
Ruthenium hydroxycyclopentadienyl N-heterocyclic carbene complexes as transfer hydrogenation catalysts†
Cristiana Cesari,
Andrea Cingolani,
Chiara Parise,
Stefano Zacchini,
Valerio Zanotti,
Maria Cristina Cassani and
Rita Mazzoni*
Dipartimento di Chimica Industriale “Toso Montanari”, viale Risorgimento 4, 40136 Bologna, Italy. E-mail: rita.mazzoni@unibo.it
First published on 27th October 2015
Abstract
A series of novel cationic hydroxycyclopentadienyl and methoxycyclopentadienyl N-heterocyclic carbene ruthenium(II) complexes have been synthesized from the corresponding neutral ruthenium(0) complexes containing both the non-innocent cyclopentadienone ligand and variously functionalized N-heterocyclic carbenes (NHCs). In particular an NHC derivative containing a pyridine group in the side chain has been designed and developed in order to evaluate the influence of a basic, potentially cooperative substituent in the catalytic activity of these complexes. All the prepared complexes were employed as selective catalysts for transfer hydrogenation reactions employing refluxing iPrOH as a hydrogen source and several ketones and aldehydes as substrates. We found that while the presence of oxidizing additives such as CAN and benzoquinone is mandatory to activate the neutral ruthenium(0) complexes, no activation is needed for the cationic Ru(II) catalysts. The catalytic activity of the latter is also influenced by the coordinating ability of the counterion, and indeed the cationic complexes having a pyridine-functionalized NHC ligand and CF3SO3− as counterion, present the best conversion (>99%) thus demonstrating the fundamental role played by the basic pyridine in the catalytic activity. With regard to the hydrogenation reaction mechanism, the release of the CO ligand was demonstrated to be the key step and the presence of hydride species has been detected at the end of the reaction.
Introduction
N-heterocyclic carbenes (NHCs) are among the most popular ancillary ligands due to a combination of unique features, such as the tunability of electronic and steric properties that influence the metal center. Moreover the synthesis of NHC ligand precursors and of the corresponding complexes is rather simple and very versatile1 allowing the rational design of transition metal catalysts and the improvement of catalytic activity.2 Several catalytic reactions such as ruthenium-catalyzed olefin metathesis,3 palladium-catalyzed cross-coupling reaction,4 iridium-catalyzed reductions and oxidations,5 and gold-catalyzed activation of π-bonds6 benefited from the introduction of NHC ligands. Apart from olefin metathesis, Ru–NHC complexes have shown catalytic activity in various redox transformations,7 including: transfer hydrogenation,8 hydrogenation of olefins9 and esters,10 asymmetric hydrogenation,11 amide synthesis from alcohols and nitriles,12 dehydrogenation of esters and imines from alcohols,13 racemization of chiral alcohols,14 oxidation of alcohols15 and water oxidation.16 Regarding the Ru–NHC complexes, most of the literature features Ru(II) complexes, while low-valent Ru(0)–NHC systems are restricted to a few examples based on either [Ru3(CO)12] or [Ru(CO)2(PPh3)3] as precursors.17Another class of versatile ligands is represented by cyclopentadienones which behave as non-innocent ligands exploiting the cyclopentadienone/hydroxycyclopentadienyl reversible transformation. The most famous example of its cooperativity with ruthenium in bifunctional catalysis is the Shvo catalyst, a well-known system employed in a plethora of catalytic applications.18 In principle by combining NHC and cyclopentadienone ligands on ruthenium complexes the properties of both ligand should be exploited allowing the design of novel catalysts finely tuning steric and electronic properties, solubility and the insertion of substituents suitable for heterogenization. Furthermore by choosing a proper substituent, NHC ligand itself could cooperate with the metal as a non-innocent species.19
With this aim in mind, a straightforward approach towards Ru(0)–NHC complexes has been recently communicated by our group,20 based on a dimeric Ru(0) cyclopentadienone dicarbonyl dimer (2). Cleavage of 2 in the presence of a silver carbene precursor provided access to a new class of ruthenium complexes in quantitative yields (some examples in Scheme 1).
 | ||
| Scheme 1 Synthesis of cyclopentadienone imidazolylidene ruthenium(0) complexes 3a–c. | ||
In this paper we first describe an extension of this synthetic methodology to a complex containing a pyridine-functionalized NHC ligand (3d: R1 = butyl; R2 = 2-pyridine) then the main body of this work concerns the transformation of the cyclopentadienone imidazolylidene ruthenium(0) complexes (3a–d) into their corresponding cationic hydroxyl- and methoxy-cyclopentadienyl ruthenium(II) derivatives [4a–d[X] (X = Cl, BF4, NO3, CF3SO3) and 5a,d[CF3SO3]], and the use of both Ru(0) and Ru(II) complexes as catalysts for transfer hydrogenation of ketones and aldehydes in refluxing isopropanol. Comparison between monodentate and pyridine-functionalized NHC ligands, has provided some insights into the reaction mechanism.
Results and discussion
Synthesis of imidazolylidene Ru complexes
The novel complex 3d (R1 = butyl; R2 = 2-pyridine) was prepared in one pot following the same procedure described in Scheme 1 and fully characterized by spectroscopic methods and elemental analysis. In particular, the 13C NMR spectrum shows the downfield shifted resonance of the carbene (δ = 173.20 ppm), whereas in the IR spectrum the CO stretching bands are consistent with those of similar complexes (νCO = 2008 cm−1, 1949 cm−1 (3d) vs. νCO = 2004 cm−1, 1945 cm−1 (3a)). Further evidences were produced by ESI-MS and X-ray diffraction analyses.The molecular structure of 3d has been determined by single crystal X-ray crystallography as its 3d·0.5CH2Cl2 solvate (Fig. 1).
 | ||
| Fig. 1 ORTEP drawing of 3d. Displacement ellipsoids are at the 30% probability level. H-atoms have been omitted for clarity. Selected bond lengths (Å): Ru(1)–C(1) 1.845(16), Ru(1)–C(2) 1.883(12), Ru(1)–C(3) 2.511(11), Ru(1)–C(4) 2.279(11), Ru(1)–C(5) 2.199(11), Ru(1)–C(6) 2.212(10), Ru(1)–C(7) 2.280(11), Ru(1)–C(34) 2.139(12), C(3)–O(3) 1.246(13). | ||
Its structure is similar to those previously reported for the analogous complexes 3a–c.20 In particular, the Ru(1)–C(3) distance [2.511(11) Å] is significantly longer than Ru(1)–C(4–7) [2.199(11)–2.280(11) Å, average 2.242(2) Å] and C(3)–O(3) [1.246(13) Å] is essentially a double bond.21 The Ru(1)–C(34) contact [2.117(3) Å] is in the typical range for the interaction between Ru(0) and a N-heterocyclic carbene.22
The reaction of 3a–d with strong acids or a methylating agent such as MeCF3SO3, leads to the quantitative formation of the hydroxycyclopentadienyl and methoxycyclopentadienyl cationic complexes 4a–d[X] (X = Cl, BF4, NO3, CF3SO3) and 5a,d[CF3SO3] (Scheme 2), in which Ru(0) is formally oxidized to Ru(II).
 | ||
| Scheme 2 Synthesis of hydroxycyclopentadienyl imidazolylidene ruthenium complexes 4a–d[X] and methoxycyclopentadienyl imidazolylidene ruthenium complexes 5a,d[CF3SO3]. | ||
Infrared spectroscopy provides a convenient technique for monitoring the progress of the protonation as, in agreement with the reduced back-donation from the metal center to the carbonyl ligands, ν–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretch vibrations undergo a significative high-energy shift upon formation of the cationic complex [e.g. νCO = 2008 cm−1, 1949 cm−1 (3d), and 2041 cm−1, 1991 cm−1 (4d[CF3SO3])]. The change in the coordination from η4 (cyclopentadienone ligand) to η5 (hydroxycyclopentadienyl ligand) is further evidenced by 13C-NMR shift of the resonance due to the endocyclic carbon involved in the ketone–alcohol transformation [δ 168.22 ppm (C
O stretch vibrations undergo a significative high-energy shift upon formation of the cationic complex [e.g. νCO = 2008 cm−1, 1949 cm−1 (3d), and 2041 cm−1, 1991 cm−1 (4d[CF3SO3])]. The change in the coordination from η4 (cyclopentadienone ligand) to η5 (hydroxycyclopentadienyl ligand) is further evidenced by 13C-NMR shift of the resonance due to the endocyclic carbon involved in the ketone–alcohol transformation [δ 168.22 ppm (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, Cp, in 3d) vs. δ 142.00 ppm (C–OH, Cp, in 4d[CF3SO3])]. Ru–carbene signals are also shifted to higher fields; from δ 173.20 ppm (3d) to δ 166.07 ppm (4d[CF3SO3]) (see Experimental section for more details).
O, Cp, in 3d) vs. δ 142.00 ppm (C–OH, Cp, in 4d[CF3SO3])]. Ru–carbene signals are also shifted to higher fields; from δ 173.20 ppm (3d) to δ 166.07 ppm (4d[CF3SO3]) (see Experimental section for more details).
With regard to the methylated complexes 5a[CF3SO3] and 5d[CF3SO3] IR spectra show a shift of 40 cm−1 from the neutral to the cationic species (e.g. νCO = 2004 cm−1, 1945 cm−1 (3a), and 2045 cm−1, 1995 cm−1 (5a[CF3SO3])) and both IR and NMR data are similar to those obtained for the hydroxycyclopentadienyl complexes 4a[CF3SO3] and 4d[CF3SO3] with the methyl group arising at δ 3.19 (1H NMR) and δ 61.93 (13C NMR) ppm.
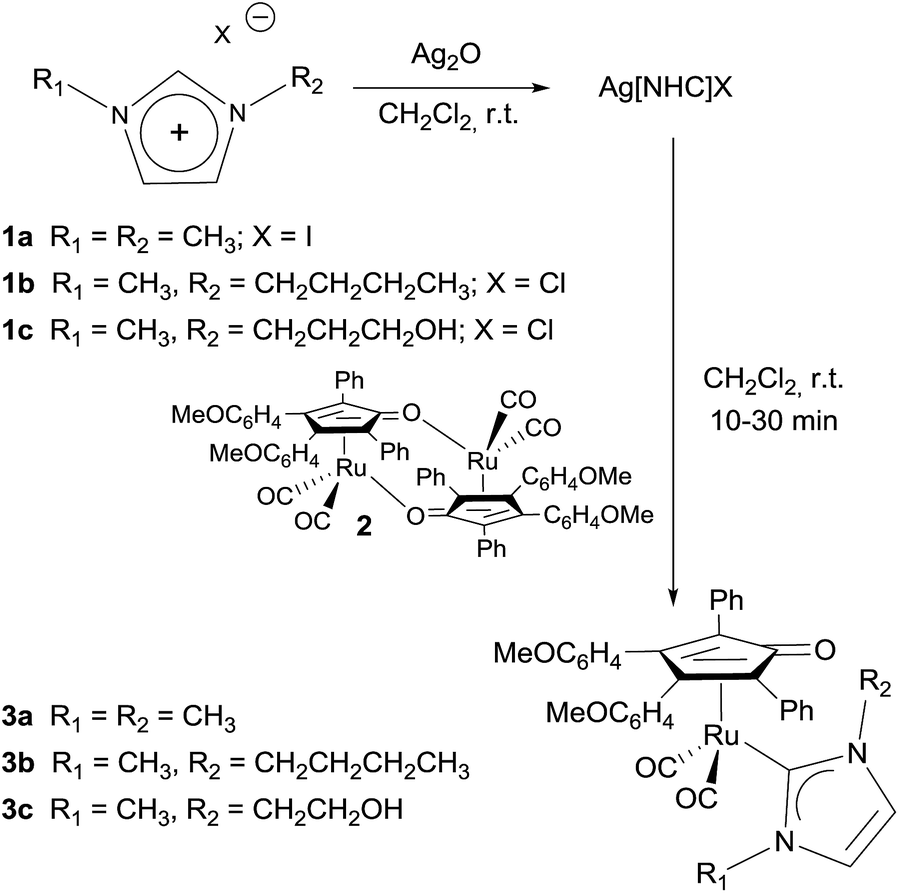
Moreover, structural evidences were obtained by X-ray diffraction analysis of single crystals of the cationic complexes [4a]+ and [4c]+ as their [4a][CF3SO3]·0.5toluene and [4c][CF3SO3]·CHCl3 solvate salts (Fig. 2). X-ray crystallography confirms that protonation has occurred on the O-atom of the cyclopentadienone ligand. As a consequence, the C(3)–O(3) interaction [1.335(6) and 1.339(9) Å for [4a]+ and [4c]+, respectively] is considerably elongated compared to the parent neutral complexes [e.g., 1.246(13) Å for 3d] and displays the character of a C(sp2)–O single bond. In addition the Ru(1)–C(3) distance [2.323(5) and 2.319(7) Å for [4a]+ and [4c]+, respectively] is shortened in respect to the parent complexes [e.g., 2.511(11) Å for 3d] and comparable to the other Ru(1)–C(4–7) distances [2.227(5)–2.290(5) Å, average 2.253(10) Å for [4a]+; 2.271(8)–2.221(7) Å, average 2.248(12) Å for [4c]+]. Thus, the protonated ligand of [4a]+ and [4c]+ is better described as a η5-cyclopentadienyl ligand, as previously reported for analogous complexes.18d,23 The O(3)–H(3) group of [4a]+ is involved in an inter-molecular H-bond with the [CF3SO3]− anion [O(3)–H(3) 0.885(10) Å, H(3)⋯O(301) 1.80(3) Å, O(3)⋯O(301) 2.626(5) Å, ∠O(3)H(3)O(301) 155(5)°]. Conversely, the O(3)–H(3) group of [4c]+ forms an intra-molecular H-bond with the O(6) atom of the side chain of the NHC ligand [O(3)–H(3) 0.84 Å, H(3)⋯O(6) 1.83 Å, O(3)⋯O(6) 2.646(7) Å, ∠O(3)H(3)O(6) 163.7°]. In addition, there is in [4c]+ also an inter-molecular H-bond between the O(6)–H(6) group of the complex and the [CF3SO3]− [O(6)–H(6) 0.84 Å, H(6)⋯O(301)#1 1.91 Å, O(6)⋯O(301)#1 2.739(9) Å, ∠O(6)H(6)O(301)#1 167.7°; symmetry transformations used to generate equivalent atoms: #1 x + 1, y, z #2 −x + 1, −y + 1, −z + 1]. The carbene interaction Ru(1)–C(34) [2.101(5) and 2.109(7) Å for [4a]+ and [4c]+, respectively] is almost unchanged compared to the parent neutral complexes [e.g., 2.117(3) Å for 3d].
 | ||
| Fig. 2 Left: ORTEP drawing of [4a]+. Displacement ellipsoids are at the 30% probability level. H-atoms have been omitted for clarity, except H(3). Selected bond lengths (Å): Ru(1)–C(1) 1.882(7), Ru(1)–C(2) 1.881(7), Ru(1)–C(3) 2.323(5), Ru(1)–C(4) 2.290(5), Ru(1)–C(5) 2.238(5), Ru(1)–C(6) 2.227(5), Ru(1)–C(7) 2.256(5), Ru(1)–C(34) 2.101(5), C(3)–O(3) 1.335(6). Right: ORTEP drawing of [4c]+. Displacement ellipsoids are at the 30% probability level. H-atoms have been omitted for clarity, except H(3) and H(6). Selected bond lengths (Å): Ru(1)–C(1) 1.876(8), Ru(1)–C(2) 1.901(9), Ru(1)–C(3) 2.319(7), Ru(1)–C(4) 2.262(7), Ru(1)–C(5) 2.240(7), Ru(1)–C(6) 2.221(7), Ru(1)–C(7) 2.271(8), Ru(1)–C(34) 2.109(7), C(3)–O(3) 1.339(9). | ||
Finally, with the aim of allowing the coordination of the pyridine to the metal centre, complex 4d[CF3SO3] has been irradiated with a UV lamp. The photolytic removal of one terminal CO led to the formation of the chelated complex 6d[CF3SO3] in good yield (Scheme 3).
 | ||
| Scheme 3 Photolytic removal of terminal CO. | ||
The IR spectrum shows the disappearance of the two absorptions due to the CO stretching of 4d[CF3SO3], and the appearance of a unique band at 1967 cm−1 (6d[CF3SO3]). Carbene resonance, in 13C NMR spectrum, is shifted to lower fields (δ 185.63 ppm (6d[CF3SO3]) vs. δ 166.07 ppm (4d[CF3SO3])).
The X-ray molecular structure of 6d[CF3SO3] further supports the characterization performed in solution (Fig. 3) and consists of a Ru center coordinated to a η5-cyclopentadienyl ligand, a terminal CO and a chelating NHC ligand bonded to Ru via the carbene and the N-atom of the pyridine side group. The Ru(1)–Cp distances [2.216(7)–2.302(8) Å, average 2.248(16) Å] are comparable to those reported above for [4a]+ and [4c]+ and analogous Ru-complexes containing protonated cyclopentadienone ligands.18d,23 As in the case of [4a]+ and [4c]+, the C(3)–O(3) interaction [1.338(9) Å] is a single bond, in view of the presence of a H atom on the oxygen, which is involved in H-bond with the triflate anion.
 | ||
| Fig. 3 ORTEP drawing of 6d[CF3SO3]. Displacement ellipsoids are at the 30% probability level. H-atoms have been omitted for clarity, except H(3). Selected bond lengths (Å): Ru(1)–C(1) 1.860(9), Ru(1)–C(3) 2.302(8), Ru(1)–C(4) 2.237(7), Ru(1)–C(5) 2.216(7), Ru(1)–C(6) 2.236(7), Ru(1)–C(7) 2.251(7), Ru(1)–C(34) 2.030(7), Ru(1)–N(3) 2.127(10), C(3)–O(3) 1.338(9). | ||
It is worth mentioning that UV irradiation (3 h) of the neutral complexes 3a and 3d leaves the reactant unaltered indicating that the formal oxidation of Ru complex, upon protonation, favor the CO removal. The same result was surprisingly obtained with the cationic complexes 4a[CF3SO3] and 5a[CF3SO3], for which CO release was expected (16 h of irradiation in iPrOH). Nonetheless irradiating 4a[CF3SO3] and 5a[CF3SO3], in the presence of a trapping agent such as pyridine, the formation of the complexes 7[CF3SO3] and 8[CF3SO3] was finally observed (Fig. 4) actually demonstrating that the CO release occurs in the cationic complexes. Indeed neutral complex 3a did not lose CO under UV irradiation even in the presence of pyridine. It is thus likely that, in the case of cationic complexes, without the help of pyridine the removal of CO is an equilibrium in which ruthenium–CO bond is reformed at the end of the reaction leading to the recovery of the precursors 4a[CF3SO3] and 5a[CF3SO3].
 | ||
| Fig. 4 Molecular structure of 7[CF3SO3] and 8[CF3SO3]. | ||
Formation of 7[CF3SO3] and 8[CF3SO3] is evidenced by the disappearance of the two carbonyl stretching bands associated to 4a[CF3SO3] (2037, 1985 cm−1) and 5a[CF3SO3] (2045, 1995 cm−1), with the concomitant appearance of a single band associated with the new complex 7[CF3SO3] (1943 cm−1) and 8[CF3SO3] (1948 cm−1) (Fig. 4). Complexes 7[CF3SO3] and 8[CF3SO3] have been found in mixture with other species and their characterization was not complete. However the bathochromic shift observed in IR spectra is in agreement with the replacement of a strong π ligand with a σ N-donor ligand such as pyridine. Furthermore ESI-MS analyses performed on complexes 7[CF3SO3] and 8[CF3SO3] showed the presence of the molecular ions.
Catalytic transfer hydrogenation
The Ru complexes 3a–d, 4a–d[X] [(X = Cl, BF4, NO3, CF3SO3)] and 5a,d[CF3SO3] were evaluated as catalyst precursors under transfer hydrogenation conditions i.e. refluxing iPrOH as hydrogen source employing 4-fluoroacetophenone as model substrate. Catalytic runs were performed to investigate the role of different additives in the activation of 3a–d, to compare the catalytic activity of the ruthenium(0) complexes 3a–d with the corresponding ruthenium(II) cationic complexes 4a–d[X] and 5a,d[CF3SO3] and finally to evaluate the influence of a pyridine substituent on the NHC side chain. Results are reported in Table 1: in all cases selectivity is complete and conversion corresponds to yield unless otherwise stated.
|
||||
|---|---|---|---|---|
| Entry | [Ru] | Additive | Conversion (%) 8 h | Conversion (%) 24 h |
| a General conditions: ruthenium complex (5 mol% Ru), iPrOH (3 mL), T = 82 °C; conversions determined by 19F NMR spectroscopy.b CAN 1 mol equiv. per ruthenium center.c Catalyst loading reduced to 1 mol% Ru.d NaIO4, BQ (benzoquinone), KOtBu and acids: 1 mol equiv. per ruthenium center.e Recycling test.f Yield ∼ 70% the co-product arising from the acid catalyzed etherification of 4-fluoroacetophenol with the solvent 2-propanol.g Yield ∼ 35% the coproduct was not identified. | ||||
| 1 | 3a | — | 0 | 0 |
| 2 | 3b | — | 0 | 0 |
| 3 | 3c | — | 0 | 0 |
| 4 | 3d | — | 0 | 0 |
| 5 | 3a | CANb | 25 | 61 |
| 6 | 3ac | CANb | 0 | 27 |
| 7 | 3b | CANb | 10 | 87 |
| 8 | 3c | CANb | 10 | 25 |
| 9 | 3d | CANb | 0 | 9 |
| 10 | 3a | NaIO4d | 0 | 0 |
| 11 | 3a | BQd | 25 | 55 |
| 12 | 4a[CF3SO3] | — | 58 | 93 |
| 13 | 4a[CF3SO3]c | — | 5 | 29 |
| 14 | 4a[CF3SO3]e | — | 64 | 87 |
| 15 | 4a[CF3SO3] | KOtBud | 14 | 35 |
| 16 | 4a[BF4] | — | 41 | 71 |
| 17 | 4a[Cl] | — | 16 | 24 |
| 18 | 3a | CF3SO3Hd | 32 | >99f |
| 19 | 3a | H2SO4d | 45 | 92 |
| 20 | 3a | HNO3d | 60 | 65 |
| 21 | 4b[CF3SO3] | — | 9 | 60 |
| 22 | 4c[CF3SO3] | — | 35 | 46g |
| 23 | 4d[CF3SO3] | — | 84 | >99 |
| 24 | 4d[Cl] | — | 0 | 5 |
| 25 | 4d[BF4] | — | 37 | 40 |
| 26 | 3d | CF3SO3Hd | 84 | >99f |
| 27 | 3d | H2SO4d | 76 | 86 |
| 28 | 3d | HNO3d | 0 | 36 |
| 29 | 5a[CF3SO3] | — | 28 | 64 |
| 30 | 5d[CF3SO3] | — | 10 | 13 |
| 31 | 6d[CF3SO3] | — | 0 | 5 |
| 32 | HCF3SO3 | — | 0 | 0 |
The neutral complexes 3a–d did not present any catalytic activity in the absence of additives (entry 1–4), as well as by adding NaIO4 (entry 10). However, we found that the addition of one equivalent of CAN (entry 5–9) results in the formation of active intermediates, likely deriving from the release of one CO ligand, favoured by oxidation as discussed in the previous section. In fact the use of benzoquinone, as oxidant additive under the same conditions (entry 11), activates the catalyst even though leading to a slightly lower conversion if compared with CAN.
Reduction of the catalyst loading to 1 mol% (3a) increases the induction time halving the conversion in 24 h (entry 5 vs. 6). In general pre-catalysts 3a–d resemble the behaviour of the triazolylidene ruthenium(0) congener, namely the dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1,3-dimethyl-4-phenyl-1,2,3-triazol-ylidene)ruthenium, previously tested as hydrogen transfer catalyst.24
The oxidation state of the metal center appears to be the key point in order to activate the catalyst and indeed, better conversions are reached with the pre-catalysts 4a–d[X] [(X = Cl, BF4, NO3, CF3SO3) (entries 12, 13, 16, 17 and 21–25)]. A recycling test has been performed with 4a[CF3SO3], the catalyst can be reused although a slight decrease in conversion from 93 to 87% was observed after 24 h (entry 14). Noteworthy also the counterion shows to influence the catalytic activity, and CF3SO3− provides the best results: for example we observed complete conversion within 24 h in the case of 4a[CF3SO3] and 4d[CF3SO3] (entries 12 and 23). Furthermore the latter pyridine-functionalized catalyst 4d[CF3SO3] shows a faster activation if compared with 4a[CF3SO3] (conversion of 84% and 58% respectively in 8 h). On the other hand, catalytic activity is suppressed in the case of 4a[Cl] and 4d[Cl] (entry 17 and 24), possibly due to the coordinating ability of Cl− counterion; on the contrary 4a[BF4] and 4d[BF4] give intermediate results (entry 16 and 25).
The in situ addition of various acids such as HCF3SO3, H2SO4 and HNO3 to 3a and 3d (entries 18–20 and 26–28) produces, in the case of triflic acid, similar conversions as for the corresponding preformed catalysts (e.g. entry 23, 4d[CF3SO3] vs. entry 26, 3d + CF3SO3H in situ). However, a lower selectivity is observed due to the acid catalyzed etherification of 4-fluorobenzylalcohol and isopropanol. Best in situ performances are accomplished by sulfuric acid (entries 19 and 27), which gives much better results than HNO3 (entry 20 and 28), confirming the detrimental effect of the coordination ability of counterions. A base, such as KOtBu, has a negative effect on the reaction (entry 15), likely due to deactivation of the pre-catalyst by deprotonation. Finally a blank experiment performed with HCF3SO3 in refluxing isopropanol rule out an exclusive role of the acid itself in the catalytic activity (entry 32).
With the aim of evaluating the role of the hydroxyl group in the catalytic reaction methylated complexes 5a[CF3SO3] and 5d[CF3SO3] have been also tested as catalysts (entry 29 and 30). Quite surprisingly, the methylated complex 5a[CF3SO3] shows a remarkable catalytic activity although in the absence of the acidic OH. Worst performances came from the pyridine-functionalized methylated complex 5d[CF3SO3], probably due to a deactivation pathway associated with the formation of stable species in the chelated form. Indeed, no conversion is observed when the chelated cationic complex 6d[CF3SO3] is employed as pre-catalyst (entry 31).
The substrate scope has been also evaluated extending the reactivity of 3a, 4a[CF3SO3] and 4d[CF3SO3] as hydrogen transfer catalysts to acetophenone, benzophenone and benzaldehyde. Data are reported in Table 2. All the substrates are converted to the corresponding alcohol by activated 3a and by the cationic complex 4a[CF3SO3]. As expected the catalysts are much more active in the case of benzaldehyde (entry 7 and 9), since aldehydes are substrates more prone to reduction than ketones. Additionally, the scale of activity is consistent with the electron withdrawing properties of the ketones examined. Again, 4d[CF3SO3] (entry 3, 6 and 9) shows a faster activation. As an example, benzaldehyde reaches complete conversion within 4 h, that becomes 8 h in the case of 4a[CF3SO3] (entry 9c vs. 8).
| Entry | Substrate | [Ru] | Additiveb | Conv. (%) 8 h | Conv. (%) 24 h |
|---|---|---|---|---|---|
| a Reaction conditions: [Ru] = 5 mol%, iPrOH (3 mL), T = 82 °C. Conversions determined by GC.b CAN 1 mol equiv. per ruthenium center.c Complete conversion is reached within 4 h. | |||||
| 1 |  |
3a | CAN | 10 | 77 |
| 2 | 4a[CF3SO3] | — | 15 | 68 | |
| 3 | 4d[CF3SO3] | — | 45 | 48 | |
| 4 |  |
3a | CAN | 0 | 42 |
| 5 | 4a[CF3SO3] | — | 0 | 36 | |
| 6 | 4d[CF3SO3] | — | 42 | 43 | |
| 7 |  |
3a | CAN | 31 | 39 |
| 8 | 4a[CF3SO3] | — | >99 | ||
| 9 | 4d[CF3SO3] | — | >99c | ||
Mechanistic insight
On the basis of the results described above, we can draw some considerations regarding the active catalytic species. First of all, the trapping experiments with pyridine (see cyclization of 4d[CF3SO3] to 6d[CF3SO3], Scheme 3 and complexes 7[CF3SO3] and 8[CF3SO3], Fig. 4) demonstrates that CO release is involved in the activation, leaving unaltered the Ru–carbene bond. Indeed, all the crude products of catalytic reactions have been analyzed by IR and NMR spectroscopy and only in a couple of cases traces of Shvo catalyst, which would results from NHC ligand release, were visible in NMR spectra, accompanied by a very bright yellow color of the solution typical of Shvo complex.18 Furthermore, an outer sphere mechanism is likely to be followed,18b in that the complex [CpRu(CO)2][CF3SO3], obtained by reaction between [Cp2Ru2(CO)4] with AgOTf, did not present any catalytic activity, supporting the role of hydroxycyclopentadienyl complex as a bifunctional catalyst. Noteworthy, in the case of 4d[CF3SO3] the pyridine itself can be involved in the catalytic cycle as a cooperative ligand through the formation of the active catalyst A in which an hydride and two different acid hydrogens should be available for the hydrogen transfer (Fig. 5).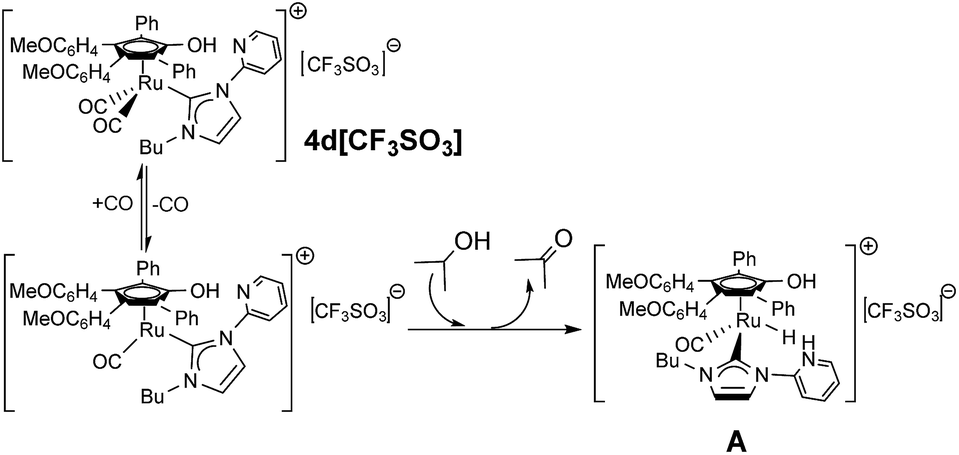 | ||
| Fig. 5 Proposed activation of the pre-catalyst 4d[CF3SO3]. | ||
Indeed, 4d[CF3SO3] results to be the best pre-catalyst. Furthermore from the crude reaction mixture at the end of the reaction some resonances in the 1H-NMR spectrum are detected in the hydride region: a singlet at δ −10.18 ppm is presumably associated with monomeric Ru–H species, while the signal at δ −14.95 ppm falls in the region of bridging hydrides (Ru–H–Ru). Even though we were not able to isolate these hydride species, in analogy with what observed in the synthesis of Shvo catalyst (δ RuHRu −18.80 ppm for the dimeric specie and δ −9.37 ppm for the monomeric one),18d,23 the formation of active hydride complexes containing the NHC ligand in place of a terminal CO reasonably occurs.
Labeling experiments on the reduction of 4-fluoroacetophenone with 4a[CF3SO3] and 4d[CF3SO3] in isopropanol-d8 (see ESI for reaction conditions and spectra†) lead to the formation of the corresponding deuterated alcohol further confirming the role of isopropanol as hydrogen source as well as the action of the catalysts in its activation.
With regard to 4c[CF3SO3], which in our experiments leads to the worst conversion after 24 h and lower selectivity (Table 1, entry 22), its molecular structure (Fig. 2, right side) evidences an hydrogen bond between the hydroxycyclopentadienyl ligand and the –OH group in the NHC side chain. Furthermore 4c[CF3SO3], upon CO release, would be prone to an intramolecular dehydrogenation, probably leading to the formation of an inactive catalyst. By comparing the behavior of 4c[CF3SO3] with that of an iridium complex containing the NHC hydroxymethyl ligand which lead to the formation of an  complex upon oxidation and cyclometallation25 we can propose a similar reactivity for our ruthenium complex in agreement with the ν(CO) and ν(CO) bands found at 1950 cm−1 and 1723 cm−1.
complex upon oxidation and cyclometallation25 we can propose a similar reactivity for our ruthenium complex in agreement with the ν(CO) and ν(CO) bands found at 1950 cm−1 and 1723 cm−1.
Concerning monodentate NHC complexes 4a–c[X] (X = Cl, BF4, NO3, CF3SO3) due to the absence of a N-containing side group able to be protonated, the active catalytic species has to be different from the proposed intermediate A. Once a vacant coordination site is formed by CO removal, proton abstraction from iPrOH should be performed by the counteranion CF3SO3− as external base, or by the oxygen of the –OH group (or –OMe group) on the catalyst itself, but at present we do not have any evidence to support such hypothesis.
Experimental
General
Solvents: dichloromethane (CH2Cl2), tetrahydrofuran (THF), diethyl ether (Et2O), petroleum ether referring to a fraction of bp 60–80 °C, acetonitrile (CH3CN) were dried and distilled prior to use. Acetone has been degassed and stored under inert atmosphere on molecular sieves. Other solvents such as ethylacetate (EtOAc), chloroform, ethanol (EtOH), methanol (MeOH), heptane, hexane, 2-propanol, CDCl3, D2O, CD3CN (Sigma Aldrich) have been employed without further purification. Reagents: triruthenium-dodecacarbonil (Ru3(CO)12) (Strem), methyl iodide, silver oxide, 1-methylimidazole, 1-chlorobutane, 1-butylimidazole, 1-chloroethanol, 1,3-diphenylacetone, 4,4′-dimethoxybenzil (Alfa Aesar), 2-bromopyridine, 4-fluoroacetophenone, acetophenone, benzophenone, benzaldehyde, dodecane, trifluoromethanesulfonic acid, tetrafluoroboric acid diethyl ether complex (Sigma Aldrich), chloridric, nitric and sulfuric acid, methyl trifluoromethanesulfonate, pyridine, cerium ammonium nitrate (CAN), benzoquinone, sodium periodate have been employed as purchased. 1,3-Dimethylimidazolium iodide (1a),26 1-methyl-3-butyl-imidazolium chloride (1b), 1-methyl-3-(2-hydroxyethyl)imidazolium chloride (1c),27 1-butyl-3-(2-pyridinyl)-imidazolium bromide (1d),28 3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone,29 dicarbonyl(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)ruthenium dimer,30 dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone-1,3(dimethyl)-imidazol-2-ylidene)ruthenium (3a), dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone-1-methyl-3-butyl-imidazol-2-ylidene)ruthenium (3b), dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone-1-methyl-3-butyl-imidazol-2-ylidene)ruthenium (3c),20 have been prepared as previously reported. The prepared derivatives were characterized by spectroscopic methods. The NMR spectra were recorded using Varian Inova 300 (1H, 300.1; 13C, 75.5 MHz), Varian Mercury Plus VX 400 (1H, 399.9; 13C, 100.6 MHz), Varian Inova 600 (1H, 599.7; 13C, 150.8 MHz) spectrometers at 298 K; chemical shifts were referenced internally to residual solvent peaks. Full 1H- and 13C-NMR assignments were done, when necessary, by gHSQC and gHMBC NMR experiments using standard Varian pulse sequences. Infrared spectra were recorded at 298 K on a Perkin-Elmer Spectrum 2000 FT-IR spectrophotometer. ESI-MS spectra were recorded on Waters Micromass ZQ 4000 with samples dissolved in MeOH or CH3CN. Elemental analyses were performed on a Thermo-Quest Flash 1112 Series EA instrument. The conversions were monitored by GC analysis (Agilent Technologies 7890A GC system) and 19F-NMR. UV irradiation was performed by using a commercial Hg lamp (365 nm, 125 W).Synthesis of dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)-1-(butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium (3d)
A mixture of 1-butyl-3-(2-pyridinyl)-1H-imidazolium bromide (1d) 0.094 g (0.33 mmol), Ag2O 0.092 g (0.40 mmol) and dicarbonyl(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)ruthenium dimer (2) 0.200 g (0.16 mmol) in dry CH2Cl2 were stirred in the dark under inert atmosphere at room temperature for 1 h. Upon filtration on a celite pad and removal of the solvent the quantitative formation of the dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)-1-(butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium (3d) was verified by 1H-NMR, 13C-NMR, ESI-MS and X-ray crystal structure. Suitable crystals were obtained by slow diffusion from toluene/hexane double layer 1H-NMR (599.7 MHz, CDCl3) δ (ppm): 8.36 (dd, 1H, CHpy), 7.60 (m, 4H, CHaryl), 7.04–6.97 (m, 10H, CHaryl + 1H, CHpy + 2H, CHNHC), 6.60–6.55 (m, 4H, CHaryl + 1H, CHpy), 3.67 (s, 6H, –OCH3), 3.54 (t, 2H, NCH2), 1.46 (m, 2H, –CH2CH2–), 1.04 (m, 2H, –CH2CH3), 0.75 (t, 3H, –CH3); 13C–{1H}NMR (150.8 MHz, CDCl3) δ (ppm): 201.77 (CO), 173.20 (Ccarbene), 168.22 (C1O, Cp), 158.41 (–COCH3), 152.16 (Cipso), 148.11 (CHpy), 138.40 (CHpy), 135.17 (Cqaryl), 133.45 (CHaryl), 129.49 (CHaryl), 127.43 (CHaryl), 125.28 (Cqaryl), 124.85 (CHaryl), 124.54 (CHNHC), 123.89 (CHpy), 122.43 (CHNHC), 121.82 (CHpy), 112.76 (CHaryl), 103.10 (C2,5, Cp), 79.69 (C3,4, Cp), 54.98 (–OCH3), 50.72 (NCH2), 33.08, 19.41 (–CH2CH2), 14.11 (–CH3); IR (CH2Cl2) ν(CO): 2008 cm−1, 1949 cm−1, ν(CO) 1583 cm−1, ν(CC) 1609 cm−1, 1518 cm−1. ESI-MS (m/z) (+) = 804 [M + H]+; 826 [M + Na]+. Anal. calcd (%) for C45H39N3O5Ru: C, 67.23; H, 4.89; N, 5.23. Found: C, 67.21; H, 4.87; N, 5.24.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1,3-dimethylimidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (4a[CF3SO3])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1,3-dimethylimidazol-2-ylidene)ruthenium complex (3a) 0.100 g (0.143 mmol) was dissolved in 10 mL of CH2Cl2 under inert atmosphere. 1.2 equivalent of HCF3SO3 (solution at 0.98% in CH2Cl2) 1.55 mL (0.172 mmol) were subsequently added. The reaction mixture was stirred for 10 minutes at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid obtained was identified as 4a[CF3SO3] by IR, 1H-NMR, 13C-NMR, 19F-NMR, ESI-MS. The yield was quantitative and suitable crystals for X-ray diffraction analysis were prepared by toluene/hexane double layer. 1H-NMR (599.7 MHz, CDCl3) δ (ppm): 7.43 (m, 4H, CHaryl), 7.27–7.01 (m, 10H, CHaryl), 6.97 (s, 2H, CHNHC), 6.65 (m, 4H, CHaryl), 3.71 (s, 6H, –OCH3), 3.27 (s, 6H, NCH3,NHC); 13C–{1H}NMR (150.8 MHz, CDCl3) δ (ppm): 199.64 (CO), 161.35 (Ccarbene), 159.88 (–COCH3), 142.77 (C–OH, Cp), 133.86 (CHaryl), 130.93 (CHaryl), 129.07 (CHaryl), 125.27 (Cqaryl), 128.38 (Cqaryl), 125.97 (CHaryl), 121.40 (CHNHC), 113.81 (CHaryl), 104.85 (C2,5, Cp), 87.90 (C3,4, Cp), 55.55 (–OCH3), 39.23 (CH3,NHC). 19F-NMR (282.4 MHz, CDCl3) δ (ppm): –78.34 (CF3SO3). IR (CH2Cl2) ν(CO): 2037 cm−1, 1985 cm−1, ν(CC) 1611 cm−1, 1520 cm−1. ESI-MS (m/z) (+) = 699 [M]+; 149 [M]−. Anal. calcd (%) for C39H33F3N2O8RuS: C, 55.18; H, 3.92; N, 3.30. Found: C, 55.14; H, 3.90; N, 3.32.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1,3-dimethylimidazol-2-ylidene)ruthenium]chloride (4a[Cl])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1,3-dimethylimidazol-2-ylidene)ruthenium complex (3a) 0.023 g (0.0330 mmol) was dissolved in 5 mL of CH2Cl2 under inert atmosphere. Two equivalent of HCl (aqueous solution at 37%, 0.005 mL, 0.0660 mmol) were subsequently added. The reaction mixture was stirred for 2 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 4a[Cl] by IR, 1H-NMR, 13C-NMR, ESI-MS obtained in quantitative yield. 1H-NMR (599.7 MHz, CDCl3) δ (ppm): 7.57 (m, 4H, CHaryl), 7.23–7.03 (m, 10H, CHaryl), 6.93 (s, 2H, CHNHC), 6.65 (m, 4H, CHaryl), 3.71 (s, 6H, –OCH3), 3.03 (s, 6H, NCH3,NHC); 13C–{1H}NMR (150.8 MHz, CDCl3) δ (ppm): 199.79 (CO), 162.42 (Ccarbene), 159.24 (–COCH3), 144.52 (C–OH, Cp), 133.47 (CHaryl), 130.95 (CHaryl), 128.60 (CHaryl), 128.18 (Cqaryl), 125.35 (CHaryl), 121.60 (CHNHC), 113.24 (CHaryl), 104.55 (C2,5, Cp), 86.61 (C3,4, Cp), 55.09 (–OCH3), 39.22 (CH3,NHC). IR (CH2Cl2) ν(CO): 2033 cm−1, 1981 cm−1, ν(CC) 1611 cm−1, 1520 cm−1. ESI-MS (m/z) (+) = 699 [M]+. Anal. calcd (%) for C38H33ClN2O5Ru: C, 62.11; H, 4.53; N, 3.81. Found: C, 62.13; H, 4.55; N, 3.79.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1,3-dimethylimidazol-2-ylidene)ruthenium]tetrafluoroborato (4a[BF4])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1,3-dimethylimidazol-2-ylidene)ruthenium complex (3a) 0.050 g (0.072 mmol) was dissolved in 10 mL of CH2Cl2 under inert atmosphere. 1.2 equivalent of tetrafluoroboric acid diethyl ether complex (0.012 mL, 0.086 mmol) were subsequently added. The reaction mixture was stirred for 1 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 4a[BF4] by IR, 1H-NMR, 13C-NMR, 19F-NMR was obtained in quantitative yield. 1H-NMR (599.7 MHz, CDCl3) δ (ppm): 7.42 (m, 4H, CHaryl), 7.31–7.03 (m, 10H, CHaryl), 7.05 (s, 2H, CHNHC), 6.68 (m, 4H, CHaryl), 3.74 (s, 6H, –OCH3), 3.29 (s, 6H, NCH3,NHC); 13C–{1H}NMR (150.8 MHz, CDCl3) δ (ppm): 199.06 (CO), 160.82 (Ccarbene), 159.57 (–COCH3), 141.82 (C–OH, Cp), 133.43 (CHaryl), 130.40 (CHaryl), 128.80 (CHaryl), 128.78 (Caryl), 127.89 (Caryl), 125.74 (CHaryl), 120.80 (CHNHC), 113.47 (CHaryl), 104.31 (C2,5, Cp), 87.51 (C3,4, Cp), 55.15 (–OCH3), 38.75 (CH3,NHC); 19F-NMR (282.4 MHz, CDCl3) δ (ppm): –152.01 (BF4). IR (CH2Cl2) ν(CO): 2038 cm−1, 1987 cm−1, ν(CC) 1611 cm−1, 1520 cm−1. Anal. calcd (%) for C38H33BF4N2O5Ru: C, 58.09; H, 4.20; N, 3.57. Found: C, 58.11; H, 4.17; N, 3.58.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1-methyl-3-butyl-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (4b[CF3SO3])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1-methyl-3-butyl-imidazol-2-ylidene)ruthenium complex (3b) 0.100 g (0.135 mmol) was dissolved in 10 mL of CH2Cl2 under inert atmosphere. 1.2 equivalent of HCF3SO3 (solution at 0.98% in CH2Cl2 1.39 mL, 0.157 mmol) were subsequently added. The reaction mixture was stirred for 1 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 4b[CF3SO3] by IR, 1H-NMR, 13C-NMR and 19F-NMR was obtained in quantitative yield. 1H-NMR (599.7 MHz, CDCl3) δ (ppm): 7.44 (m, 4H, CHaryl), 7.28–7.02 (m, 10H, CHaryl), 7.05 (2H, CHNHC), 6.66 (m, 4H, CHaryl), 3.72 (s, 6H, –OCH3), 3.70 (m, 2H, –NCH2), 3.32 (s, 3H, NCH3,NHC), 1.49 (m, 2H, –CH2CH2–), 0.97 (m, 2H, –CH2CH3), 0.77 (m, 3H, –CH3); 13C–{1H}NMR (150.8 MHz, CDCl3) δ (ppm): 199.27 (CO), 160.57 (Ccarbene), 159.47 (–COCH3), 142.83 (C–OH, Cp), 133.43 (CHaryl), 130.55 (CHaryl), 128.60 (CHaryl), 128.09 (Caryl), 126.24 (Caryl), 123.02 (CHaryl), 121.01 (CHNHC), 113.39 (CHaryl), 104.42 (C2,5, Cp), 87.45 (C3,4, Cp), 55.13 (–OCH3), 50.81 (–NCH2), 38.91 (–NCH3), 32.97 (–CH2CH2–), 19.48 (–CH2CH3), 13.65 (–CH3); 19F-NMR (282.4 MHz, CDCl3) δ (ppm): –78.37. IR (CH2Cl2) ν(CO): 2035 cm−1, 1984 cm−1, ν(CC) 1610 cm−1, 1520 cm−1. ESI-MS (m/z) (+) = 741 [M]+, 149 [M]−. Anal. calcd (%) for C42H39F3N2O8RuS: C, 56.62; H, 4.38; N, 3.15. Found: C, 56.59; H, 4.37; N, 3.16.
Synthesis of [dicarbonyl-η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1-methyl-3-(2-hydroxyethyl-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (4c[CF3SO3])
Dicarbonyl-η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1-methyl-3-(2-hydroxyethyl-imidazol-2-ylidene)ruthenium complex (3c) 0.050 g (0.069 mmol) was dissolved in 10 mL of CH2Cl2 under inert atmosphere. 1.2 equivalent of HCF3SO3 (solution at 0.98% in CH2Cl2 0.73 mL, 0.082 mmol) were subsequently added. The reaction mixture was stirred for 1 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 4c[CF3SO3] by IR, 1H-NMR, 13C-NMR, 19F-NMR, ESI-MS was obtained in quantitative yield. Suitable crystals for X-ray diffraction analysis were prepared by slow diffusion from CH2Cl2/Et2O double layer. 1H-NMR (399.9 MHz, CDCl3) δ (ppm): 7.46 (m, 4H, CHaryl), 7.30–7.03 (m, 10H, CHaryl), 7.40 (s, 1H, CHNHC), 7.01 (s, 1H, CHNHC), 6.68 (m, 4H, CHaryl), CH2 not detected, (m, 2H, –NCH2), 3.73 (s, 6H, –OCH3), 3.24 (s, 3H, –NCH3); 13C-NMR (100.6 MHz, CDCl3) δ (ppm): 206.97 (CO), 162.98 (Ccarbene), 159.58 (–COCH3), 142.23 (C–OH, Cp), 133.41 (CHaryl), 130.49 (CHaryl), 128.84 (CHaryl) 128.62 (Caryl), 126.08 (Caryl), 123.02 (CHaryl), 120.75 (CHNHC), 113.48 (CHaryl), Caryl, Cp not detected, 58.43 (–NCH2), 55.14 (–OCH3), 51.82 (–CH2OH), 38.45 (–NCH3); 19F-NMR (282.4 MHz, CDCl3) δ (ppm): –78.37. IR (CH2Cl2) ν(CO): 2038 cm−1, 1988 cm−1, ν(CC) 1610 cm−1, 1520 cm−1. ESI-MS (m/z) (+) = 729 [M]+, 149 [M]−. Anal. calcd (%) for C40H35F3N2O9RuS: C, 54.67; H, 3.85; N, 3.19. Found: C, 54.68; H, 3.83; N, 3.22.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (4d[CF3SO3])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium complex (3d) 0.100 g (0.124 mmol) was dissolved in 10 mL of CH2Cl2 under inert atmosphere. 1.2 equivalent of HCF3SO3 (solution at 0.98% in CH2Cl2 1.35 mL, 0.149 mmol) were subsequently added. The reaction mixture was stirred for 15 minutes at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly orange solid identified as 4d[CF3SO3] by IR, 1H-NMR, 13C-NMR, 19F-NMR, ESI-MS was obtained in quantitative yield. 1H-NMR (599.7 MHz, CD3CN) δ (ppm): 7.88 (m, 1H, CHpy), 7.38 (m, 1H, CHpy), 7.34 (m, 1H, CHpy), 7.28 (d, 1H, CHNHC), 7.25 (d, 1H, CHNHC), 7.23 (m, 1H, CHpy), 7.17–6.99 (m, 10H, CHaryl), 6.78 (m, 4H, CHaryl), 6.48 (m, 4H, CHaryl), 3.70 (t, 2H, NCH2), 3.68 (s, 6H, –OCH3), 1.72 (m, 2H, –CH2CH2–), 1.19 (m, 2H, –CH2CH3), 0.88 (t, 3H, –CH3); 13C–{1H}NMR (150.8 MHz, CD3CN) δ (ppm): 199.59 (CO), 166.73 (Ccarbene), 160.80 (–COCH3), 149.27 (Cipso), 147.43 (CHpy), 144.94 (CHpy), 136.56 (C1–OH, Cp), 134.52–114.29 (Caryl), 127.93 (CHNHC), 125.71 (CHpy), 125.13 (CHpy), 121.95 (CHNHC), 104.10 (C2,5, Cp), 94.05 (C3,4, Cp), 55.98 (–OCH3), 52.87 (–NCH2), 33.72 (–CH2CH2), 20.42 (–CH2CH3), 13.93 (–CH3); 19F-NMR (282.4 MHz, CD3CN) δ (ppm): –79.33 (CF3SO3). IR (CH2Cl2) ν(CO): 2041 cm−1, 1991 cm−1, ν(CC) 1610 cm−1, 1520 cm−1. ESI-MS (m/z) (+) = 804 [M]+, 149 [M]−. Anal. calcd (%) for C46H40F3N3O8RuS: C, 57.91; H, 4.23; N, 4.41. Found: C, 57.89; H, 4.22; N, 4.38.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium]chloride (4d[Cl])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium complex (3d) 0.025 g (0.031 mmol) was dissolved in 5 mL of CH2Cl2 under inert atmosphere. Two equivalent of HCl (aqueous solution at 37%, 0.005 mL, 0.061 mmol) were subsequently added. The reaction mixture was stirred for 1 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 4d[Cl] by IR, 1H-NMR, 13C-NMR obtained in quantitative yield. 1H-NMR (599.7 MHz, CD3CN) δ (ppm): 8.10 (m, 1H, CHpy), 7.69 (m, 1H, CHpy), 7.48 (d, 2H, CHNHC), 7.45 (d, 2H, CHNHC), 7.41–7.19 (m, 10H, CHaryl + 2H, CHpy), 6.98 (m, 4H, CHaryl), 6.65 (m, 4H, CHaryl), 3.68 (t, 2H, –NCH2), 3.69 (s, 6H, –OCH3), 1.70 (m, 2H, –CH2CH2–), 1.14 (m, 2H, –CH2CH3), 0.84 (t, 3H, –CH3); 13C–{1H}NMR (150.8 MHz, CD3CN) δ (ppm): 199.73 (CO), 166.09 (Ccarbene), 160.67 (–COCH3), 150.34 (Cipso), 147.87 (CHpy), 143.52 (CHpy), 136.47 (C1–OH, Cp), 134.62–114.23 (Caryl + CHpy), 124.71 (CHNHC), 122.13 (CHNHC), 102.43 (C2,5, Cp), 87.44 (C3,4, Cp), 55.95 (–OCH3), 52.65 (–NCH2), 33.24 (–CH2CH2), 20.40 (–CH2CH3), 13.93 (–CH3); IR (CH2Cl2) ν(CO): 2041 cm−1, 1992 cm−1, ν(CC) 1609 cm−1, 1521 cm−1. Anal. calcd (%) for C45H39ClN3O5Ru: C, 64.43; H, 4.69; N, 5.01. Found: C, 64.40; H, 4.66; N, 5.02.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium]tetrafluoroborate (4d[BF4])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium complex (3d) 0.050 g (0.062 mmol) was dissolved in 5 mL of CH2Cl2 under inert atmosphere. 1.2 equivalent of tetrafluoroboric acid diethyl ether complex (0.012 mL, 0.086 mmol) were subsequently added. The reaction mixture was stirred for 1 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 4d[BF4] by IR, 1H-NMR, 19F-NMR, 13C-NMR, ESI-MS was obtained in quantitative yield. 1H-NMR (599.7 MHz, CD3CN) δ (ppm): 7.95 (m, 1H, CHpy), 7.49–7.16 (m, 10H, CHaryl + 3H, CHpy + 2H, CHNHC), 6.98 (m, 4H, CHaryl), 6.69 (m, 4H, CHaryl), 3.69 (s, 6H, –OCH3), 3.67 (t, 2H, NCH2), 1.75 (m, 2H, –CH2CH2–), 1.25 (m, 2H, –CH2CH3), 0.79 (t, 3H, -CH3); 13C–{1H}NMR (150.8 MHz, CD3CN) δ (ppm): 199.65 (CO), 166.30 (Ccarbene), 160.78 (–COCH3), 151.52 (Cipso), 149.07 (CHpy), 142.05 (CHpy), 134.63 (C1–OH, Cp), 134.06–114.30 (Caryl), 126.82 (CHNHC), 124.74 (CHpy), 124.25 (CHpy), 122.20 (CHNHC), 104.16 (C2,5, Cp), 87.35 (C3,4, Cp), 56.00 (–OCH3), 52.84 (NCH2), 33.21 (–CH2CH2–), 20.55 (–CH2CH3), 13.91 (–CH3); 19F-NMR (282.4 MHz, CD3CN) δ (ppm): −151.63 (BF4). IR (CH2Cl2) ν(CO): 2041 cm−1, 1992 cm−1, ν(CC) 1609 cm−1, 1521 cm−1. Anal. calcd (%) for C45H39BF4N3O5Ru: C, 60.66; H, 4.41; N, 4.71. Found: C, 60.63; H, 4.40; N, 4.69.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylmethoxycyclopentadienyl)(1,3-dimethyl-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (5a[CF3SO3])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1,3-dimethylimidazol-2-ylidene)ruthenium complex (3a) 0.100 g (0.143 mmol) was dissolved in 10 mL of CH2Cl2 under inert atmosphere. Three equivalent of MeCF3SO3 (0.048 mL, 0.429 mmol) were subsequently added. The reaction mixture was stirred for 1 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 5a[CF3SO3] by IR, 1H-NMR, 13C-NMR, 19F-NMR, ESI-MS was obtained in quantitative yield. 1H-NMR (599.7 MHz, CD3CN) δ (ppm): 7.40–7.31 (m, 10H, CHaryl), 7.13 (s, 2H, CHNHC), 7.04 (m, 4H, CHaryl), 6.66 (m, 4H, CHaryl), 3.68 (s, 6H, –OCH3), 3.43 (s, 6H, NCH3), 2.99 (s, 3H, –OCH3); 13C-NMR (150.8 MHz, CD3CN) δ (ppm): 199.52 (CO), 161.90 (Ccarbene), 160.59 (–COCH3), 141.45 (C–OCH3, Cp), 134.53 (CHaryl), 132.34 (CHaryl), 129.66 (CHaryl), 129.28 (Caryl), 128.24 (Caryl), 126.22 (CHaryl), 121.71 (CHNHC), 114.04 (CHaryl), 104.86 (C2,5, Cp), 93.24 (C3,4, Cp), 62.47 (–OCH3, Cp), 55.87 (–OCH3), 39.71 (–NCH3); 19F-NMR (282.4 MHz, CD3CN) δ (ppm): −78.23. IR (CH2Cl2) ν(CO): 2045 cm−1, 1995 cm−1; ν(CC) 1610 cm−1, 1521 cm−1. ESI-MS (m/z) (+) = 713 [M]+; 149 [M]−. Anal. calcd (%) for C40H35F3N2O8RuS: C, 55.68; H, 4.09; N 3.25. Found: C, 55.65; H, 4.07; N 3.27.
Synthesis of [dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylmethoxycyclopentadienyl1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (5d[CF3SO3])
Dicarbonyl-(η4-3,4-bis(4-methoxyphenyl)-2,5-diphenylcyclopenta-2,4-dienone)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium complex (3d) 0.050 g (0.072 mmol) was dissolved in 10 mL of CH2Cl2 under inert atmosphere. Three equivalent of MeCF3SO3 (0.023 mL, 0.216 mmol) were subsequently added. The reaction mixture was stirred for 1 h at room temperature, then the solvent was removed under vacuum and the crude washed twice with 10 mL of Et2O. The slightly brown solid identified as 5d[CF3SO3] by IR and ESI-MS was obtained in quantitative yield. 1H-NMR (599.7 MHz, CD3CN) δ (ppm): 8.47 (m, 1H, CHpy), 7.87 (m, 1H, CHpy), 7.71 (m, 1H, CHpy), 7.65 (d, 1H, CHNHC), 7.56 (d, 1H, CHNHC), 7.46–6.59 (m, 18H, CHaryl + 1H, CHpy), 3.69 (s, 6H, –OCH3), 3.63 (t, 2H, NCH2), 3.15 (s, 6H, –OCH3, Cp), 1.52 (m, 2H, –CH2CH2–), 1.11 (m, 2H, –CH2CH3), 0.77 (t, 3H, –CH3); 13C–{1H}NMR (150.8 MHz, CD3CN) δ (ppm): 199.85 (CO), 167.40 (Ccarbene), 160.61 (–COCH3), 150.52 (Cipso), 149.56 (CHpy), 145.70 (CHpy), 140.66 (C1–OCH3, Cp), 134.60–114.14 (Caryl), 127.56 (CHNHC), 124.19 (CHpy), 121.64 (CHpy), 120.73 (CHNHC), Caryl, Cp not detected, 63.36 (–OCH3, Cp), 55.93 (–OCH3), 52.14 (–NCH2), 33.04 (–CH2CH2), 20.15 (–CH2CH3), 14.01 (–CH3); 19F-NMR (282.4 MHz, CD3CN) δ (ppm): –79.32 (CF3SO3). IR (CH2Cl2) ν(CO): 2045 cm−1, 1996 cm−1, ν(CC) 1611 cm−1, 1520 cm−1. ESI-MS (m/z) (+) = 818 [M]+; 149 [M]−. Anal. calcd (%) for C47H42F3N3O8RuS: C, 58.32; H, 4.34; N, 4.34. Found: C, 58.30; H, 4.32; N, 4.35.
Synthesis of [carbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylmethoxycyclopentadienyl)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (6d[CF3SO3])
[Dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1-butyl-3-(2-pyridinyl)-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (4d[CF3SO3]) was dissolved in 25 mL of iPrOH. The reaction mixture was irradiated under stirring for 3 h, then the solvent was removed under vacuum and the crude washed twice with 20 mL of hexane. The brown solid was identified as 6 and obtained with an yield of 78%. 6d[CF3SO3] has been completely characterized by IR, 1H-NMR, 13C-NMR, 19F-NMR, ESI-MS. Suitable crystals for X-ray diffraction analysis were prepared by slow diffusion from CH2Cl2/hexane double layer. 1H-NMR (599.7 MHz, CDCl3) δ (ppm): 8.33 (d, 1H, CHpy), 8.27 (m, 1H, CHpy), 8.09 (m, 1H, CHpy), 7.04 (m, 4H, CHaryl), 7.30–6.66 (m, 2H, CHNHC + 10H, CHaryl), 3.86 (m, 2H, NCH2), 3.79 (s, 3H, –OCH3), 3.74 (s, 3H, –OCH3), 1.47 (m, 2H, –CH2CH2), 0.97 (m, 2H, –CH2CH3), 0.72 (t, 3H, –CH3); 13C–{1H}NMR (150.8 MHz, CDCl3) δ (ppm): 199.57 (CO), 185.63 (Ccarbene), 159.30, 159.08 (–COCH3), 154.60 (Cipso), 153.34 (CHpy), 141.53 (C1–OH, Cp), 138.08 (CHpy), 133.31–112.74 (Caryl + CHNHC), 97.38 (C2,5, Cp), 84.34 (C3,4, Cp), 55.21 (–OCH3), 50.67 (NCH2), 32.66, 19.35 (–CH2CH2), 13.56 (–CH3); 19F-NMR (282.4 MHz, CD3CN) δ (ppm): −78.19 (CF3SO3); IR (CH2Cl2) ν(CO): 1967 cm−1, ν(CC) 1609 cm−1, 1521 cm−1; ESI-MS (m/z) (+) = 776 [M]+, 149 [M]−. Anal. calcd (%) for C45H40F3N3O7RuS: C, 58.37; H, 4.36; N, 4.54. Found: C, 58.35; H, 4.35; N, 4.51.
Synthesis of [carbonyl-pyridin-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1,3-dimethylimidazol-2-ylidene)ruthenium] trifluoromethanesulfonate (7[CF3SO3])
[Dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylhydroxycyclopentadienyl)(1,3-dimethylimidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (4a[CF3SO3]) 0.060 g (0.071 mmol) and pyridine 0.023 mL (0.283 mmol) were dissolved in iPrOH (25 mL). The reaction mixture was irradiated under stirring for 6 h and 7[CF3SO3] identified from the reaction mixture by IR and ESI-MS. IR (CH2Cl2) ν(CO): 1943 cm−1, ν(CC) 1610 cm−1, 1520 cm−1. ESI-MS (m/z) (+) (CH3CN) = 671 [M − py]+, 643 [M − py − CO]+.
Synthesis of [carbonyl-pyridin-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylmethoxycyclopentadienyl)(1,3-dimethylimidazol-2-ylidene)ruthenium] trifluoromethanesulfonate (8[CF3SO3])
[Dicarbonyl-(η5-3,4-bis(4-methoxyphenyl)-2,5-diphenylmethoxycyclopentadienyl)(1,3-dimethyl-imidazol-2-ylidene)ruthenium]trifluoromethanesulfonate (5a[CF3SO3]) 0.050 g (0.058 mmol) and pyridine 0.023 mL (0.232 mmol) were dissolved in toluene (25 mL). The reaction mixture was irradiated under stirring for 18 h and 8[CF3SO3] identified from the reaction mixture by IR and ESI-MS. IR (CH2Cl2) ν(CO): 1948 cm−1, ν(CC) 1610 cm−1, 1520 cm−1. ESI-MS (m/z) (+) (MeOH) = 764 [M]+; 149 [M]−.
General procedure for transfer hydrogenation
Complex (15 μmol, 5% mol), additive (1 eq. when needed) and iPrOH (3 mL) were stirred at reflux for 15 min. Then 4-fluoroacetophenone (36 μL, 300 μmol) was added and samples were taken at regular intervals. Aliquots (ca. 0.05 mL) were diluted with CDCl3 (0.5 mL) and conversions were determined by 19F-NMR spectroscopy and GC analysis.X-ray crystallography
Crystal data and collection details for 3d·0.5CH2Cl2, [4a][CF3SO3]·0.5toluene, [4c][CF3SO3]·CHCl3 and [6d][CF3SO3] are reported in Table S1.† The diffraction experiments were carried out on a Bruker APEX II diffractometer equipped with a CCD detector using Mo-Kα radiation. Data were corrected for Lorentz polarization and absorption effects (empirical absorption correction SADABS).31 Structures were solved by direct methods and refined by full-matrix least-squares based on all data using F2.32 All hydrogen atoms were fixed at calculated positions and refined by a riding model, unless otherwise stated. All non-hydrogen atoms were refined with anisotropic displacement parameters.Conclusions
A series of cationic hydroxy- or methoxy-cyclopentadienyl ruthenium(II) N-heterocyclic carbene complexes have been prepared in quantitative yield upon protonation with strong acids or methylation with MeCF3SO3 of neutral ruthenium(0) complexes containing both the non-innocent cyclopentadienone ligand and variously functionalized NHCs. These novel complexes have been successfully employed as selective catalysts in transfer hydrogenation. The reaction is general and several substrates such as various ketones and aldehydes can be selectively reduced to the corresponding alcohol. The catalytic activity can thus be tuned by the proper choice of both substituents on NHC ligand and counterions. Neutral Ru(0) complexes 3a–d need an oxidizing agent such as CAN or benzoquinone in order to be activated, while the corresponding cationic complexes 4a–d[X] (X = Cl, BF4, NO3, CF3SO3) and 5a,d[CF3SO3], formally containing a Ru(II) center are more prone to release a CO ligand and do not need any additives to be activated. In the cationic complexes the counterion plays a non-innocent role and CF3SO3− showed the best outcome due to its coordinating ability.Noteworthy the insertion of a pyridine substituent on the NHC side chain further improves the catalytic activity due to the presence of a second cooperative ligand containing a basic nitrogen.
With regard to the reaction mechanism CO release and its key role in the catalyst activation was demonstrated by trapping experiment with pyridine as well as by the UV-mediated formation of the chelated complex 6d[CF3SO3]. Hydride species has been identified in the reaction mixture of the pyridine functionalized complex 4d[CF3SO3] and the cooperative function of nitrogen- (pyridine in 4d[X] and 5d[CF3SO3]) and oxygen-containing (CF3SO3− or –OH and –OMe in 4a–c[X] and 5a[CF3SO3]) moieties with the vacancy on ruthenium prone to accept an hydride species has been drawn as consideration in order to explain iPrOH activation and subsequent active catalyst formation.
The versatility of NHC ligands which can be variously functionalized, together with the presence of the non-innocent cyclopentadienone open the way to the rational design of novel metal-ligand bifunctional catalysts liable to heterogenization and tunable solubility.
Acknowledgements
Authors wish to thank the University of Bologna and the FARB Project “Catalytic transformation of biomass-derived materials into high added-value chemicals”, 2014–2015.Notes and references
- For reviews, see: (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed; (c) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed; (d) L. A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (c) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS; (d) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677 CrossRef CAS PubMed; (e) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef CAS PubMed; (f) L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903 RSC; (g) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (h) S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, Wiley VCH, 2014 Search PubMed.
- (a) C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708 CrossRef CAS PubMed; (b) S. Monfette and D. E. Fogg, Chem. Rev., 2009, 109, 3783 CrossRef CAS PubMed; (c) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS PubMed.
- (a) E. A. B. Kantchev, C. J. O'Brien and M. G. Organ, Angew. Chem., Int. Ed., 2007, 46, 2768 CrossRef CAS PubMed; (b) G. C. Fortman and S. P. Nolan, Chem. Soc. Rev., 2011, 40, 5151 RSC.
- For examples, see: (a) R. Corberan and E. Peris, Organometallics, 2008, 27, 1954 CrossRef CAS; (b) A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218 RSC; (c) A. Petronilho, J. A. Woods, H. Mueller-Bunz, S. Bernhard and M. Albrecht, Chem.–Eur. J., 2014, 20, 15775 CrossRef CAS PubMed.
- (a) A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed; (b) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC; (c) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2008, 47, 6754 CrossRef CAS PubMed.
- (a) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef CAS PubMed; (b) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032 RSC.
- (a) J. Witt, A. Pothig, F. E. Kuhn and W. Baratta, Organometallics, 2013, 32, 4042 CrossRef CAS; (b) N. Gurbuz, E. O. Ozcan, I. Ozdemir, B. Cetinkaya, O. Sahin and O. Buyukgungor, Dalton Trans., 2012, 41, 2330 RSC; (c) S. Horn, C. Gandolfi and M. Albrecht, Eur. J. Inorg. Chem., 2011, 2863 CrossRef CAS; (d) S. Kuwata and T. Ikariya, Chem.–Eur. J., 2011, 17, 3542 CrossRef CAS PubMed; (e) W. W. N. O, A. J. Lough and R. H. Morris, Organometallics, 2012, 31, 2137 CrossRef CAS.
- (a) C. Gandolfi, M. Heckenroth, A. Neels, G. Laurenczy and M. Albrecht, Organometallics, 2009, 28, 5112 CrossRef CAS; (b) T. Wang, C. Pranckevicius, C. L. Lund, M. J. Sgro and D. W. Stephan, Organometallics, 2013, 32, 2168 CrossRef CAS; (c) B. Bagh and D. W. Stephan, Dalton Trans., 2014, 43, 15638 RSC; via transfer hydrogenation: (d) S. Horn and M. Albrecht, Chem. Commun., 2011, 47, 8802 RSC.
- E. Fogler, E. Balaraman, Y. Ben-David, G. Leitus, L. J. W. Shimon and D. Milstein, Organometallics, 2011, 30, 3826 CrossRef CAS.
- (a) S. Urban, B. Beiring, N. Ortega, D. Paul and F. Glorius, J. Am. Chem. Soc., 2012, 134, 1541 CrossRef PubMed; (b) J. Wysocki, N. Ortega and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 8751 CrossRef CAS PubMed.
- B. Kang, Z. Fu and S. H. Hong, J. Am. Chem. Soc., 2013, 135, 11704 CrossRef CAS PubMed.
- I. S. Makarov and R. Madsen, J. Org. Chem., 2013, 78, 6593 CrossRef CAS PubMed.
- J. Balogh, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2012, 31, 3259 CrossRef CAS.
- (a) A. Prades, E. Peris and M. Albrecht, Organometallics, 2011, 30, 1162 CrossRef CAS; (b) A. Solvhoj and R. Madsen, Organometallics, 2011, 30, 6044 CrossRef; (c) D. Canseco-Gonzalez and M. Albrecht, Dalton Trans., 2013, 7424 RSC; (d) M. Delgado-Rebollo, D. Canseco-Gonzalez, M. Hollering, H. Müller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462 RSC; (e) B. Bagh, A. M. McKinty, A. J. Lough and D. W. Stephan, Dalton Trans., 2014, 43, 12842 RSC.
- L. Bernet, R. Larlempuia, W. Ghattas, H. Mueller-Bunz, L. Vigara, A. Llobet and M. Albrecht, Chem. Commun., 2011, 47, 8058 RSC.
- (a) J. A. Cabeza, M. Damonte and E. Perez-Carreno, Organometallics, 2012, 31, 8355 CrossRef CAS; (b) J. A. Cabeza, M. Damonte, P. Garcia-Alvarez and E. Perez-Carreno, Chem. Commun., 2013, 49, 2813 RSC; (c) C. E. Ellul, M. F. Mahon, O. Saker and M. K. Whittlesey, Angew. Chem., Int. Ed., 2007, 46, 6343 CrossRef CAS PubMed; (d) L. Benhamou, J. Wolf, V. Cesar, A. Labande, R. Poli, N. Lugan and G. Lavigne, Organometallics, 2009, 28, 6981 CrossRef CAS.
- (a) M. C. Warner, C. P. Casey and J.-E. Backvall, Top. Organomet. Chem., 2011, 37, 85 CrossRef CAS; (b) B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294 CrossRef CAS PubMed; (c) T. Pasini, G. Solinas, V. Zanotti, S. Albonetti, F. Cavani, A. Vaccari, A. Mazzanti, S. Ranieri and R. Mazzoni, Dalton Trans., 2014, 43, 10224 RSC; (d) C. Cesari, L. Sambri, S. Zacchini, V. Zanotti and R. Mazzoni, Organometallics, 2014, 33, 2814 CrossRef CAS and reference cited therein.
- H. Ohara, W. W. N. O, A. J. Lough and R. H. Morris, Dalton Trans., 2012, 41, 8797 RSC.
- C. Cesari, S. Conti, S. Zacchini, V. Zanotti, M. C. Cassani and R. Mazzoni, Dalton Trans., 2014, 43, 17240 RSC.
- H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- J. A. Cabeza and P. Garcia-Alvarez, Chem. Soc. Rev., 2011, 40, 5389 RSC.
- (a) Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400 CrossRef CAS; (b) C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123, 1090 CrossRef CAS PubMed; (c) L. K. Thalen, D. Zhao, J. B. Sortais, J. Paetzold, C. Hoben and J.-E. Backwall, Chem.–Eur. J., 2009, 15, 3402 CrossRef PubMed.
- C. Cesari, R. Mazzoni, H. Müller-Bunz and M. Albrecht, J. Organomet. Chem., 2015, 793, 256 CrossRef CAS.
- M. Benìtez, E. Mas-Marzà, J. A. Mata and E. Peris, Chem.–Eur. J., 2011, 17, 10453 CrossRef PubMed.
- (a) A. M. Oertel, V. Ritleng, L. Burr, C. Harwig and M. J. Chetcuti, Organometallics, 2011, 30, 6685 CrossRef CAS; (b) L. B. Benac, E. M. Burgess, L. Burr and A. J. Arduengo, Org. Synth., 1990, 7, 195 Search PubMed ; 1986, 64, 92.
- L. C. Branco, J. N. Rosa, J. J. Moura Ramos and C. A. M. Afonso, Chem.–Eur. J., 2002, 8, 3671 CrossRef CAS.
- J. A. Loch, M. Albrecht, E. Peris, J. Mata, J. W. Faller and R. H. Crabtree, Organometallics, 2002, 21, 700 CrossRef CAS.
- M. Boiani, A. Baschieri, C. Cesari, R. Mazzoni, S. Stagni, S. Zacchini and L. Sambri, New J. Chem., 2012, 36, 1469 RSC.
- Y. Mum, D. Czarkle, Y. Rahamlm and Y. Shvo, Organometallics, 1985, 4, 1461 CrossRef.
- G. M. Sheldrick, SADABS, Program for empirical absorption correction, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELX97, Program for crystal structure determination, University of Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of novel complexes; selected ESI-MS; crystal data and experimental details; catalytic time–conversion profiles. CCDC 1421543–1421546. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra18345f |
| This journal is © The Royal Society of Chemistry 2015 |