DOI:
10.1039/C4RA08025D
(Paper)
RSC Adv., 2014,
4, 53520-53530
Influence of anionic co-ligands on the structural diversity and catecholase activity of copper(II) complexes with 2-methoxy-6-(8-iminoquinolinylmethyl)phenol†
Received
2nd August 2014
, Accepted 9th October 2014
First published on 10th October 2014
Abstract
A novel dinuclear copper(II) complex, [Cu2L2(μ1,1-N3)2] (1), and a mononuclear copper(II) complex, [CuL(NCO)] (2), have been synthesized from a planar tridentate ligand 2-methoxy-6-(8-iminoquinolinylmethyl)phenol (HL) together with pseudohalides as coligands, and the solid state structures were determined by X-ray crystallography. Structural characterizations reveal that the geometry of centrosymmetrically related copper(II) centers in 1 is square pyramidal while it is square planar in 2. The impact of the structural diversity was found on their catechol oxidase mimicking activity. Strongly bridging azide ions being substitutionally inert mean complex 1 is inactive towards the catecholase activity, while mononuclear analogue 2 exhibits moderately strong catechol oxidase activity. The ESI-MS positive spectrum of a mixture of complex 2 and 3,5-DTBCH2 shows a peak corresponding to both superoxo and substrate bound species, Na[CuL(O2)(3,5-DTBCH)]+, suggesting that both the dioxygen and substrate simultaneously coordinated to the metal center in the catalytic cycle. Most importantly, complex 2 not only represents the mononuclear class of copper(II) compounds that are rarely visited for the study of catecholase mimicking activity but also the first example of a mononuclear square planar complex exhibiting catechol oxidase activity.
Introduction
Dioxygen represents the ultimate in “green” oxidants as it is atom economic and freely available in the environment. Metalloenzymes that activate molecular dioxygen are of great interest because they facilitate spin forbidden interactions between molecular dioxygen and organic substances.1–3 The study of model complexes of such enzymes not only helps to understand the biochemical phenomena in the natural systems but also provides clues to the development of small molecule catalysts for specific oxidation reactions under mild conditions.4–6 Therefore, most of the focus of ongoing biomimetic and bioinspired chemistry is on the synthesis and reactivity studies of transition metal-based model complexes for metalloenzymes that show oxidase or oxygenase activity.7,8
One of the major enzymes that utilizes molecular dioxygen is catechol oxidase, a lesser explored member of the type-III copper proteins.9 This enzyme belongs to the class of polyphenol oxidases that catalyzes exclusively the oxidation of a wide range of o-diphenols (catechols) to the corresponding o-quinones in a process known as catecholase activity.10–12 Quinones are highly reactive compounds which undergo auto polymerization to produce melanin, a brown colored pigment, and this process is most likely responsible for protecting damaged tissues against pathogens and insects.13 The structural determination of the catechol oxidase has encouraged an extensive investigation of model compounds to understand the structure–property relationship.9,14–18 As the active site of the structure consists of a dicopper(II) moiety, several dinuclear copper(II) complexes derived from N/O donor dinucleating ligands have been mainly employed for this purpose.9,14–30 In addition to the dicopper(II) systems, a few mononuclear copper(II) complexes31–35 and even a few copper(II) clusters and polymers have been found as active catalysts.36 Although few structure–property correlations have been performed, they do not cover a wide range of compounds but only applicable for a set of limited compounds. All these facts indicate that exploration of the possibility of exhibiting catecholase activity by new types of compounds in terms of any of the following deserves importance: metal–metal distance, flexibility of the ligand, type of exogenous ligand and coordination geometry around the metal ion.
We have recently reported several transition metal complexes those were found to be efficient functional models for phenoxazinone synthase37–40 and catechol oxidase.41 Close inspection of our previous results suggests that both the substitutionally labile coordination sites and the electronic environment of the metal center play important role for the biomimetic catalytic activity.37–41 From the reports in the literature on various copper(II) model compounds14–30 and from the crystal structure of catechol oxidase,9 it is also clear that the active site of functional models must have free coordination sites available for substrate binding. Therefore, the ligands having lesser number of donor atoms are more promising. As a part of our ongoing study in the development of functional model complexes of various oxidase metalloenzymes,37–41 we report herein the synthesis and crystal structures of a new dinuclear copper(II) complex, [Cu2L2(μ1,1-N3)2] (1), and a mononuclear copper(II) complex, [CuL(NCO)] (2), derived from a tridentate Schiff-base ligand, 2-methoxy-6-(8-iminoquinolinylmethyl)phenol (HL) (Scheme 1) together with exogenous pseudohalide coligands (N3− and NCO−, respectively). Role of the exogenous pseudohalide coligands on the structural diversity and on the relative catecholase activity of the complexes have also been explored.
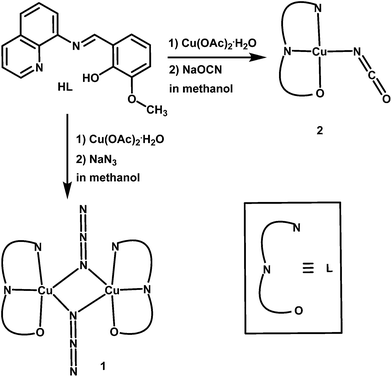 |
| | Scheme 1 Route to the syntheses of complexes 1 and 2. | |
Experimental section
Materials and physical measurements
All reagent or analytical grade chemicals and solvents were purchased from commercial sources and used as received. Elemental analyses for C, H and N were carried out using a Perkin-Elmer 240 elemental analyzer. Infrared spectra (400–4000 cm−1) were recorded from KBr pellets on a Nickolet Magna IR 750 series-II FTIR spectrophotometer. Absorption spectra were measured using a Shimadzu UV-2450 spectrophotometer with a 1 cm path-length quartz-cell. Electrochemical data were collected on an EG & G Princeton Applied Research potentiostat model 263A with a Pt working electrode, Pt auxiliary electrode and Ag/AgCl reference electrode. Electrospray ionization (ESI-MS positive) mass spectrometric studies were performed using Micromass Q-tof-Micro Quadruple and Q-tof-Micro YA263 mass spectrophotometers.
Synthesis of Schiff base ligand HL
The tridentate Schiff base ligand (HL) was prepared by the standard method.42 Briefly, 1.0 mmol of 8-aminoquinoline (145 mg) were mixed with 1.0 mmol of o-vanillin (152 mg) in 20 mL of methanol. The resulting solutions were heated to reflux for ca. 1 h, and allowed to cool. The dark orange methanol solution was used directly for complex formation.
Synthesis of complex [Cu2L2(μ1,1-N3)2] (1)
A 20 mL methanol solution of HL (1.0 mmol) was combined with a solution of Cu(OAc)2·H2O (202 mg, 1.0 mmol) and another 5 mL aqueous solution of sodium azide (65 mg, 1.0 mmol). The resulting mixture was heated to reflux for 1 h during which time color of the solution changed to dark brown. The reaction mixture was then filtered and kept at room temperature. Analytically pure dark-brown crystals suitable for X-ray diffraction were obtained from the solution after several days, which was collected by filtration and washed with methanol/ether and air dried. Yield: 272 mg (71%). Anal. calcd for C34H26N10O4Cu2: C 53.33%, H 3.42%, N 18.29%. Found: C 53.54%, H 3.35%, N 17.96%. IR (cm−1, KBr): 3041w, 2925w, 2046vs, 1603m, 1539w, 1504w, 1429s, 1404w, 1334w, 1245m, 1215s, 1174m, 1090w, 980w, 831w, 765w, 734m, 671w. UV-Vis λmax nm−1 (ε dm−3 mol−1 cm−1) in DMF–methanol: 353 (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 975), 462 (9986), 615 (92).
975), 462 (9986), 615 (92).
Synthesis of complex [CuL(NCO)] (2)
Complex 2 was synthesized following the very similar procedure as described for complex 1 except that sodium cyanate was used instead of sodium azide. Color: dark brown, yield: 306 mg (80%). Anal. Calcd. for C18H13N3O3Cu: C 56.47%, H 3.51%, N 10.97%. Found: C 56.56%, H 3.65%, N 10.72%. IR (cm−1, KBr): 3056w, 2960w, 2835w; 2204vs, 1608vs, 1602w, 1553w, 1508w, 1460s, 1414w, 1401w, 1290s, 1249s, 1240s, 1101m, 1005m, 982w, 855w, 758w, 744m, 684w. UV-Vis λmax nm−1 (ε dm−3 mol−1 cm−1) in DMF–methanol: 351 (11890), 456 (5520), 627 (121).
X-ray crystallography
Single crystal X-ray data of complexes 1 and 2 were collected on a Bruker SMART APEX-II CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at room temperature. Data processing, structure solution, and refinement were performed using Bruker Apex-II suite program. All available reflections to 2θmax were harvested and corrected for Lorentz and polarization factors with Bruker SAINT plus.43 Absorption corrections, inter-frame scaling, and other systematic errors were performed with SADABS.43 The structures were solved by the direct methods and refined by means of full matrix least-square technique based on F2 with SHELX-97 software package.44 All the nonhydrogen atoms were refined with anisotropic thermal parameters. All the hydrogen atoms belonging to carbon were placed in their geometrically idealized positions, and all of them were constrained to ride on their parent atoms. Crystal data and details of data collection and refinement for 1 and 2 are summarized in Table 1.
Table 1 Crystal data and structure refinement of 1 and 2
| Compound |
1 |
2 |
| Empirical formula |
C34H26N10O4Cu2 |
C18H13N3O3Cu |
| Formula weight |
765.73 |
382.85 |
| Temperature (K) |
298(2) |
298(2) |
| Wavelength (Å) |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Triclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) |
9.0785(6) |
7.2980(2) |
| b (Å) |
22.8156(16) |
14.4856(5) |
| c (Å) |
8.3584(5) |
14.9016(5) |
| α (°) |
90 |
85.634(2) |
| β (°) |
115.520(4) |
83.851(2) |
| γ (°) |
90 |
78.042(2) |
| Volume (Å3) |
1562.37(18) |
1529.93(8) |
| Z |
2 |
4 |
| Dcalc (g cm−3) |
1.628 |
1.662 |
| Absorption coefficient (mm−1) |
1.420 |
1.451 |
| F(000) |
780 |
780 |
| θ range for data collection (°) |
1.79–26.43 |
1.94–27.24 |
| Reflections collected |
23810 |
24661 |
| Independent reflection/Rint |
1573/0.1356 |
4653/0.0425 |
| Data/restraints/parameters |
3211/0/226 |
6762/0/453 |
| Goodness-of-fit on F2 |
0.962 |
1.020 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0547, wR2 = 0.0988 |
R1 = 0.0428, wR2 = 0.0940 |
| R indices (all data) |
R1 = 0.1424, wR2 = 0.1231 |
R1 = 0.0704, wR2 = 0.1045 |
| Largest diff. Peak/hole (e Å−3) |
0.318/−0.356 |
0.472/−0.311 |
Computational method
All the theoretical calculations were carried out by DFT method implemented in Gaussian 09 software.45 Ground states of the ligand and complex 2 were optimized by DFT method with B3LYP exchange correlation functional in methanol (MeOH) media associated with PCM model (Table S1†). The absorption spectra in methanol were calculated by time dependent density functional theory (TDDFT) method, while for the complex split basis set was used. The 6-31g (d) basis set was used for H, C, N and O atoms and LANL2DZ was used for Cu atom (Table S1†). For all the intermediates and transition state UB3LYP functional was used for mechanistic interpretation (Table S1†). The frontier molecular orbitals were prepared by using GaussView 5.0 software package and the molecular orbital contribution from groups or atoms was calculated by GaussSum 3.0 program.
Catalytic oxidation of 3,5-DTBCH2
The catecholase activity of the complexes was studied by the reaction of 200 equivalents of 3,5-di-tert-butylcatechol (3,5-DTBCH2) with 2 × 10−5 M solutions of the complexes under aerobic conditions at 25 °C in methanol–DMF (50:1 v/v). The reaction was followed spectrophotometrically by monitoring increase in the absorbance as a function of time at ca. 400 nm which is characteristic of o-quinone formation. To determine the dependence of rate of the reaction on substrate concentration and various kinetic parameters, 2 × 10−5 M solutions of the complexes were treated with at least 10 equivalents of substrate to follow pseudo-first order condition. To check the rate dependency on catalyst concentrations similar set of experiments were performed at fixed concentration of substrate with various catalyst concentrations. The initial rate method was applied to determine the rate of a reaction, and the average initial rate over three independent measurements was recorded.
Results and discussion
Syntheses and general characterizations
The synthetic route employed for the preparation of complexes 1 and 2 is depicted in Scheme 1. The tridentate ligand (HL) was synthesized by the Schiff base condensation of equimolecular mixture of o-vanillin and 8-aminoquinoline in methanol under reflux. Reaction of Cu(OAc)2·H2O, Schiff base ligand (HL), and a coligand (azide ion) in 1:1:2 molar ratio in methanol–water medium afforded 1 in high yield. Very similar procedure but using sodium cyanate instead of azide as a coligand produced only 2. The nature of the solid crystalline products was examined for both the compounds, and no visible difference in the crystalline state was observed, indicating that the solids are not mixtures of different compounds. The results of elemental analyses also confirm that the compositions of the complexes are eventually same as those obtained from X-ray diffractions.
Both the compounds were characterized by the FT-IR spectroscopy and important infrared bands of them are given in the Experimental section. The IR spectra of these complexes are very similar to each other with regards to mono-anionic tridentate L ligand. IR spectra show strong and sharp bands for ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) at 1603 and 1608 cm−1 for complexes 1 and 2, respectively, indicating the presence of Schiff base ligand. In the spectrum of complex 2, a strong band at 2204 cm−1 is attributed to the presence of N-coordinated cyanate group. Whereas the IR spectrum shows only a single sharp absorption band in 1, consistent with the presence of only one type of azide (bridging azide) in the structure and it appears at lower frequency, 2046 cm−1, which is in agreement with the asymmetric bridging.46–51 Both the complexes are electrochemically active which is the signature of the transition metal complexes. The cyclic voltammograms of the complexes show an electrochemically irreversible peak at −0.80 V and −0.66 V for complexes 1 and 2, respectively, which are assigned to the reduction from copper(II) to copper(1) centers.
N) at 1603 and 1608 cm−1 for complexes 1 and 2, respectively, indicating the presence of Schiff base ligand. In the spectrum of complex 2, a strong band at 2204 cm−1 is attributed to the presence of N-coordinated cyanate group. Whereas the IR spectrum shows only a single sharp absorption band in 1, consistent with the presence of only one type of azide (bridging azide) in the structure and it appears at lower frequency, 2046 cm−1, which is in agreement with the asymmetric bridging.46–51 Both the complexes are electrochemically active which is the signature of the transition metal complexes. The cyclic voltammograms of the complexes show an electrochemically irreversible peak at −0.80 V and −0.66 V for complexes 1 and 2, respectively, which are assigned to the reduction from copper(II) to copper(1) centers.
UV-vis spectroscopy and DFT study
Both the complexes are practically insoluble in acetonitrile and partly soluble in methanol, while they are sufficiently soluble in DMF medium. Therefore, to keep consistency, solution studies of the complexes including catecholase activity (vide infra) were carried out in methanol–DMF (50:1 v/v) mixture. Electronic spectra of the complexes display broad absorption bands at 615 and 627 nm for complexes 1 and 2, respectively. The positions of these bands are in agreement with the d–d transition of copper(II) ion found in similar complexes.52,53 Additionally, two distinct higher energy bands in the spectra of both the complexes appear at 353 and 462 nm for 1, and 351 and 456 nm for 2. Interestingly, the absorption spectrum of the Schiff base ligand alone shows only single π → π* transition band at 339 nm which undoubtedly confirms that the bands at 462 nm for 1 and 456 nm for 2 are due the charge transfer (CT) transition. In order to understand origin of the charge transfer transition, a base titration of the ligand alone was conducted, and the resultant spectra are depicted in Fig. S1.† This spectral titration shows that with addition of increasing amount of base a new band is appeared at 430 nm, and as expected π → π* transition band of the ligand at 339 nm is red-shifted about 6 nm due to deprotonation of the phenolic-OH. These results prompt us to conclude that the metal center does not participate in the charge transition in these complexes but it helps to deprotonate the phenolic-OH group thereby favoring the purely ligand-based charge transfer transition.
In order to confirm donor and accepter parts of the CT transition, DFT study is carried out both for the ligand and a representative complex 2. The calculated absorption peak at 350 nm is overlapped with another band at 387 nm for the ligand (Fig. S2†). The oscillator strengths of these transitions are 0.438 and 0.139, respectively. The calculation of orbital contribution for these absorptions shows that 387 nm peak is due to HOMO to LUMO transition, while peak at 350 nm arises from HOMO − 1 to LUMO (major) and HOMO − 2 to LUMO (minor) transitions. The orbital contribution from groups in the ground state of the ligand shows that HOMO (−5.67 eV) is composed of 88% phenol moiety and 12% quinoline moiety, whereas HOMO − 1 (−5.99 eV) is composed of 35% phenol moiety and 65% quinoline moiety, and HOMO − 2 (−6.77 eV) is composed of 51% phenol and 49% quinoline moiety. The experimental absorption peak observed at 339 nm is in very good agreement with the calculated peak. The orbital contributions also suggest that these two calculated absorption bands are inter ligand charge transfer (ILCT) character. The TDDFT calculation of the complex shows two bands at 487 and 372 nm in which oscillator strengths are 0.08 and 0.368, respectively (Fig. 1). The 487 nm peak can be assigned to the HOMO → LUMO transition and the peak at 372 nm can be attributed to HOMO − 1 → LUMO and HOMO − 1 → LUMO + 2. The orbital contribution also indicates that these transitions are ILCT character, and these transitions occur from phenol moiety as a donor to quinoline moiety as an acceptor.
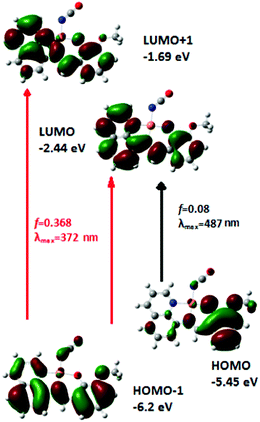 |
| | Fig. 1 Frontier molecular orbitals involved in UV-Vis absorption of complex 2. | |
Description of crystal structures of 1 and 2
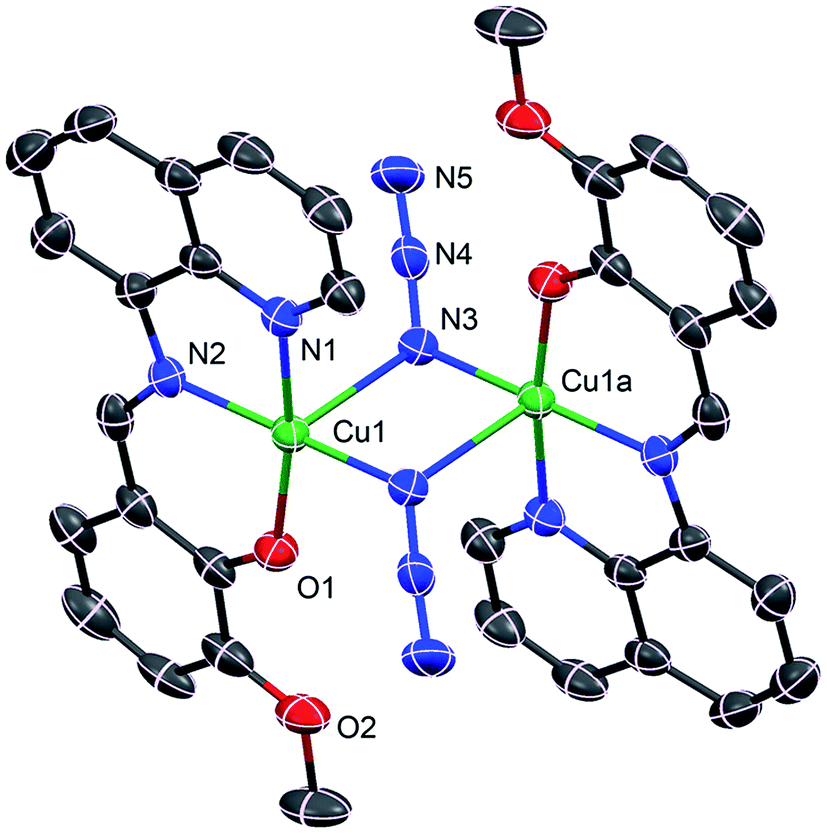
Complex 1 crystallizes in the monoclinic P21/c space group, where the asymmetric unit corresponds to half of the molecule. The perspective view of 1 along with atom numbering scheme at metal coordination sphere is depicted in Fig. 2, while important bond distances and angles are given in Table 2. This cetrosymmetric complex is formed by a bis(μ1,1-N3)-bridged copper(II) dimer in which each of the copper(II) center is five-coordinate being bonded to three donor atoms from one mono-negative Schiff base ligand together with two nitrogen atoms (N3 and N3a) from two bridging azide groups. The coordination geometry of copper(II) ion can be best described as the ideal square pyramid as indicated by the Addison parameter (τ) of 0.052. Four atoms in the equatorial plane consist of three donor atoms from the tridentate ligand together with a nitrogen atom from a bridging azide with bond lengths of Cu1–O1 1.900(3) Å, Cu1–N1 1.993(4) Å, Cu1–N2 1.964(4) Å for tridentate ligand L and Cu1–N3 1.987(4) Å for azide ion. A second nitrogen atom (N3a) from centrosymmetrically related bridging azide coordinates an apical position at a rather long distance [2.505(4) Å], furnishing an elongated square-pyramidal geometry for each copper(II) center which is consistent with d9 electronic configuration. The basal–apical bridging ultimately leads to an asymmetric μ1,1 bridging mode of azide ions in 1. The bridging Cu1–N3–Cu1a angle is 89.7(1)° with metal–metal separation of 3.389(1) Å. The bridging N3− anions are quasi-linear with N–N–N angles of 176.6(5)°. All the bond lengths and angles are within the range of those found in similar end-on azido-bridged copper(II) complexes of tridentate Schiff bases.54,55 Molecular packing of 1 is primarily stabilized by the π–π staking interactions between the adjacent quinoline rings as shown in Fig. 3.
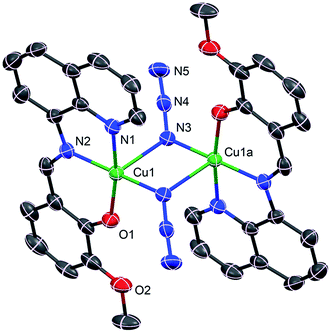 |
| | Fig. 2 Crystal structure of 1 showing the atom numbering scheme. Ellipsoids are drawn at 30% probability level. H atoms are omitted for clarity. Symmetry code: a = 2 − x, −y, 1 − z. | |
Table 2 Selected bond lengths (Å) and bond angles (°) for 1 and 2a
| 1 |
2 |
| Symmetry code: a = 2 − x, −y, 1 − z. |
| Cu1–O1 |
1.900(3) |
Cu1–O1 |
1.891(2) |
| Cu1–N1 |
1.993(4) |
Cu1–N1 |
1.989(2) |
| Cu1–N2 |
1.964(4) |
Cu1–N2 |
1.949(2) |
| Cu1–N3 |
1.987(4) |
Cu1–N3 |
1.931(3) |
| Cu1–N3a |
2.505(4) |
Cu2–O4 |
1.915(2) |
| |
|
Cu2–N4 |
2.012(2) |
| |
|
Cu2–N5 |
1.954(2) |
| |
|
Cu2–N6 |
1.924(2) |
| N2–Cu1–N1 |
81.96(17) |
N2–Cu1–N1 |
82.51(9) |
| O1–Cu1–N2 |
92.75(16) |
O1–Cu1–N2 |
92.75(16) |
| N2–Cu1–N3 |
172.94(15) |
O1–Cu1–N1 |
169.82(14) |
| O1–Cu1–N1 |
169.82(14) |
N2–Cu1–N3 |
172.94(15) |
| N5–N4–N3 |
176.6(5) |
N5–Cu2–N4 |
82.12(9) |
| Cu1–N3–Cu1a |
97.3(2) |
O4–Cu2–N5 |
92.84(8) |
| |
|
O4–Cu2–N4 |
174.87(8) |
| |
|
N6–Cu2–N5 |
174.51(10) |
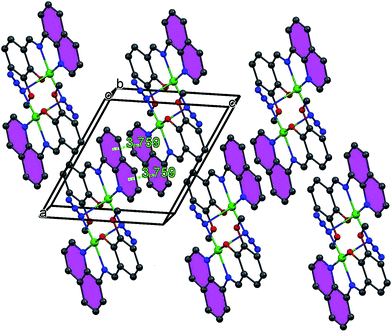 |
| | Fig. 3 Crystal packing of complex 1 showing π–π stacking interactions. | |
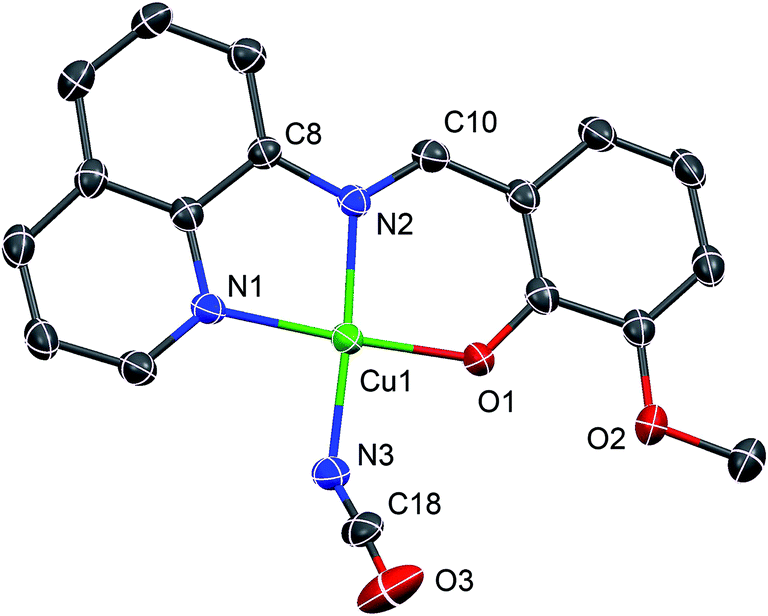
Complex 2 crystallizes in the triclinic P space group such that the asymmetric unit contains two crystallographically independent molecules. A representative molecular structure along with selected atom numbering scheme is depicted in Fig. 4. X-ray crystallography reveals that copper(II) ion is four coordinate in square-planar environment being bonded with three donor atoms from mono-negative tridentate Schiff base ligand and a nitrogen atom from cyanate group. Further, average displacement of the metal centers from the respective N3O least-square planes is found to be 0.026 Å, which indicates that coordination environment of the copper(II) ions in 2 is merely square-planar. Cu–N bond lengths vary in the range 1.924(2) to 2.012(2) Å, while Cu–O bond distances [1.891(2) and 1.915(2) Å] are close for two crystallographically independent molecules.54,55 The mono-anionic tridentate L ligand being planar the entire structure of complex 2 is also nearly planar although the cyanate group is coordinated to central Cu atom by the bent fashion at an angle C–N–Cu of 138.5(3)/148.0(3)°. This planarity helps to stabilize crystal packing by the strong π–π interactions between the adjacent molecules as depicted in Fig. 5.
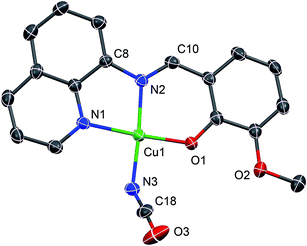 |
| | Fig. 4 A representative ORTEP-type plot of the structure of 2 with selected atom numbering scheme. Ellipsoids are drawn at 30% probability level and H atoms are omitted for clarity. | |
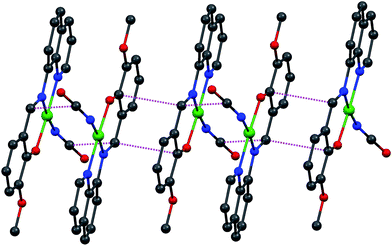 |
| | Fig. 5 A part of molecular packing of 2 showing different kinds of π–π interactions. | |
From the crystal structures of complexes 1 and 2, it is clear that in both the cases mono-negative Schiff base ligand binds the metal center in the planar arrangement which is consistent with the electronic structure of the ligand. But the diverse structural patterns in these complexes mainly arises from the mode of coordination of pseudohalides ligands. Azide being stronger bridging ligand bridges two metal centers in an end-on fashion, resulting as a formation of a dinuclear copper(II) complex 1, while its congener cyanate ion only acts as a terminal ligand in 2.
Catecholase activity
Catecholase activity is usually studied with 3,5-di-tert-butylcatechol (3,5-DTBCH2) as a model substrate because of its low redox potential that facilitates ease of oxidation of 3,5-DTBCH2 to its corresponding quinone, 3,5-di-tert-butylquinone (3,5-DTBQ), later of which has a characteristic transition at about 400 nm. In order to check ability of the complexes to behave as catalysts for the catecholase activity, 2.0 × 10−5 M solutions of the complexes were treated with 200-fold concentrated solution of 3,5-DTBCH2, and the spectra were recorded up to 2 h in dioxygen saturated methanol–DMF (50:1 v/v) at 25 °C. The time dependent spectral changes for a period of 2 h after the addition of 3,5-DTBCH2 are shown in Fig. S3 (in ESI†) & Fig. 6 for 1 and 2, respectively. The spectral scan reveals the development of a strong absorption band at ca. 400 nm characteristic of quinone chromophore for 2, while significant spectral growth was not observed for 1 in an identical condition. These spectral behaviors prompt us to conclude that complex 2 is reactive towards the catecholase activity, while complex 1 is almost inactive. It is worthy to note that after addition of substrate 3,5-DTBCH2 to the solutions of the catalysts CT bands of the complexes did not disappear although progressive increase of quinone band along with the CT bands especially for 2 is observed. This observation suggests that destruction of the complex in terms of loss of coordinated Schiff base ligand in the presence of excess substrate is not happened, which not only proves the much stronger chelating ability of ligand L than that of the substrate but also rules out the possibility of any decomposition of active catalyst in terms of loss of coordinated ligands during the catalytic cycle.
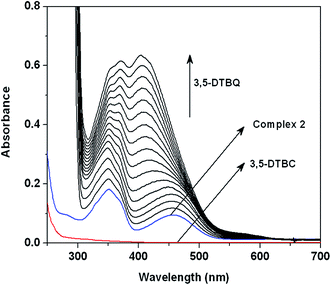 |
| | Fig. 6 UV-vis spectral profile showing the growth of quinone band at 405 nm after the addition of 200-fold 3,5-DTBCH2 to a solution containing complex 2 (2 × 10−5 M) in dioxygen saturated MeOH–DMF (50:1) at 25 °C. The spectra were recorded for the period of 2 h. | |
Kinetic studies were performed to understand the extent of the catalytic efficiency of complex 2 as it exhibits reasonably good catecholase activity. For this purpose, 2 × 10−5 M solutions of the complex were treated with at least 10-fold concentrated substrate solution to maintain the pseudo-first-order condition. For a particular complex–substrate mixture, time scan at the maximum of quinone band was carried out for a period of 20 min, and initial rate was determined by linear regression from slope of the absorbance versus time. The initial rate of the reactions verses concentration of the substrate data show that rate is first order at the region of low concentrations of 3,5-DTBCH2 but zero order at the higher concentrations (Fig. 7). This observation suggests that quinone formation proceeds through a relatively stable intermediate, a complex–substrate adduct, followed by redox decomposition of the intermediate at the rate determining step. This type of saturation rate dependency on the concentration of the substrate may be explained by considering the Michaelis–Menten model, originally developed for the enzyme kinetics, which on linearization gives double reciprocal Lineweaver–Burk plot to analyze values of the parameters Vmax, KM, and Kcat. The observed and simulated initial rates versus substrate concentration plot and the Lineweaver–Burk plot for complex 2 are shown in Fig. 7. Analysis of the experimental data yielded Michaelis binding constant (KM) value of 2.27 × 10−3 M−1 and Vmax value of 1.31 × 10−7 M−1. The turnover number (Kcat) value is obtained by dividing the Vmax by the concentration of the catalyst, and is found to be 23.58 h−1. Moreover, for a particular substrate concentration, varying the complex concentration, a linear relationship for the initial rates was obtained, which shows a first-order dependency on the catalyst concentration (Fig. S4†).
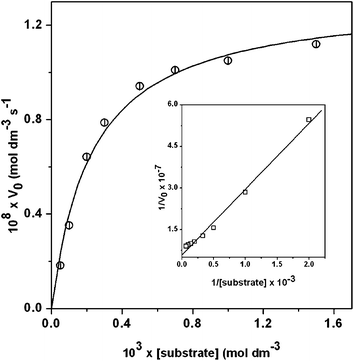 |
| | Fig. 7 Plot of the initial rates versus substrate concentration for the oxidation reaction catalyzed by complex 2 at a fixed concentration in dioxygen-saturated MeOH–DMF (50:1) at 25 °C. Inset shows Lineweaver–Burk plot. Symbols and solid lines represent the experimental and simulated profiles, respectively. | |
Electrospray ionization mass spectral study
As compound 2 shows only catecholase activity, the electrospray ionization mass spectrum (ESI-MS positive) of methanol–DMF solution of compound 2 was recorded and is shown in Fig. S5.† Compound 2 exhibits five assignable peaks at m/z = 339.83, 404.78, 420.74, 721.63 and 786.55 with line-to-line separation of 1.0. The peaks at m/z = 339.83, 404.78 and 420.74 arise from the monomeric entity of the complex and are assigned to [CuIIL]+, [NaCuIIL(NCO)]+ and [KCuIIL(NCO)]+, respectively, while low intense peaks at m/z = 721.63 and 786.55, 302 appear due to the dimeric aggregate of the complex and these can be assigned to [CuII2L2(NCO)]+ and [NaCuII2L2(NCO)2]+, respectively. All these monopositive species are well matched with the simulated isotropic distribution pattern as shown in Fig. S5.†
In order to get further insight into the nature of possible complex–substrate intermediates, ESI-MS positive spectrum of a mixture of complex 2 and 3,5-DTBCH2 in 1:50 molar ratio was recorded after 5 min of mixing in methanol–DMF. The observed and simulated patterns are presented in Fig. 8. As can be seen from the figure, the most abundant species found at m/z 340.05 in the mass spectrum corresponds to the original compound 2 and it can assigned to [CuIIL]+. The peak at 243.16 is well assignable to the quinone-sodium aggregate [3,5-DTBQ-Na]+. The remaining peak at m/z = 584.19 including two minor peaks at m/z = 616.13 and 923.21 are quite interesting because the peak position, line-to-line separation, and matching of the isotopic distributions of the observed and the simulated patterns (Fig. 8) clearly indicate that these peaks arise from complex–substrate aggregates. More specifically these peaks at m/z = 584.19, 616.13 and 923.21 can be assigned to [NaCuIIL(3,5-DTBCH)]+, [NaCuL(O2)(3,5-DTBCH)]+ and [NaCu2IIL2(3,5-DTBC)]+, respectively. Although mass spectroscopy does not necessarily mirror the species present in the solution state yet it provides significant information based on that one could get a mechanistic inference of a catalytic process.
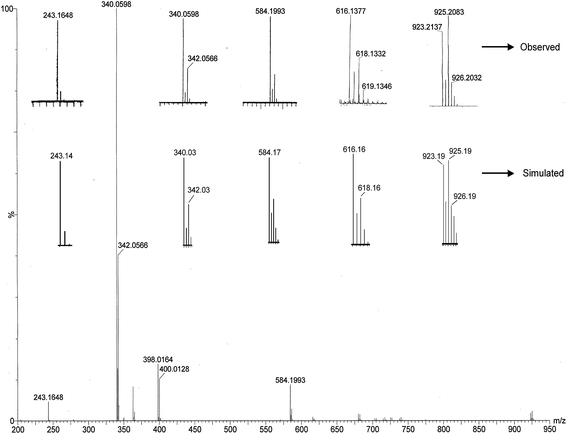 |
| | Fig. 8 Electrospray ionization mass spectrum (ESI-MS positive) of a 1:50 mixture of [CuL(NCO)] (2) and 3,5-DTBCH2 in methanol–DMF, recorded after 5 min of mixing. Both the observed and simulated isotopic distribution patterns are shown. | |
Plausible mechanism and DFT studies
At this juncture, it is to be mentioned that the catechol oxidase activity depends on several factors namely Cu⋯Cu distance, geometry around the metal center, nature of the exogenous ligand, and flexibility of the primary ligand. The metal–metal separation (3.398 Å) in complex 1 lies in the range where catecholase activity of several systems is found to be very efficient.14–27 Complex 1 is EPR silent at 77 K which ensures that the dimer preserves its integrity in acetonitrile–DMF solution. Thus the lack of activity of complex 1 towards the catecholase activity may be due to the fact that the doubly bridging dicopper(II) system is substitutionally inert for incoming substrate.56,57 Inactivity of the complex further suggests that although one of the apical position is available but copper(II) center is reluctant to increase its coordination number up to six. It is also interesting to note that as the geometry of the copper(II) center is a trigonal pyramidal in the crystal structure of met form of the enzyme, most of the efficient models reported in the literature are found to be non-planar structures.14–17,19–27 Only limited numbers of square planar complexes are known which show catecholase activity.18 In the present case, the moderately strong activity as suggested by the turnover number for mononuclear complex 2 would definitely be a significant contribution towards the modeling of the catechol oxidase. Either of the vacant apical positions or substitutionally labile cyanate ion favors the binding of the substrate with copper(II) center thereby leading to exhibit moderately strong catecholase activity.
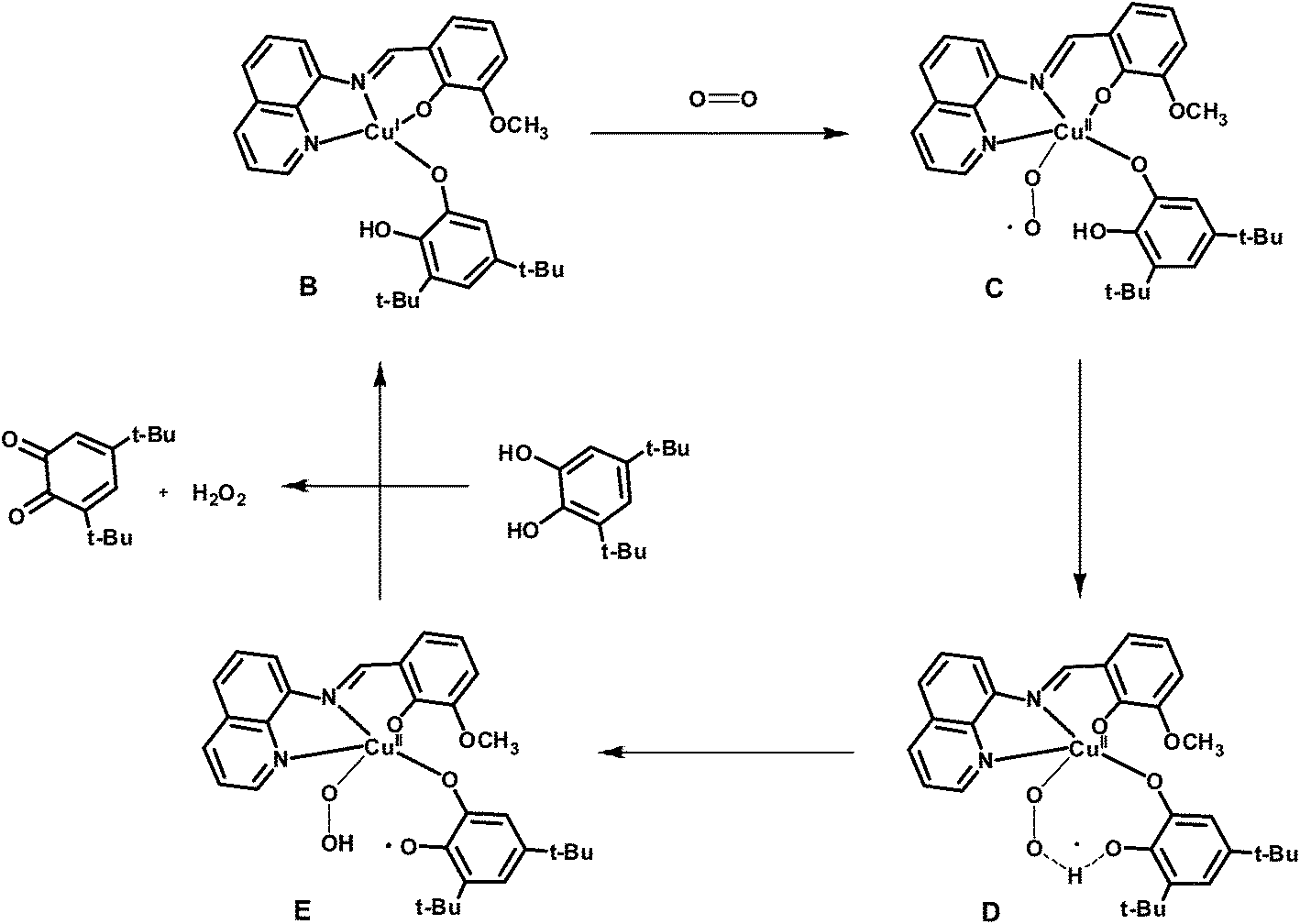
The EPR spectrum of complex 2 is typical for mononuclear copper(II) complexes (Fig. S6†), but on addition of 3,5-DTBCH2, the characteristic signal for copper(II) ion was disappeared which suggests that copper(II) center is reduced to copper(I) state in the catalytic cycle. It is now well known that catecholate oxidation by dicopper(II) complexes takes place by the transfer of couple of electrons from substrate to copper(II) centers. But mononuclear copper(II) complex in the present case could only act as single electron oxidant, and thus it could be possible that 3,5-DTBCH2 bridges two neighboring mononuclear copper(II) centers followed by the electron transfer to copper(II) centers in the rate determining step. Although a species of formula [NaCu2IIL2(3,5-DTBC)]+ is identified in the mass spectroscopy, unimolecular rate of the reaction with respect to complex 2 clearly precludes the possibility of involvement of two copper(II) centers in the main catalytic cycle. The possibility of involvement of phenolate radical as a second oxidant can also be neglected as the electrochemical study suggests that the oxidation of phenolate ion is quite difficult. Therefore, the dioxygen-bound copper species could probably be the active catalyst for the oxidation of 3,5-DTBCH2. Both the substrate and dioxygen bound species, Na[CuL(O2)(3,5-DTBCH)]+, identified in the mass spectroscopy supports the above fact. We are at the stage to tempt a plausible pathway in which at the first stage active catalyst (B) is generated by the reaction of 3,5-DTBCH2 with two equivalents of complex 2 (Scheme 2). That catecholate bound copper(I) complex (B) is supposed to act as the active catalyst to generate the superoxide intermediate (C) by the reaction with aerial oxygen in which copper(I) center in B undergoes an oxidation to a copper(II) center in C (Scheme 3). In the catalytic cycle, next step involves the intramolecular proton transfer process from oxygen atom of catechol moiety to the superoxide oxygen atom in intermediate C via a transition state D to produce a peroxo-intermediate E. Finally, the last step involves the binding of another molecule of substrate, 3,5-DTBCH2, to the copper center in E regenerating the starting B species with concomitant release of the desired 3,5-DTBQ.
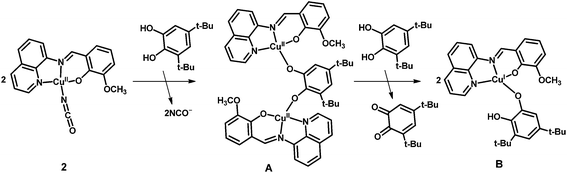 |
| | Scheme 2 Probable pathway for the generation of the precatalyst. | |
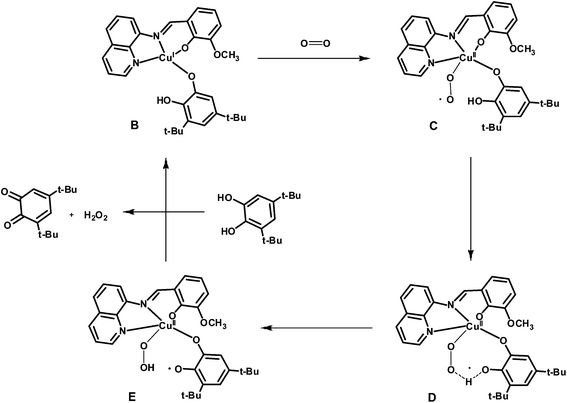 |
| | Scheme 3 Probable catalytic cycle for the oxidation of 3,5-DTBCH2 by complex 2. | |
The proposed mechanistic pathway for the catechol oxidation by molecular dioxygen in the presence of complex 2 as a catalyst is further supported by the density functional theory (DFT) study. As expected theoretical calculations show that the Mulliken charge on Cu center in C is changed to 0.490 from 0.315 in B, and the bond length of molecular oxygen (1.21 Å) is changed to superoxide (1.31 Å) in C (Table S1†). The O–O bond in the transition state D has been elongated to 1.34 Å compared with 1.31 Å in C. The Mulliken charge of Cu in D transition state is slightly increased to 0.490 in comparison to 0.452 in C. Whereas the Mulliken charge on superoxide-O atoms (−0.253 and −0.251) in intermediate C changed to −0.302 and −0.341 in D transition state, respectively. This change of charge density suggests proton transfer occurs from catecholate-O to the superoxide-O atom. As expected the elongated O–O peroxide bond distance of 1.42 A is observed in peroxide intermediate state E. Consequently the semiquinone character of the catechol moiety is observed in the intermediate state E having shorter C–O bond lengths (1.26 and 1.28 Å) compared to catecholate functionality (1.35 and 1.31 Å) in superoxo intermediate C.58,59 The proton transfer is also corroborated by shifting of the Mulliken charge of peroxide (in intermediate E) oxygen atoms −0.525 and −0.561 compared to that of the superoxide oxygen atoms −0.253 and −0.251 in intermediate C. Therefore both the experimental facts and theoretical study (Table S1 and Fig. S7†) agree well with the proposed mechanistic pathway for the catalytic oxidation of 3,5-DTBCH2 by molecular dioxygen in the presence of complex 2 as a catalyst.
Conclusion
Two new di- and mono-nuclear copper(II) complexes have been synthesized from a planar tridentate ligand 2-methoxy-6-(8-iminoquinolinylmethyl)phenol (HL) together with pseudohalides as coligands. Structural characterizations reveal that complex 1 is a centrosymmetric bis(azide)-bridged dimer in which copper(II) centers reside in the square pyramidal environment while it is square planar in 2. From the crystal structure it is clear that pseudohalide coligands have diverse impact on the nuclearity of those complexes – azide ions bind the metal centers in an end-on asymmetric bridging fashion in 1, whereas cyanate ion simply acts as a monodentate terminal ligand in 2. Although the crystal structure of the native met form of catechol oxidase suggested a dicopper(II) system, but in the present investigation dinuclear compound 1 is found to be almost inactive. On the other hand, mononuclear compound 2 exhibits significant catecholase activity which is probably due to the presence of labile cyanate ion together with unsaturated coordination number that favors binding of substrate to the metal center. The ESI-MS positive spectrum of the mixture of complex 2 and 3,5-DTBCH2 shows a peak corresponding to Na[CuL(O2)(3,5-DTBCH)]+, suggesting simultaneous coordination of the substrate and molecular dioxygen to the metal center in the catalytic cycle. Interestingly, complex 2 not only represents the mononuclear class of copper(II) compounds that rarely investigated for the catecholase mimicking activity but also the first example of a mononuclear square planar complex exhibiting catechol oxidase activity.
Acknowledgements
M. S. thanks the University Grant Commission, India for a Fellowship. A. S. gratefully acknowledges the financial support of this work by the DST, India (Sanction no. SB/EMEQ-159/2013 dated 10/10/2013). A. P. also gratefully acknowledges the financial support of this work by the University Grant Commission, India (Sanction no. PSW-173/11-12). The ESI-MS spectra facility was availed from the Indian Association for the Cultivation of Science, India. Crystallography was performed at the DST-FIST, India, funded single crystal diffractometer facility at the Department of Chemistry, Jadavpur University, India.
References
- E. I. Solomon, R. Sarangi, J. S. Woertink, A. J. Augustine, J. Yoon and S. Ghosh, Acc. Chem. Res., 2007, 40, 581 CrossRef CAS PubMed.
- P. C. A. Bruijnincx, G. van Koten and R. J. M. Klein Gebbink, Chem. Soc. Rev., 2008, 37, 2716 RSC.
- S. Friedle, E. Reisner and S. J. Lippard, Chem. Soc. Rev., 2010, 39, 2768 RSC.
- R. Hage and A. Lienke, Angew. Chem., 2006, 118, 212 CrossRef.
- N. Duran and E. Esposito, Appl. Catal., B, 2000, 28, 83 CrossRef CAS.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939 CrossRef CAS PubMed.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333 CrossRef CAS PubMed.
- E. B. Meunier, Biomimetic Oxidations Catalyzed by Transition Metal Complexes, Imperial College Press, London, 2000 Search PubMed.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084 CrossRef CAS PubMed.
- I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814 RSC.
- K. Selmeczi, M. Reglier, M. Giorgi and G. Speier, Coord. Chem. Rev., 2003, 245, 191 CrossRef CAS.
- R. Than, A. A. Feldmann and B. Krebs, Coord. Chem. Rev., 1999, 182, 211 CrossRef.
- B. J. Dervall, Nature, 1961, 189, 311 CrossRef.
- N. A. Rey, A. Neves, A. J. C. T. Bortoluzzi, C.T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348 CrossRef CAS PubMed.
- C. Belle, C. Beguin, I. Gautier-Luneau, S. Hamman, C. Philouze, J. L. Pierre, F. Thomas and S. Torelli, Inorg. Chem., 2002, 41, 479 CrossRef CAS PubMed.
- J. Anekwea, A. Hammerschmidta, A. Rompelb and B. Krebs, Z. Anorg. Allg. Chem., 2006, 632, 1057 CrossRef.
- J. Ackermann, F. Meyer, E. Kaifer and H. Pritzkow, Chem.–Eur. J., 2002, 8, 247 CrossRef CAS.
- J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429 CrossRef CAS.
- M. Merkel, N. Mçller, M. Piacenza, S. Grimme, A. Rompel and B. Krebs, Chem.–Eur. J., 2005, 11, 1201 CrossRef CAS PubMed.
- C. Fernandes, A. Neves, J. Bortoluzzi, A. S. Mangrich, E. Rentschler, B. Szpoganicz and E. Schwingel, Inorg. Chim. Acta, 2001, 320, 12 CrossRef CAS.
- A. Neves, L. M. Rossi, A. J. Bortoluzzi, A. S. Mangrich, W. Haase and R. Werner, J. Braz. Chem. Soc., 2001, 12, 747 CrossRef CAS.
- P. K. Nanda, V. Bertolasi, G. Aromí and D. Ray, Polyhedron, 2009, 27, 987 CrossRef.
- R. Gupta, S. Mukherjee and R. Mukherjee, Polyhedron, 2000, 19, 1429 CrossRef CAS.
- A. Majumder, S. Goswami, S. R. Batten, M. S. El Fallah, J. Ribas and S. Mitra, Inorg. Chim. Acta, 2006, 359, 2375 CrossRef CAS.
- C.-H. Kao, H.-H. Wei, Y.-H. Liu, G.-H. Lee, Y. Wang and C.-J. Lee, J. Inorg. Biochem., 2001, 84, 171 CrossRef CAS PubMed.
- M. Thirumavalavan, P. Akilan, M. Kandaswamy, G. Chinnakali, G. Senthil Kumar and H. K. Fun, Inorg. Chem., 2003, 42, 3308 CrossRef CAS PubMed.
- S. J. Smith, C. J. Noble, R. C. Palmer, G. R. Hanson, G. Schenk, L. R. Gahan and M. Riley, J. Biol. Inorg. Chem., 2008, 13, 499 CrossRef CAS PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563 CrossRef CAS PubMed.
- N. Kitajima and Y. Moro-oka, Chem. Rev., 1994, 94, 737 CrossRef CAS.
- C. Eicken, B. Krebs and J. C. Sacchettini, Curr. Opin. Struct. Biol., 1999, 9, 677 CrossRef CAS PubMed.
- M. K. Panda, M. M. Shaikh, R. J. Butcher and P. Ghosh, Inorg. Chim. Acta, 2011, 372, 145 CrossRef CAS.
- G. Grigoropoulou, K. C. Christoforidis, M. Louloudi and Y. Deligiannakis, Langmuir, 2007, 23, 10407 CrossRef CAS PubMed.
- G. Speier, J. Mol. Catal., 1986, 37, 259 CrossRef CAS.
- Z.-F. Chen, Z.-R. Liao, D.-F. Li, W.-K. Li and X.-G. Meng, J. Inorg. Biochem., 2004, 98, 1315 CrossRef CAS PubMed.
- Á. Kupán, J. Kaizer, G. Speier, M. Giorgi, M. Réglier and F. Pollreisz, J. Inorg. Biochem., 2009, 103, 389 CrossRef PubMed.
- S. Majumder, S. Sarkar, S. Sasmal, E. C. Sañudo and S. Mohanta, Inorg. Chem., 2011, 50, 7540 CrossRef CAS PubMed.
- A. Panja, M. Shyamal, A. Saha and T. K. Mandal, Dalton Trans., 2014, 43, 5443 RSC.
- A. Panja, Dalton Trans., 2014, 43, 7760 RSC.
- A. Panja and P. Guionneau, Dalton Trans., 2013, 42, 5068 RSC.
- A. Panja, RSC Adv., 2014, 4, 37085 RSC.
- A. Panja, S. Goswami, N. Shaikh, P. Roy, M. Manassero, R. J. Butcher and P. Banerjee, Polyhedron, 2005, 24, 2921 CrossRef CAS.
- S. Nayak, P. Gamez, B. Kozlevčar, A. Pevec, O. Roubeau, S. Dehnen and J. Reedijk, Polyhedron, 2010, 29, 2291 CrossRef CAS.
- G. M. Sheldrick, SAINT, Version 6.02, SADABS, Version 2.03, Bruker AXS Inc, Madison, Wisconsin, 2002 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Crystal Structure Refinement Program, University of Gottingen, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. Manikandan, R. Muthukumaran, K. R. Justin Thomas, B. Varghese, G. V. R. Chandramouli and P. T. Manoharan, Inorg. Chem., 2001, 40, 2378 CrossRef CAS PubMed.
- S. Koner, S. Saha, K.-I. Okamoto and J.-P. Tuchagues, Inorg. Chem., 2003, 42, 4668 CrossRef CAS PubMed.
- S. Koner, S. Saha, T. Mallah and K. I. Okamoto, Inorg. Chem., 2004, 43, 840 CrossRef CAS PubMed.
- A. Escuer, M. F. Bardía, S. S. Massoud, F. A. Mautner, E. Peñalba, X. Solans and R. Vicente, New J. Chem., 2004, 28, 681 RSC.
- J. D. Woodward, R. V. Backov, K. A. Abboud, D. Dai, H.-J. Koo, M.-H. Whangbo, M. W. Meisel and D. R. Talham, Inorg. Chem., 2005, 44, 638 CrossRef CAS PubMed.
- M. Zbiri, S. Saha, C. Adhikary, S. C. Daul and S. Koner, Inorg. Chim. Acta, 2006, 359, 1193 CrossRef CAS.
- M. Dolai, T. Mistri, A. Panja and M. Ali, Inorg. Chim. Acta, 2013, 399, 95 CrossRef CAS.
- C.-T. Yang, M. Vetrichelvan, X. Yang, B. Moubaraki, K. S. Murray and J. J. Vittal, Dalton Trans., 2004, 113 RSC.
- M. S. Ray, A. Ghosh, S. Chaudhuri, M. G. B. Drew and J. Ribas, Eur. J. Inorg. Chem., 2004, 3110 CrossRef CAS.
- S. Naiya, S. Biswas, M. G. B. Drew, C. J. Gómez-García and A. Ghosh, Inorg. Chim. Acta, 2011, 377, 26–33 CrossRef CAS.
- A. Banerjee, R. Singh, E. Colacio and K. K. Rajak, Eur. J. Inorg. Chem., 2009, 277 CrossRef CAS.
- S. Sarkar, S. Majumder, S. Sasmal, L. Carrella, E. Rentschler and S. Mohanta, Polyhedron, 2013, 50, 270 CrossRef CAS.
- N. Shaikh, S. Goswami, A. Panja, X.-Y. Wang, S. Gao, R. J. Butchler and P. Banerjee, Inorg. Chem., 2004, 43, 5908 CrossRef CAS PubMed.
- A. Panja, RSC Adv., 2013, 3, 4954 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 and Table S1. CCDC 956080 and 956081. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra08025d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.