations.31–33 Where, when interacting with biological surfaces, proteins remain stable.32 Thus, this new experimental approach is also appropriate for investigating the long-standing unsolved questions around the unusual physicochemical behavior of proteins when interacting with artificial surfaces (e.g., bionic implants).30 This is quite important for allowing further development of protein-based sensors or other technologies that necessitate surfaces that do not alter protein structure and function.32
Cross-linked protein brush strategy
Next, we will describe a new reagentless protein-based biosensor that has been reported recently and which is aimed at the detection of cancer biomarkers directly in human blood. The difference between this new approach and the direct protein-folding biosensor is that instead of attaching the protein recognition element to the electrode surface using alkanethiol chemistry, it uses a second recombinant protein as a surface linker as well as a multifunctional component. The limitation of using alkanethiol chemistry is that only noble metal electrodes (e.g. gold) can be used as electrode substrate to design the sensing interface. This new reagentless protein-based biosensor comprised a peanut agglutinin (PNA) functionalized with a methylene blue redox reporter, which was further attached to the electrode surface via a glycoprotein called lubricin (LUB), the surface linker in this case.34 This is facilitated by the natural binding affinities of LUB and PNA.34 PNA is a plant-based lectin, which possesses a highly specific carbohydrate binding site that permits interactions with its carbohydrate-binding targets, cancer T and Tn antigens.35,36 T and Tn antigens are cancer biomarkers that are expressed at the surface of cancer cells and not expressed by healthy cells.35The self-assembly of LUB onto different electrode materials (e.g. carbon-based substrates, metal surfaces and conducting polymers) and its excellent performance as an antifouling coating for biosensors and bioelectronics applications have been extensively investigated by our research group.37–44 LUB is a flexible protein, which when fully extended, displays a length of roughly 200 nm, and it basically comprises three domains; the central mucin domain and two end domains. The mucin domain presents a high glycosylation degree containing polar galactose (encompassing ≈33% of the glycans) and negatively charged sialic acid (encompassing ≈66% of the overall glycans).40,44 Attached to each side of the middle mucin domain are two globular end-domains, which contain sub-domains similar to two globular proteins, hemopexin and somatomedin B. The end-domains of LUB are very adhesive, and therefore it provides LUB with the capability to self-assemble on different surfaces.34 When self-assembled onto a surface, LUB adopts a telechelic brush configuration. This brush configuration displays two main characteristics that are very attractive for electrochemical sensing applications. First, when self-assembled on a surface, LUB is fully extended even at low surface graft density (≈9 nm between end domains).45 Second, the LUB brush presents a very diffuse nature (>95% water), creating hence a flexible brush, which is required for electrochemical sensors that are based on the principle of electronic transfer between surface tethered redox reporters and the electrode surface.34,45
Therefore in this reagentless protein-based sensing architecture, LUB present multi-functionalities: it anchors the methylene blue-PNA to the electrode surface (referred to as LUB–PNA interface), it works as an antifouling agent, and it also creates a dynamic harmonic motion which is central to the sensing transduction mechanism.34Fig. 4 shows the LUB–PNA sensing mechanism, where in the absence of the Tn antigen target, the LUB brush is cross-linked with the PNA. This cross-linked brush structure is elastic in nature, creating a dynamic fluctuation motion in which the methylene blue collides with the electrode surface at a given rate. In this way, a high faradaic current can be measured at the redox potential expected for methylene blue using voltammetric techniques. When the LUB–PNA sensor is exposed to a solution containing the target antigens, it breaks the LUB–PNA cross-links as the antigens binds to the PNA. This break in the cross-linked LUB–PNA brush leads to isolated LUB molecules, and small island clusters of coupled LUB become severed and sequestered from the larger, cross-linked network. This causes the dynamic motion of the brush to fluctuate at an altered rate, which consequently diminishes the efficiency with which the attached redox reporter, methylene blue, transfers electrons to the electrode surface. Resulting hence in a reduced measured current signal.34
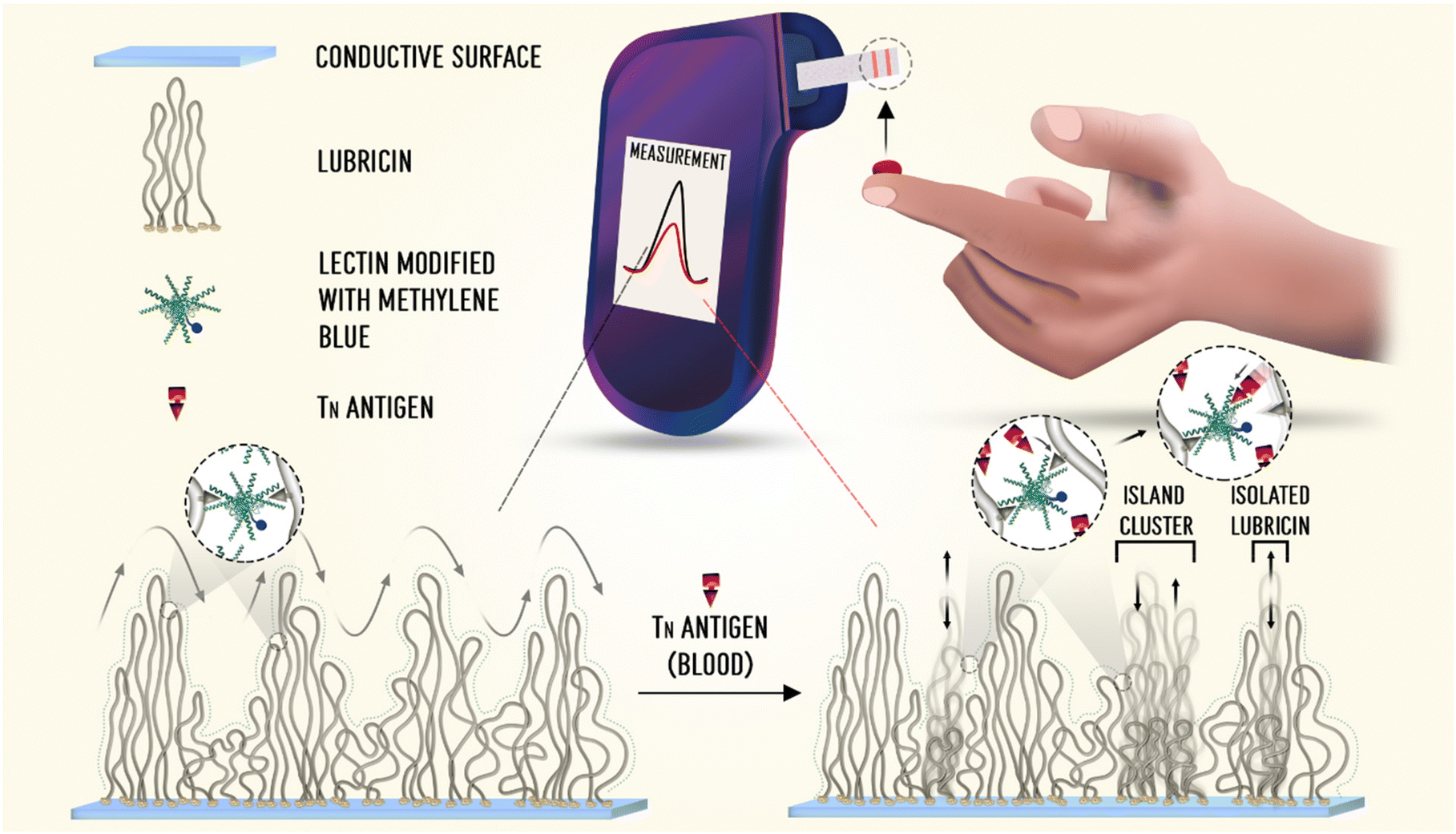 | ||
| Fig. 4 Schematic illustration of the LUB–PNA sensor for the detection of Tn antigen in whole blood. Figure reproduced from ref. 36, with permission from American Chemical Society, Copyright © 2022. | ||
The LUB–PNA sensor presented a great analytical performance for the detection of Tn antigen. It was capable of detecting Tn antigen directly in whole blood in concentrations as low as 54 pM.34 The versatility of this sensor was tested by producing the surface chemistry on different substrates, carbon screen-printed and gold electrodes. The LUB–PNA sensor response was also tested in retrospective clinical samples, where the sensor could differentiate the response arising from plasma samples collected from healthy donors from response measured in plasma samples from patients with breast, colorectal, and prostate cancer.34 This new reagentless, protein-based sensor offered an easy, fast, and precise means of creating an ultrasensitive point-of-care diagnostic tool for the detection of tumour associated antigens. The time for sample analysis can be as quick as a few minutes, it necessitates low-cost supporting electronics, and it can be produced on a large scale, using similar manufacturing technologies to those used for the production of glucose meter test strips. The challenges faced with this sensor configuration for future translation and commercialization will be the essential improvement of the lectin recognition element's selectivity for specific cancer biomarkers and the requirement of sensor multiplexing capability (detection of multiple target analytes simultaneously) for an effective diagnostic tool. Next, we will discuss the cutting-edge reagentless protein-based sensors called the molecular pendulum.
Molecular pendulum: using antibodies as recognition elements
Aiming to solve the long-standing challenges around how to monitor a broad range of relevant biomarkers using a methodology that is compatible with continuous in vivo monitoring, Shana Kelley and co-workers recently developed a new electrochemical concept called inverted molecular pendulum.46,47 The new molecular pendulum uses antibodies as the recognition element. Antibodies have been used extensively to develop biosensors as they provide high sensitivity and specificity for biomolecular antibody-target biomolecule interactions. However, one of the main challenges that have impeded antibody-based electrochemical biosensors to reach sensors translation and commercialization, is that antibodies do not change their shape or conformation easily upon binding their target species. This limitation has now been overcome with the molecular pendulum that translates the binding with the target analyte into an electrochemical readout. The inverted molecular pendulum approach uses a double-stranded DNA linker covalently immobilized on an electrode surface. The double-stranded DNA contains on its distal end an antibody that recognizes the target analyte as well as a ferrocene redox reporter that generates an electrochemical measurable current response (Fig. 5a).46 By applying a positive potential to the sensor interface, the negatively charged DNA pendulum is pulled down towards the electrode surface, where the motion dynamics are dictated by the hydrodynamics of the DNA pendulum. When the target analyte binds to the antibody, the pendulum kinetics slows down, increasing hence the hydrodynamic radius of the pendulum.48 Thus, by using ferrocene redox reporter and the inverted molecular pendulum complex it is possible to attain time-resolved electrochemical measurements of the electron transfer kinetics relating intrinsically to the occupancy of the antibody with its target analyte. | ||
| Fig. 5 (a) Molecular pendulum signal generation mechanism. The inverted molecular pendulum strategy translates the binding of an antibody to its target species into an electrochemical response, which is reversible and does not require extra reagents. The pendulum is comprised of a short double-stranded DNA linker that is covalently attached to an electrode and functionalized on its distal end with both an antibody and the ferrocene redox reporter. When a positive potential is applied to the electrode surface, the negatively charged molecular pendulum is attracted closer to the surface, but the kinetics with which it approximates to the electrodes are controlled by the hydrodynamic size of the target. In simpler words, in the absence of the target the free molecular pendulum moves to the surface more rapidly than when there is no target analyte present. Therefore, the electronic transfer rate between ferrocene redox reporter and electrode surface depends on receptor occupancy. (b) An illustrative example of a time-resolved chronoamperometry measurement used to monitor analyte binding. | ||
Using this new sensing technology, the detection of Cardiac troponin I (an important clinical biomarker of heart failure)46 and SARS-CoV-2 spike protein47 has been reported in two separate works. The modularity for sensing different target species is achieved by simply replacing the antibody recognition element that is a conjugated double-stranded DNA linker. Chronoamperometry measurements are used to determine the target concentration through the binding-caused change in the current decay rate (Fig. 5b). The current decay rate for an unbound (absence of target analyte) inverted molecular pendulum is fast, but it slows significantly when the target is present, as the mass change leads to an increased drag force and slower current decay rates. Concentration-dependent signal change for troponin I, SARS-CoV-2 viral particles, and SARS-CoV-2 N protein, reported in these works demonstrates the quantitative detection features and satisfactory sensitivity of the molecular pendulum biosensor.46,47
Overall, more than 10 different proteins, including viral proteins and biomarkers with varying charges, sizes, and molecular weights, have been detected using the molecular pendulum sensor.46 All proteins were detectable using the same mechanism by simply changing the recognition antibody, indicating that the kinetic measurements of molecular pendulum transport behavior provide a universal method for protein biomarker detection.
Although many protein-based sensors can maintain good selectivity in biological media, their performance can be inconsistent across different media types as the electrolyte (ionic strength), buffering condition and viscosity may differ. The molecular pendulum sensor of troponin I has also been studied in various bodily fluids, including saliva, urine, tear fluid, blood, and sweat. Although small fluctuations of transients and statistically significant changes were observed against the type of fluid, the molecular pendulum was capable of performing well in all tested bodily fluids.46 Encouraged by these and based on the fact that the molecular pendulum transduction mechanism is reversible, Kelley's team further challenged the sensor for continuous, real-time in-vivo measurements. The molecular pendulum sensor was positioned in the mouths of anesthetized mice, where continuous monitoring of troponin I was attained directly in saliva. The studied mice received intravenous injections of drugs that provoked cardiac damage, resulting in abnormal levels of troponin I.46 The molecular pendulum also shows excellent performance for the detection of SARS-CoV-2 in direct saliva testing (Fig. 6a and b).47
 | ||
| Fig. 6 Molecular pendulum sensor performance for the detection of N-protein (a) and viral load (b) in healthy and Covid-19 patient saliva samples. Figure reproduced from ref. 47, with permission from American Chemical Society, Copyright © 2022. | ||
In a more recent work reported by Shana Kelley and co-workers, the molecular pendulum sensing technology was explored for the detection of two analytes simultaneously, demonstrating the multiplexing capability.49 For multiplexing, the authors used two working sensors in an array configuration, where each working electrode contained a molecular pendulum constructed with different target binding aptamers. It was possible to detect B-type natriuretic peptide and N-terminal pro B-type natriuretic peptide simultaneously in a single clinical sample.49 It was also shown that cross-reactivity of the molecular pendulums with the two peptides binding aptamers was not an issue.49
These results indicate the promising future for molecular pendulum-based sensors for real-time and continuous monitoring of proteins in complex biological media. This is substantial progress in our capability to detect molecular markers in health and disease.
Future outlook and challenges
The molecular level sensing approaches described here have presented extraordinary analytical performances, such as allowing sensors to work in complex biological matrices and even presenting the capability of continuous monitoring of molecular species, in real-time, in-vivo. This is an incredible accomplishment that embodies a paradigm shift in what electrochemical biosensors have been capable to do. The reagentless protein-based biosensors, through close collaborations between academic researchers and diagnostic companies, are moving towards translation and commercialization for diverse health applications including rapid detection of infectious diseases, point-of-care detection of cardiac biomarkers which are indicators of heart failure progression, point-of-care detection of insulin levels in diabetic patients, and point-of-care monitoring of cancer related antigens levels in cancer patients to monitor disease progression and treatment efficacies. While such technologies are progressing towards translation, challenges are faced in different stages of the process, and some considerations should be taken into account for further development of such sensors.In this mini-review we discussed the cutting-edge developments of reagentless protein-based electrochemical biosensors that can monitor relevant biomarkers directly in biofluids. Given recent advances in the development of portable monitoring electrochemical devices, these innovative sensors concept will find broad applicability, especially in clinical diagnostics.
Abbreviations
| LUB | Lubricin |
| PNA | Peanut agglutinin |
| CheY | Bacterial chemotaxis protein |
| CheA-P | P2 domain of the chemotaxis kinase |
| FliM | 16-Residue target region of the flagellar switch protein |
| EAB | Electrochemical aptamer-based. |
Author contributions
S. M. S, M. L., A. X. M., S. E. M jointly wrote this review article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ARC Centre of Excellence for Electromaterials Science (ACES) and the Aikenhead Centre for Medical Discovery – St Vincent's Hospital.References
- E. D. Bojescu, D. Prim, M. E. Pfeifer and J.-M. Segura, Anal. Chim. Acta, 2022, 1225, 340240 CrossRef CAS PubMed.
- M. Chen, B. Wan, W. Du, H. Hu, L. Zeng, X. Duan, J. Liu, Z. Wei, L. Tang and Y. Peng, RSC Adv., 2020, 10, 21789–21794 RSC.
- J. W. Choi, B. M. K. Vasamsetti, J. Choo and H. Y. Kim, Analyst, 2020, 145, 3222–3228 RSC.
- C. Y. Lee, I. Degani, J. Cheong, J. H. Lee, H. J. Choi, J. Cheon and H. Lee, Biosens. Bioelectron., 2021, 178, 113049 CrossRef CAS PubMed.
- K. Nishiyama, Y. Takeda, M. Maeki, A. Ishida, H. Tani, K. Shigemura, A. Hibara, Y. Yonezawa, K. Imai, H. Ogawa and M. Tokeshi, Sens. Actuators, B, 2020, 316, 128160 CrossRef CAS PubMed.
- O. Pashchenko, T. Shelby, T. Banerjee and S. Santra, ACS Infect. Dis., 2018, 4, 1162–1178 CrossRef CAS PubMed.
- O. D. Hendrickson, N. A. Taranova, A. V. Zherdev, B. B. Dzantiev and S. A. Eremin, Sensors, 2020, 20, 7132 CrossRef CAS PubMed.
- A. A. Goulko, Q. Zhao, J. W. Guthrie, H. Zou and X. C. Le, in Standardization and Quality Assurance in Fluorescence Measurements I: Techniques, ed. U. Resch-Genger, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 303–322. DOI:10.1007/4243_2008_021.
- A. M. Rossi and C. W. Taylor, Nat. Protoc., 2011, 6, 365–387 CrossRef CAS PubMed.
- K. Nielsen and D. Gall, J. Immunoassay Immunochem., 2001, 22, 183–201 CrossRef CAS PubMed.
- R. S. Gaster, D. A. Hall, C. H. Nielsen, S. J. Osterfeld, H. Yu, K. E. Mach, R. J. Wilson, B. Murmann, J. C. Liao, S. S. Gambhir and S. X. Wang, Nat. Med., 2009, 15, 1327–1332 CrossRef CAS PubMed.
- D. A. Giljohann and C. A. Mirkin, Nature, 2009, 462, 461–464 CrossRef CAS PubMed.
- G. Rong, S. R. Corrie and H. A. Clark, ACS Sens., 2017, 2, 327–338 CrossRef CAS PubMed.
- D. Kang, S. Sun, M. Kurnik, D. Morales, F. W. Dahlquist and K. W. Plaxco, J. Am. Chem. Soc., 2017, 139, 12113–12116 CrossRef CAS PubMed.
- D. Kang, C. Parolo, S. Sun, N. E. Ogden, F. W. Dahlquist and K. W. Plaxco, ACS Sens., 2018, 3, 1271–1275 CrossRef CAS PubMed.
- S. L. Bidinger, S. T. Keene, S. Han, K. W. Plaxco, G. G. Malliaras and T. Hasan, Sci. Adv., 2022, 8, eadd4111 CrossRef CAS PubMed.
- A. Idili, C. Parolo, G. Ortega and K. W. Plaxco, ACS Sens., 2019, 4, 3227–3233 CrossRef CAS PubMed.
- S. M. Silva, S. Hoque, V. R. Gonçales and J. J. Gooding, Electroanalysis, 2018, 30, 1529–1535 CrossRef CAS.
- S. M. Silva, R. Tavallaie, V. R. Goncales, R. H. Utama, M. B. Kashi, D. B. Hibbert, R. D. Tilley and J. J. Gooding, Langmuir, 2018, 34, 1249–1255 CrossRef CAS PubMed.
- A. Chamorro-Garcia, J. Gerson, C. Flatebo, L. Fetter, A. M. Downs, N. Emmons, H. L. Ennis, N. Milosavic, K. Yang, M. Stojanovic, F. Ricci, T. E. Kippin and K. W. Plaxco, ACS Sens., 2023, 8, 150–157 CrossRef CAS.
- A. M. Downs and K. W. Plaxco, ACS Sens., 2022, 7, 2823–2832 CrossRef CAS PubMed.
- P. Dauphin-Ducharme, K. Yang, N. Arroyo-Curras, K. L. Ploense, Y. Zhang, J. Gerson, M. Kurnik, T. E. Kippin, M. N. Stojanovic and K. W. Plaxco, ACS Sens., 2019, 4, 2832–2837 CrossRef CAS PubMed.
- N. Arroyo-Currás, P. Dauphin-Ducharme, K. Scida and J. L. Chávez, Anal. Methods, 2020, 12, 1288–1310 RSC.
- A. A. Lubin and K. W. Plaxco, Acc. Chem. Res., 2010, 43, 496–505 CrossRef CAS PubMed.
- F. Ricci, A. Vallee-Belisle, A. J. Simon, A. Porchetta and K. W. Plaxco, Acc. Chem. Res., 2016, 49, 1884–1892 CrossRef CAS PubMed.
- L. R. Schoukroun-Barnes, F. C. Macazo, B. Gutierrez, J. Lottermoser, J. Liu and R. J. White, Annu. Rev. Anal. Chem., 2016, 9, 163–181 CrossRef CAS PubMed.
- M. Gerstein and W. Krebs, Nucleic Acids Res., 1998, 26, 4280–4290 CrossRef CAS PubMed.
- A. Vallee-Belisle and K. W. Plaxco, Curr. Opin. Struct. Biol., 2010, 20, 518–526 CrossRef CAS.
- M. Kurnik, E. Z. Pang and K. W. Plaxco, Angew. Chem., Int. Ed., 2020, 59, 18442–18445 CrossRef CAS PubMed.
- M. Kurnik, G. Ortega, P. Dauphin-Ducharme, H. Li, A. Caceres and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8352–8357 CrossRef CAS PubMed.
- G. Ortega, M. A. Aguilar, B. K. Gautam and K. W. Plaxco, Protein Sci., 2021, 30, 2408–2417 CrossRef CAS PubMed.
- G. Ortega, M. Kurnik, P. Dauphin-Ducharme, H. Li, N. Arroyo-Curras, A. Caceres and K. W. Plaxco, Angew. Chem., Int. Ed., 2019, 58, 1714–1718 CrossRef CAS PubMed.
- G. Ortega, M. Kurnik, B. K. Gautam and K. W. Plaxco, J. Am. Chem. Soc., 2020, 142, 15349–15354 CrossRef CAS PubMed.
- M. S. Silva, D. P. Langley, L. R. Cossins, A. N. Samudra, A. F. Quigley, R. M. I. Kapsa, R. W. Tothill, G. W. Greene and S. E. Moulton, ACS Sens., 2022, 7, 3379–3388 CrossRef PubMed.
- G. Poiroux, A. Barre, E. J. M. Van Damme, H. Benoist and P. Rougé, Int. J. Mol. Sci., 2017, 18, 1232 CrossRef PubMed.
- X.-P. He, X.-W. Wang, X.-P. Jin, H. Zhou, X.-X. Shi, G.-R. Chen and Y.-T. Long, J. Am. Chem. Soc., 2011, 133, 3649–3657 CrossRef CAS PubMed.
- P. E. Desroches, S. M. Silva, S. W. Gietman, A. F. Quigley, R. M. I. Kapsa, S. E. Moulton and G. W. Greene, ACS Appl. Bio Mater., 2020, 3, 8032–8039 CrossRef CAS PubMed.
- M. Han, J. D. Berry, S. M. Silva, M. L. P. Vidallon, W. Lei, A. F. Quigley, R. M. I. Kapsa, S. E. Moulton, R. Tabor and G. W. Greene, ACS Appl. Nano Mater., 2020, 3, 11527–11542 CrossRef CAS.
- M. Y. Han, S. M. Silva, W. W. Lei, A. Quigley, R. M. I. Kapsa, S. E. Moulton and G. W. Greene, Langmuir, 2019, 35, 15834–15848 CrossRef CAS PubMed.
- D. M. Khwannimit, S. Silva, P. E. Desroches, A. F. Quigley, R. M. I. Kapsa, G. W. Greene and S. E. Moulton, Langmuir, 2021, 37, 11188–11193 CrossRef CAS PubMed.
- M. J. Russo, M. Han, P. E. Desroches, C. S. Manasa, J. Dennaoui, A. F. Quigley, R. M. I. Kapsa, S. E. Moulton, R. M. Guijt, G. W. Greene and S. M. Silva, ACS Sens., 2021, 6, 1482–1507 CrossRef CAS PubMed.
- M. J. Russo, M. Y. Han, A. F. Quigley, R. M. I. Kapsa, S. E. Moulton, E. Doeven, R. Guijt, S. M. Silva and G. W. Greene, Electrochim. Acta, 2020, 333, 135574 CrossRef CAS.
- M. J. Russo, A. F. Quigley, R. M. I. Kapsa, S. E. Moulton, R. Guijt, S. M. Silva and G. W. Greene, ChemElectroChem, 2020, 7, 2851–2858 CrossRef CAS.
- S. M. Silva, A. F. Quigley, R. M. I. Kapsa, G. W. Greene and S. E. Moulton, Chemelectrochem, 2019, 6, 1939–1943 CrossRef CAS.
- G. W. Greene, R. Thapa, S. A. Holt, X. Wang, C. J. Garvey and R. F. Tabor, Langmuir, 2017, 33, 2559–2570 CrossRef CAS PubMed.
- J. Das, S. Gomis, J. B. Chen, H. Yousefi, S. Ahmed, A. Mahmud, W. Zhou, E. H. Sargent and S. O. Kelley, Nat. Chem., 2021, 13, 428–434 CrossRef CAS PubMed.
- H. Zargartalebi, H. Yousefi, C. D. Flynn, S. Gomis, J. Das, T. L. Young, E. Chien, S. Mubareka, A. McGeer, H. Wang, E. H. Sargent, A. S. Nezhad and S. O. Kelley, J. Am. Chem. Soc., 2022, 144, 18338–18349 CrossRef CAS PubMed.
- K. J. Cash and K. W. Plaxco, Nat. Chem., 2021, 13, 392–393 CrossRef CAS PubMed.
- A. Mahmud, D. Chang, J. Das, S. Gomis, F. Foroutan, J. B. Chen, L. Pandey, C. D. Flynn, H. Yousefi, A. Geraili, H. J. Ross, E. H. Sargent and S. O. Kelley, Angew. Chem., Int. Ed., 2023, e202213567 Search PubMed.
- C. Jiang, G. Wang, R. Hein, N. Liu, X. Luo and J. J. Davis, Chem. Rev., 2020, 120, 3852–3889 CrossRef CAS PubMed.
- S. Campuzano, M. Pedrero, P. Yanez-Sedeno and J. M. Pingarron, Int. J. Mol. Sci., 2019, 20, 423 CrossRef PubMed.
- S. S. Timilsina, P. Jolly, N. Durr, M. Yafia and D. E. Ingber, Acc. Chem. Res., 2021, 54, 3529–3539 CrossRef CAS PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- A. Barfidokht and J. J. Gooding, Electroanalysis, 2014, 26, 1182–1196 CrossRef CAS.
- N. Liu, Z. Xu, A. Morrin and X. Luo, Anal. Methods, 2019, 11, 702–711 RSC.
- G. P. Sakala and M. Reches, Adv. Mater. Interfaces, 2018, 5, 1800073 CrossRef.
- L. Zhou, X. Li, B. Zhu and B. Su, Electroanalysis, 2022, 34, 966–975 CrossRef CAS.
- N. Hermann, K. Dressen, L. Schroeder, M. Debald, F. A. Schildberg, G. Walgenbach-Bruenagel, K. Hettwer, S. Uhlig, W. Kuhn, G. Hartmann and S. Holdenrieder, Tumor Biol., 2017, 39, 1010428317711381 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |