DOI:
10.1039/C4RA05680A
(Paper)
RSC Adv., 2014,
4, 53531-53542
Synthesis and photovoltaic properties of new donor–acceptor (D–A) copolymers based on benzo[1,2-b:3,4-b′:6,5-b′′] trithiophene donor and different acceptor units (P1 and P2)†
Received
21st June 2014
, Accepted 7th October 2014
First published on 10th October 2014
Abstract
Two new conjugated D–A copolymers P1 (PBTT-TQ) and P2 (PBTT-TPz) based on the same benzo[1,2-b:3,4-b′:6,5-b′′] trithiophene (BTT) donor and different acceptor monomers 4,9-bis(5-bromithiophen-2-yl)-6,7-bis(2-ethylhexyl)[1,2,5] thiadiazolo[3,4-g] quinoxaline (TQ) and 5,7-bis(5-bromothiophen-2-yl)-2,3-bis(5-octylthiophen-2-yl) thieno[3,4-b] pyrazine (TPy), respectively, were synthesized by Stille cross-coupling reaction and characterized by gel permeation chromatography (GPC), 1H NMR, UV-Vis absorption, thermal analysis and electrochemical cyclic voltammetry (CV) tests. The optical bandgaps of P1 and P2 measured from the onset of optical absorption of these copolymers in thin films were 1.14 eV and 1.38 eV, respectively. Photovoltaic properties of the bulk heterojunction (BHJ) devices were studied using P1 and P2 as the donor and PC61BM or PC71BM as the acceptor with weight ratios of polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC61BM of 1:1, 1:2 and 1:3. The optimized photovoltaic devices fabricated with active blend layers of P1:PC71BM (1:2) and P2:PC71BM (1:2) cast from o-diclorobenzene (o-DCB) showed PCEs of 2.05% and 3.14%, respectively, whereas P2:PC61BM and P2:PC71BM yielded PCEs of 1.44% and 2.11%, respectively. The PCEs of BHJ solar cells based on P1:PC71BM and P2:PC71BM processed from 1-chloronapthanene (CN)/o-DCB solvent were further improved up to 5.28% and 2.6%, respectively.
:PC61BM of 1:1, 1:2 and 1:3. The optimized photovoltaic devices fabricated with active blend layers of P1:PC71BM (1:2) and P2:PC71BM (1:2) cast from o-diclorobenzene (o-DCB) showed PCEs of 2.05% and 3.14%, respectively, whereas P2:PC61BM and P2:PC71BM yielded PCEs of 1.44% and 2.11%, respectively. The PCEs of BHJ solar cells based on P1:PC71BM and P2:PC71BM processed from 1-chloronapthanene (CN)/o-DCB solvent were further improved up to 5.28% and 2.6%, respectively.
Introduction
Bulk heterojunction (BHJ) solar cells based on blends of conjugated polymer donors and fullerene derivative acceptors have been of interest as a promising technology for renewable energy because of their unique advantages such as flexibility, low weight and cost effectiveness of large area roll to roll manufacturing techniques.1 In the last ten years, significant progress has been made in BHJ polymer solar cells with the power conversion efficiency (PCE) improving from 2% (ref. 2) to more than 9%.3 This progress has mainly benefited from the development of photoactive materials, particularly electron donating conjugated polymers, and device optimization.4 Indeed, as improvements in stability and processability in these solar cells continue and their PCEs also continue to increase, the polymer BHJ solar cells are beginning to find application in the commercial market.5 However, a PCE of more than 10% is required for commercialization. An ideal conjugated polymer for BHJ polymer solar cells (PSCs) needs to possess broad and strong absorption in the visible and near infrared regions, suitable highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) energy levels and appropriate miscibility with the fullerene derivatives for the formation of a bi-continuous network with large interface area for efficient exciton dissociation, and high mobility for efficient transportation of photogenerated charge carriers.4,6 The most commonly used approach to obtain suitable donor materials with a broad absorption band of the solar spectrum and the corresponding energy levels is the donor–acceptor (D–A) approach.7 The advantage of this class of copolymers is that the photoelectronic properties such as absorption spectra and energy levels can be easily tuned by combining different electron rich (D) and electron deficient (A) units as well as by introducing different side chains. The incorporation of alternating electron donating units (push) and electron withdrawing units (pull) within the polymer backbone results in intramolecular charge transfer (ICT) which lowers the bandgap of the resulting copolymers. The combination of already existing structures and development of new donor and acceptor fragments to form new low band gap donor–acceptor copolymers for improving the understanding of the physical processes and the efficiency of solar cells are very important. Benzotrithiophenes (BTT) have emerged as attractive donor units for the design of D–A copolymers and show promising performance in both organic field effect transistors and PSCs.8 BTT donors possess high coplanarity and extended π-conjugation which promote intermolecular π-stacking and charge transport in BTT-containing copolymers. In this study we report new electron donor benzotrithiophene (BTT) and electron acceptor BTT structures, which are not only promising donor and acceptor blocks for the donor–acceptor copolymers, but can also be used as a hole-transport copolymer in organic field-effect transistors. The BTT-structure is planar and leads to efficient intramolecular packing in the solid state. Copolymers based on BTT are highly ordered, have partially crystalline structures and exhibit good mobility of the charge carriers. However, very few studies have been devoted to the benzotrithiophene structures.9 In this study new low bandgap conjugated copolymers P1 and P2 consisting of the same benzotrithiophene (BTT) donor unit and different 4,9-bis(5-bromothiophene-2-yl)-6,7-bis(2-ethylhexyl)[1,2,5] thiadiazolo[3,4-g] quinoxaline (TQ) and 5,7-bis(5-bromothiophene-2-yl)-2,3-bis(5-octylthiophen-2-yl)-thieno[3,4-b] pyrazine (TPz) acceptors, respectively, as promising “building blocks”, were developed and used as donor components along with fullerene derivatives (PC61BM and PC71BM) as electron acceptors for the fabrication of BHJ polymer solar cells. The BHJ solar cells with P1 and P2 as donor along with PC71BM as electron acceptor cast from o-diclorobenzene (o-DCB) showed overall PCEs of 3.14% and 2.11%, respectively. Additionally, the PCEs of P1:PC71BM and P2:PC71BM have been enhanced up to 5.28% and 2.60%, respectively, when the active layer is processed from 1-chloronapthanene (CN)/o-DCB solvent.
Results and discussion
1. Synthesis and characterization of the copolymers
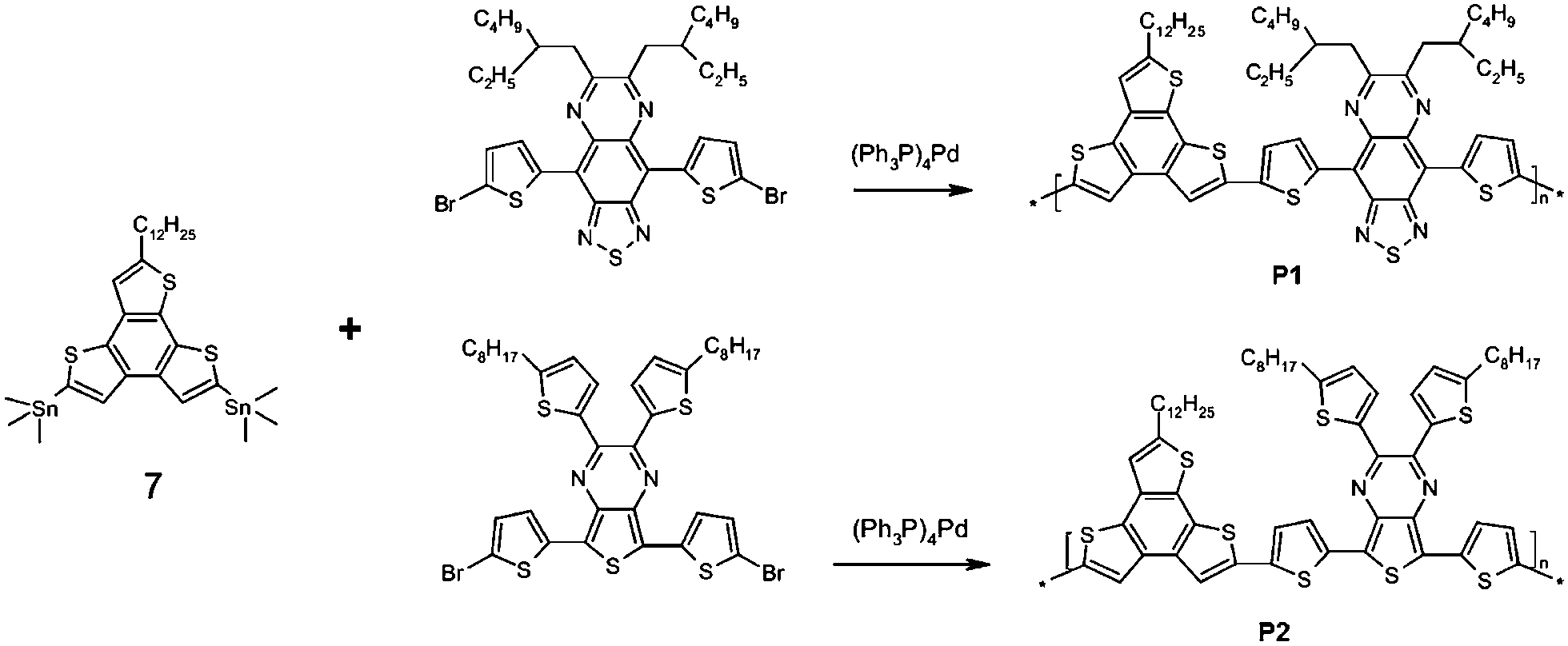
New monomers 5,8-dibromo-2-dodecanoylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (5) and 5,8-bis(trimethylstannyl)-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′] trithiophene (7) have been developed by sequential reactions according Scheme 1 involving bromination of 3-bromothiophene followed by acylation of obtained 2,3-dibromothiophene (1) to form intermediate compound 2,3-dibromo-5-dodecanoylthiophene (2) with 89% yield. 5-Dodecanoyl-2,3-bis(thiophen-2-yl)thiophene derivative (3) has been synthesized by interaction of intermediate compound 2 with 2 moles of thiophene-2-boronic acid using Suzuki reaction conditions with palladium catalyst leading to the 63% yield, subsequent bromination with NBS and cyclization with FeCl3 have led to the intermediate compound 5-dodecanoyl-2,3-bis(5-bromotiophen-2-yl) thiophene (4) and 5,8-dibromo-2-dodecanoylbenzo[1,2-b:3,4-b′:6,5-b′′] trithiophene (5) respectively with high yields. The last have been reduced by hydrazine hydrate in the base conditions. The resulting compound, 5,8-dibromo-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (6) has been transformed into desired compound 7 by halogen exchange reaction at α-positions of thiophenes and by subsequent alkylation with the trimethylstannyl electrofile.
 |
| | Scheme 1 | |
The composition and structure of the intermediate compounds 1–4, 6 and the desired compounds 5 and 7 have been confirmed by elemental analysis, 1H and 13C NMR spectroscopy. In particular, typical signals of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group have been observed on the 13C NMR spectrum of compounds 2, 3, 4 and 5 in the area 193–195 ppm but the same signal have disappeared on the 13C NMR spectrum of compounds 6 monomers 7. There are three singlets in the areas 7.84–7.41 ppm, 7.53–7.03 ppm, 7.87–7.31 ppm related to the protons of thiophene moieties and signals in the area 3.03–0.90 ppm related to the aliphatic protons in the 1H NMR spectrum of compounds 5–7 respectively. Also there is multiplet related to the eighteen protons of trimethylstannyl group in the area 0.50 ppm in the 1H NMR spectrum of desired product 7. The ratio of the integrated intensity of the aromatic and aliphatic protons for all intermediate compounds and desired product corresponds to the presumed structures (Fig. S1 ESI†).
O group have been observed on the 13C NMR spectrum of compounds 2, 3, 4 and 5 in the area 193–195 ppm but the same signal have disappeared on the 13C NMR spectrum of compounds 6 monomers 7. There are three singlets in the areas 7.84–7.41 ppm, 7.53–7.03 ppm, 7.87–7.31 ppm related to the protons of thiophene moieties and signals in the area 3.03–0.90 ppm related to the aliphatic protons in the 1H NMR spectrum of compounds 5–7 respectively. Also there is multiplet related to the eighteen protons of trimethylstannyl group in the area 0.50 ppm in the 1H NMR spectrum of desired product 7. The ratio of the integrated intensity of the aromatic and aliphatic protons for all intermediate compounds and desired product corresponds to the presumed structures (Fig. S1 ESI†).
New donor–acceptor low bandgap copolymers P1 and P2 on the basis of the synthesized heteroaromatic thiophene-containing monomer 7 has been developed in Stille cross-coupling reaction conditions according to the Scheme 2.
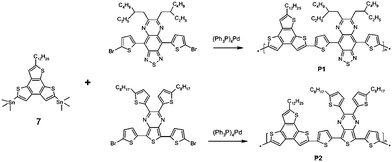 |
| | Scheme 2 | |
5,8-Bis(trimethylstannyl)-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (7) is the electron donor moiety in the structure of both P1 and P2. Monomers 4,9-bis(5-bromothiophene-2-yl)-6,7-bis(2-ethylhexyl)[1,2,5]thiadiazolo[3,4-g] quinoxaline (TQ) and 5,7-bis(5-bromothiophene-2-yl)-2,3-bis(5-octylthiophen-2-yl)-thieno[3,4-b] pyrazine (TPy) the electron acceptor fragments in P1 and P2, respectively. Polycondensation was carried out in argon atmosphere in a mixture of toluene and DMF at 115 °C for 48 h, using tetrakis(triphenylphosphine) palladium as a catalyst. The resulting copolymers P1 and P2 were purified from metal complex catalyst residues, organotin and low molecular weight impurities by double reprecipitation from solution in methanol and sequential extraction with methanol, hexane, acetone and chloroform in a Soxhlet apparatus. The yield of copolymers P1 and P2 was in the range 69–85%. The copolymers are soluble in common organic solvents such as DMF, N-methylpyrrolidone, chloroform, THF, o-dichlorobenzene.
The composition and structure of copolymers P1 and P2 were confirmed by 1H NMR spectroscopy. In particular, multiplet signals that correspond to aromatic protons of fused thiophene fragments are presented in the 1H NMR spectrum of copolymer P1 in the area δH = 8.5–6.5 ppm. Broad resonance signals related to protons of CH2 groups which are bounded with N have been obtained in the range δH = 3.5–2.9 ppm. Signals located in the range δH = 2.7–0.56 ppm correspond to the rest protons of the alkyl substituents of the polymer unit (Fig. S2†). 1H NMR spectrum of polymers are quite complex, however the ratio of the integrated intensity of the aromatic and aliphatic protons for both polymers corresponds to the presumed structures of the macromolecules. The weight-average molecular weight (Mw) and polydispersity index (Mw/Mn) were measured by gel permeation chromatography (GPC) using 1,2,4-trichlorobenzene as eluent at 150 °C and polystyrene as the internal standard. The weight-average molecular weight (Mw) of both copolymers is in the range of 9700–12000 with corresponding polydispersity index of 2.5 and 1.8 for P1 and P2, respectively. The related data are summarized in Table 1.
Table 1 Molecular weights and the thermal properties polymers P1 and P2
| Copolymer |
Mn (kDa) |
Mw (kDa) |
Mw/Mn |
Tg (°C) |
T5% (°C) |
| P1 |
4.7 |
11.9 |
2.5 |
301 |
343 |
| P2 |
6.3 |
12.0 |
1.8 |
292 |
352 |
2. Thermal properties
Thermal properties of copolymers were characterized by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere. Both the P1 and P2 copolymers exhibited good thermal stability with 5% weight-loss temperature (T5%) 343 °C and 352 °C, respectively (Table 1). The glass transition temperature of copolymers P1 and P2 is 301 °C and 292 °C, respectively. The good thermal stability of the resulting polymers retards the deformation of the polymer morphology and the degradation of the active layer at elevated temperature which are desirable for copolymer in PSCs.
3. Optical properties
The photophysical properties of the copolymers were investigated both in o-dichlorobenzene dilute solution (10−5 mol L−1) and in thin film and are shown in Fig. 1 and the corresponding absorption data are summarized in Table 2. In solution copolymers P1 and P2 showed two absorption peaks which is a common feature of D–A copolymers. The absorption peaks at shorter wavelength (434 and 419 nm for P1 and P2, respectively) originated from π–π* transition of their conjugated backbone, while the absorption peaks at longer wavelength (800 nm and 661 for P1 and P2) could be attributed to the strong intramolecular charge transfer (ICT) interaction between donor and acceptor units present in the polymers. The molar extinction coefficient in longer wavelength region for copolymer P1 is much higher than that for P2. Moreover, in solution, P1 exhibited clearly redshifted ICT absorbance peak compared to P2, which may be attributed to stronger ICT interactions between donor and acceptor units in P1 as compared to P2.
 |
| | Fig. 1 Absorption spectra of copolymer (a) P1 and (b) P2 in dilute o-DCB solution (black) and in the film state (red). | |
Table 2 Optical and electrochemical properties of the P1 and P2 copolymers
| Copolymer |
λsolmax (nm) |
λfilmmax (nm) |
Eoptg (eV) |
Eecox (V vs. NHE) |
HOMO (eV) |
Eechred (V vs. NHE) |
LUMO (eV) |
Eecg (eV) |
| P1 |
434 and 800 |
443 and 823 |
1.14 |
0.44 |
−5.24 |
−1.12 |
−3.68 |
1.56 |
| P2 |
419 and 661 |
437 and 695 |
1.38 |
0.56 |
−5.36 |
−1.06 |
−3.74 |
1.62 |
The UV-visible spectra of copolymers P1 and P2 as thin film are shown in Fig. 1. The thin film absorption spectra are generally similar in shape to those in dilute solution. The thin film absorption spectra of all both polymers have much more broad absorption range than solution absorption spectra. In thin film absorption spectra, P1 displayed two absorption peaks located at 443 nm (molar extinction coefficient = 3.4 × 104 M−1 cm−1) and 823 nm (molar extinction coefficient = 1.8 × 104 M−1 cm−1) and P2 showed two absorption peaks located at 437 nm (molar extinction coefficient = 2.8 × 104 M−1 cm−1) and 695 nm (molar extinction coefficient = 1.23 × 104 M−1 cm−1). The broadening and red-shifting of the film absorption spectra are due to the thin film shift to the interchain interaction between the polymeric chains in the copolymer thin film.10 The optical bandgap was calculated to be 1.14 eV (absorption edge ∼1088 nm) for P1 and 1.42 eV for P2 (absorption edge ∼864 nm). It can be seen form the absorption spectra of these copolymers that P1 shows higher absorption coefficients than P2 over the whole wavelength range. This is beneficial because a high absorption coefficient of a conjugated copolymer means that it can harvest photons more efficiently, and therefore achieve a large short circuit photocurrent in the PSCs. The optical absorption spectra clearly shows that the photophysical properties and band gap of those copolymers can be easily tuned by using 5,8-bis(trimethylstannyl)-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′] trithiophene (7) as donor, and the fragment of 5,7-bis (5-bromothiophene-2-yl)-2,3-bis(5-octylthiophen-2-yl)-thieno[3,4-b] pyrazine, 4,9-bis(5-bromothiophene-2-yl)-6,7-bis(2 ethylhexyl)[1,2,5]thiadiazolo[3,4-g] quinoxaline and 5,8-dibromo-2-dodecanoylbenzo[1,2-b:3,4-b′:6,5-b′′] trithiophene as the acceptors. Moreover, the P1 and P2 have different molar mass which means different number of conjugated units involved in absorption therefore; the electronic properties of the two polymers are different as the acceptor units and the molar mass differ.
4. Electrochemical properties
The electrochemical behaviors of the copolymers were investigated by cyclic voltammetry (CV). We used CV to investigate the redox behavior on the copolymers and obtain its HOMO and LUMO energy levels. HOMO and LUMO energy levels are key parameters for the application of the conjugated copolymers in PSCs. Therefore we measured the HOMO and LUMO energy levels of these two copolymers by cyclic voltammetry. The cyclic voltammograms of the copolymers P1 and P2 are displayed in Fig. 2. Both copolymers showed stable one reversible oxidation and one reversible reduction peaks, which are good signs for high structural stability in the charged state. From the onset oxidation potentials (Eonsetox) and the onset reduction potentials (Eonsetred) of the copolymers HOMO and LUMO energy level as well as the energy gaps were calculated according to the following equations
| EHOMO (eV) = −q(Eonsetox + 4.8) |
| ELUMO (eV) = −q(Eonsetred + 4.8) |
| Eelecg (eV) = EHOMO − ELUMO |
ehere Eonsetox and Eonsetred are the measured onset potentials versus Fc/Fc+ (4.8 eV below vacuum level). HOMO energy levels for copolymers P1 and P2 were −5.24 and −5.36 eV, respectively implying that they varied with respect to the modulated ICT strengths resulting from the presence of electron acceptors with various electron-accepting abilities. The LUMO levels of P1 and P2 were −3.68 and −3.74 eV, estimated from the onset reduction potentials Eonsetred of polymers, and were significantly greater than that of PC61BM or PC71BM (ca 4.0 eV) thus we would expect efficient charge transfer/dissociation to occur in their corresponding devices. In addition the electrochemical band gap Eelecg of P1 and P2 estimated from the difference between the onset potentials for oxidation and reduction were 1.56 eV and 1.62 eV, respectively. i.e., they were larger than the corresponding optical bandgap Eoptg. The discrepancy between the electrochemical and optical band gaps presumably resulted from the exciton binding energies of the copolymer and/or the interface barrier for charge injection.11 Electrochemical band gaps were higher than the optical band gap, presumably because of the interface energy barrier between the polymer film and the electrode surface.12
 |
| | Fig. 2 Cyclic voltammogram of the copolymers P1 and P2 in solid film. | |
Theoretical calculations. We have additionally theoretically examined the P1 and P2 monomers within the framework of density functional theory (DFT) and time-dependent density functional theory (TD-DFT). At the first stage of calculations the structures were optimized employing the gradient corrected functional PBE13 of Perdew, Burke and Ernzerhof. The geometry optimizations were performed initially using the SVP basis set14 followed by a subsequent optimization using the triple-ζ quality TZVP basis set.15 At this stage the resolution of the identity method16 was used for the treatment of the two-electron integrals to increase the computational efficiency (without loss in accuracy). The resulting structures were further optimized using the hybrid exchange–correlation functional B3LYP,17 and the same basis set. Tight convergence criteria were placed for the SCF energy (up to 10−7 Eh) and the one-electron density (rms of the density matrix up to 10−8) as well as for the norm of the Cartesian gradient (residual forces both average and maximum smaller than 1.5 × 10−5 a.u.) and residual displacements (both average and maximum smaller than 6 × 10−5 a.u.). Solvent effects were included for o-dichlorobenzene using the integral equation formalism variant of the Polarizable Continuum Model (IEFPCM), as implemented in the Gaussian package.18The excited state calculations have been performed in the framework of time-dependent DFT to calculate the optical gaps and the UV/Vis spectra of the monomer structures using the B3LYP functional on the corresponding ground state structures. All of the calculations were performed using the Gaussian package.18
For each of the two monomer structures we have calculated the corresponding optical gaps, taken here to be the energetically lowest allowed vertical electronic excitation, employing the PBE and B3LYP functionals. In Table 3 we give the HOMO and LUMO energy levels, the HOMO–LUMO gap, the optical gap along with the main contributions to the first (allowed) excitation. We also provide the wavelengths of the first excitation and the excitation with the highest oscillator strength for comparison with the corresponding experimental values. The calculated wavelengths (B3LYP) for the first and the highest peaks are in good agreement with the corresponding experimental ones, overestimated by about 4–20 nm. The transitions that are shown in Table 3 are those with a contribution over 4% to the first excitation. The first excitation for both structures exhibits a clear single-configuration HOMO to LUMO character.
Table 3 Calculated properties of the P1 and P2 monomers. Specifically HOMO and LUMO energies (eV), HOMO–LUMO gap (eV), HL, optical gap (eV, nm), OG, with corresponding oscillator strengths, and dipole moment (D), μ
| |
HOMO (eV) |
LUMO (eV) |
HL (eV) |
OG (eV) |
λ1st/max (nm) |
F |
Main contributions |
μ (D) |
| Values when solvent effects are taken into account for o-dichlorobenzene. |
| P1 |
| PBE |
−4.40 |
−3.67 |
0.72 |
1.12 |
1103/517 |
0.414 |
H → L 92.9%; H − 1→ L 4.1% |
2.62 |
| B3LYP |
−5.06 |
−3.37 |
1.69 |
1.51 |
823/427 |
0.525 |
H → L 98.5% |
2.46 |
| −5.11a |
−3.39a |
1.72a |
1.44a |
859a/442a |
0.713a |
2.79a |
|
| P2 |
| PBE |
−4.23 |
−3.18 |
1.05 |
1.40 |
887/567 |
0.327 |
H → L 89.7%; H − 1→ L 4.0% |
3.18 |
| B3LYP |
−4.90 |
−2.86 |
2.05 |
1.83 |
676/461 |
0.510 |
H → L 97.1% |
3.34 |
| −5.01a |
−2.92a |
2.08a |
1.75a |
710a/479a |
0.685a |
3.61a |
In Fig. 3 we have plotted the iso-surfaces (isovalue = 0.02, B3LYP) of the HOMO and LUMO for P1 and P2. For both monomers, we additionally show MOs down to HOMO − 2 for which a clear localization appears over the benzotrithiophene moiety for P1 and over both the donor and acceptor moieties for P2. The LUMOs are highly localized over the acceptor moieties; however the HOMOs are extended on the backbone of the structures over regions that include portions of both the donor and acceptor moieties. The first excitation, dominated by the HOMO → LUMO contribution, exhibits a charge transfer character which is somewhat quenched by this delocalization of the HOMO. To quantify the contributions of the moieties to the frontier orbitals we have calculated the total and partial density of states (PDOS). The PDOS for the P1 and P2 structures are shown in Fig. SI3.† We denote the benzotrithiophene, thiophene-quinoxalothiadiazole and thiophene-pyrazothiophene moieties as BtTP, TP-QTD and TP-PTP, respectively. We further partition the remaining parts of the structures to the central thiophene and aliphatic parts. The insets of the figures are zoomed in the near frontier orbital region. For the P1 monomer the HOMO has similar contributions from the BtTP, TP-QTD moieties and the central thiophene of the structure's backbone, with a percentage of 32.0%, 41.6% and 25.3% respectively. For the HOMO − 1 the respective contributions are 55.1%, 37.7% and 5.4% with a noticeable drop from the central thiophene. The HOMO − 2 is highly localized on the BtTP that contributes 94.2%. For the P2 monomer the HOMO has varying contributions from the BtTP, TP-PTP moieties and the central thiophene of the structure's backbone, with a percentage of 23.4%, 51.5% and 24.4% respectively. For the HOMO − 1 the respective contributions are 53.4%, 39.0% and 5.7%, again in this case with a noticeable drop from the central thiophene. In contrast to P1, for the P2 monomer the HOMO − 2 exhibits only a 24.1% contribution from the BtTP moiety, with a 71.3% from the TP-QTD. In both monomers the LUMO has negligible contributions from the BtTP moiety (also apparent from the contour plots). Significant contributions from the aliphatic moieties are noticed only energetically much further down starting around −8.5 eV (and below).
 |
| | Fig. 3 Frontier and near frontier orbitals of the (left) P1 and (right) P2 monomers. | |
Fig. 4 shows the UV/visible absorption spectrum of the P1 and P2 monomers calculated at the TD-DFT/B3LYP level of theory. The spectra have been produced by convoluting Gaussian functions with HWHM = 0.25 eV centered at the excitation wave numbers. The UV/spectra of the two monomers exhibit similarities and nicely reproduce the experimental spectra. In the region between 350–1400 nm, that is also available experimentally, there are two prominent bands. The high intensity band peaks at 442 nm and 475 nm, for P1 and P2 respectively, and a somewhat lower intensity band at 861 nm and 709 nm, for P1 and P2 respectively. At shorter wave numbers we note a significant band with a peak at 323 nm for P1, and with two sub peaks at 278 nm and 353 nm for P2.
 |
| | Fig. 4 Theoretical UV/Vis absorption spectrum (using the B3LYP functional) of the (top) P1 and (bottom) P2 monomers. (HWHM = 0.25 eV). | |
5. Photovoltaic properties
Photovoltaic properties of the copolymers were investigated in solar cells with device structure of ITO/PEDOT:PSS/P1 or P2:PC61BM or PC71BM/Al. The blends of copolymer (P1 or P2): fullerene derivative (PC61BM or PC71BM) in o-DCB were tested in weight ratios of 1:1, 1:2 and 1:3. A blend ratio of 1:2 was found to give the best results for both P1 and P2. Fig. 5 shows the current–voltage (J–V) curves of the PSCs based on optimized weight ratio, under illumination of AM 1.5 solar irradiance (100 mW cm−2) and the corresponding photovoltaic data are summarized in Table 3. PSC fabricated from P1:PC61BM showed a VOC of 0.68 V, a JSC of 6.56 mA cm−2 and a FF of 0.46, leading to an overall PCE of 2.05%. Under same conditions, the P2:PC61BM device showed a PCE of 1.44%, with a VOC of 0.82 V, a JSC of 4.38 mA cm−2 and a FF of 0.40. The higher value of VOC for the device based on P2 conjugated copolymer donor is attributed to the deeper value of HOMO energy level of P2 as compared to P1, since the VOC of BHJ PSC is directly related to the energy difference between donor and acceptor employed in active layer. However, the JSC of the device based on P1 device is higher than that for device based on P2. The high JSC of the P1 based device appeared to have resulted from the broader absorption profile as well as higher optical absorption coefficient.
 |
| | Fig. 5 J–V characteristics of polymer solar cells based on different blends, under illumination intensity of 100 mW cm−2. | |
Since PC61BM has poor absorption profile in visible region and does not contribute in the exciton generation process thereby leads to a low value of JSC. In order to improve the PCE further, we have used PC71BM as acceptor material to improve JSC because of its higher absorption coefficient in the visible region as compared to PC61BM.19 The J–V characteristics of the device based on blend of copolymers and PC71BM acceptor are shown in Fig. 5 and the photovoltaic parameters are summarized in Table 4. When PC71BM was used as acceptor the devices based on P1 and P2 conjugated donors showed JSC up to 9.62 mA cm−2 and 5.84 mA cm−2, with the same value of VOC, leading to an overall PCE of 3.14% and 2.11%, respectively. The higher value of the PCE for devices based on P1 as donor with either PC61BM or PC71BM as compared to P2 is attributed mainly to the enhanced value of JSC. This enhancement could be due to the higher energy offset value of LUMO for P1 (0.42 eV) and fullerene as compared to P2 and fullerene (0.28 eV). Therefore, the exciton splitting and photoinduced charge separation is more efficient in the P1: fullerene active layer than P2: fullerene active layer. However, the FF was still low 0.48 and 0.44 for P1:PC71BM and P2:PC71BM based devices, respectively, which might be related to the poor morphology of the active layer. The low value of JSC may also be related to the insufficient nanoscale morphology of the BHJ active layer. In order to improve the photovoltaic performance of the device based on P1:PC71BM and P2:PC71BM through the optimization of its blend nano-morphology, a 3% (v/v) of CN was added to the active layer solution. The J–V characteristics of the device with active layer P1:PC71BM and P2:PC71BM processed from CN/o-DCB solvent is shown in Fig. 5a and b. As a result of solvent additive, the JSC and FF of the P1:PC71BM based device was increased up to 13.48 mA cm−2 and 0.60, respectively, resulting enhanced over all PCE of 5.26%. The device based on P2:PC71BM cast from the CN/o-DCB also showed an improved PCE of 2.89% with JSC of 7.14 mA cm−2 and FF of 0.52.
Table 4 Photovoltaic properties polymers P1 and P2
| Blend |
VOC (V) |
JSC (mA cm−2) |
FF (%) |
PCE (%) |
| o-DCB cast. CN/o-DCB cast. |
| P1:PC61BM (1:2)a |
0.68 |
6.56 |
46 |
2.05 |
| P1:PC71BM (1:2)a |
0.68 |
9.62 |
48 |
3.14 |
| P2:PC61BM (1:2)a |
0.82 |
4.38 |
40 |
1.44 |
| P2:PC71BM (1:2)a |
0.82 |
5.84 |
44 |
2.11 |
| P1:PC71BM (1:2)b |
0.65 |
13.48 |
60 |
5.26 |
| P2:PC71BM (1:2)b |
0.78 |
7.14 |
0.52 |
2.89 |
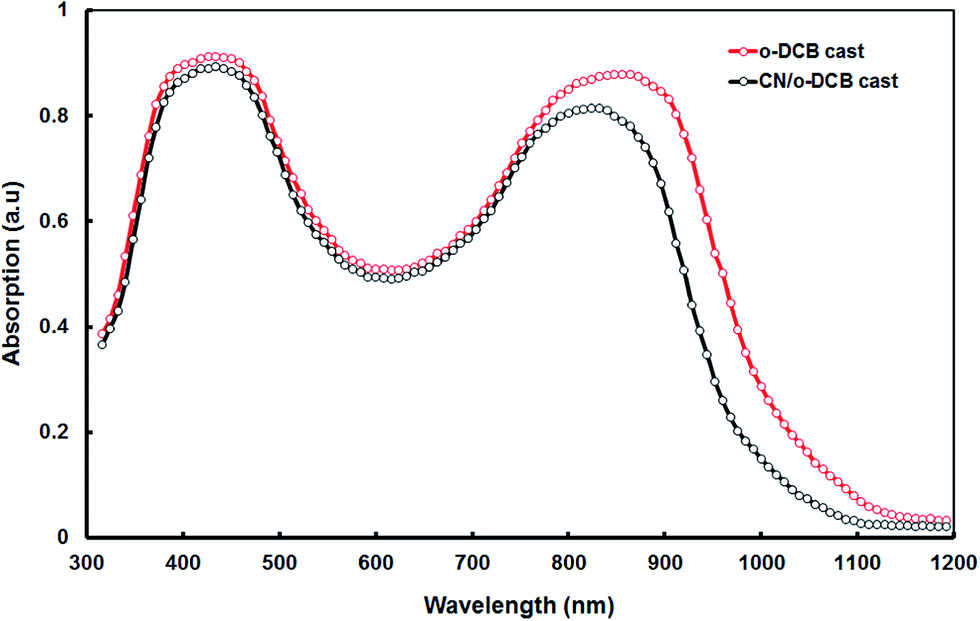
Fig. 6 displays the UV-visible absorption spectra for P1:PC71BM blend films processed with and without CN additive. The P1:PC71BM blend processed from o-DCB displays two main absorption bands having peaks one at 390 nm and other at 825 nm. Both P1 and PC71BM contribute to absorption for the absorption band for shorter absorption band while the absorption band in longer wavelength region contributes exclusively related to P1. The introduction of the processing CN additive induces a redshift of about 20 nm in the absorption band in longer wavelength region and also the absorption also broadened. This indicates that the addition of CN induces stronger P1 chain interaction and enhanced local over within the polymer phase.20 The packing of P1 chains is disrupted by blending with PC71BM. The selective solubility of the co-solvent allows for a rearrangement of the P1 chain packing, such that it more close.
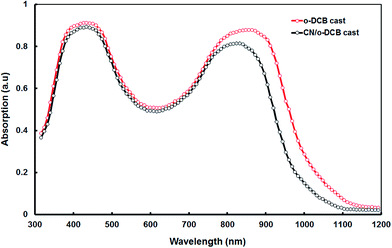 |
| | Fig. 6 Optical absorption spectra of P1:PC71BM (1:2) blend thin film cast from o-DCB and CN/o-DCB solvents. | |
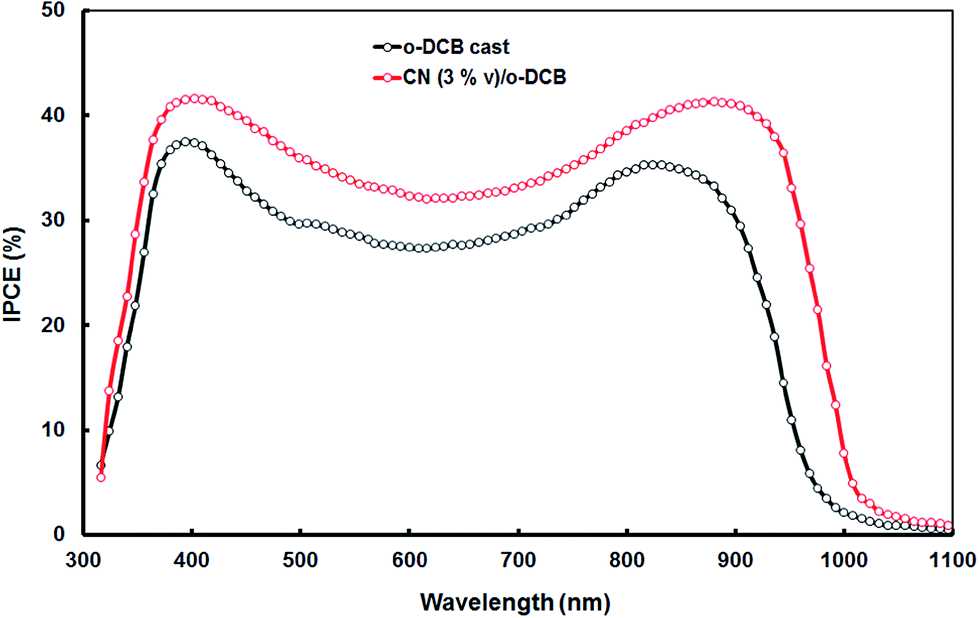
To address the origin of the enhanced device performance, we have measured the IPCE spectra for the P1:PC71BM (o-DCB cast) and P1:PC71BM (CN/o-DCB cast) BHJ devices (Fig. 7). The shape of IPCE spectra of the device based on o-DCB and CN/o-DCB processed active layers closely follows the optical absorption profile. The device processed with CN/o-DCB displays significantly improved IPCE values as compared to the device processed with o-DCB. Additionally, the IPCE spectrum of the device based on solvent additive active layer is redshifted having a peak about 835 nm. This improved performance is owned partly to the increased absorption as shown in Fig. 6. Apart from improved absorption, IPCE enhancement may also be attributed to increased charge carrier mobility for the device processed with additive.21 The more ordered nano-morphology of active film processed with additives can reduce the binding energy of charge transfer excitons at the interface,22 as well as reducing the decay rate of photogenerated excitons, which improves the efficiency of exciton dissociation.23
 |
| | Fig. 7 IPCE spectra of ITO/PEDOT:PSS/P1:PC71BM (1:2)/Al devices cast from o-DCB and CN/o-DCB solvents. | |
The higher values of IPCE values for the P1:PC71BM (CN/o-DCB cast) device relatively to P1:PC71BM (o-DCB cast) device confirm the higher photocurrent generation efficiencies for the device made from P1:PC71BM (CN/o-DCB cast). The JSC values obtained from IPCE spectra reached about 9.78 mA cm−2 and 14.12 mA cm−2, for the devices based on P1:PC71BM (o-DCB cast) and P1:PC71BM (CN/o-DCB cast), respectively which are consistent with the values obtained from J–V characteristics.
We found that when the concentration of additive CN increased beyond 3%, the IPCE values of device across all the wavelengths decreased and the corresponding overall PCE also drops. This is due primarily to a reduction in absorption. It is also possible that films with high concentration of additive, the film was not dried completely, leading reduction to charge carrier mobility.24
The film morphology of the films P1:PC71BM cast from the o-DCB and CN/o-DCB was investigated by transmission electron microscopy (TEM) and shown in Fig. 8. As shown in the TEM images, the bright regions and dark regions can be attributed to the donor's domains and PC71BM domains, respectively. The dark region for PC71BM may be due to its high electron scattering density.25 The film cast from the o-DCB displayed a ribbon type pattern with sizes about 70 nm and a dark area with average diameter of about 40 nm. Compared to the film cast from o-DCB, the film cast from CN/o-DCB showed a typical fibrillar structure and the both donor and acceptor phase sizes were decreases up to 20 nm, which is very close to exciton diffusion length, contributing to enhancing the exciton diffusion and separation efficiency.26 Moreover, the fibers like structure also help for efficient transport of free charge carriers, enhances the FF of the device.26
 |
| | Fig. 8 TEM images of P1:PC71BM cast for o-DCB and CN/o-DCB solvents. Scale bar is 200 nm. | |
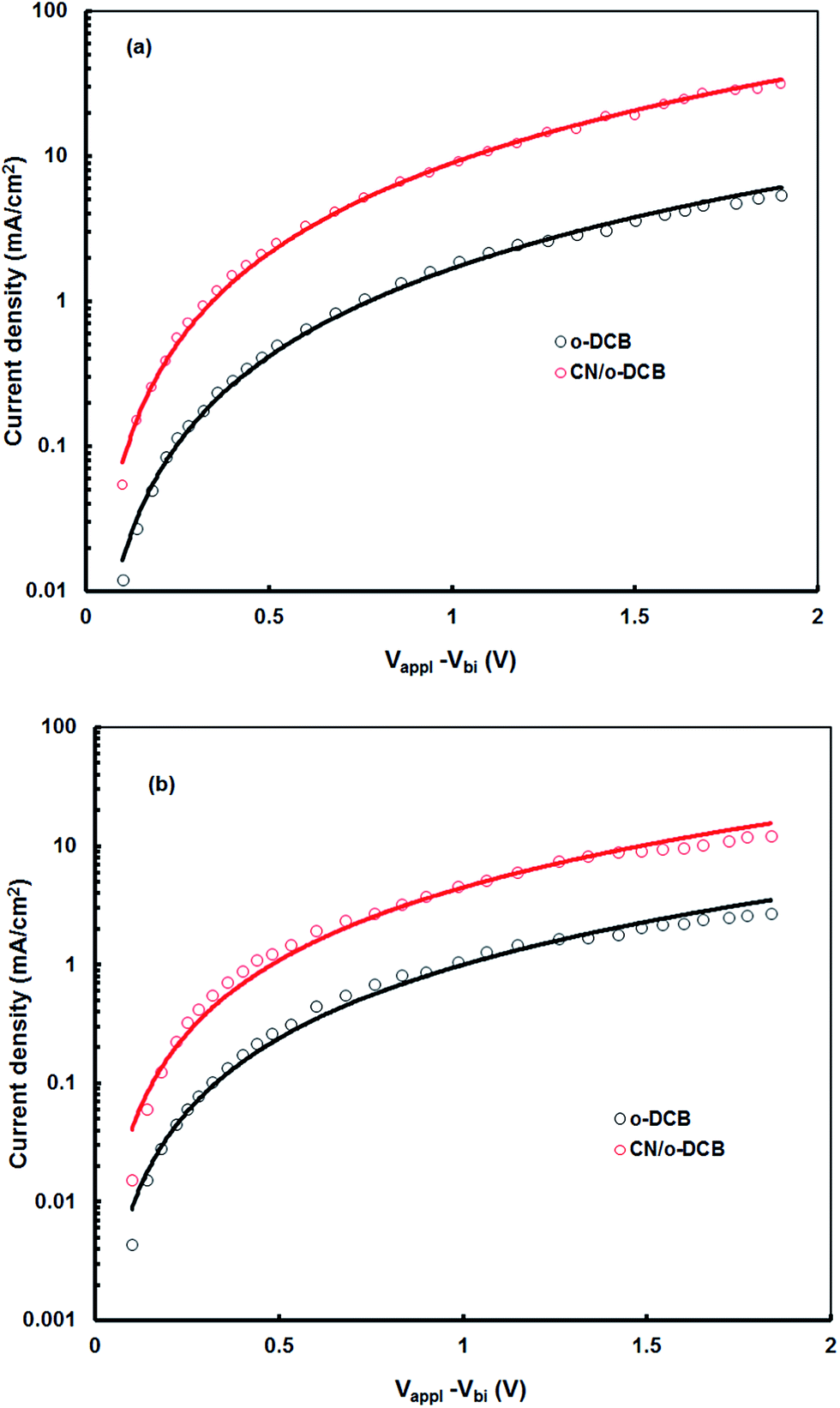
The balanced charge transport in the device also influences the JSC of the polymer solar cells and is decided by electron to hole mobilities ratio of in the BHJ active layer. We have measured the hole and electron mobilities in the active layer by the space charge limited current (SCLC) model with the ITO/PEDOT:PSS/BHJ layer/Au hole-only and Al/BHJ layer/Al electron-only devices, respectively, in dark.27 The J–V characteristics of the hole only devices in dark based on P1:PC71B blend and P2:PC71BM processed with and without additives are shown in Fig. 9. The hole mobility of P1:PC71BM active layer cast for CN/o-DCB is about 8.56 × 10−5 cm2 V−1 s−1 which is about 26 time higher than that cast from o-DCB (3.24 × 10−6 cm2 V−1 s−1). Similarly the hole mobility for P2:PC71BM processed with and without CN additive is about 5.43 × 10−5 cm2 V−1 s−1 and 2.2 × 10−6 cm2 V−1 s−1, respectively. However, the electron mobility estimated from the similar J–V characteristics using electron only devices, remains almost same (2.38 × 10−4 and 2.46 × 10−4 cm2 V−1 s−1, for o-DCB and CN/o-DCB cast film, respectively). The increase in the hole mobility reduces the ratio between electron and hole mobilities significantly in the active layer (73 to 2.87 for P1:PC71BM and 108 to 4.53 for P2:PC71BM, respectively), indicates a more balanced charge transport in the device based on the blend cast from CN/o-DCB solvent relative to that for the o-DCB cast blend. The balanced charge transport within the active layer resulted in high JSC and FF.
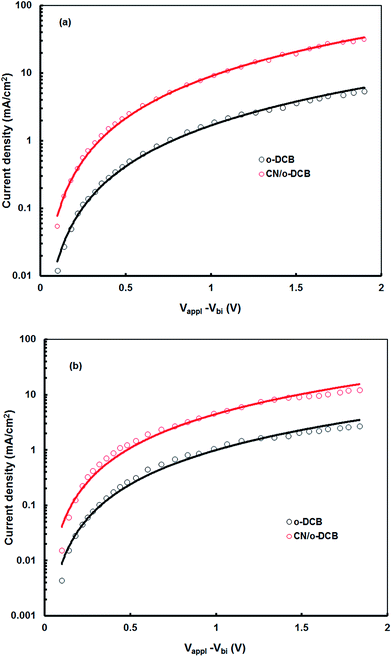 |
| | Fig. 9 Current–voltage (J–V) characteristics of the hole only devices based on (a) P1:PC71BM and (b) P2:PC71BM blend active layers processed from o-DCB and CN/o-DCB solvents. Solid lines are SCLC fits. | |
Conclusion
Two new conjugated D–A polymers P1 and P2 based on benzo[1,2-b:3,4-b′:6,5-b′′] trithiophene were synthesized by Stille cross-coupling reaction and characterized by gel permeation chromatography (GPC), 1H NMR, UV-Vis absorption, thermal analysis and electrochemical cyclic voltammetry (CV) tests. The weight-average molecular weight (Mw) of P1 and P2 are in the range of 9700–12000 with corresponding polydispersity index in the range of 1.5–2.9. All copolymer exhibited good thermal stability with 5% weight-loss temperature (T5%) in the range 323–352 °C respectively. The glass transition temperature for both P1 and P2 is 152 °C. HOMO energy levels P1 and P2 were −5.03 and −5.13 eV, respectively, implying that they varied with respect to the modulated ICT strengths resulting from the presence of electron acceptors with various electron-accepting abilities. The LUMO level was about −3.68 eV and −3.82 eV for P1 and P2, respectively, estimated from the onset reduction potentials (Eonsetred) of polymers, and were greater than that of PC61BM (ca. 4.0 eV) thus we would expect efficient charge transfer/dissociation to occur in their corresponding devices. Photovoltaic properties of the polymers were studied by using the polymers as donor and fullerene derivatives, i.e. PC61BM and PC71BM as acceptor with a weight ratio of polymer–fullerene (1:1, 1:2, 1:3 and 1:4). The optimized photovoltaic device was fabricated with an active layer of a blend P1:PC61BM, P2:PC61BM, P1:PC71BM and P2:PC71BM blends cast from o-DCB showed overall PCE of 2.05%, 1.44%, 3.14% and 2.11%, respectively. The PCE of the photovoltaic device based on P1:PC71BM and P2:PC71BM active layer processed from the CN/o-DCB solvent increased up to 5.26% and 2.60%, respectively, attributed to the more controlled favorable nanomorphology for exciton dissociation and charge transport within the active layer.
Experimental
1. Instrument
UV-vis absorption spectra was tested on a Shimadzu UV-3600 UV-vis-NIR spectrometer. The bandgap was calculated according to the onset absorption of UV-vis spectra (Eg = 1240/λonset eV). High temperature GPC analysis was conducted on a PL-GPC 220 system using polystyrene as standard and 1,2,4-trichlorobenzene as eluent at 150 °C. CV of films was performed on a CHI660a electrochemical analyzer with a three-electrode cell at a scan rate of 100 mV s−1 (glassy carbon electrode, Pt wire and saturated calomel electrode). Bu4NPF6 (0.1 mol L−1) was used as electrolyte and anhydrous acetonitrile (drying with CaH2) was used as the solvent. Transmission electron microscopy (TEM) of the polymers blend films was performed on JEOL JEM-1011 transmission electron microscope operated at an acceleration voltage of 100 kV. Polymers/PCBM films for TEM were prepared in identical conditions to those prepared for devices fabrication on PEDOT:PSS-coated ITO substrates. Then samples were immersed in water, and the floating active layer onto water surface was transferred to a TEM grid.
2. Materials
All reagent, unless otherwise specified, were obtained from Aldrich and across and used as received. All the solvents were freshly distilled prior to use.
3. Fabrication and characterization of polymer solar cell
The devices with the structure of ITO/PEDOT:PSS/P1 or P2:PC71BM/Al were fabricated as follow: the ITO substrate was cleaned with de-ionized water, acetone, and isopropyl alcohol successively, then dried in oven at 120 °C overnight. After treating with UV/ozone at room temperature for 30 min, 40 nm PEDOT:PSS was spin-coated on the ITO substrate and dried at 100 °C for 30 min. After that, the ITO substrate was transferred into glove-box for spin-coating the active layer. The concentration of the blend solution was 8 mg ml−1 in o-diclorobenzene (o-DCB) or 1-chloronapthanene (CN)/o-DCB (for solvent additive). Finally, the device was completed by evaporating aluminum (Al) with an area of 0.20 cm2. The current–voltage curves were measured under 100 mW cm−2 standard AM 1.5G spectrum using An Oriel 150 W solar simulator, the light intensity was determined by silicon diode with KG-5 visible colour filter. The current–voltage (J–V) characteristics of the devices were obtained with a Keithley source meter. We have kept a mask on the device while illumination. The incident photon to current efficiency (IPCE) of the devices was recorded by illuminating the device through the monochromator coupled with a xenon lamp (illumination intensity at each wavelength was kept constant) and resulting photocurrent was measured with the Keithley electrometer under short circuit conditions. The IPCE was estimated at each wavelength using following expression.
where JSC is the short circuit photocurrent density, λ (nm) is the mono-chromatic wavelength of incident light and Pin is the incident light intensity per unit area.
4. Synthesis of monomers and polymers
2,3-Dibromothiophene (1). 3-Bromothiophene (16.3 g; 100 mmol) was added to a suspension of NBS (17.8 g; 100 mmol) in hexane (50 ml) followed by HClO4 (70% solution in H2O; 0.7 ml; 5 mol%) addition. The reaction mixture was stirred at room temperature for 24 h and than K2CO3 (200 mg) was added. Reaction mixture was filtrated, solids were washed with hexane. Organic phases were concentrated, and residue was distilled in vacuo. Yield is 89%. Found, %: C 19.60, H 0.88, S 13.53, Br 65.88; calcd, %: for C4H2Br2S, C 19.86, H 0.83, S 13.25, Br 66.05. 1H NMR (400.13 MHz; CDCl3; δ, ppm): 7.24 (s, 1H); 6.90 (s, 1H).
2,3-Dibromo-5-dodecanoylthiophene (2). AlCl3 (7.142 g; 53.5 mmol) was added by small portions during 20–30 min to a cooled solution of 2,3-dibromothiophene (10 g; 41.33 mmol) and dodecanoyl chloride (10 g; 45.56 mmol) in CH2Cl2 (100 ml) at 0 °C under argon. Reaction mixture was stirred at 0 °C during 2 h and than a cooled hydrochloric acid (2 M, 160 ml) was slowly added. Phases were separated, water layer was extracted with CH2Cl2 (2 × 100 ml), organic solution was dried with MgSO4, filtrated and concentrated in vacuo. Residue was purified on SiO2 column with hexane as eluent. After solvent evaporation the title compound was obtained with 90% yield as orange oil, which crystallized upon standing. Found, %: C 45.20, H 5.48, S 7.23, Br 37.19; calcd % for C16H24Br2OS, C 45.30, H 5.70, S 7.56, Br 37.67. 1H NMR (400 MHz; CDCl3; δ, ppm.): 7.46 (s, 1H); 2.79 (br.s, 2H); 1.70 (br.s, 2H); 1.24 (m, 16H); 0.87 (br.s, 3H).
5-Dodecanoyl-2,3-bis(thiophene-2-yl)thiophene (3). Thiophenyl-2-boronic acid (1.811 g; 14.143 mmol), 2,3-dibromo-5-dodecanoylthiophene (2) (2.5 g; 5.893 mmol), DMF (125 ml) and aqueous solution of Na2CO3 (20 ml, 1 M, 20 mmol) were loaded into flask (250 ml), and the mixture were degassed with argon during 40 min with stirring. Than Pd(PPh3)4 (300 mg; 0.259 mmol) was added and the mixture was heated at 125 °C for 20 h under argon. After cooling, the reaction mixture was treated with H2O (150 ml) and extracted with CHCl3 (3 × 100 ml). Organic phases were dried with MgSO4 and evaporated to dryness. After purification of the residue by chromatography on SiO2 (hexane–toluene = 2:1 as eluent) and recrystallisation from hexane, the title compound was obtained. The yield is 1.63 g (63, 4%). 1H NMR (600.13 MHz; CDCl3; δ, ppm): 7.70 (s, 1H); 7.37 (dd, 1H); 7.35 (dd, 1H); 7.22 (dd, 1H); 7.09 (m, 1H); 7.07 (m, 1H); 7.03 (dd, 1H); 2.90 (t, 2H); 1.78 (m, 2H); 1.28 (m, 16H); 0.9 (t, 3H). 13C NMR (150.93 MHz; CDCl3; δ, ppm): 193.33; 141.39; 140.45; 136.09; 134.34; 134.29; 132.13; 128.37; 127.88; 127.54; 127.45; 127.36; 126.37; 39.09; 31.93; 29.63; 29.50; 29.44; 29.36; 24.84; 22.71; 14.15. Found, %: C 67.01; H 7.15; calcd, %: for C24H30S3O, C 66.93, H 7.02.
5-Dodecanoyl-2,3-bis(5-bromothiophen-2-yl)thiophene (4). To a cooled solution of 5-dodecanoyl-2,3-bis(thiophene-2-yl)thiophene (3) (5.890 g; 13.676 mmol) in DMF (100 ml) a solution of NBS (5.400 g; 30.000 mmol) in DMF (40 ml) was added dropwise at 0 °C, and the reaction mixture was stirred at room temperature overnight. After completion of the reaction, the reaction mixture was poured onto ice (200 g), extracted with CH2Cl2 (3 × 100 ml), dried with MgSO4 and evaporated at 50 °C. The residue was extracted with hot hexane, filtrated and evaporated. The yield of title compound is 7.1 g (88%). 1H NMR (600.13 MHz; CDCl3; δ, ppm): 7.61 (s, 1H); 7.04 (d, 1H); 7.01 (dd, 2H); 6.85 (d, 1H); 7.09 (m, 1H); 2.97 (t, 2H); 1.76 (m, 2H); 1.32 (m, 16H); 0.90 (t, 3H). Found, %: C 48.70, H 4.68, S 16.03, Br 26.87; calcd, %: for C24H28Br2OS3, C 48.98, H 4.80, S 16.35, Br 27.16.
5,8-Dibromo-2-dodecanoylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (5). A solution of FeCl3 (5.190 g; 32 mmol) in nitromethane (70 ml) was added dropwise to a solution of 5-dodecanoyl-2,3-bis(5-bromothiophen-2-yl)thiophene (4) (8.1 g; 13.764 mmol) in dichloromethane (300 ml) at room temperature, and the resulting mixture was stirred overnight. After the completion of the reaction, ethanol (300 ml) was added and the mixture was concentrated to the volume of 300 ml and cooled to −15 °C. The solids were filtrated and dried. After purification on SiO2 column (toluene–hexane – 1:1 as eluent) and recrystallization from this mixture, the yield of the title compound is 6.6 g (81.8%). 1H NMR (600.13 MHz; CDCl3; δ, ppm): 7.84 (s, 1H); 7.42 (s, 1H); 7.41 (s, 1H), 3.03 (t, 2H); 1.83 (m, 2H); 1.32 (m, 16H); 0.90 (t, 3H). 13C NMR (150 MHz; CDCl3; δ, ppm): 193.89; 143.18; 134.01; 133.12; 132.94; 131.70; 131.44; 129.78; 129.05; 128.24; 125.64; 125.31; 125.11; 115.78; 113.93; 39.35; 31.95; 29.68; 29.67; 29.55, 29.51; 29.39–29.38 (br); 24.61; 22.73; 14.17. Found, %: C 49.00, H 4.38, S 16.13, Br 26.83; calcd, %: for C24H26Br2OS3, C 49.15, H 4.47, S 16.40, Br 27.25.
5,8-Dibromo-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (6). A mixture of 5,8-dibromo-2-dodecanoylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (5) (3.5 g; 5.973 mmol), hydrazine-hydrate (15 ml; 0.3 mol) and KOH (9 g; 153.9 mmol) in ethylene glycol (100 ml) was stirred at 190–200 °C for 16 h. Cooled reaction mixture poured into water, extracted with CH2Cl2 and evaporated in vacuo. The orange solid residue was extracted with hot hexane and filtrated through SiO2 column. The colorless filtrate was evaporated to give 462 mg (13.5%) of the desired product. 1H NMR (600.13 MHz; CDCl3; δ, ppm): 7.53 (s, 1H); 7.52 (s, 1H); 7.03 (s, 1H), 2.95 (t, 2H); 1.80 (m, 2H); 1.23 (m, 18H); 0.90 (t, 3H). 13C NMR (150 MHz; CDCl3; δ, ppm): 147.15; 132.65; 132.12; 131.01; 130.70; 130.43; 128.56; 125.25; 125.16; 118.49; 113.05; 112.52; 31.94; 31.27; 30.77; 29.68; 29.65 (br); 29.54, 29.37; 29.15; 22.71; 14.15. Found, %: C 50.20, H 4.88, S 16.53, Br 27.48; calcd, %: for C24H28Br2S3, C 50.35, H 4.93, S 16.80, Br 27.91.
5,8-Bis(trimethylstannyl)-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (7). n-BuLi solution (1.6 M in hexanes; 1.2 ml; 2.046 mmol) was added dropwise to a cooled solution of 5,8-dibromo-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (6) (0.46 g; 0.8035 mmol) in THF (30 ml) at −78 °C, and the reaction mixture was stirred at −78 °C for 30 min. Then chlorotrimethyltin solution (1 M in hexane; 4.1 ml; 4.1 mmol) was added dropwise and the reaction mixture was warmed to room temperature, treated with water (75 ml), and extracted with hexane (3 × 50 ml). Combined organic extracts were dried with MgSO4, filtrated and evaporated to dryness. The residue was recrystallized from hot isopropanol to give 505 mg (85% yield) of the title compound. 1H NMR (400.13 MHz; CDCl3; δ, ppm): 7.87 (s, 1H); 7.86 (s, 1H); 7.31 (s, 1H); 2.99 (t, 2H); 1.62 (m, 2H); 1.50–1.20 (m, 18H); 0.90 (t, 3H); 0.50 (s, 18H). Found, %: C 48.40, H 6.18, S 12.53; for C30H46S3Sn2, calcd, %: C 48.68, H 6.26, S 12.99.
Synthesis of P1. 5,8-Bis(trimethylstannyl)-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (7) (0.6119 g; 0.8265 mmol), 4,9-bis(5-bromithiophen-2-yl)-6,7-bis(2-ethylhexyl)[1,2,5]thiadiazolo[3,4-g]quinoxaline (0.8265 mmol), Pd(Ph3P)4 (0.065 g; 0.056 mmol), dry toluene (15 ml) and dry DMF (2 ml) were placed in a 25 ml flask equipped with reflux condenser and magnetic stirrer. After stirring at 115 °C for 48 h under argon, 2-bromothiophene (0.02 g; 0.123 mmol) and 2-(tributylstannyl)thiophene (0.02 g; 0.054 mmol) was added to the mixture and reaction was continued for 5 h. The reaction mixture was cooled to room temperature, poured into methanol and filtered. The polymer was dissolved in CHCl3, precipitated from methanol, extracted in Soxlet apparatus with methanol, hexane and chlorophorm and dried in vacuo. The yield is 67%. 1H NMR (CDCl3, 400 MHz, δ, ppm): 8.5–6.5 (m, 7H), 3.5–2.9 (m, 6H), 2.7–0.56 (m, 53H).
Synthesis of P2. Copolymer P2 was prepared similarly as P1, from 5,8-bis(trimethylstannyl)-2-dodecylbenzo[1,2-b:3,4-b′:6,5-b′′]trithiophene (7) as donor monomer and 5,7-bis(5-bromothiophen-2-yl)-2,3-bis(5-octylthiophen-2-yl)thieno[3,4-b]pyrazine as acceptor. The yield is 64%. 1H NMR (CDCl3, 400 MHz, δ, ppm): 8.5–6.0 (m, 11H), 3.5–2.8 (m, 6H), 2.34–0.33 (m, 53H).
Acknowledgements
M. L .K. and S. A. K. thank Russian Science Foundation (grant number 14-13-01444) and G. D. Sharma thanks to Department of Science and Technology (DST), Government of India for financial support through DST-RFBR project.
References
- N. Espinosa, M. Hosel, D. Angmo and F. C. Krebs, Energy Environ. Sci., 2012, 5, 5117 CAS.
- M. Svensson, F. L. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersson, Adv. Mater., 2003, 15, 988 Search PubMed.
-
(a) M. J. Zhang, X. Guo, S. Q. Zhang and J. H. Hou, Adv. Mater., 2014, 26, 1118 Search PubMed;
(b) Z. C. He, C. Zhong, X. Huang, W. Y. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636 Search PubMed;
(c) L. T. Dou, J. B. You, J. Yang, C. C. Chen, Y. J. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180 CrossRef CAS;
(d) M. J. Zhang, Y. Gu, X. Guo, F. Liu, S. Q. Zhang, L. J. Huo, T. P. Russell and J. H. Hou, Adv. Mater., 2013, 25, 4944 Search PubMed;
(e) Z. C. He, C. M. Zhong, S. J. Su, M. Xu, H. B. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591 Search PubMed.
-
(a) Y. F. Li, Acc. Chem. Res., 2012, 45, 723 Search PubMed;
(b) P. M. Beaujuge and J. M. Frechet, J. Am. Chem. Soc., 2011, 133, 20009 Search PubMed;
(c) S. Genes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 Search PubMed;
(d) Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868 Search PubMed.
- http://www.heliatek.com/newscenter/latest_news/neuer-weltre-kord-fur-organische-solarzellen-heliatek-behauptet-sich-mit-12-zell-effizienz-als-technologiefuhrer/?lang=en, accessed February 2014.
-
(a) R. F. Service, Science, 2011, 332, 293 Search PubMed;
(b) T. Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J. R. Pouliot, J. Zhou, A. Najari, M. Leclerc and Y. Tao, Adv. Funct. Mater., 2012, 22, 2345 Search PubMed;
(c) N. D. Treat, M. A. Brady, G. Smith, M. F. Toney, E. J. Kramer, C. J. Hawker and M. L. Chabinyc, Adv. Energy Mater., 2011, 1, 82 Search PubMed.
-
(a) L. Dou, J. You, J. Yang, C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180 Search PubMed;
(b) Y. Li, Acc. Chem. Res., 2012, 45, 723 Search PubMed;
(c) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 Search PubMed;
(d) R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 743 Search PubMed;
(e) S. C. Lan, P. A. Yang, M. J. Zhu, C. M. Yu, J. M. Jiang and K. H. Wei, Polym. Chem., 2013, 4, 1132 Search PubMed;
(f) X. Zhao, D. Yang, H. Lv, L. Yin and X. Yang, Polym. Chem., 2013, 4, 57 Search PubMed;
(g) M. L. Keshtov, D. V. Marochkin, V. S. Kochurov, A. R. Khokhlov, E. N. Koukaras and G. D. Sharma, Polym. Chem., 2013, 4, 4033–4044 Search PubMed.
-
(a) X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula, K. Müllen and J. Am, Chem. Soc., 2012, 134, 8404 Search PubMed;
(b) X. Zhao, D. Yang, H. Lv, L. Yin and X. Yang, Polym. Chem., 2013, 4, 57 Search PubMed;
(c) S.-C. Lan, P.-A. Yang, M.-J. Zhu, C.-M. Yu, J.-M. Jiang and K.-H. Wei, Polym. Chem., 2013, 4, 1132 RSC;
(d) M. Kashiki, I. Kohara, E. Osaka, K. Miyazaki and J. Takimiya, Org. Chem., 2011, 76, 4061 Search PubMed;
(e) C. B. Nielsen, R. S. Ashraf, B. C. Schroeder, P. D'Angelo, S. E. Watkins, K. Song, T. D. Anthopoulos and I. McCulloch, Chem. Commun., 2012, 48, 5832 Search PubMed;
(f) X. Guo, S. R. Puniredd, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2013, 25, 5467 Search PubMed.
-
(a) C. B. Nielson, J. M. Fraser, B. C. Schroeder, J. Du, A. J. P. White, W. Zhang and I. McCulloch, Org. Lett., 2011, 13, 2414 CrossRef PubMed;
(b) T. Taerum, O. Lukoyanova, R. G. Wylie and D. F. Perepichka, Org. Lett., 2009, 11, 3230 Search PubMed.
-
(a) R. Steyrleuthner, M. Schubert, I. Howard, B. Klaumünzer, K. Schilling, Z. Chen, P. Saalfrank, F. Laquai, A. Facchetti and D. Neher, J. Am. Chem. Soc., 2012, 134, 18303 Search PubMed;
(b) J. K. Lee, W. L. Ma, C. J. Brabec, J. Yuen, J. S. Moon, J. Y. Kim, K. Lee, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2008, 130, 3619 Search PubMed.
- P.-T. Wu, F. S. Kim, R. D. Champion and S. A. Jenekhe, Macromolecules, 2008, 41, 7021 CrossRef CAS.
- E. Wang, M. Wang, L. Wang, C. Duan, J. Zhang, W. Cai, C. He, H. Wu and Y. Cao, Macromolecules, 2009, 42, 4410 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 Search PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 Search PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 Search PubMed.
- K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji M. Caricato X. Li H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 03, revision C.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
-
(a) Z. G. Zhang and J. Z. Wang, J. Mater. Chem., 2012, 22, 4178 Search PubMed;
(b) H. J. Son, F. He, B. Carsten and L. P. Yu, J. Mater. Chem., 2011, 21, 18934 Search PubMed;
(c) W. L. Zhang, M. Bolognesi, M. Seri, P. Henriksson, D. Gedefaw, R. Kroon, M. Jarvid, A. Lundin, E. G. Wang, M. Muccini and M. R. Andersson, Macromolecules, 2013, 46, 8488 Search PubMed.
- J. Peet, J. Y. Kim, N. E. Coastes, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497 Search PubMed.
- S. Albrecht, W. Schinaller, J. Kurpiers, J. Kniepert, J. C. Blakesley, I. Dumsch, A. Allard, K. Fostiropoulos, U. Scherf, D. Neher and J. Phys, Chem. Lett., 2012, 23, 640 Search PubMed.
- M. C. Scharber, C. Lungenschmied, H. J. Egelhaaf, G. Matt, M. Bednorz, T. Fromherz, J. Gao, D. Jarzeb and M. A. Loi, Energy Environ. Sci., 2011, 4, 5011 Search PubMed.
- D. J. D. Moet, M. Lenes, M. Morana, H. Azimi, C. J. Brabec and P. W. M. Blom, Appl. Phys. Lett., 2010, 96, 213506 Search PubMed.
- S. Cho, J. K. Lee, J. S. Moon, J. Yuen, K. Lee and A. J. Heeger, Org. Electron., 2008, 9, 1107 Search PubMed.
- X. N. Yang, J. Loos, S. C. Veenstra, W. J. H. Verhees, M. M. Wienk, J. M. Kroon, M. A. J. Michels and R. A. J. Janssen, Nano Lett., 2005, 5, 579 Search PubMed.
-
(a) T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736 Search PubMed;
(b) W. W. Li, K. H. Hendriks, A. Furlan, W. S. C. Roelofs, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 18942 Search PubMed.
-
(a) C. Melzer, E. J. Koop, V. D. Mihailetchi and P. W. M. Blom, Adv. Funct. Mater., 2004, 14, 865 Search PubMed;
(b) V. D. Mihailetchi, L. J. A. Koster, P. W. M. Blom, C. Melzer, B. De Boer, J. K. J. VanDuren and R. A. J. Janssen, Adv. Funct. Mater., 2005, 15, 795 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05680a |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.