
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst total synthesis of versicotide A, B and C†
Laura Posada ab,
Danilo Davyta and
Gloria Serra*a
ab,
Danilo Davyta and
Gloria Serra*a
aQuímica Farmacéutica, Departamento de Química Orgánica, Facultad de Química, Universidad de la República, General Flores 2124, CC1157, Montevideo, Uruguay. E-mail: gserra@fq.edu.uy
bGraduate Program in Chemistry, Facultad de Química, Universidad de la República, Uruguay
First published on 9th December 2020
Abstract
The syntheses of versicotides A–C, natural products containing anthranilic acid and NMe-Ala, were achieved by solid phase peptide synthesis on 2-chlorotrityl resin followed by solution phase macrocyclization. Using an oxyma additive, the difficult coupling reactions to the deactivated aromatic amine of o-aminobenzoic acid, were performed in high yield, avoiding anthranilic rearrangements or side reactions.
1. Introduction
In the past years, a significant number of macrocyclic peptides, with very interesting biological activities, have been described in the literature.1 They are known to be promising candidates in the search for new drugs as they show enhanced metabolic stability in comparison with their corresponding linear peptides, and could bind to receptors with high selectivity.2 Moreover, chemoinformatic exploratory analyses of synthetic peptide combinatorial data sets have suggested that N-methylation and cyclization shifts their position toward the chemical space defined by sets of FDA approved drugs. Therefore, this modifications could result in compounds with biological activity and suitable physicochemical properties.3 In addition, it was demonstrated that multiple backbone N-methylation of cyclic peptides remarkably improves their cell permeability and therefore can be utilized in the design of new orally available drugs.4 Recent studies suggest that because of their conformational flexibility, cyclopeptides behave as molecular chameleons as they adjust their conformations and polar surface areas according with the properties of surrounding environment.5 On the other hand, constrained structures are expected to present a minor loss of conformational entropy upon binding, resulting in higher target affinity.6 Thus, many groups investigate the addition of further constraints to cyclopeptides in order to obtain a potential bioactive conformer.7Natural products are a continuous source of inspiration for the discovery and development of new drug candidates.8 Those proceeding from marine environments, have been attracting special interest for their unexplored structural diversity.9 Particularly, marine fungi are subjected to many factors that contribute to gene modifications and to adaptive processes producing unique secondary metabolites. Aspergillus genus is an important source of bioactive secondary metabolites with high structural diversity.10 Versicotides are natural products produced by fungi of the Aspergillus family. Versicotides A (1) and B (2), Fig. 1, were first isolated by Li and co-workers from Aspergillus versicolor ZLN-60 extracts of the Yellow Sea sediment.11 Later, the same group employed the OSMAC (one strain many compounds) approach on this fungus, resulting in the isolation and characterization of a new compound with structural similarities named versicotide C (3),12 the first natural cyclic hexapeptide containing two anthranilic acids (Anth). Further on, three new cyclopeptides were isolated from Aspergillus versicolor LZD-14-1 namely versicotides D–F,13 along with the previously known versicotides A and B.
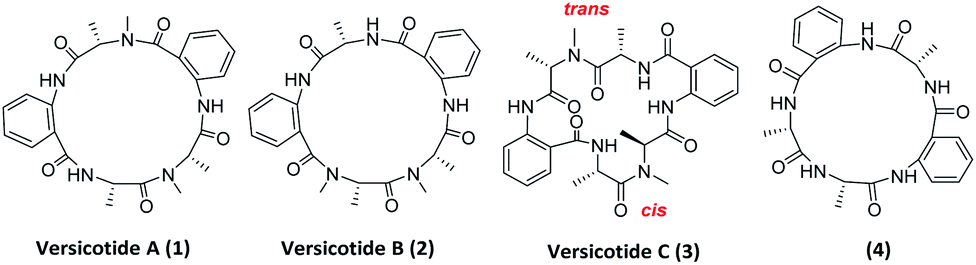 | ||
| Fig. 1 Versicotides A–C and analogue 4. | ||
The presence of two rigid β-amino acids, Anth units, and two N-methyl-alanine (NMe-Ala) in 1–3 add further constraints to these cyclopeptides. In the case of the reported isolation of versicotide C (3), the authors stated that, in DMSO-d6, it presents one trans amide bond and one cis amide bond on its structure.12
Regarding their biological activity, 1 and 2 showed no cytotoxicity against murine leukemia P388, human hepatoma cell line BEL-7402 and human leukemia cell HL-60; versicotides A–C showed no activity against cancer cell lines A-549, HeLa and SMMC-7721. In addition, versicotides D–F exerted lipid lowering effects through the regulation of cholesterol efflux to HDL in RAW264.7 macrophages, while 1–3 do not exhibit this effect.
Recently, as part of a general program to obtain antiplasmodial candidates,14 our group reported the synthesis of versicotide D and analogues and their evaluation as antimalarials against the chloroquine sensitive strain P. falciparum 3D7, where versicotide D showed EC50 = 1.5 μM.15
There are few examples in the literature related to the synthesis of cyclopeptide natural products containing anthranilic acid,16 and they are all broadly in line to the fact that amide bond formation involving the amine of Anth is a challenge not only because of the deactivation of the aromatic amine via resonance, but also due to rearrangements in which the Anth motif can be involved.17 Frequently, long, complicated, multi-stage and low yielding routes have been used to achieve the synthesis of natural products containing this motif. An example is the total synthesis of deoxo-solomonamide B in which Anth motif was masked with an indole group that is cleaved by ozonolysis in the last steps of the synthetic route.18 Therefore, a simpler methodology to obtain compounds with Anth in good yield is a chemical challenge. In addition, as many cyclopeptides containing Anth, present diverse bioactivities such as increasing blood pressure,16b lipid lowering effect,12 anti-proliferative,16a,16d anti-inflammatory,18 insecticide,16e the interest on this issue is enhanced. Here, we report the total synthesis of versicotides A–C and analogue 4. The structures of the obtained compounds were confirmed by MS, NMR and, in the case of versicotide A (1), by X-ray analysis. A deep analysis of NMR data (1H, 13C, COSY, HSQC and HMBC) was done in order to determine the adopted conformations in the used solvents, along with temperature variation studies.
2. Results and discussion
2.1. Solution phase approach
First, we decided to prepare the cyclo[Ala-Anth-Ala-Anth-Ala] (4), analogue of 1 and 2, and to investigate the solution phase synthesis of the peptide precursor containing only Ala and Anth. The in situ acyl chloride formation using triphosgene19 made it possible to couple Ala to the amine of methyl anthranilate in 51% yield, Scheme 1. No racemization was observed by 1H-NMR analysis. However, advanced intermediates could not be prepared using triphosgene. In an attempt to circumvent this, we tried to apply the methodology described by Xin and Burguess,20 which employed a combination of EDCI and HOBt. As HOBt and HOAt present restrictions for their overseas shipment, we decided to substitute HOBt by Cl-HOBt.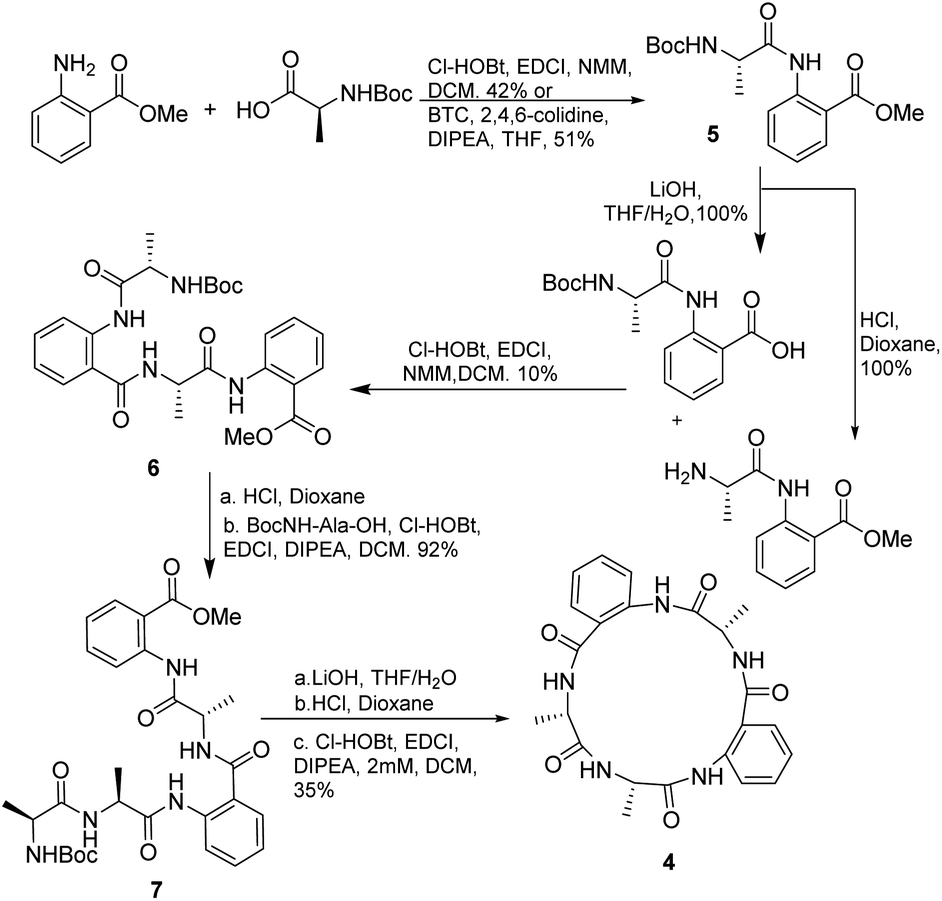 | ||
| Scheme 1 Solution synthesis of cyclopentapeptide analogue 4. | ||
Dipeptide 5 and tetrapeptide 6 were obtained in 42% and 10% yield, respectively. No racemization during the coupling reaction steps was detected according with the NMR spectra of 6. However, in the case of the synthesis of 6 many by-products, as fragments of the desired compound, were observed. Two of the detected by-products were methyl anthranilate and the tripeptide NH2-Anth-Ala-Anth-OMe, derived from fragmentation of the labile Anth amide bond according with Hamada et al. results.17b The preparation of pentapeptide 7 from 6 resulted in a high yielding process (92%). Finally, deprotection and cyclization in diluted conditions (2 mM) rendered analogue 4 in 35% yield.
Attempts to apply this strategy for the synthesis of versicotide A were not successful. The coupling reaction between BocNMeAla-Anth (8) and Ala-Anth(OMe) using EDCI and Cl-HOBt rendered the corresponding tetrapeptide intermediate in very low yield along with the benzoxazinone 9, Scheme 2. According with previous results,21 this heterocycle is derived from the cyclodehydration reaction of 8.
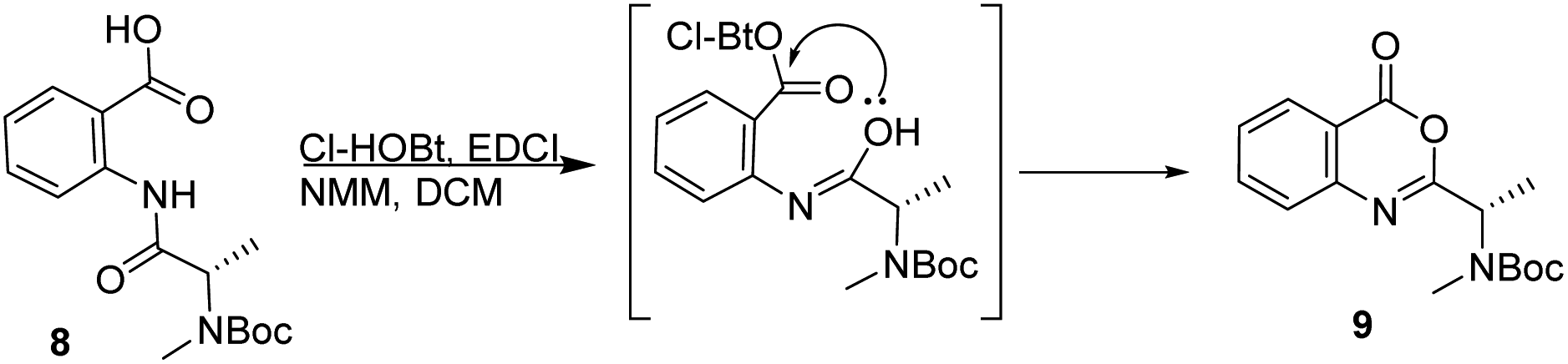 | ||
| Scheme 2 Synthesis of benzoxazinone 9 from BocNMeAla-Anth. | ||
2.2. SPPS synthesis of linear precursors and solution phase cyclization
In view of the limited achievements of the solution phase synthetic methodology, we decided to explore the synthesis of the linear precursors of versicotides A–C on solid phase and cyclize them on solution phase. The linear sequence of each one was chosen taking into account the characteristics of Anth explained before. Based on this, Anth amine was not chosen as the N-terminal in any case. Furthermore, we realized in early steps of this work that using Anth as the C terminal on solid phase peptide synthesis (SPPS) was not a good choice either, as it leads to very low resin loadings and it could give a great steric hindrance for the first coupling to occur. Because of this, the linear sequences chosen were: NHMeAla-Anth-NMeAla-Anth-Ala-OH for 1, NHMe-Ala-Anth-Ala-Anth-NMeAla-OH for 2 and NMeAla-Anth-Ala-NMeAla-Anth-Ala for 3.Our first efforts to obtain the linear precursor of versicotide A, 10, by SPPS on 2-chlorotrityl chloride resin (2-CTC), were not successful. The use of HBTU, HATU, or DIC/Cl-HOBt for the coupling involving the amine of Anth led to incomplete or no coupling at all. Doi and co-workers described the SPPS of Anth containing peptides on the 2-CTC resin by generating an acyl chloride of the corresponding amino acid with triphosgene and adding it to the resin without previous purification.22 During attempts to apply this methodology to obtain the desired peptide, it was possible to prepare the tripeptide FmocNMeAla-Anth-Ala, but subsequent coupling reactions produced cleavage of the growing peptide from the resin.
Albericio et al. described the impressive coupling efficiency of oxyma (ethylcianoacetate oxime), in combination with DIC,23 surpassing HOAt coupling agent in more demanding peptide models. Taking into account their results, we decided to select it as an excellent option to overcome those difficulties. Consequently, the use of oxyma for the coupling involving the amine of Anth allowed us to prepare the dipeptide FmocNMeAla-Anth-OH by SPPS, Scheme 3, in excellent yield. Moreover, the synthesis of the open precursor of versicotide A (10) was achieved in 90% yield, based on the determination of the resin loading. Completion of each coupling step was verified by Kaiser or chloranil test, for primary or secondary amines, respectively. These colorimetric assays were not useful for monitoring couplings to anthranilic acid amine group because we notice this throws false negative results as an incomplete coupling was observed by HPLC-MS analysis of a small sample cleaved from the resin. Nevertheless, with oxyma/DIC, we confirmed optimal coupling after 60 min of reaction time by HPLC and 1H-NMR analysis. For the solution phase cyclization, HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate was employed as coupling reagent and dilution of the linear peptide was taken to 5 mM concentration. After purification, 1 was obtained in 49% yield and no dimer formation was observed.
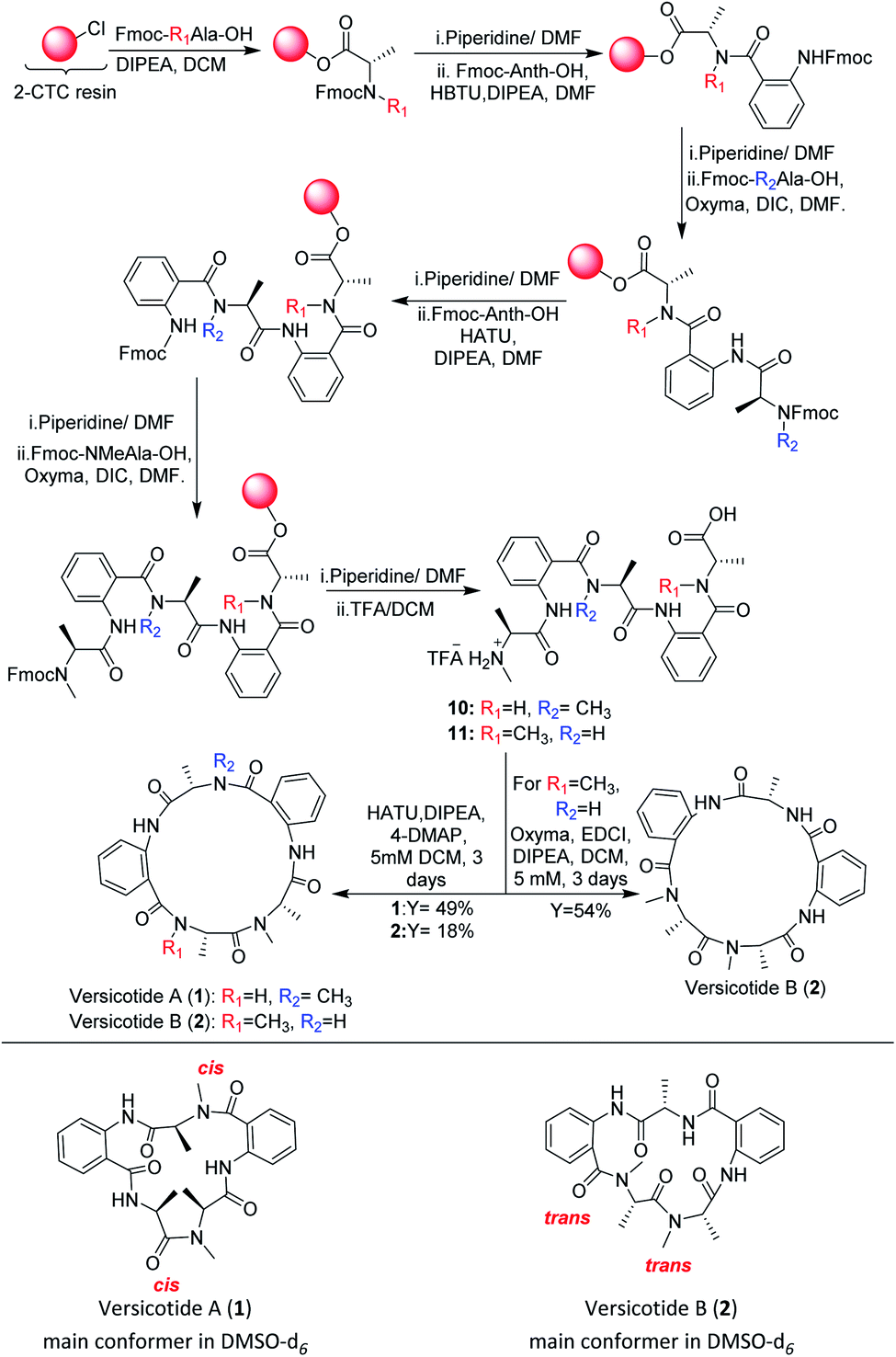 | ||
| Scheme 3 SPPS and solution macrocyclization of versicotides A (1) and B (2). | ||
Chromatographically pure versicotide A (1) showed three different conformers in the NMR spectra using DMSO-d6 as solvent. To discard epimerization, 1H-NMR studies using temperature gradient were performed. The spectra showed variations of NMe and α methine peak integrations arising from conformers in slow exchange (see ESI†). The amide of NMe-amino acids can present cis or trans configuration that depends, in many cases, on the used solvent. Taking into account that the N-methyl resonance of the amides in trans configuration are shifted downfield relative to the cis because of the shielding effect of the carbonyl group,24 we observed that the major conformer, presents the two N-methyl amide bonds in cis configuration, Scheme 3. The chemical shifts of this conformer match with the reported ones for the isolated natural product. The other two conformers would be in (cis, trans) and (trans, trans) configuration (see ESI†). X-ray crystallographic structure of versicotide A, Fig. 2, obtained from a crystal grown in methanol, also agrees with previously described data for the natural product, and presents both NMe-amides in cis configuration.
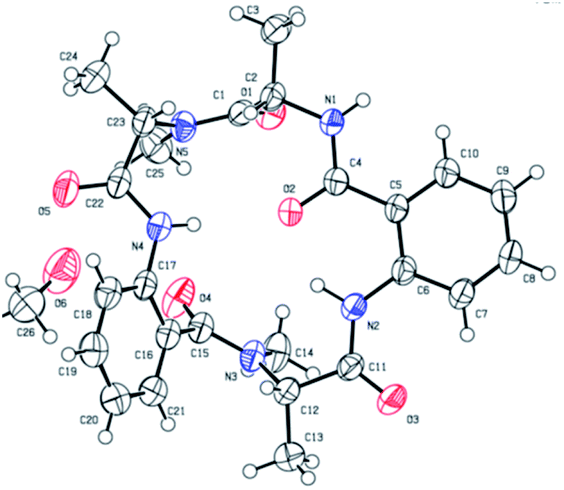 | ||
| Fig. 2 X-ray crystallographic structure of 1.25 | ||
Following the same procedure, the linear precursor of versicotide B (11), Scheme 3, was successfully synthesized in 87% global yield (based on the determination of the resin loading). However, cyclization using HATU in diluted conditions (5 mM), led to a small amount of 2 (18% yield). The macrolactamization reaction was repeated using EDCI/oxyma and 2 was purified by flash chromatography to obtain 54% yield. The product presented a single spot by TLC in different mobile phases and two peaks by HPLC. The analysis of both peaks by FIA-LC-ESI-MS showed the signal of m/z 480.3 corresponding to [M + H]+ ion of versicotide B (2). The 1H-NMR of each sample in DMSO-d6 showed a greater number of signals than expected. Finally, a thermal gradient of the 1H-NMR in DMSO-d6 showed that some signals corresponding to NMe and α methine change their proportion at higher temperatures. This fact allowed us to discard epimerization and to conclude that both samples contain a mixture of conformers of 2; in one of them both NMe-amides are in trans configuration, Scheme 3, and in the other conformer, one amide is in cis and the other in trans configuration, (see ESI†) according with the literature.24
In a similar way, the synthesis of the linear precursor of versicotide C (12), Scheme 4, was successfully achieved employing Oxyma + DIC in key steps, leading to 98% overall yield (based on the determination of the resin loading).
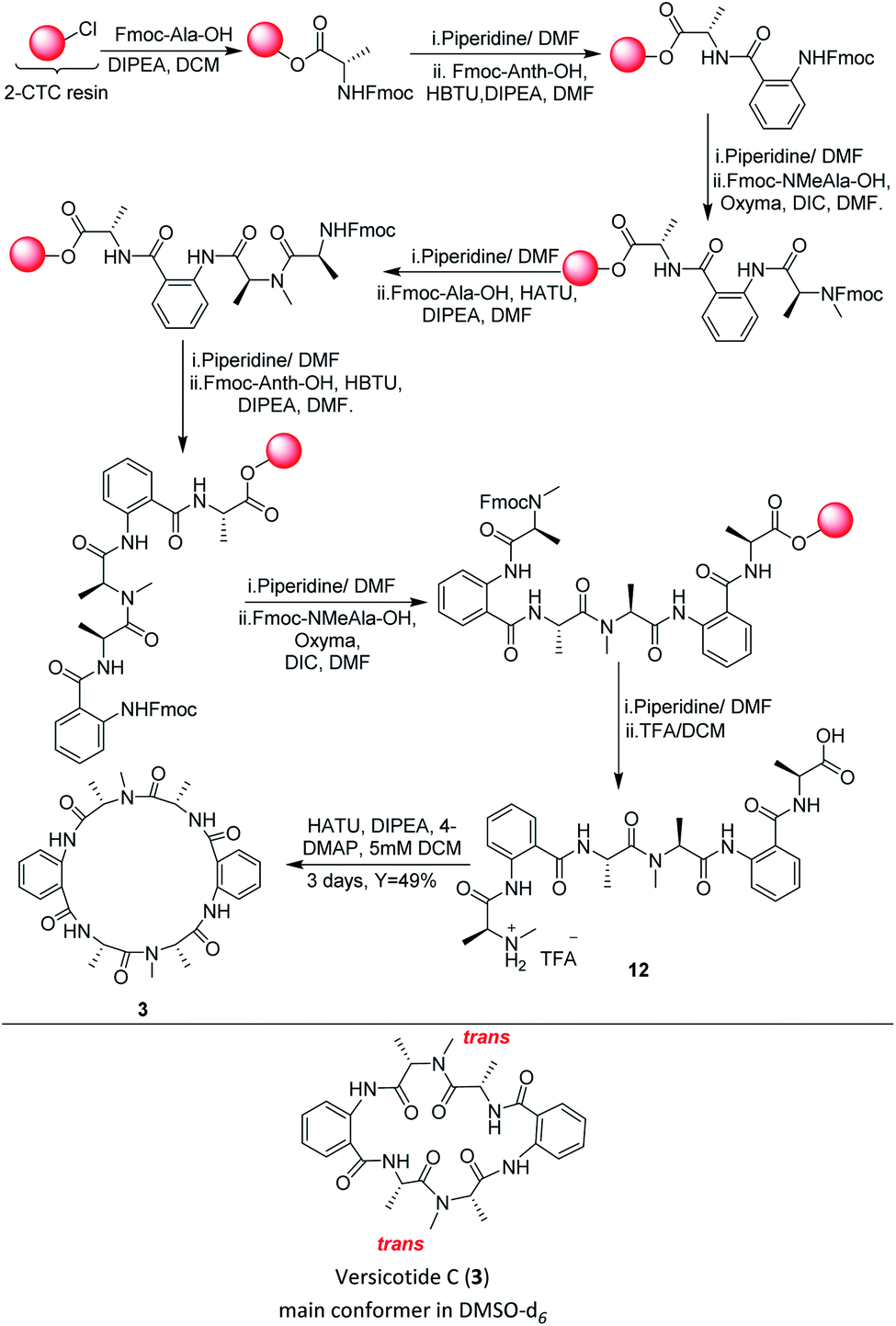 | ||
| Scheme 4 SPPS and solution macrocyclization of versicotide C (3) | ||
Then, macrocyclization in solution phase, using HATU in DCM (5 mM), rendered versicotide C (3) in 49% yield after purification and no dimer formation was observed. NMR spectra of synthetic versicotide C in DMSO-d6, match those of the isolated natural product, even though the authors informed a single conformer with one NMe amide bond in cis and the other one in trans configuration.12 A deep analysis of NMR spectra, with special regard to signal integrations, lead us to conclude that two conformers of 3, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 ratio, are present in both acetone-d6 and DMSO-d6. The conformers, which present symmetrical structure, were easily distinguished by the NMR signals of the two N-Me groups. The two corresponding N-methyl amide bonds of one conformer are in trans configuration, Scheme 4, and of the other conformer, in cis configuration. The presence of these two conformers were further confirmed by a 1H-NMR experiment of 3 at 80 °C which showed that the peaks assignable to the (cis, cis) conformer at 25 °C, disappeared at this temperature (see ESI†).
:0.2 ratio, are present in both acetone-d6 and DMSO-d6. The conformers, which present symmetrical structure, were easily distinguished by the NMR signals of the two N-Me groups. The two corresponding N-methyl amide bonds of one conformer are in trans configuration, Scheme 4, and of the other conformer, in cis configuration. The presence of these two conformers were further confirmed by a 1H-NMR experiment of 3 at 80 °C which showed that the peaks assignable to the (cis, cis) conformer at 25 °C, disappeared at this temperature (see ESI†).
3. Experimental section
3.1. General procedure. Solid phase peptide synthesis
2-Chlorotrityl chloride resin (2-CTC) (100–300 mesh, 1.20 mmol g−1) was added to a syringe peptide synthesis vessel. The resin was swelled in CH2Cl2 (3 × 5 min). A solution of first protected amino acid Fmoc-AA-OH (1 eq. for 0.8 mmol g−1 loading) and DIPEA (3 eq.) in CH2Cl2 was added and the resin was shaken 10 minutes. Then, an extra 7.0 eq. of DIPEA were added and shaking was continued for 50 min. MeOH (0.8 mLg−1 of resin) was added to the previous mixture in order to cap unreacted functional groups on the resin, and shaken for 10 min. After filtering, the resin was washed with CH2Cl2 (×3), MeOH (×3), CH2Cl2 (×3), DMF (×3). The resin was washed with DMF (×3) and Fmoc protecting group was removed by treating the resin with piperidine–DMF solution (1:4) for 1, 5 and 5 minutes successively. In exceptional cases deprotection step was accomplish by a single treatment with piperidine–DMF solution for 5 minutes, in order to prevent side reactions. Then, the resin was washed with DMF (×3), CH2Cl2 (×3) and DMF (×3). A solution of Fmoc-AA-OH or Fmoc-Anth-OH (3 eq.) and DIPEA (6 eq.) in DMF was added to the resin, followed by a solution of HBTU, for coupling to primary amines, or HATU (2.9 eq.) in DMF, in case of coupling to an N-methylated amino acid. The mixture was stirred for 60 min. After the coupling was completed, the resin was washed with DMF (×3) and CH2Cl2 (×3). Completion of the coupling was monitored by colorimetric assays; Kaiser test in case of primary amines and Chloranil test for secondary amines. Coupling procedure was repeated in case of positive results. For the coupling of subsequent Fmoc-AA to anthranilic acid, a solution of Fmoc-AA-OH (5 eq.), Oxyma Pure (5 eq.), and DIC (5 eq.) was added to the vessel. The mixture was stirred for 60 min. Then, the resin was washed with DMF (×3) and CH2Cl2 (×3). Deprotection and coupling cycles were repeated with the appropriate amino acids or Anth to provide the desired compound. The peptide was cleaved from the resin by treatment with 1% TFA in CH2Cl2 for 2–3 minutes at room temperature followed by filtration and collection of the filtrate in MeOH. The treatment was repeated three times and then the resin washed with CH2Cl2 (×5) and MeOH (×3). Solvents were removed in vacuo to obtain the crude peptide. LC-MS was used to identify the product.
3.2. Solution phase synthesis
3.2.1.1. Method I. EDC·HCl (1.5 eq.), Cl-HOBt (1.5 eq.) and DIPEA (2.0 eq.) were added to a solution of Boc protected amino acid (Boc-AA-OH, 1.0 eq.) in DCM at 0 °C under N2 atmosphere. N-Terminus deprotected linear peptide or amino acid ester (NH2-AA-COOEt) was added and the reaction mixture was stirred at 0 °C for 10 min and then at room temperature, overnight.
DCM was removed under vacuum and AcOEt was added. The organic phase was washed with 0.1 M HCl aqueous solution (30 mL × 2), brine (10 mL), saturated NaHCO3 solution (30 mL × 2) and brine (10 mL), dried over MgSO4 and filtered. The solvent was removed under vacuum to give the crude material. The crude material was purified by flash chromatography.
3.2.1.2. Method II. Bis(trichloromethyl)carbonate (0.33 eq.) was added to a solution of Boc-Ala-OH (1 eq.) in dry THF under N2 atmosphere at 0 °C. 2,4,6-Colidine (2.6 eq.) were added to the solution and a white suspension was formed. The reaction mixture was stirred for 5 minutes and the suspension was added to a solution of N-terminus deprotected linear peptide or amino acid ester (NH2-AA-COOEt) (1 eq.) in dry THF, followed by DIPEA (1 eq.). The reaction mixture was stirred overnight and concentrated under vacuum. 30 mL of AcOEt were added and the organic phase was washed with HCL 5% (3 × 5 mL), brine (3 × 5 mL) and NaHCO3 (3 × 5 mL), dried with Na2SO4, filtered and concentrated. The crude material was purified by flash chromatography.
3.3. Macrocyclization in solution phase
3.3.2.1. Cyclo-[NHMeAla-Anth-NMeAla-Anth-Ala] (versicotide A) (1). The trifluoroacetate salt of NHMeAla-Anth-NMeAla-Anth-Ala-OH was obtained as a white solid following the general SPPS procedure. The global yield, based on the determination of the resin loading, was 90% (352 mg, 0.58 mmol). ESI-MS m/z calc. for C25H32N5O6 ([M + H]+) 498.2, found 498.4. Macrocyclization reaction was performed following the general procedure method I (dilution 5 mM, 3 days), starting from the trifluoroacetate salt of linear peptide NHMe-Ala-Anth-Ala-NMe-Ala-Anth-Ala-OH (200 mg, 0.33 mmol), using HATU as coupling agent. Further purification by flash chromatography using AcOEt as mobile phase, rendered the desired macrocycle in 50% yield (78 mg, 0.16 mmol). HPLC analysis employing H2O
:MeCN and 0.1% formic acid in a 90:10 to 0:100 linear gradient showed two peaks of retention times 7.9 and 8.1 min in 1:1.4 ratio. Thermal analysis by NMR, proved them to be conformers. 85% total purity.
3.3.2.2. Cyclo-[NMeAla-Anth-NMeAla-Anth-Ala] (1). White solid (Y = 50%). Rf = 0.32 (AcOEt). [α]25D = −215.7 (c 0.275, CHCl3). 1H NMR (400 MHz, DMSO-d6) Three conformers were present: conformer A
:conformer B:conformer C in 1:0.4:0.1 ratio. δ 1.30–1.37 (m, 3Ha, 6Hb, 6Hc), 1.41 (d, J = 6.8 Hz, 3Ha, 3Hb), 1.53 (d, J = 6.7 Hz, 3Ha), 1.58 (d, J = 6.9 Hz, 3Hc), 2.70 (s, 3Ha), 2.75 (s, 3Hb), 2.77 (s, 3Hb), 2.84 (s, 3Ha), 2.87 (s, 3Hc), 3.23 (s, 3Hc), 4.68–4.80 (m, 1Ha, 2Hc), 4.81–4.92 (m, 1Ha), 5.00–5.08 (m, 1Hb), 5.20 (q, J = 6.8 Hz, 1Ha), 5.26–5.34 (m, 1Hc), 5.35–5.44 (m, 2Hb, 1Hc), 7.14–7.20 (m, 1Ha, 1Hb), 7.22–7.28 (m, 1Ha, 2Hc), 7.40–7.58 (m, 3Ha, 1Hb, 3Hc), 7.77 (d, J = 8.2 Hz, 1Hb), 7.86 (dd, J = 8.0, 0.9 Hz, 1Hb), 7.98–8.09 (m, 2Ha, 1Hc), 8.48 (dd, J = 8.3, 0.7 Hz, 1Hc), 8.54 (dd, J = 8.4, 0.4 Hz, 1Hb), 8.60 (dd, J = 8.4, 0.7 Hz, 1Ha), 8.73 (s, 1Hc), 9.04 (s, 1Hb), 9.27 (d, J = 3.7 Hz, 1Hb), 9.40 (d, J = 4.2 Hz, 1Ha), 9.51 (s, 1Ha), 11.43 (s, 1Hb), 12.22 (s, 1Ha), 12.67 (s, 1Hc). 13C NMR (100 MHz, DMSO-d6) δ 13.5 (Cb), 15.0 (Ca), 15.3 (Ca), 15.5 (Cb), 16.6 (Ca), 17.5 (Cb), 28.7 (Cb), 29.4 (Ca), 29.8 (Ca), 38.7 (Cb), 45.5 (Cb), 46.7 (Ca), 55.7 (Cb), 56.2 (Ca), 59.2 (Ca), 116.9 (Ca), 119.4 (Cb), 119.5 (Ca), 119.7 (Cb), 122.9 (Ca), 123.0 (Cb), 124.4 (Ca), 124.8 (Cb), 125.4 (Ca), 125.5 (Ca), 126.2 (Cb), 126.6 (Cb), 127.5 (Ca), 129.4 (Cb), 129.6 (Ca), 130.6 (Cb), 130.7 (Ca), 133.1 (Cb), 133.8 (Ca), 136.0 (Cb), 136.2 (Cb), 136.7 (Ca), 139.2 (Cb), 140.3 (Ca), 167.9 (Ca), 168.5 (Ca), 168.8 (2Cb), 169.3 (Cb), 169.5 (Ca), 169.6 (Ca), 169.9 (Cb), 170.1 (Cb), 173.0 (Ca), 173.1 (Cb). ESI-MS m/z calc. for C25H30N5O6 ([M + H]+) 480.2, found 480.3.
3.3.2.3. Cyclo-[NHMe-Ala-Anth-Ala-Anth-NMeAla] (versicotide B) (2). The trifluoroacetate salt of NHMe-Ala-Anth-Ala-Anth-NMeAla-OH was obtained as a white solid following the general SPPS procedure. The global yield based on the determination of the resin loading, was 87% (350 mg, 0.57 mmol). ESI-MS m/z calc. for C25H32N5O6 ([M + H]+) 498.2, found 498.4. Macrocyclization reaction was performed following the general procedure method I or II (dilution 5 mM, 3 days), starting from the trifluoroacetate salt of linear peptide NHMe-Ala-Anth-Ala-NMe-Ala-Anth-Ala-OH (200 mg, 0.33 mmol). Cyclization method I, using HATU as coupling agent, rendered the desired macrocycle in 18% yield (28 mg, 0.06 mmol) after purification by flash chromatography using AcOEt as mobile phase. HPLC analysis employing H2O
:MeCN and 0.1% formic acid in a 90:10 to 0:100 linear gradient showed two peaks tr = 7.5 and 7.8 min. Cyclization method II, using Oxyma and EDCI as coupling reagents rendered the desired macrocycle in 54% yield (65 mg, 0.13 mmol) after purification by flash chromatography using AcOEt:EP (3:2) as mobile phase. HPLC analysis employing H2O:MeCN and 0.1% formic acid in a 90:10 to 0:100 linear gradient in 15 min showed two peaks tr = 7.5 and 7.8 min in a 1.4:1 ratio. Collection of the peaks at the end of the column and LC-MS analysis revealed both peaks have the same mass as the natural product versicotide B. Thermal analysis by NMR proved them to be conformers. 91% total purity.
3.3.2.4. Cyclo-[NMeAla-Anth-Ala-Anth-NMeAla] (2). Yellow solid (Y = 54%). ESI-MS m/z calc. for C25H30N5O6 ([M + H]+) 480.2, found 480.6. 1H NMR (400 MHz, DMSO-d6) two conformers A
:B in 1:1.3 ratio. δ 1.13 (d, J = 6.6 Hz, 3Ha), 1.26 (d, J = 6.7 Hz, 3Hb), 1.40–1.44 (m, 6Ha, 3Hb), 1.47 (d, J = 6.5 Hz, 3Hb), 2.24 (s, 3Ha), 2.96 (s, 3Hb), 2.99 (s, 3Hb), 3.06 (s, 3Ha), 4.14–4.25 (m, 1Ha), 4.52–4.68 (m, 1Hb), 4.70–4.87 (m, 1Ha), 5.08–5.17 (m, 1Hb), 5.17–5.26 (m, 1Ha), 5.30–5.41 (m, 1Hb), 7.12–7.22 (m, 1Ha + 1Hb), 7.23–7.31 (m, 1Ha), 7.34 (d, J = 6.7 Hz, 1Hb), 7.39–7.58 (m, 2Ha, 2Hb), 7.60 (m, 2Ha), 7.81 (d, J = 6.1 Hz, 1Hb), 8.07 (d, J = 7.6 Hz, 1Hb), 8.16 (d, J = 8.2 Hz, 1Ha), 8.44–8.5 (m, 2Ha,+ 2Hb), 9.07–9.20 (m, 2Ha), 9.24 (s, 1Hb), 9.36 (s, 1Hb), 10.58 (s, 1Ha), 10.92 (s, 1Hb). 13C NMR (100 MHz, DMSO-d6) δ 13.5 (Ca), 13.8 (Cb), 14.8 (Ca), 16.7 (Ca), 17.1 (Cb), 17.5 (Cb), 28.7 (Ca), 30.3 (Cb), 30.8 (Ca), 34.3 (Cb), 49.8 (Cb), 50.3 (Cb), 52.1 (Ca), 52.5 (Ca), 52.6 (Cb), 54.5 (Ca), 120.3, 120.6, 122.1, 123.1, 123.7, 124.3, 124.6, 125.6, 126.1, 127.3, 128.6, 129.2, 129.4 (Ca), 130.2, 130.6, 132.1 (Ca), 132.9, 135.3, 136.2, 137.4 (Ca), 139.8, 141.6, 145.7, 168.4 (Cb), 168.6 (Ca), 169.6 (2Ca,b), 169.8 (Ca), 170.3 (Cb), 170.9 (Cb), 171.3 (Ca), 171.9 (Ca), 172.3 (Cb).
3.3.2.5. Cyclo-[NHMe-Ala-Anth-Ala-NMe-Ala-Anth-Ala] (versicotide C) (3). The trifluoroacetate salt of NHMe-Ala-Anth-Ala-NMe-Ala-Anth-Ala-OH was obtained as a white solid following the general SPPS procedure. The global yield, based on the determination of the resin loading, was 98% (240 mg, 0.35 mmol). ESI-MS m/z calc. for C28H37N6O7 ([M + H]+) 569.3, found 569.3. Macrocyclization reaction was performed following the general procedure method I (dilution 5 mM, 3 days), starting from the trifluoroacetate salt of linear peptide NHMe-Ala-Anth-Ala-NMe-Ala-Anth-Ala-OH (200 mg, 0.30 mmol), using HATU as coupling agent. Further purification by flash chromatography using AcOEt as mobile phase, rendered the desired macrocycle in 49% yield (77 mg, 0.15 mmol). HPLC analysis employing H2O
:MeCN and 0.1% formic acid in a 90:10 to 0:100 linear gradient showed 93% purity, tr = 6.88 min.
3.3.2.6. Cyclo-[NMe-Ala-Anth-Ala-NMe-Ala-Anth-Ala] (3). White solid (Y = 49%). Rf = 0.25 (AcOEt). [α]D25 = −35.83 (c 0.25, MeOH). 1H NMR (400 MHz, (CD3)2CO) two conformers were present: A (trans, trans)-conformer and B (cis, cis)-conformer in 1
:0.2 ratio. (trans, trans) Conformer: δ 1.43 (d, J = 7.0 Hz, 3H), 1.50 (d, J = 6.8 Hz, 3H), 3.53 (s, 3H), 3.93 (q, J = 6.8 Hz, 1H), 5.26–5.36 (m, 1H), 6.91–6.99 (m, 1H), 7.34–7.41 (m, 1H), 7.67 (d, J = 7.2 Hz, 1H), 7.75 (dd, J = 7.9, 1.4 Hz, 1H), 8.69 (dd, J = 8.4, 0.9 Hz, 1H), 11.88 (s, 1H). (cis, cis) Conformer: δ 1.34 (d, J = 6.6 Hz, 1H), 1.67 (d, J = 7.2 Hz, 1H), 2.73 (s, 3H), 4.69 (q, J = 7.2 Hz, 1H), 5.26–5.36 (m, 1H), 6.86–6.91 (m, 1H), 7.05–7.11 (m, 1H), 7.88 (dd, J = 8.1, 1.2 Hz, 1H), 8.07 (dd, J = 8.4, 0.8 Hz, 1H), 8.15 (d, J = 9.5 Hz, 1H), 12.29 (s, 1H). 13C NMR (100 MHz, (CD3)2CO) (trans, trans) conformer: δ 12.7(2C), 18.1(2C), 38.4(2C), 46.7(2C), 63.1(2C), 119.5(2C), 119.9(2C), 122.6(2C), 128.6(2C), 133.2(2C), 141.6(2C), 168.3(2C), 169.8(2C), 172.3(2C). (cis, cis) Conformer: δ 17.1(2C), 18.1(2C), 28.4(2C), 46.1(2C), 56.4(2C), 119.8(2C), 120.1(2C), 128.1(2C), 133.3(2C), 141.6(2C), 142.9(2C), 169.7(2C), 170.7(2C), 171.1(2C). ESI-MS m/z calc. for C28H35N6O6 ([M + H]+) 551.3, found 551.3.
3.3.2.7. Cyclo-[NH2Ala-Ala-Anth-Ala-Anth] (4). Compound 5 was prepared by solution phase synthesis, either by coupling method I or II (yield 42% and 51% respectively). Half of dipeptide 5 was deprotected on the C terminal following the general ester hydrolysis procedure. The other half was deprotected on the N terminal by Boc deprotection procedure. The resulting dipeptides were coupled following coupling method I to obtain 6 in a 10% yield. 6 was subjected to Boc deprotection conditions and then Boc-Ala-OH was coupled with coupling method I to obtain 7. 7 was deprotected first on the C terminal with ester hydrolysis procedure, and then at the N terminal with Boc deprotection procedure. The resulting peptide was cyclized following macrocyclization method II to render cyclopeptide 4 in 35% yield.
3.3.2.8. Cyclo-[Ala-Ala-Antr-Ala-Antr] (4). Yellow solid Rf = 0.41 (AcOEt
:EP, 4:1). 1H-NMR (400 MHz, (CD3)2CO) δ 1.27–1.36 (m, 3H), 1.53–1.62 (m, 6H), 4.46–4.58 (m, 1H), 4.63–4.75 (m, 1H), 4.80–4.95 (m, 1H), 7.08–7.19 (m, 2H), 7.72 (dd, J = 7.6, 1.0 Hz, 1H), 7.48 (m, 2H), 7.85 (d, J = 7.9 Hz, 1H), 7.98–8.06 (m, 1H), 8.10–8.19 (m, 1H), 8.20–8.26 (m, 1H), 8.35 (d, J = 8.3 Hz, 1H), 8.62 (d, J = 8.7 Hz, 1H), 11.05 (s, 1H), 11.31 (s, 1H). 13C NMR (100 MHz, (CD3)2CO) δ (ppm) 15.5, 17.1, 17.4, 49.6, 50.4, 51.2, 121.1, 121.2, 121.4, 121.5, 122.6, 122.8, 123.7, 125.3, 127.4, 128.6, 131.4, 132.1, 168.3, 170.2, 171.2, 173.0, 173.1. MS m/z calc. for C23H25N5O5 ([M + H]+) 452.19, found 452.2.See ESI† for HPLC chromatograms, MS and NMR spectra data of the cyclopeptides.
4. Conclusions
In conclusion, the syntheses of three natural products, versicotides A–C, containing Anth, were accomplished in very good yields and purity after searching for the most adequate methodology and coupling reagents. The combination of oxyma and DIC, provided a simple methodology to achieve difficult couplings involving the aromatic amine of Anth, in short times, to avoid possible rearrangements or side reactions, and with very good yields. This methodology could be applied to the synthesis of interesting compounds containing this motif.Macrocyclization of versicotides A and C was achieved with HATU and no epimerization was detected. For macrocyclcization of versicotide B (2), Oxyma and EDCI provided higher yield. In the versicotides A–C NMR spectra, the presence of well-defined peaks for each conformer shows that these molecules are rigid enough for the conversion from one conformer into another to be slower than NMR time scale. A deep analysis of versicotide C NMR spectra led us to conclude that a mixture of (cis, cis) and (trans, trans) symmetrical conformers is present in DMSO-d6 rather than a single (cis, trans) conformer as previously reported for the natural product. Biological evaluation against P. falciparum of compounds 1–4 will be performed and reported in the due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the collaboration of Leopoldo Suescun and Alejandro Peixoto to obtain the versicotide A X-ray crystallographic data. This work was supported by Grants from CSIC Grupos 2006 (Universidad de la República) and PEDECIBA (Uruguay). The authors acknowledge a postgraduate fellowship from CAP (UdelaR) (Laura Posada).Notes and references
- (a) F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57, 278 CrossRef CAS; (b) S. Peña, L. Scarone and G. Serra, Future Med. Chem., 2015, 7, 355 CrossRef.
- (a) J. Mallinson and I. Collins, Future Med. Chem., 2012, 4, 1409 CrossRef CAS; (b) J. E. Bock, J. Gavenonis and J. A. Kritzer, ACS Chem. Biol., 2013, 8, 488 CrossRef CAS; (c) A. Bhat, L. R. Roberts and J. J. Dwyer, Eur. J. Med. Chem., 2015, 94, 471 CrossRef CAS; (d) A. A. Vinogradov, Y. Yi and H. Suga, J. Am. Chem. Soc., 2019, 141(10), 4167 CrossRef CAS.
- B. I. Díaz-Eufracio, O. Palomino-Hernández, R. A. Houghten and J. L. Medina-Franco, Mol. Diversity, 2018, 22, 259 CrossRef.
- (a) W. M. Hewitt, S. S. F. Leung, C. R. Pye, A. R. Ponkey, M. Bednarek, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2015, 137, 715 CrossRef CAS; (b) L. Doedens, F. Opperer, M. Cai, J. G. Beck, M. Dedek, E. Palmer, V. J. Hruby and H. Kessler, J. Am. Chem. Soc., 2010, 132, 8115 CrossRef CAS; (c) O. Ovadia, S. Greenberg, J. Chatterjee, B. Laufer, F. Opperer, H. Kessler, C. Gilon and _A. Hoffman, Mol. Pharmaceutics, 2011, 8, 479 CrossRef CAS.
- (a) A. Alex, D. S. Millan, M. Perez, F. Wakenhut and G. A. Whitlock, MedChemComm, 2011, 2, 669 RSC; (b) A. Whitty, M. Zhong, L. Viarengo, D. Beglov, D. R. Hall and S. Vajda, Drug Discovery Today, 2016, 21, 712 CrossRef CAS; (c) P. Matsson and J. Kihlberg, J. Med. Chem., 2017, 60, 1662 CrossRef CAS; (d) G. Caron and G. Ermondi, Drug Discovery Today, 2017, 22, 835 CrossRef CAS; (e) M. Rossi Sebastiano, B. C. Doak, M. Backlund, V. Poongavanam, B. Over, G. Ermondi, G. Caron, P. Matsson and J. Kihlberg, J. Med. Chem., 2018, 61, 4189 CrossRef CAS.
- D. W. Carney, K. R. Schmitz, J. V. Truong, R. T. Sauer and J. K. Sello, J. Am. Chem. Soc., 2014, 136, 1922 CrossRef CAS.
- L. M. De Leon Rodriguez, E. T. Williams and M. A. Brimble, Chemistry, 2018, 24, 17869 CrossRef CAS.
- (a) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef CAS; (b) J. W. Li and J. C. Vederas, Science, 2009, 325(5937), 161 CrossRef.
- (a) C. Jiménez, ACS Med. Chem. Lett., 2018, 9(10), 959 CrossRef; (b) T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2008, 8, 69 CrossRef.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010 RSC.
- L. N. Zhou, H. Q. Gao, X. S. Cai, T. J. Zhu, Q. Q. Gu and D. H. Li, Helv. Chim. Acta, 2011, 94, 1065 CrossRef CAS.
- J. Peng, H. Gao, X. Zhang, S. Wang, C. Wu, Q. Gu, P. Guo, T. Zhu and D. Li, J. Nat. Prod., 2014, 77, 2218 CrossRef CAS.
- R. Chen, Z. Cheng, J. Huang, L. Dong, C. Wu, P. Guo and W. Lin, RSC Adv., 2017, 7, 49235 RSC.
- (a) S. Peña, C. Fagundez, A. Medeiros, M. Comini, L. Scarone, D. Sellanes, E. Manta, J. Tulla-Puche, F. Albericio, L. Stewart, V. Yardley and G. Serra, MedChemComm, 2014, 5, 1309 RSC; (b) C. Fagundez, D. Sellanes and G. Serra, ACS Comb. Sci., 2018, 20, 212 CrossRef CAS; (c) C. Fagundez, D. Sellanes, S. Peña, L. Scarone, A. C. C. Aguiar, J. de Souza, R. V. C. Guido, L. Stewart, V. Yardley, S. Ottilie, E. A. Winzeler, F.-J. Gamo, L. M. Sanz and G. L. Serra, ACS Med. Chem. Lett., 2019, 10, 137 CrossRef CAS.
- L. Posada and G. Serra, Tetrahedron Lett., 2019, 60(48), 151281 CrossRef CAS.
- (a) S. T. Y. Ngen, H. Kaur, P. A. Hume, D. P. Fukert and M. A. Brimble, J. Org. Chem., 2016, 81, 7635 CrossRef CAS; (b) K. Nakao, Y. Hamada and T. Shioiri, Chem. Pharm. Bull., 1989, 37, 930 CrossRef CAS; (c) M. C. Tseng, H. Y. Yang and Y. H. Chu, Org. Biomol. Chem., 2010, 8, 419 RSC; (d) S. K. Chettu, R. B. Madhu, G. B. Raolji, K. R. Babu, N. S. K. Rao, S. Gopalakrishnan, A. Ismail, G. B. Reddyc and S. Shafi, RSC Adv., 2016, 6, 61555 RSC; (e) P. Lewer, P. R. Graupner, D. R. Hahn, L. L. Karr, D. O. Duebelbeis, J. M. Lira, P. B. Anzeveno, S. C. Fields, J. R. Gilbert and C. Pearce, J. Nat. Prod., 2006, 69, 1506 CrossRef CAS.
- (a) R. Costil, H. J. A Dale, N. Fey, G. Whitcombe, J. V Matlock and J. Clayden, Angew. Chem., Int. Ed., 2017, 56, 12533 CrossRef CAS; (b) J. Noguchi, M. Kawai and M. Hamada, Isr. J. Chem., 1974, 12, 87 CrossRef CAS.
- N. Vasudevan, K. Kashinath and D. S. Reddy, Org. Lett., 2014, 16(23), 6148 CrossRef CAS.
- E. Falb, T. Yechezkel, Y. Salitra and C. Gilon, J. Pept. Res., 1999, 53, 507 CrossRef CAS.
- D. Xin and K. Burgess, Org. Biomol. Chem., 2016, 14, 5049 RSC.
- (a) S. B. Baravkar, M. A. Wagh, D. Paul, M. Santra and G. J. Sanjayan, Tetrahedron Lett., 2018, 59, 3473 CrossRef CAS; (b) S. B. Baravkar, A. Roy, R. L. Gawade, V. G. Puranik and G. J. Sanjayan, Synth. Commun., 2014, 44, 2955 CrossRef CAS; (c) L. A. Errede, J. Org. Chem., 1976, 41, 1763 CrossRef CAS; (d) L. A. Errede, J. J. McBrady and H. T Oien, J. Org. Chem., 1977, 42, 656 CrossRef CAS.
- Y. Masuda, R. Tanaka, K. Kai, A. Ganesan and T. Doi, J. Org. Chem., 2014, 79(17), 7844 CrossRef CAS.
- R. Subirós-Funosas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Chem.–Eur. J., 2009, 15, 9394 CrossRef.
- (a) D. E. Dorman and F. A. Bovey, J. Org. Chem., 1973, 38, 1719 CrossRef CAS; (b) R. Quintanilla-Licea, J. F. Colunga-Valladares, A. Caballero-Quintero, C. Rodríguez-Padilla, R. Tamez-Guerra, R. Gómez-Flores and N. Waksman, Molecules, 2002, 7, 662 CrossRef CAS.
- X ray CDCC reference number of compound 1: 2023773†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2023773. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09635k |
| This journal is © The Royal Society of Chemistry 2020 |