
New bifunctional-pullulan-based micelles with good biocompatibility for efficient co-delivery of cancer-suppressing p53 gene and doxorubicin to cancer cells
Lili Chenb,
Xiaohong Wangab,
Fangling Jia,
Yongming Baob,
Jingyun Wang*ab,
Xianwu Wangb,
Lianying Guoc and
Yachen Li*c
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, P.R. China. E-mail: wangjingyun67@dlut.edu.cn; Fax: +86-41184706365; Tel: +86-41184706356
bSchool of Life Science and Biotechnology, Dalian University of Technology, Dalian, 116024, P.R. China
cDepartment of Environmental Health and Toxicology, School of Public Health, Dalian Medical University, Dalian 116044, P.R. China. E-mail: liy76@yahoo.com; Fax: +86-41186110329; Tel: +86-41186110330
First published on 27th October 2015
Abstract
Combined treatment of drugs and therapeutic genes has emerged as a new modality of anticancer therapy. In this study, a new amphiphilic bifunctional pullulan derivative (named as PSP) containing stearic acid and low-molecular weight (1 kDa) branched polyethylenimine was prepared and evaluated as a nano-carrier for the co-delivery of a drug and gene for potential cancer therapy. The amphiphilic PSP could self-assemble into cationic core–shell nano-micelles in water, with a critical micelle concentration of around 58.9 mg L−1. PSP nanomicelles had an average size of 188.75 ± 3.18 nm, and a positive zeta potential of 17.83 ± 0.75 mV. The drug loading content and encapsulation efficiency of the PSP nanomicelles for doxorubicin (DOX), an anti-tumor drug, were about 5.10% and 56.07%, respectively, and DOX in PSP nanomicelles showed sustained release. The flow cytometry and confocal laser scanning microscopy showed that PSP/DOX nanomicelles could be successfully internalized by MCF-7 cells. The in vitro IC50 of PSP/DOX nanomicelles was slightly lower than that of free DOX against MCF-7 cells. Additionally, PSP nanomicelles condensed DNA efficiently to form compact structures, and induced comparable GFP gene expression level to Lipo2000 at N/P = 10 in gene transfection studies. In comparison with single DOX or p53 delivery, the co-delivery of DOX and therapy gene p53 using PSP micelles displayed higher cytotoxicity and induced a higher apoptosis rate of tumor cells in vitro. Moreover, PSP exhibited good blood compatibility and low cytotoxicity in the hemolysis and MTT assays, respectively. Altogether, PSP nanomicelles have a great potential in delivering hydrophobic anticancer drugs and therapeutic genes simultaneously for improved cancer therapy.
Introduction
Chemotherapy remains the main conventional option to treat cancer. But the majority of chemotherapeutic agents damage healthy tissues leading to systemic toxicity and adverse side effects.1 Moreover, the complexity of their signaling network allows tumor cells to develop multiple pathways to escape from death induced by chemotherapeutics.2 Therefore, new combination therapy strategies for cancer treatment taking the advantages of the co-delivery of more than one therapeutic agent in one delivery system, have recently been shown to be more effective than monotherapies through providing potential synergistic effects of different treatment mechanisms.3,4 Recently, the co-delivery of nucleic acids and chemotherapeutics has been proposed to achieve a synergistic/combined effect of gene therapy with chemotherapy.5,6 The construction of highly efficient new nanocarriers with multifunctionality has drawn increasing attention and represents a growing area of research in drug-delivery systems.7Up to now, attempts have been made to deliver drugs and DNA/RNA simultaneously into cancer cells using polymers,8–10 liposomes,11–13 dendrimers,14,15 mesoporoussilica nanoparticles,16,17 quantum-dot (QD)-based nanoparticles,18 polysaccharides19 and so on. Amongst them, biodegradable amphiphilic cationic copolymers have received considerable attention recently. Because they can easily self-assemble into core–shell nanoparticles (NPs) with a hydrophobic core and a cationic hydrophilic shell in an aqueous solution, and load hydrophobic antitumor drugs and polyanionic genes simultaneously in a single-component carrier.20,21 For example, Zheng et al.,22 synthesized the amphiphilic triblock copolymer poly(ethylene glycol)-b-poly(L-lysine)-b-poly(L-leucine) polypeptide to co-deliver docetaxel and siRNA-Bcl-2 to overcome drug resistance in MCF-7 cells. Ma et al.,23 reported a novel cyclodextrin derivative (CD-PLLD) for the co-delivery of docetaxel (DOC) and siRNA plasmid targeting MMP-9 (pMR3). Their data demonstrated that CD-PPLD/DOC/pMR3 complexes could induce a more significant apoptosis of HNE-1 cells than DOC or pMR3 used only.
Pulluan is a natural non-ionic and linear homopolysaccharide formed by repeating units of maltotriose condensed through α-1,6-linkages.24 Due to its non-immunogenic, non-toxic, non-carcinogenic, and non-mutagenic nature, and to the possibility to easily modify it, pullulan has gained considerable attention in various pharmaceutical and biomedical applications such as platform materials for drug delivery25–27 and gene-delivery systems.28,29 For instance, Seogjin Seo et al.30 reported the physicochemical characterization and in vitro release of doxorubicin from self-organized nanogels of pullulan-g-poly(L-lactide) copolymers. Thakor et al.31 achieved an efficient in vitro gene expression in rapidly proliferating tumor cells by conjugating the naturally-occurring pullulan and polyamine spermine. Rekha,29,32 modified pullulan with different amounts of cationic groups by reacting glycidyltrimethyl ammonium chloride or polyethylenimine (PEI, 25 kDa) with pullulan to make new gene-delivery systems. However, to our knowledge, few researches have concerned the application of amphiphilic pullulan derivatives as co-delivery systems of drug and gene.
In previous studies, low-molecular-weight branched PEI (1 kDa)-grafted pullulan nanoparticles were developed as a safe and effective delivery system for gene.33 The amphiphilic hydrophobically-modified pullulan derivatives also were reported to promote the encapsulation and cellular uptake of hydrophobic antitumor drugs.30 Therefore, in our present study, a new amphiphilic pullulan derivative (called as PSP) consisting of stearic acid (SA) and low-molecular-weight branched PEI (1 kDa) was synthesized to form micellar carriers for the efficient co-delivery of drug and gene to cancer cells. Herein, the PEI part would act as a complexing site with DNA through electrostatic interactions as well as the hydrophilic shell of the self-assembled micelles, while SA constructed a hydrophobic core to encapsulate hydrophobic anticancer drugs. Micelles self-assembled from PSP polymer were characterized by their critical micelle concentration (CMC), hydrodynamic particle size, and zeta potential. Doxorubicin (DOX) was chosen as a model drug since DOX is not only one of the most effective chemotherapeutics for many cancer treatments such as ovarian and breast cancers, but also can emit detectable fluorescence, which may allow studying cellular uptake with a flow cytometer and a confocal laser-scanning microscope (CLSM). Drug loading capacity of PSP micelles and in vitro drug release from the micelles were investigated. The cytotoxicity of DOX-loaded PSP micelles (PSP/DOX) was evaluated against MCF-7 cells in comparison with that of free DOX. The DNA binding ability of PSP micelles, DNase I protection of DNA by the micelles were studied through gel-retardation assay, and the possibility of using these PSP micelles to transfect gene into COS-7 cells was also explored with green fluorescent protein reporter gene. The cellular uptake and intracellular distribution of PSP/DOX were detected through a flow cytometer and a confocal laser-scanning microscope. Furthermore, in order to evaluate the feasibility of using the PSP micelles as drug and gene co-delivery carriers for cancer treatment, the antitumor efficacy of the co-delivery of DOX and tumor suppressor gene p53 using these PSP micelles was assayed by MTT and annexin V-FITC/PI apoptosis assay in vitro. The tumor suppressor gene p53 has a pivotal role in inducing apoptosis in response to cellular damage. p53 mutations occur in almost half of all soft tissue sarcomas, which also contribute to drug resistance. It was reported that p53-gene delivery could sensitize cancer cells towards anticancer drugs.34 We expected that the co-delivery of DOX and p53 by PSP micelles would show the potential to achieve synergistic or combined effects in inducing cancer cell death. Additionally, the biocompatibility of PSP micelles was also evaluated by hemocompatibility assay and MTT assay.
Experimental
Materials and reagents
Pullulan (Mw = 100 kDa) was purchased from Shandong Zhongqing Biotechnology Company. Stearic acid (SA) was obtained from Tianjin Fuchen Chemical Reagents Factory. Branched polyethylenimine (PEI, Mw = 1 kDa) was purchased from Wuhan Qianglong New Chemical Materials Company. 1-Ethyl-3-(3-dimethyllaminopropyl) carbodiimide hydrochloride (EDC·HCl), 4-dimethylaminopyridine (DMAP), N-hydroxysuccinimide (NHS), propidium iodide (PI) were obtained from Aladdin Industrial Corporation. Lipofectamine™ 2000 was obtained from Invitrogen. Plasmid encoding enhanced green fluorescent protein (pGFP) was provided by Professor Li wenli (Dalian University of Technology, China). Plasmid encoding tumor suppressor wild-type p53 protein driven by CMV promoter (pCMV-p53-wt) was from Shanghai Jiaotong University micro-nano science and technology research institute. All other reagents were of analytical grade and used without further purification.Human breast cancer cell (MCF-7), African green monkey kidney cell (COS-7) and human embryonic kidney cell (HEK293) lines were purchased from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, SIBS, CAS and cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (50 U mL−1) and streptomycin (50 U mL−1) under 5% CO2 at 37 °C. All experiments were performed on cells in the logarithmic phase of growth.
Preparation and chemical characterization of amphiphilic pullulan derivative PSP
PSP was synthesized in a three-step reaction as shown in Scheme 1 according to the methods described in literatures33,35 with some modifications. For the synthesis of pullulan-SA, 0.57 g (2 mmol) SA, 0.61 g (2 mmol) DMAP and 0.96 g (2.4 mmol) EDC were dissolved in 5 mL anhydrous dimethyl sulfoxide (DMSO), and the carboxyl group of SA was activated by stirring the mixture for 1 h at room temperature. Then, the mixture was added dropwise to pullulan (1.64 g, 10 mmol)/DMSO solution, and the reactant mixture was further stirred at room temperature for 48 h. After reaction completion, the reaction mixture was added to 500 mL of absolute ethyl alcohol to precipitate pullulan-SA. Pullulan-SA obtained by centrifugation was redissolved in 10 mL of deionized water and dialyzed against deionized water (MWCO, 10 kDa) for 3 d with change of water at specific intervals, and then pure pullulan-SA was obtained by freeze drying in lyophilizer. | ||
| Scheme 1 Synthesis scheme of PSP. | ||
For the synthesis of pullulan-SA-COOH, pullulan-SA (0.95 g, 5 mmol) was dissolved in anhydrous DMSO (20 mL), then added with succinic anhydride (0.75 g, 7.5 mmol) and DMAP (0.12 g, 1 mmol). The solution was stirred at 50 °C for 18 h. Then, the reaction mixture was poured into anhydrous ethanol to precipitate pullulan-SA-COOH. The precipitate was further dissolved in distilled water and dialyzed (MWCO, 10 kDa) against deionized water for 72 h. The pullulan-SA-COOH solution was lyophilized and stored at 4 °C until further use. The degree of succinoylation was calculated by acid–base titration against 0.1 M NaOH with phenolphthalein as an indicator.
Finally, for the preparation of PSP, pulluan-SA-COOH (0.22 g, n–COOH = 0.65 mmol) was dissolved in 0.2 M PBS (pH 7.8), and EDC (0.187 g, 0.975 mmol) and NHS (0.112 g, 0.975 mmol) were added to this solution. The mixture was stirred at 4 °C for 4 h and then, PEI (0.171 g, n–NH2 = 0.650 mmol) was added dropwise. The reaction was carried out at 25 °C for 24 h under stirring. Then, the reaction mixture was dialyzed against deionized water for 3 d. The resultant solution was freeze dried to yield white polymer PSP.
Fourier transform infrared (FT-IR), 1H NMR were employed to confirm the chemical structures of PSP and all intermediates. FT-IR spectra were recorded on a nicolet 6700 instrument using KBr method, and 1H NMR were measured by 1H NMR at 400 MHz (Varian INOVA400, USA) in D2O or DMSO-d6.
Determination of critical micelle concentration (CMC)
The CMC of PSP was determined with fluorescent spectroscopy. Pyrene was used as a extrinsic fluorescent probe to prove the potential of micelle formation.36 Briefly, 30 μL of 6.0 × 10−5 M pyrene solution in methanol was added to glass vials, and then methanol was evaporated. Subsequently, 3 mL of different concentrations of PSP solution (1 × 10−4 mg mL−1 to 1 mg mL−1) was added to the glass vials and left to stand in darkness for 1 h. The solutions were incubated at 37 °C in darkness for another 1 h, followed by sonication for 1 h. Fluorescence spectra were then recorded using a microplate reader (Thermo Fisher Scientific, Shanghai, China) with an excitation wavelength of 334 nm and emission wavelength changed from 350 nm to 450 nm. The excitation and emission bandwidths were both 2.5 nm. The intensity ratios of I372 to I383 were plotted as a function of the logarithm of PSP concentration. The CMC value was obtained as the crossover point of the two tangents of the curves.Preparation of blank PSP micelles and DOX-loaded PSP micelles (PSP/DOX)
Blank PSP nanomicelles were prepared by dialysis method. Exactly 10 mg of PSP amphiphilic copolymers was dissolved in 2 mL of DMSO, and the solution was dialyzed against 200 mL distilled water for 48 h (MWCO, 10 kDa). Deionized water was replaced every 4 h for the first 12 h and then every 6 h thereafter. Subsequently, the solution was lyophilized, and the blank PSP micelles were obtained in powder and stored prior to use.PSP/DOX micelles were also prepared with a dialysis method. Briefly, DOX·HCl was dissolved in DMSO in the presence of triethylamine (2.0 times molar quantity of DOX) to form a free DOX containing solution, and then mixed with PSP/DMSO solution at a drug/carrier feeding weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. The resulting solution was transferred into a dialysis bag (MWCO, 10 kDa) and subjected to dialysis against 200 mL distilled water for 72 h to remove non-encapsulated drug and organic solvent. The dialysis medium was changed every 8 h and then freeze-dried to obtain PSP/DOX micelles. The DOX loading content (LC) and encapsulation efficiency (EE) were calculated as: LC% = (mass of DOX encapsulated in micelles)/(mass of PSP/DOX micelles) × 100% and EE% = (mass of DOX encapsulated in micelles)/(mass of DOX added) × 100%.
:10. The resulting solution was transferred into a dialysis bag (MWCO, 10 kDa) and subjected to dialysis against 200 mL distilled water for 72 h to remove non-encapsulated drug and organic solvent. The dialysis medium was changed every 8 h and then freeze-dried to obtain PSP/DOX micelles. The DOX loading content (LC) and encapsulation efficiency (EE) were calculated as: LC% = (mass of DOX encapsulated in micelles)/(mass of PSP/DOX micelles) × 100% and EE% = (mass of DOX encapsulated in micelles)/(mass of DOX added) × 100%.
In vitro release of DOX from PSP/DOX micelles
The in vitro release of DOX from PSP/DOX micelles was studied using a dialysis method.37 Lyophilized PSP/DOX micelles containing 0.2 mg DOX were suspended in 2 mL of PBS (10 mM) with different pH (5.0, 6.5 and 7.4) and placed in a dialysis tube (MWCO, 10 kDa). Then, the tube was immersed in the corresponding release media (200 mL) in a shaking water bath at 37 °C. At predetermined time intervals, 3 mL of release media was withdrawn and replaced with 3 mL of the corresponding fresh buffer solution. The amount of DOX release was determined by fluorescence measurement (excitation at 485 nm).Formation of PSP/pDNA and PSP/DOX/pDNA complexes
The plasmidic DNA (pDNA) was used in the formation of complexes between DNA and PSP micelles. Briefly, a series of PSP micelle or PSP/DOX micelle solutions with varying micelle concentration were prepared by dissolving micelles in deionized water first. After adding a calculated amount of DNA to each vial, the mixture was vortexed for 15 s and kept still at room temperature for 30 min. As a result, a series of PSP/pDNA and PSP/DOX/pDNA complexes with different N/P ratios (weight of PSP/weight of pDNA) were formed.Particle size, zeta potential measurements and morphology
The particle size and zeta potential of the PSP micelles, PSP/pDNA complexes, as well as PSP/DOX/pDNA complexes were measured by dynamic light scattering (DLS, Brookhaven Instrument Corp., Holtsville, NY, USA) and Zetasizer (Malvern Instrument Ltd., Worchestershire, UK) respectively. All the particle size measurements were performed with a He–Ne laser beam at 658 nm and a scattering angle of 90. The zeta potential data were calculated automatically using the software from the electrophoretic mobility based on Smoluchowski's formula. For each sample, data obtained from three measurements were averaged to yield the mean particle size and zeta potential. The morphology of PSP, PSP/DOX, PSP/pDNA and PSP/DOX/pDNA micelles was observed by transmission electron microscopy (TEM, Tecnai G2 Spirit 120 kV) after counter stained for 2 min with uranyl acetate.Gel-retardation assay
PSP/pDNA or PSP/DOX/pDNA complexes at various weight ratio of PSP to pDNA (N/P ratio) were prepared. Subsequently, 5 μL of the pDNA complexes (pDNA = 1 μg per lane) and naked pDNA were electrophoresed on a 1% agarose gel containing 0.2 μg mL−1 ethidium bromide in Tris-acetate–ethylene diaminetetraacetic acid buffer at 140 V for 40 min. The gels were visualized and imaged under an ultraviolet transilluminator (G: BOX, Gene Company, Hong Kong, China).For DNase resistance assay, 5 μL PSP/pDNA containing 1 μg pDNA with different N/P ratios was incubated with 1 unit of DNase I in 2 μL buffer containing 40 mM Tris–HCl, 10 mM MgCl2, and 1 mM CaCl2 (pH 8.0) at 37 °C for 30 min or 4 h. The digestion was terminated by the addition of 7 μL of 10 mM EDTA solution. The mixtures were then heated in a water bath at 65 °C for 10 min to denature DNase I. To dissociate pDNA from the PSP polymers, 7 μL of heparin solution (240 mg mL−1) was added, and the mixtures were further incubated at 37 °C for 2 h. Subsequently, 18 μL of the solution was electrophoresed on 1.0% agarose gel to examine the integrity of the recovered pDNA.
In vitro transfection
The plasmid pGFP was employed to examine transfection efficiency of PSP in vitro. Briefly, COS-7 cells were seeded onto 6-well culture plates (1 × 105 cells per well) in DMEM containing 10% FBS and incubated for 24 h at 37 °C in a humidified atmosphere with 5% CO2 in air prior to transfection. Then, the medium was removed and replaced with 2 mL serum-free medium containing PSP/pGFP at different N/P ratios (2.5, 5, 10, 20) with 4 μg of pGFP. After 4 h incubation, the medium was removed and 2 mL of fresh DMEM culture medium with 10% serum was added. Then, after a further 44 h of incubation, the cells were analyzed for green fluorescence protein (GFP) expression with a fluorescence microscope (OLYMPUS, Japan). Moreover, a flow cytometer (FACSCanto, BD, USA) was used to determine the transfection efficiency of PSP with the percentage of cells expressing GFP. Naked pGFP and Lipo2000/pGFP were respectively utilized as negative and positive controls.Cellular uptake and distribution of DOX
As for the cellular uptake of free DOX and PSP/DOX nanomicelles, MCF-7 cells were seeded onto 6-well culture plates (1 × 105 cells per well) in DMEM containing 10% FBS and incubated for 24 h at 37 °C in a humidified atmosphere with 5% CO2 in air. Then, the cells were incubated with free DOX and PSP/DOX (2.5 μg mL−1 DOX-equivalent) for 0.5 h and 2 h respectively. After incubation, the cells were rinsed with PBS three times and trypsinized. The cell pellets were collected using centrifugation, re-suspended in 200 μL ice PBS, and the fluorescence intensity of DOX was analyzed by a flow cytometer.The intracellular distribution of free DOX and PSP/DOX was examined by confocal laser scanning microscopy (CLSM). MCF-7 cells were grown on glass bottom dishes in DMEM containing 10% FBS and incubated at 37 °C for 24 h in a humidified atmosphere with 5% CO2 in air. Then cells were cultured with free DOX and PSP/DOX nanomicelles at a DOX concentration of 2.5 μg mL−1 for 0.5 h or 2 h. After incubation, the cells were washed with PBS three times. Then the nuclei was stained with Hoechst 33342 with a final concentration of 10 μg mL−1 for 30 min. Fluorescence images of cells were obtained on an OLYMPUS FV 1000 (OLYMPUS, Japan).
Cellular toxicity assay
The cytotoxicities of free DOX, PSP/DOX and PSP/DOX/p53 were examined in vitro using MTT assay. Briefly, MCF-7 cells were cultured onto a 96-well plate (1 × 104 cells per well) in DMEM containing 10% FBS and incubated for 24 h (37 °C, 5% CO2). Then, the old medium was replaced with the medium containing different concentrations of the tested samples. Untreated cells in growth medium were used as the blank control. After 24 or 48 h of culture, the medium in each well was replaced with 100 μL fresh medium. 20 μL MTT reagents (5 mg mL−1) was added to each well and the cells were incubated for another 4 h at 37 °C. Subsequently, the medium was removed, the resulting formazan product was dissolved with 200 μL DMSO, and the absorbance was measured at 570 and 630 nm using a microplate reader (Thermo Fisher Scientific). Cell viability was expressed as a percentage of the control culture value.Apoptosis assay
MCF-7 cells were seeded onto a 6-well plate at a density of 1 × 105 cells per well in DMEM containing 10% FBS (37 °C, 5% CO2). After 24 h, the cells were exposed to blank PSP, free p53, free DOX, free p53 + free DOX, Lipo2000/p53, PSP/p53, PSP/DOX, PSP/DOX/p53 (0.5 μg mL−1 of DOX and 0.95 μg mL−1 of p53 at N/P = 10) at 37 °C for 24 h. Then, all cells were trypsinized, collected and resuspended in 200 μL of binding buffer. Thereafter, 5 μL of annexin V-FITC and 10 μL of PI were added and mixed for 15 min in the dark. The stained cells were analyzed using a flow cytometer. The untreated cells were used as control.Hemocompatibility assay and cell viability assay
1 mL of 2% red blood cells (RBCs) suspensions in PBS from sheep blood were added to PSP solution and the final concentrations of PSP were 0.5, 1, 2 mg mL−1 respectively. The mixture was incubated at 37 °C for 1 h, and then centrifuged at 1500 rpm for 10 min to remove intact RBCs. The absorbance of the supernatant was determined for the release of hemoglobin at 545 nm using a microplate reader. All measurements were performed in triplicate. PBS and deionized water were used as negative and positive control respectively. The hemolysis rate (%) was calculated as:| HR (%) = (ODsample − ODnegative control)/(ODpositive control − ODnegative control) × 100%. |
For cell viability assay, MCF-7 cells (COS-7, and HEK293 cells) were cultured onto a 96-well plate at a density of 1 × 104 cells per well in DMEM containing 10% FBS and incubated for 24 h (37 °C, 5% CO2). Then, the old medium was replaced with the medium containing different concentrations of PSP (2.5, 5, 10, 40 μg mL−1). After 48 h of culture, the medium in each well was replaced with 100 μL fresh medium. 20 μL MTT reagents (5 mg mL−1) was added to each well and the cells were incubated for another 4 h at 37 °C. Subsequently, the medium was removed, the resulting formazan product was dissolved with 200 μL DMSO, and the absorbance was measured at 570 and 630 nm using a microplate reader (Thermo Fisher Scientific). The untreated cells were used as control. Cell viability was expressed as a percentage of the control culture value.
Statistical analysis
All the experiments were performed three times and the data were presented as means ± standard deviation (S.D.). Statistical analysis was performed according to the Student's t-test and one-way ANOVA analysis by Origin version 8.5 software (Northampton, USA). The difference was considered to be statistically significant when p value < 0.05 (*) and p value < 0.01 (**).Results and discussion
Synthesis and characterization of PSP copolymers
The synthesis scheme of PSP polymer is shown in Scheme 1. In order to prepare the pullulan-based bifunctional PSP polymer as the co-delivery system for drug and gene, SA was grafted onto pullulan backbone through reaction between the carboxyl group of SA and hydroxyl linked to the C6 of sugar ring in pullulan to obtain pullulan-SA. Then, low-molecular weight branched PEI was further grafted on pullulan-SA with succinic anhydride as a spacer to produce PSP polymer finally.The chemical structure of PSP was first confirmed using FT-IR spectra, as shown in Fig. 1. In comparison with the spectrum of pullulan (Fig. 1A), the new absorption band at 1717 cm−1 in Fig. 1B belonged to the ester carbonyl of pullulan-SA, which was absent in pullulan and the enhanced characteristic peaks at around 2926 cm−1 of pullulan-SA were attributed to the symmetric or asymmetric stretching vibrations of CH2 and CH3 in SA. Moreover, the FT-IR spectrum of PSP (Fig. 1C) revealed a characteristic amide peak of PEI at 1561 cm−1.29
 | ||
| Fig. 1 FT-IR spectra obtained for (A) pullulan, (B) pullulan-SA and (C) PSP. FT-IR spectra were recorded on a nicolet 6700 instrument using KBr method. | ||
1H NMR was further used to indentify the chemical structure of PSP, and the 1H NMR spectra of pullulan, pullulan-SA, and PSP are displayed in Fig. 2. In comparison with the 1H NMR spectrum of pullulan (Fig. 2A), the new chemical shifts for protons at the range of 0.80–1.3 ppm in Fig. 2B were associated with methyl protons (–CH3) and methylene protons (–CH2–) of stearic acid, which demonstrating that SA has been grafted on pullulan backbone successfully. In Fig. 2C, the appearance of characteristic resonance proton peaks of PEI (–NHCH2CH2–) at 2.43–3.05 ppm confirmed that PEI was introduced to pullulan-SA in PSP, but the signals of SA could not be detected, which indicating PSP copolymer maybe have spontaneously self-assembled into micellar structure in D2O, with stearic acid in the core, pullulan and PEI in the shell. However, the 1H NMR of PSP in DMSO (Fig. 2D), the signals of SA were recovered. Therefore, all the results demonstrated that SA and PEI were grafted onto the backbone of pullulan polysaccharide and PSP polymer was successfully synthesized.
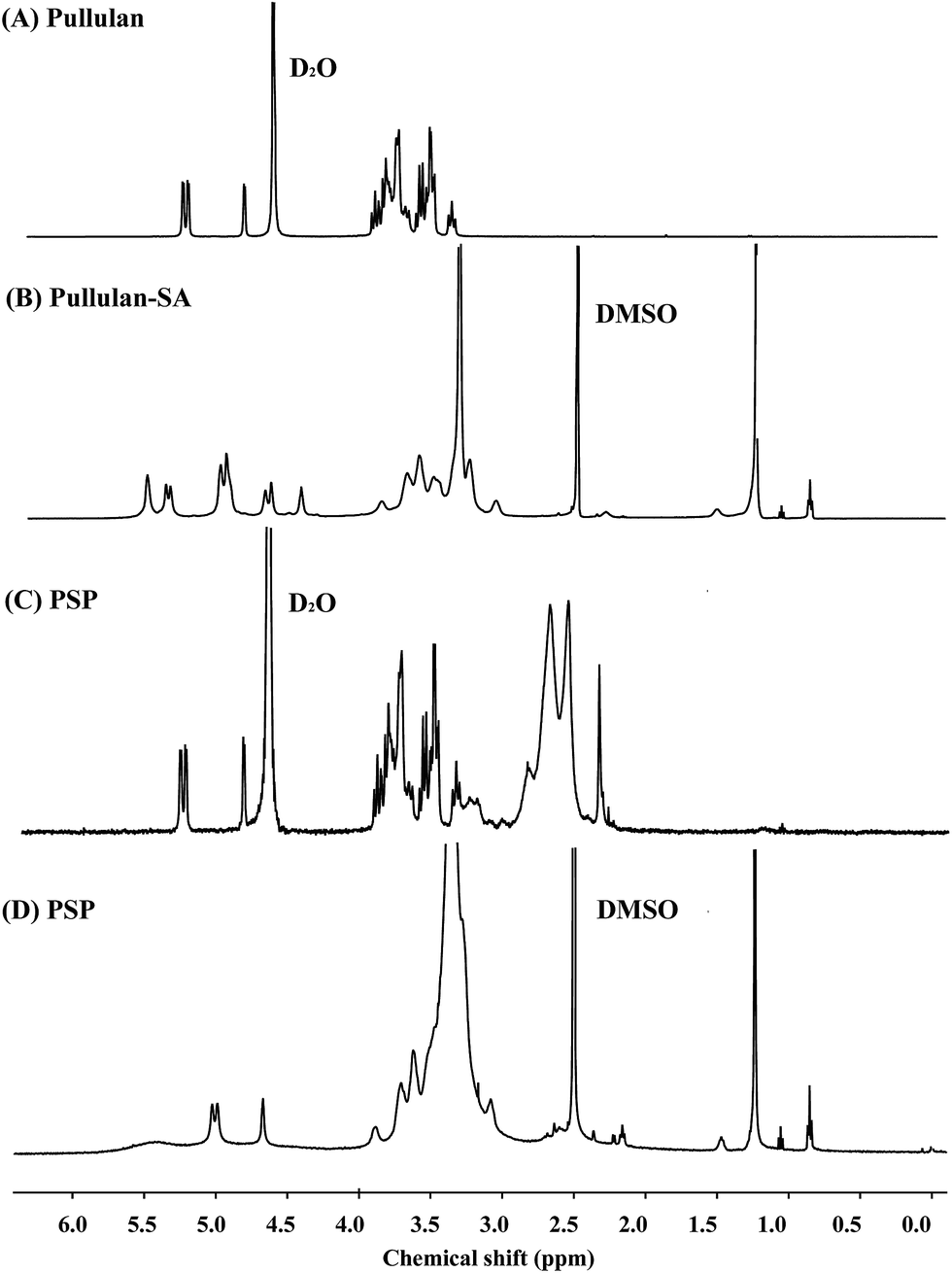 | ||
| Fig. 2 1H NMR spectra of pullulan and its derivatives. | ||
Self-assembly of PSP, drug loading and characterization of blank and PSP/DOX micelles
The amphiphilic nature of PSP bearing hydrophobic SA and hydrophilic pullulan and PEI enables its easy formation of micelles in water. CMC is an important parameter of polymeric micelles in drug-delivery applications and defines the thermodynamic stability of micelles. Amphiphilic polymers can form self-assembled micelles when their concentration in aqueous solution is above the CMC.38 Pyrene is usually used as a fluorescence probe to investigate the self-assembly behaviors of amphiphilic polymers at the molecular level.39 In the present study, the CMC of PSP micelles was 58.9 mg L−1, determined from the intersection of the fluorescence intensity ratio I372/I383 at low and high sample concentrations using pyrene as the fluorescence probe (Fig. 3A). The relative low CMC value ensured that the PSP micelles retained their stable structure despite of the extensive dilution after administration, which was necessary for their drug-delivery application.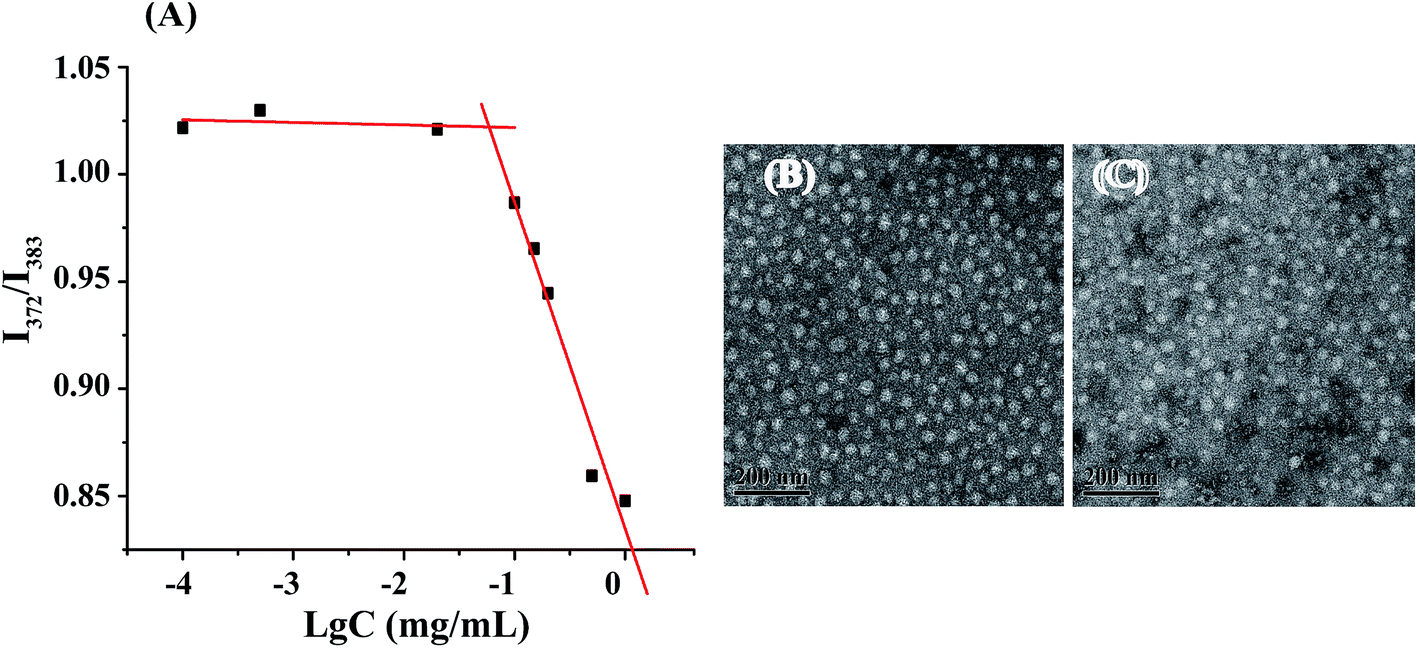 | ||
| Fig. 3 (A) Critical micelle concentration of PSP nanomicelles measured using pyrene as a fluorescence probe. Transmission electron microscopy images of PSP micelles (B) and PSP/DOX micelles (C). | ||
Drug-loaded, cationic PSP micelles was successfully fabricated using DOX as the model chemotherapeutic agent by dialysis method. Due to the intrinsic fluorescence of DOX, it was primarily selected to evaluate uptake and intracellular localization of anticancer drugs delivered by PSP micelles. In our present study, when the mass ratio of drug to PSP was 1:10, the drug loading content and encapsulation efficiency of the PSP nanomicelles for DOX were 5.10 ± 0.16% and 56.07 ± 1.80%, respectively.
Data measured by DLS further showed that the PSP polymer self-assembled to form micelles and the particle size of the blank micelles was around 188 nm. Moreover, the relatively low polydispersity of the blank PSP micelles (PDI = 0.298) demonstrated that PSP nanomicelles were uniformly distributed. On the other hand, the size of PSP/DOX micelles was about 151.60 nm, smaller than that of blank micelles, which might be caused by the hydrophobic interaction between DOX and SA core. The morphologies of the self-assembled micelles were also observed by TEM. As shown in Fig. 3(B and C), blank and PSP/DOX micelles were uniformly dispersed and regularly spherical, and the size of PSP/DOX micelles was slightly smaller than that of blank ones, in accordance with the DLS results. The size measured from TEM was smaller than that from DLS because of different sample preparation technologies.40,41 Additionally, the blank PSP micelles and PSP/DOX micelles revealed positive surface zeta potential of 17.83 ± 0.76 mV and 20.57 ± 1.42 mV, respectively, due to the presence of PEI on the surface of micelles. The positive zeta potential rendered the PSP and PSP/DOX micelles ability to bind negatively-charged gene further for the co-delivery of drug and gene in one carrier system.
In vitro drug release and cytotoxicity of PSP/DOX micelles
The in vitro release of DOX from PSP nanomicelles was evaluated in PBS solutions with pH values of 7.4, 6.5, 5.0, respectively, and free DOX was used as the control. As shown in Fig. 4A, PSP/DOX micelles had sustained release of DOX over 80 h, but 100% free DOX was released within 7 h, indicating that DOX molecules were well encapsulated within the PSP micelles. Moreover, the release of DOX from PSP micelles was also influenced by the pH of medium used for drug release. As displayed in Fig. 4A, at pH 7.4, only 52% of total DOX was released within 80 h, while when pH was decreased to 5.0, 73% of the total drug was released under the same conditions. It is generally accepted that the hydrolysis of carboxylic acid esters can take place under acidic or alkali condition. Therefore, the possible mechanism for pH sensitivity of PSP micelles is that the ester bond between the pullulan and SA was likely to be hydrolyzed at lower pH medium, resulting in the destruction of PSP/DOX micelles and a pH-sensitive release of DOX from PSP/DOX micelles. Cancer cells were reported to have lower extracellular pH (pH 4.5–7.2),40 thus, the pH-sensitive release behavior of DOX from PSP nanomicelles is advantageous for cancer treatment. However, in our present study, not 100% of DOX was released. This might be because that under our experimental conditions, not all PSP micelles were destroyed, or PSP has a long chain, resulting in the tangle of pullulan chain which prevented partial amount of DOX from escaping from PSP. Moreover, the hydrophobic interaction between DOX and SA on the backbone of pullulan also might prevent the release of DOX.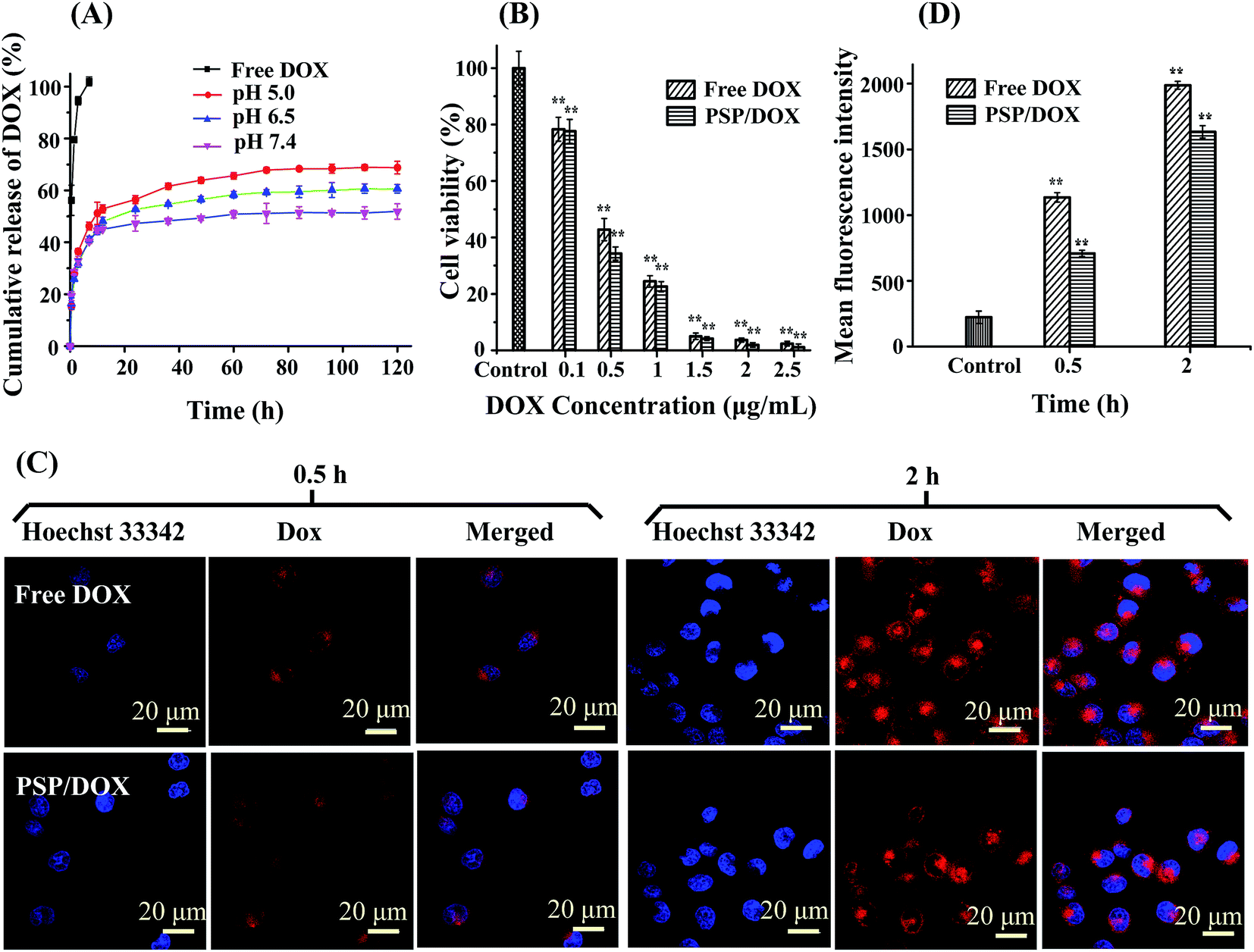 | ||
| Fig. 4 (A) In vitro release profiles of DOX from PSP nanomicelles in PBS at pH 5.0, 6.5 and 7.4. (B) Cytotoxicity of free DOX and PSP/DOX nanomicelles against MCF-7 cells for 48 h. MCF-7 cells were treated with free DOX or PSP/DOX at different equivalent concentrations of DOX (0.1, 0.5, 1.0, 1.5, 2.0, 2.5 μg mL−1) for 48 h at 37 °C. Cell viability was measured by the MTT method as described in materials and methods. Data are shown as mean ± SD values obtained from three separate experiments. Statistical analysis was performed according to the Student's t-test and one-way ANOVA analysis. (*) p < 0.05 and (**) p < 0.01 compared with the control group. (C) Cellular localization of DOX in MCF-7 cells observed by CLSM. Blue represents nuclei and red represents DOX. DOX dosage was at 2.5 μg mL−1. (D) Investigation of PSP/DOX cellular uptake by MCF-7 cells by a flow cytometer. DOX dosage was at 2.5 μg mL−1. | ||
The in vitro anticancer cytotoxic activity of free DOX and PSP/DOX micelles against MCF-7 cells was shown in Fig. 4B. After 48 h incubation, both free DOX and PSP/DOX micelles displayed a concentration-dependent cytotoxicity against MCF-7 cells. In comparison with free DOX, PSP/DOX micelles displayed slightly higher cytotoxicity to MCF-7 cells, and IC50 of PSP/DOX was calculated as 0.36 μg mL−1 of DOX, which was slightly lower than that of free DOX (0.38 μg mL−1).
Cellular uptake and distribution of PSP/DOX nanomicelles
Fig. 4C displayed confocal images of MCF-7 cells treated with free DOX or PSP/DOX micelles at DOX concentration of 2.5 μg mL−1 for 0.5 h and 2 h respectively. Free DOX and PSP/DOX micelles were successfully uptaken by MCF-7 cells, and with prolonged time, the fluorescence intensity of DOX in cells increased, demonstrating more free DOX molecules or PSP/DOX micelles entered MCF-7 cells. Stronger fluorescent signals were observed in the cells treated with free DOX as compared to those treated with PSP/DOX micelles, indicating that the cells might take up more free DOX molecules. In addition, free DOX molecules delivered without carrier were localized in the nucleus of the cells after 2 h of incubation, where DOX could exhibit its antimitotic activity, this can be explained that DOX is a small molecule and it can diffuse into cells and enter nuclei by passive diffusion quickly.41 On the other hand, DOX molecules delivered by PSP micelles were mostly found in the cytoplasm of the cells during the same incubation period. Due to the large size of PSP/DOX micelles, PSP/DOX micelles couldn't enter the nucleus directly, therefore only a small portion of DOX molecules was released from these micelles during the first 2 h of incubation, resulting in smaller available amount of free DOX molecules in the cell's cytoplasm to diffuse to its nucleus.Furthermore, quantitative analysis of cellular uptake of DOX was further measured by a flow cytometer. The results were shown in Fig. 4D. It was clear that the fluorescence intensity in MCF-7 cells treated with free DOX was higher than that of cells treated with PSP/DOX micelles at both 0.5 h and 2 h, which is well in accordance with the CLSM observation.
Formation and characterization of PSP/pDNA complexes
The appropriate size and positive surface charge of a gene carrier are very significant for its effective cellular uptake, and cells preferably uptake nanoparticles ranging from 50 to several hundred nanometers.42 Thus, in our present study, the particle size and zeta potential of PSP/pDNA complexes were measured at various weight ratios of PSP to DNA (N/P) as shown in Fig. 5A. The particle size of PSP/pDNA complexes was 80–180 nm at weight ratio of PSP/DNA ranging from 1.25 to 20, which is a prerequisite for efficient cellular uptake. Moreover, the particle size of PSP/pDNA complexes decreased with the increase of N/P ratio, as previously reported by most research groups. The morphologies of PSP/pDNA and PSP/DOX/pDNA were also regularly spherical observed by TEM (Fig. 5B and C). Meanwhile, the zeta potential of PSP/pDNA complexes increased rapidly with the increment of N/P, and showed positive charge, which suggesting the complete condensation of PSP to DNA. The net positive surface charge of PSP/pDNA micelles provided desirable properties for easy cellular uptake and gene/drug delivery. | ||
| Fig. 5 Particle size and zeta potential of PSP/pDNA complexes at different N/P ratios (A). Transmission electron microscopy images of PSP/pDNA (B) and PSP/DOX/pDNA (C). Agarose gel electrophoresis retardation assay of pDNA at various N/P by PSP (D) and PSP/DOX (E). The protection of pDNA by PSP against DNase I degradation at varying N/P ratios for 30 min (F) and 4 h (G). Results were expressed as mean ± S.D. (n = 3). | ||
Gel retardation assay and DNase I protection assay
The ability of the positively charged PSP to condense pDNA was analyzed using DNA retardation assay by agarose gel electrophoresis at different N/P ratios. As demonstrated in Fig. 5D, at lower N/P ratios, blank PSP could not condense pDNA completely and the pDNA migration could be found. But when N/P was bigger than 2.5, blank PSP could completely retard the migration of pDNA. On the other hand, as illustrated in Fig. 5E, the drug-loading in PSP didn't change the ability of PSP to condense DNA greatly, and PSP/DOX also could condense pDNA completely and retard the migration of pDNA at N/P > 2.5.A successfully effective gene vector is required to protect DNA from degradation by DNase I in blood and tissues to ensure delivery of undamaged DNA to target cells. Therefore, the susceptibility of the pDNA combined by PSP to enzymatic degradation in the presence of DNase I was investigated. As demonstrated in Fig. 5F and G, the naked pDNA treated with 1 unit DNase I was almost completely degraded within 30 min (lane 5, Fig. 5F). However, pDNA which was complexed with PSP at various N/P ratios displayed no significant degradation within 30 min. After incubation with DNase I for up to 4 h (Fig. 5G), the pDNA complexed with PSP at the N/P ratio of 10 was still not digested and kept in good integrity, indicating that PSP could effectively bind pDNA and protect pDNA from degradation by DNase I. Therefore, the results aforementioned implied that PSP might also be a good candidate carrier for gene delivery.
In vitro transfection
The in vitro transfection capacity of PSP was evaluated in COS-7 cells using pGFP as a reporter gene at four N/P ratios (2.5, 5, 10, and 20). The control groups were naked pGFP and Lipofectamine 2000™/pGFP at its optimal N/P ratio of 3. As displayed in Fig. 6A, when N/P ratio increased from 2.5 to 10, the transfection efficiency of PSP/pGFP in COS-7 cells also increased from 14.10 ± 1.64% to 64.33 ± 4.29%. But the transfection efficiency of PSP/pGFP decreased as the N/P ratio was bigger than 10. The highest transfection efficiency of PSP/pGFP observed at an N/P ratio of 10 was slightly lower than that for Lipo2000. In addition, the gene expression of GFP mediated by the PSP in COS-7 cells was also observed under a fluorescence microscope to directly visualize the gene delivery capability of PSP. Green fluorescent protein expression can be clearly observed in Fig. 6B, indicating that pGFP was transported into COS-7 cells and GFP was successfully expressed. The strongest green fluorescence spots were also observed at a N/P ratio of 10 for PSP. This result was consistent with that obtained from the GFP system by a flow cytometer. | ||
| Fig. 6 (A) Transfection efficiency of PSP/pGFP in COS-7 cells defined as the percentage of GFP positive cells determined by a flow cytometer. Results were expressed as mean ± S.D. (n = 3). (B) Transfection effect of PSP/pGFP in COS-7 cells at different N/P ratios observed by confocal micrographs. Naked pGFP and Lipo2000/pGFP at a N/P ratio of 3 as negative and positive controls, respectively. | ||
Co-delivery of drug and p53 gene
The experimental results above-mentioned showed that amphiphilic cationic PSP polymer could successfully deliver drug and gene respectively. Herein, we then further demonstrated the potential use of PSP micelles as carriers for the co-delivery of a therapeutic gene (p53-encoding plasmid) and DOX, and investigated the synergistic/combined effect of the two therapeutics at endpoint cytotoxicity level against MCF-7 cells, as well as apoptosis level of the same cell line. The p53 gene is a tumor suppressor gene, which can inhibit cell proliferation through apoptosis and/or G1-cell cycle arrest in mammalian cells, and was reported to sensitized cancer cells to anti-cancer drug.43,44 As illustrated in Fig. 7A, at N/P 10, p53 gene transfection mediated by PSP indeed reduced the viability of MCF-7 cells in comparison with control and free p53 groups. As an efficient anti-tumor drug, the free DOX showed higher cytotoxicity against MCF-cells than those of free p53 and PSP/p53 groups. The free p53/free DOX group displayed similar cytotoxicity to that of free DOX group, because it was difficult for large amount of the free p53 to enter MCF-7 cells, resulting in the lower expression level of exogenous p53. However, the co-delivery of p53 gene and DOX using PSP micelles showed significantly higher cytotoxicity against MCF-7 cells at N/P = 10 when compared to PSP/p53 and PSP/DOX treatment alone and free p53/free DOX. It was reported that the increased production of reactive oxygen was associated with DOX-treated cells.45 These cellular stresses can activate the expression of endogenous p53 and halt cell cycle progression. Therefore, our data implied that the co-delivery of p53 gene and DOX using PSP micelles could achieved a synergistic/combined effect in suppressing the proliferation of MCF-7 cells.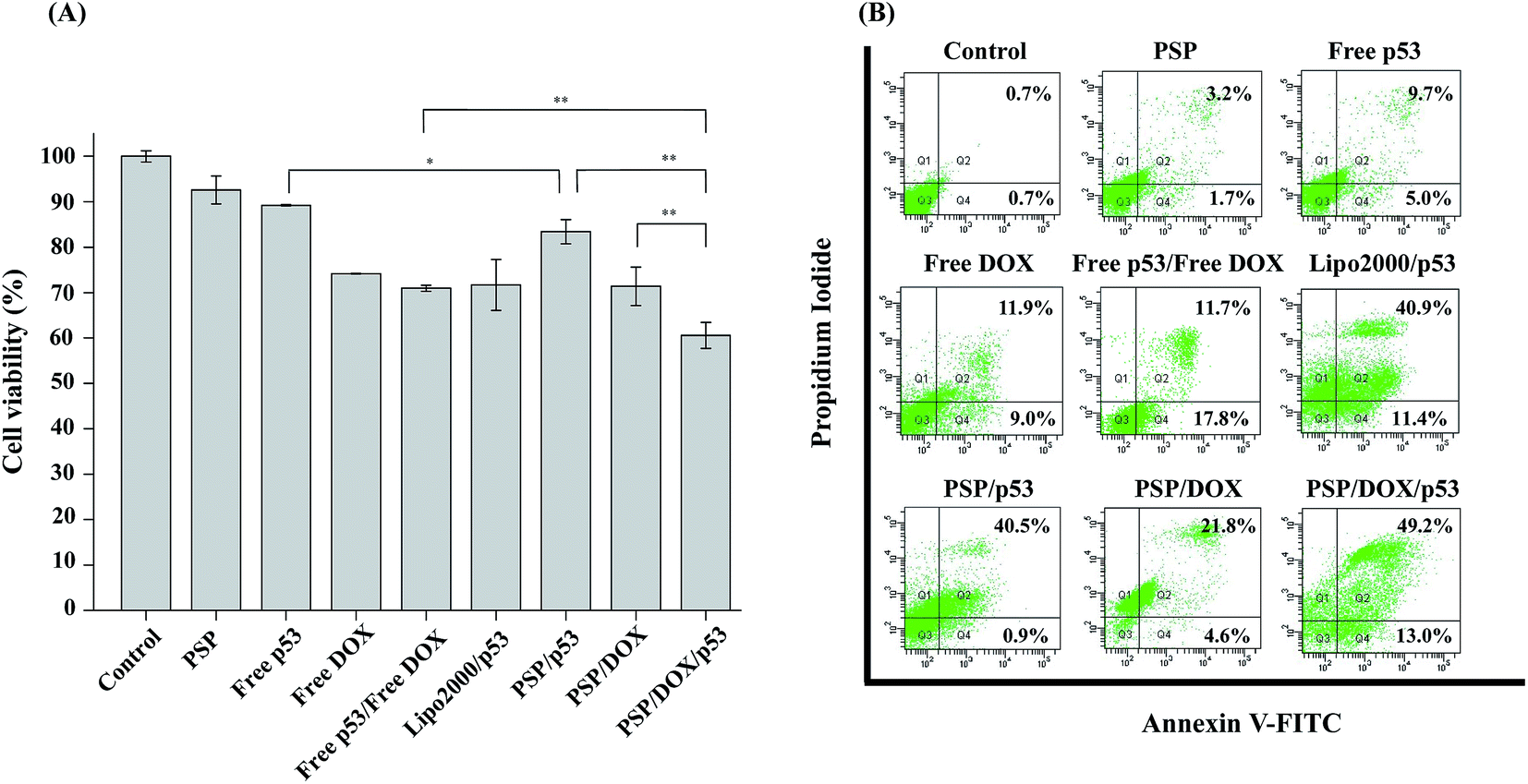 | ||
| Fig. 7 Cell viability assayed by MTT (A) and apoptosis analysis (B) by a flow cytometer after MCF-7 cells were incubated with various samples for 24 h. In experiments, DOX concentration was set at 0.5 μg mL−1; p53 concentration was at 0.95 μg mL−1; N/P = 10. Cell viability was measured by the MTT method as described in experimental section. Data are shown as mean ± S.D. Values were obtained from three separate experiments. Statistical analysis was performed according to the Student's t-test and one-way ANOVA analysis. (**) p < 0.01 compared with the control group. | ||
Furthermore, the effect of PSP/DOX/p53 on apoptosis of MCF-7 was investigated by annexin V-FITC and PI double staining and analyzed via a flow cytometer. As shown in Fig. 7B, MCF-7 cells exposed to blank PSP micelles did not show any obvious apoptosis after 24 h incubation. Similarly, there were only total 20.9% of apoptotic and dead cells induced by free DOX. However, PSP/DOX micelles enhanced the susceptibility of cancer cells to DOX and induced more apoptotic and dead cell percentage (26.4%). The p53 gene was reported to show important role in inhibiting tumor cell proliferation by promoting apoptotic cell death, and this was further confirmed by the higher apoptotic and necrotic rates (41.4%) in MCF-7 cells displayed by the PSP/p53 transfection group in our work. Furthermore, the co-delivery of the p53 gene and DOX drug led to a total 62% of apoptotic and dead MCF-7 cells, which was significantly higher than those of all control groups. By contrary, the free p53/free DOX group only induced 29.5% apoptotic and dead MCF-7 cells. Because free p53 and free DOX couldn't enter the same cells simultaneously. In summary, the results presented above demonstrated that the micelles formed by amphiphilic PSP copolymer could efficiently transfer tumor suppressor p53 gene and deliver anti-cancer drug DOX into MCF-7 cells at the same time and jointly cause higher cell apoptosis and necrosis rates.
Biocompatibility
The biocompatibility of PSP micelles was first evaluated by blood compatibility. In order to evaluate the blood compatibility of PSP micelles, the hemolysis test was performed. The level of hemolysis of PSP micelles was compared with that of PEI 20k, as shown in Fig. 8A, PEI 20k induced less than 5% hemolysis when its concentration was 0.5 mg mL−1 and incubation time was 1 h. However, when the concentration of PEI 20k increased to 1 mg mL−1 and 2 mg mL−1, PEI caused 23.6% and 41.4% hemolysis rate respectively. In contrast, the hemolysis caused by PSP micelles were all below 5% at the same concentration range, which was significantly different from PEI 20k (p < 0.01). Because up to 5% hemolysis rate is acceptable for biomaterials;28 thus, PSP micelles have shown good blood compatibility. | ||
| Fig. 8 (A) Hemolysis test results of PSP and PEI 20k at different concentrations of 0.5, 1.0, and 2.0 mg mL−1. (B) Viability of different cells assayed by MTT after the cells were treated with PSP at different concentrations of 2.5, 5, 10, and 40 μg mL−1 for 48 h. Data are shown as mean ± SD values obtained from three separate experiments. Statistical analysis was performed according to the student's t-test and one-way ANOVA analysis. (**) p < 0.01 compared with the control group. | ||
Furthermore, the biocompatibility of PSP micelles was evaluated by the cytotoxicity of PSP micelles against HEK293, COS-7, MCF-7 cells after incubation of 48 h. As shown in Fig. 8B, with the concentrations of PSP ranged from 2.5 to 40 μg mL−1, the cell viability of three kinds of cell lines decreased. However, it should be noted that PSP kept over 60% cell viability for all three cell lines at a high concentration of 40 μg mL−1, and when the concentration of PSP was lower than 10 μg mL−1, the cell viability for all the cell lines was over 85%, indicating that PSP cationic micelles have low cytotoxicity.
Conclusions
In summary, in this study, a novel bifunctional pullulan-based amphiphilic copolymer PSP was designed and successfully synthesized by introducing stearic acid and PEI onto pullulan backbone. PSP showed lower cytotoxicity and excellent hemocompatibility. PSP could easily self-assemble into nanomicelles in water with low CMC of 58.9 mg L−1. The micelles could entrap hydrophobic DOX, and showed strong DNA condensation ability and protected DNA from degradation by DNase I efficiently. FCM and CLSM showed that PSP/DOX micelles could be successfully internalized in MCF-7 cells. The in vitro IC50 of PSP/DOX micelles was slightly lower than that of free DOX in MCF-7 cells. In addition, PSP showed the highest transfection efficiency in COS-7 cells at N/P ratio of 10. More importantly, the micelles formed by PSP could co-deliver tumor-suppressor p53 gene and anticancer drug DOX to MCF-7 cells, resulting in higher tumor cell apoptosis and cytotoxicity compared to PSP/p53 and PSP/DOX treatment alone. Altogether, the results demonstrated that amphiphilic pullulan-based copolymer PSP may be used as a promising carrier system for the co-delivery of drug and gene in future cancer therapy.Acknowledgements
This work was funded by the Natural Science Foundation of China (grant no. 21376039).References
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- C. Bock and T. Lengauer, Nat. Rev. Cancer, 2012, 12, 494–501 CrossRef CAS PubMed.
- R. Salehi, S. Rasouli and H. Hamishehkar, Int. J. Pharm., 2015, 487, 274–284 CrossRef CAS PubMed.
- N. S. Gandhi, R. K. Tekade and M. B. Chougule, J. Controlled Release, 2014, 194, 238–256 CrossRef CAS PubMed.
- J. Li, Y. Wang, Y. Zhu and D. Oupicky, J. Controlled Release, 2013, 172, 589–600 CrossRef CAS PubMed.
- D. Dong, W. Gao, Y. Liu and X. R. Qi, Cancer Lett., 2015, 359, 178–186 CrossRef CAS PubMed.
- V. Tsouris, M. K. Joo, S. H. Kim, I. C. Kwon and Y. Y. Won, Biotechnol. Adv., 2014, 32, 1037–1050 CrossRef CAS PubMed.
- C. K. Chen, W. C. Law, R. Aalinkeel, Y. Yu, B. Nair, J. Wu, S. Mahajan, J. L. Reynolds, Y. Li, C. K. Lai, E. S. Tzanakakis, S. A. Schwartz, P. N. Prasad and C. Cheng, Nanoscale, 2014, 6, 1567–1572 RSC.
- N. Cao, D. Cheng, S. Zou, H. Ai, J. Gao and X. Shuai, Biomaterials, 2011, 32, 2222–2232 CrossRef CAS PubMed.
- Y. Li, B. Xu, T. Bai and W. Liu, Biomaterials, 2015, 55, 12–23 CrossRef PubMed.
- Y. H. Yu, E. Kim, D. E. Park, G. Shim, S. Lee, Y. B. Kim, C. W. Kim and Y. K. Oh, Eur. J. Pharm. Biopharm., 2012, 80, 268–273 CrossRef CAS PubMed.
- R. S. Chang, M. S. Suh, S. Kim, G. Shim, S. Lee, S. S. Han, K. E. Lee, H. Jeon, H. G. Choi, Y. Choi, C. W. Kim and Y. K. Oh, Biomaterials, 2011, 32, 9785–9795 CrossRef CAS PubMed.
- O. Taratula, A. Kuzmov, M. Shah, O. B. Garbuzenko and T. Minko, J. Controlled Release, 2013, 171, 349–357 CrossRef CAS PubMed.
- L. Han, R. Huang, J. Li, S. Liu, S. Huang and C. Jiang, Biomaterials, 2011, 32, 1242–1252 CrossRef CAS PubMed.
- S. Biswas, P. P. Deshpande, G. Navarro, N. S. Dodwadkar and V. P. Torchilin, Biomaterials, 2013, 34, 1289–1301 CrossRef CAS PubMed.
- H. Meng, M. Liong, T. Xia, Z. Li, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2010, 4, 4539–4550 CrossRef CAS PubMed.
- H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 994–1005 CrossRef CAS PubMed.
- J. M. Li, Y. Y. Wang, M. X. Zhao, C. P. Tan, Y. Q. Li, X. Y. Le, L. N. Ji and Z. W. Mao, Biomaterials, 2012, 33, 2780–2790 CrossRef CAS PubMed.
- X. Deng, M. Cao, J. Zhang, K. Hu, Z. Yin, Z. Zhou, X. Xiao, Y. Yang, W. Sheng, Y. Wu and Y. Zeng, Biomaterials, 2014, 35, 4333–4344 CrossRef CAS PubMed.
- N. R. Ko, J. Cheong, A. Noronha, C. J. Wilds and J. K. Oh, Colloids Surf., B, 2015, 126, 178–187 CrossRef CAS PubMed.
- Y. Wang, S. Gao, W. H. Ye, H. S. Yoon and Y. Y. Yang, Nat. Mater., 2006, 5, 791–796 CrossRef CAS PubMed.
- C. Zheng, M. Zheng, P. Gong, J. Deng, H. Yi, P. Zhang, Y. Zhang, P. Liu, Y. Ma and L. Cai, Biomaterials, 2013, 34, 3431–3438 CrossRef CAS PubMed.
- T. Liu, W. Xue, B. Ke, M. Q. Xie and D. Ma, Biomaterials, 2014, 35, 3865–3872 CrossRef CAS PubMed.
- R. S. Singh, N. Kaur and J. F. Kennedy, Carbohydr. Polym., 2015, 123, 190–207 CrossRef CAS PubMed.
- Y. Wang, H. Chen, Y. Liu, J. Wu, P. Zhou, Y. Wang, R. Li, X. Yang and N. Zhang, Biomaterials, 2013, 34, 7181–7190 CrossRef CAS PubMed.
- Y. S. Wang, Y. Liu, Y. Y. Liu, Y. Wang, J. Wu, R. S. Li, J. R. Yang and N. Zhang, Polym. Chem., 2014, 5, 423–432 RSC.
- B. C. Bae and K. Na, Biomaterials, 2010, 31, 6325–6335 CrossRef CAS PubMed.
- X. C. Yang, Y. L. Niu, N. N. Zhao, C. Mao and F. J. Xu, Biomaterials, 2014, 35, 3873–3884 CrossRef CAS PubMed.
- M. R. Rekha and C. P. Sharma, Acta Biomater., 2011, 7, 370–379 CrossRef CAS PubMed.
- S. J. Seo, C. S. Lee, Y. S. Jung and K. Na, Carbohydr. Polym., 2012, 87, 1105–1111 CrossRef CAS.
- D. K. Thakor, Y. D. Teng and Y. Tabata, Biomaterials, 2009, 30, 1815–1826 CrossRef CAS PubMed.
- M. R. Rekha and C. P. Sharma, Biomaterials, 2009, 30, 6655–6664 CrossRef CAS PubMed.
- J. Y. Wang, S. Cui, Y. M. Bao, J. S. Xing and W. B. Hao, Mater. Sci. Eng., C, 2014, 43, 614–621 CrossRef CAS PubMed.
- X. Lu, Q. Q. Wang, F. J. Xu, G. P. Tang and W. T. Yang, Biomaterials, 2011, 32, 4849–4856 CrossRef CAS PubMed.
- J. Y. Wang, B. R. Dou and Y. M. Rao, Mater. Sci. Eng., C, 2014, 34, 98–109 CrossRef PubMed.
- L. Zhu, F. Perche, T. Wang and V. P. Torchilin, Biomaterials, 2014, 35, 4213–4222 CrossRef CAS PubMed.
- Y. S. Chen, Q. J. Wu, L. J. Song, T. He, Y. C. Li, L. Li, W. J. Su, L. Liu, Z. Y. Qian and C. Y. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 534–542 CAS.
- Y. Song, L. Zhang, W. Gan, J. Zhou and L. Zhang, Colloids Surf., B, 2011, 83, 313–320 CrossRef CAS PubMed.
- L. Shi, C. Tang and C. Yin, Biomaterials, 2012, 33, 7594–7604 CrossRef CAS PubMed.
- W. She, N. Li, K. Luo, C. Guo, G. Wang, Y. Geng and Z. Gu, Biomaterials, 2013, 34, 2252–2264 CrossRef CAS PubMed.
- X. Zhang, K. Achazi, D. Steinhilber, F. Kratz, J. Dernedde and R. Haag, J. Controlled Release, 2014, 174, 209–216 CrossRef CAS PubMed.
- S. Nimesh, M. M. Thibault, M. Lavertu and M. D. Buschmann, Mol. Biotechnol., 2010, 46, 182–196 CrossRef CAS PubMed.
- Y. Shen and E. White, Adv. Cancer Res., 2001, 82, 55–84 CrossRef CAS PubMed.
- L. Xu, K. F. Pirollo and E. H. Chang, J. Controlled Release, 2001, 74, 115–128 CrossRef CAS PubMed.
- W. P. Tsang, S. P. Chau, S. K. Kong, K. P. Fung and T. T. Kwok, Life Sci., 2003, 73, 2047–2058 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |