Synthesis and characterization of well-defined thermo- and light-responsive diblock copolymers by atom transfer radical polymerization and click chemistry
Xiangdong
Tao
a,
Zhengguo
Gao
a,
Toshifumi
Satoh
b,
Yuan
Cui
a,
Toyoji
Kakuchi
b and
Qian
Duan
*a
aSchool of Materials Science and Engineering, Changchun University of Science and Technology, Changchun, 130022, China. E-mail: duanqian88@hotmail.com; Fax: +86 431 85306769; Tel: +86 431 85583105
bDivision of Biotechnology and Macromolecular Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo, 060-8628, Japan
First published on 9th June 2011
Abstract
We synthesized a series of well-defined, stimuli-responsive diblock copolymers composed of poly(N-isopropylacrylamide) and poly(6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate) by atom transfer radical polymerization and click chemistry. The diblock copolymers were then characterized by nuclear magnetic resonance spectroscopy, Fourier transform infrared spectroscopy, and gel permeation chromatography. When the azobenzene content reached 4.5 mol%, the diblock copolymer became insoluble in water. The aqueous solutions of the polymers exhibited a lower critical solution temperature (LCST) that depended on the amount of incorporated azobenzene. Higher LCSTs were observed after UV irradiation, with a maximum difference of 4.1 °C for the copolymer containing 1.4 mol% azobenzene groups. The photochemical properties of the polymer were also studied by UV-vis spectroscopy.
1. Introduction
Block copolymers have drawn considerable attention because of their capacity for self-assembling. Self-assembled materials have many potential applications, both as dispersions and as solids, because of their ability to form versatile and functional morphology.1 Block copolymer systems currently being developed have found numerous applications in biomedical devices,2–5 biomaterials,3,6,7 thermoplastic elastomers,8 fuel cells,9,10 and electronics.11,12Poly(N-isopropylacrylamide), or PNIPAM, has been the most frequently studied thermosensitive polymer because of its lower critical solution temperature (LCST) of 32 °C, which is close to the temperature of the human body.13,14 Investigations on the phase transition of PNIPAM have revealed that its macromolecules experience dehydration and collapse from a hydrated, extended coil to a hydrophobic globule; raising the temperature through the cloud point ultimately results in intermolecular aggregation.15 Functional PNIPAMs have been synthesized and combined with various hydrophobic polymer blocks including polystyrene16,17 and tetrahydrofuran-protected 2-hydroxyethyl methacrylate,18 as well as alkyl groups,19 alkyl chain transfer agents,20 and chromophores.15
Polymers are no longer responsive to only a single stimulus but show responsive behavior to multiple stimuli. Polymers that are responsive to light and temperature are of special importance. Several reports on thermo-responsive polymers, i.e., polymers containing a light-responsive azobenzene moiety, have been published.21–29 In all these studies, appropriate azobenzene-containing monomers have been copolymerized with either NIPAM or N,N-dimethylacrylamide yielding light- and thermo-responsive copolymers. In such polymers, the polar cis form of azobenzene exhibits increased solubility in water compared with the relatively nonpolar trans form because a balance between polar and nonpolar moieties is important in LCST control.21,30–32 Previous studies have also reported the synthesis of a polymer that contains NIPAM and azobenzene moieties by free-radical copolymerization22,23,27 or polymer analogous reaction.29 However, the diblock copolymers synthesized by these methods generally have higher polydispersity indexes. Click chemistry,33 (Huisgen's 1,3-dipolar cycloaddition34 between azido and alkynyl groups catalyzed by copper salts) introduced by Sharpless, has recently been proved a very powerful synthetic tool in polymer science. Numerous studies have demonstrated the applicability of this approach in the synthesis of block copolymers with linear-linear35–42 macromolecular architecture. Atom transfer radical polymerization (ATRP) and click chemistry are employed in the synthesis of diblock copolymers with narrow polydispersity indexes and controlled macromolecular architecture. First, ATRP enables good control over the molecular weight of the polymer and end group, so that precisely defined reactive polymer structures are obtained. Second, click chemistry has been used extensively because of its quantitative yields, high tolerance of functional groups, and insensitivity of the reaction to solvents. Additionally, this method enables the production of copolymers from monomers that would otherwise be impossible to copolymerize.
In this study, we report the preparation of well-defined diblock copolymers containing NIPAM and azobenzene moieties by ATRP and click chemistry. These copolymers are responsive to light and temperature. In contrast to the copolymers produced in previous reports, those in the current work have lower polydispersity indexes (1.13–1.20). The LCST exhibited by the resultant polymers at different amounts of azobenzene is also investigated.
2. Experimental
2.1 Materials
N-Isopropylacrylamide (Aldrich, 98%) was recrystallized twice from a hexane/benzene mixture (3/2, v/v). Anisole (SCRC, 99%), as the solvent used in solution polymerization, was purified by distillation from sodium with benzophenone. CuCl (Aldrich, 99%) and CuBr (SCRC, 98.5%) catalysts were washed successively with acetic acid and ether, dried, and then stored under a nitrogen atmosphere. Tris(2-(dimethylamino)ethyl)amine (Me6TREN) was synthesized from tris(2-amino) ethyl amine (TREN, Aldrich, 99%), according to reported methods.43 Propargyl 2-chloropropionate (PCP) was synthesized from propargyl alcohol (SCRC, 99%), according to previous studies.44 All other chemicals were obtained commercially and used as received, unless otherwise stated.2.2 Characterization instruments
The 1H nuclear magnetic resonance (NMR) spectra of monomers and polymers in CDCl3 were obtained on a Varian 300 MHz FT-NMR spectrometer. Molecular weights (Mn) and polydispersity (Mw/Mn) were measured on a gel permeation chromatograph (GPC) using a Waters 510 pump and a Model 410 differential refractometer at 25 °C. THF was used as a mobile phase at a flow rate of 1.0 mL min−1. The UV-vis absorption spectra of the polymers were determined on a Shimadzu-1240 spectrophotometer. Infrared spectra were obtained by measuring samples in KBr disks on a Shimadzu IR-8400S spectrometer.2.3 Synthesis of 6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate (AzoMA)
The monomer, 6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate (AzoMA), was synthesized following a previous method (Scheme 1).451H NMR(CDCl3, ppm): 7.87 (m, 4H), 6.99 (m, 4H), 6.10 (s, 1H), 5.55 (s, 1H), 4.17 (t, 2H), 4.04 (t, 2H), 3.88 (s, 3H), 1.95 (s, 3H), 1.87–1.80 (m, 2H), 1.77–1.70(m, 2H), 1.58–1.45 (m, 4H). | ||
| Scheme 1 Synthetic route of the monomer AzoMA. | ||
2.4 Synthesis of 2-azidoethyl 2-chloropropanoate (1)
A solution of 2-azidoethanol (871 mg, 10 mmol) and triethylamine (3.48 mL, 25 mmol) in Et2O (10 mL) was cooled to 0 °C in an ice bath, after which 2-chloropropionyl chloride (1.90 g, 15 mmol) was added dropwise. After being stirred for 1 h at 0 °C, the solution was stirred further for 20 h at room temperature. The resultant white suspension was filtered, and the filtrate was washed three times with 50 mL of saturated aqueous NaHCO3 solution. The organic layer was dried over Na2SO4, filtered, and then evaporated to obtain a yellow oily crude residue. The obtained oil was purified by flash chromatography (silica column, petroleum ether/Et2O 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain a colorless oil (1.26 g, 71% yield). 1H NMR (CDCl3, ppm): 4.48 (q, 1H), 4.37 (t, 2H), 3.56 (t, 2H), 1.75 (d, 3H).
:1) to obtain a colorless oil (1.26 g, 71% yield). 1H NMR (CDCl3, ppm): 4.48 (q, 1H), 4.37 (t, 2H), 3.56 (t, 2H), 1.75 (d, 3H).
2.5 Synthesis of poly(6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate (PAzoMA) (2)
PAzoMA was synthesized by using a previously reported method (Scheme 2).46 A mixture of CuBr (22 mg, 0.15 mmol) and N,N,N′,N′,N′-pentamethyldiethylenetriamine (PMDETA) (33 μL, 0.15 mmol) in anisole (0.8 mL) was placed on one side of an H-shaped glass ampoule and stirred at room temperature. AzoMA (300 mg, 0.75 mmol) and PCP (29 mg, 0.15 mmol) in anisole (1.5 mL) were placed on the other side of the ampoule. Nitrogen was bubbled through both mixtures for 5 min to remove any oxygen. Three freeze-pump-thaw cycles were then performed to degas the solution. Both mixtures were mixed and placed in an oil bath thermostated at 80 °C for 20 h. The polymerization was terminated by exposure to air. The reaction mixture was diluted with THF and passed through a neutral Al2O3 column using THF as eluent to remove the copper complex. After precipitation by adding the polymer solution of THF into methanol three times, yellow product 2 was collected by filtration and dried in a vacuum oven overnight (153 mg, 47% yield). Mn = 2100, Mw/Mn = 1.07.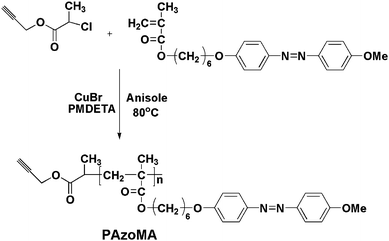 | ||
| Scheme 2 Synthesis of PAzoMA by ATRP. | ||
2.6 Synthesis of N3-PNIPAM (3)
Four precursors of end-functionalized PNIPAM containing the azide group (Table 1) were synthesized (Scheme 3). The procedure for N3-PNIPAM production is described as follows. A mixture of CuCl (10.0 mg, 0.1 mmol) and Me6TREN (24 μL, 0.1 mmol) in 1:1 (v/v) DMF/H2O (1.0 mL) was placed on one side of an H-shaped glass ampoule and stirred at room temperature. NIPAM (1.13 g, 10 mmol) and 1 (17.8 mg, 0.1 mmol) in 1:1 (v/v) DMF/H2O (1.5 mL) were placed on the other side of the ampoule. Nitrogen was bubbled through both mixtures for 5 min to remove any oxygen. Three freeze-pump-thaw cycles were then performed to degas the solution. Both mixtures were mixed and left to stand at 25 °C for 30 min. The polymerization was terminated by exposure to air. The reaction mixture was diluted with THF and passed through a neutral Al2O3 column using THF as eluent to remove the copper complex. After precipitation by adding the polymer solution of THF into hexane three times, white polymer 3a was collected by filtration and dried in a vacuum oven overnight (0.98 g, 87% yield). Mn = 10600, Mw/Mn = 1.13.
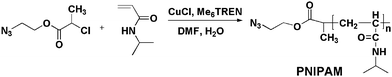 | ||
| Scheme 3 Synthesis of PNIPAM by ATRP. | ||
2.7 Synthesis of PAzoMA-b-PNIPAM by click chemistry
A Schlenk flask was charged with alkyne-functionalized 2 (51 mg, 0.024 mmol), 3a (127 mg, 0.012 mol), and CuBr (6.9 mg, 0.048 mmol). The flask was closed with a rubber septum, evacuated, then backfilled with nitrogen several times. PMDETA (10 μL, 0.048 mmol) and deoxygenated DMF (3 mL) were added using a nitrogen-purged syringe; the flask was degassed further by three freeze-pump-thaw cycles. The reaction mixture was stirred under a nitrogen atmosphere at 40 °C for 24 h. The mixture was diluted with THF and then passed through a neutral Al2O3 column using THF as eluent to remove copper salts. The solvent was evaporated and the resultant mixture was dialyzed in DMF using a cellophane tube (Spectra/Por 6 Membrane; MWCO:10000). Finally, the solvent was evaporated and a yellow product was obtained (130 mg, 85% yield). Mn = 13000, Mw/Mn = 1.22. The synthesis of PAzoMA-b-PNIPAM copolymer is shown in Scheme 4.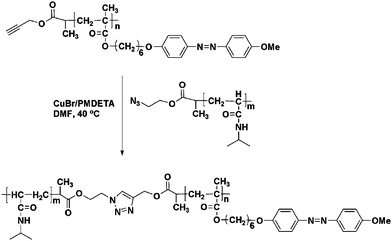 | ||
| Scheme 4 Synthesis of PAzoMA-b-PNIPAM by click chemistry. | ||
3. Results and discussion
3.1 Synthesis and characterization
To prepare a well-defined diblock polymer, synthesizing the two precursors at a narrow molecular weight distribution is necessary. These distributions are normally realized by the ATRP method. 2-Azidoethyl 2-chloropropanoate was used as the initiator for ATRP of NIPAM in DMF/H2O solution at room temperature, yielding the azide group-ended PNIPAM. The Mn values of the four products were determined by GPC using polystyrene as the standard. The results are presented in Table 1. The data in the table show that all the samples had narrow molecular weight distributions in the range 1.09–1.13. Alkyne-functionalized PAzoMA was successfully prepared in anisole using the PCP/CuBr/PMDETA system at 80 °C for 20 h. Because of the high hydrophobicity of the azobenzene moiety, only diblock copolymers with a long hydrophilic NIPAM block and a short azo block (<10 monomer units) were soluble in pure water.The click reaction between alkyne-functionalized PAzoMA and PNIPAM was carried out in DMF at 40 °C using CuBr/PMDETA complex as the catalytic system (Scheme 4). Excess alkyne-functionalized PAzoMA was used to facilitate the completion of the click reactions. Excess PAzoMA was removed by dialysis, and a yellow solid product was obtained.
The obtained block copolymers were characterized by 1H NMR, GPC, and Fourier transform infrared (FT-IR) techniques, all of which can confirm the occurrence of the click reaction between the alkyne and azide groups. Fig. 1 shows the 1H NMR spectra of PNIPAM with the azido end-group, PAzoMA with alkyne, and the resultant product. Fig. 1(A) shows that the methylene protons next to the alkyne group exhibited a signal at ∼4.6 ppm. This characteristic signal was not observed in the click product but a new signal at ∼5.3 ppm appeared in Fig. 1(C). This signal is attributed to methylene protons next to 1,2,3-triazole. Fig. 1(B) shows the 1H NMR spectrum of PNIPAM. The signal of the methylene protons adjacent to the azido group at ∼3.5 ppm completely disappeared, as shown in Fig. 1(C).
 | ||
| Fig. 1 1H NMR spectra in CDCl3 of the polymers PAzoMA, PNIPAM, and PAzoMA-b-PNIPAM. | ||
The IR spectra reveal that the characteristic absorption band of the azide group at ∼2100 cm−1 of PNIPAM was missing in the product (Fig. 2). A representative GPC trace showed a lower elution time for the block copolymer compared with the two precursors, indicating the increase in the molecular weight of the former (Fig. 3). However, the molecular weight distribution did not change appreciably. All the results indicate that the product was the targeted PNIPAM-b-PAzoMA with a triazole ring.
 | ||
| Fig. 2 FT-IR spectra obtained for (A) PAzoMA, (B) PNIPAM-N3, and (C) PAzoMA-b-PNIPAM. | ||
 | ||
| Fig. 3 GPC traces of precursors (2 and 3a) and the resultant product P1. | ||
3.2 Thermo- and light-responsive characterization
Aqueous PNIPAM solutions show a LCST at 32 °C. This phenomenon has been attributed to the association of polymer molecules; this association creates large aggregates through intermolecular hydrogen bonds and non-polar bonds. When PNIPAM contains different amounts of azobenzene moieties, LCST changes. The isomerization of azobenzene always entails a change in the dipole moment of the molecular structure. Azobenzene groups have a dipole moment of 0 Debye in trans-configuration, whereas azobenzene molecules in cis-configuration have a dipole moment of 3 Debye;47 this indicates that the LCST shifted during UV irradiation in the copolymers containing azobenzene groups. Because of the increased dipole moment of cis-azobenzene, the respective copolymers showed higher LCSTs for cis-azobenzene-containing polymers than copolymers containing trans-azobenzene.The LCSTs of the copolymer solutions were determined by turbidimetry; the optical transmittance of a light beam (600 nm) through the sample cell in the photospectrometer was monitored as a function of temperature. The concentration of all the copolymer solutions was 0.5 mg mL−1 and the heating rate was 1 °C min−1. The cloud points were measured before and after irradiation with UV light at 365 nm. LCST is defined as the temperature at which 90% transmission was observed. The results are shown in Table 2. At 4.5 mol% azobenzene moiety, the polymer was insoluble in water. The LCSTs of the aqueous solutions of the copolymers were dependent on the content of azobenzene moieties. In all cases, higher LCST values were observed after irradiation with UV light. From the copolymer P2–P4, the highest LCST shift was observed (4.1 °C) for 1.4 mol% azobenzene (P3) compared with the LCST shift of 0.6 °C (2.2 mol% azobenzene) for P2 and 1.9 °C (0.97 mol% azobenzene) for P4. At an azobenzene content higher than 1.4 mol%, the temperature difference decreased. This phenomenon was previously described and reported by Irie and Kungwatchakun.27 By incorporating azobenzene chromophores into the polymer, the hydrophobic interaction was induced, and the solubility of the polymer decreased. In one case, the azobenzene concentration range displaying a photoinduced LCST change was quite narrow; in another, the LCSTs of all the copolymers decreased with increasing azobenzene.
| Sample | M n,GPC | PDIGPC | Amount of azobenzene (mol%) | LCST before irradiation (°C) | LCST after irradiation (°C) | ΔLCST (°C) | |
|---|---|---|---|---|---|---|---|
| P1 | PAzoMA5-b- PNIPAM105 | 13000 | 1.22 | 4.5 | |||
| P2 | PAzoMA5-b- PNIPAM226 | 25800 | 1.15 | 2.2 | 19.4 | 20.0 | 0.6 |
| P3 | PAzoMA5-b- PNIPAM357 | 37800 | 1.17 | 1.4 | 23.6 | 27.7 | 4.1 |
| P4 | PAzoMA5-b- PNIPAM510 | 53500 | 1.18 | 0.97 | 27.5 | 29.4 | 1.9 |
The UV-vis spectra of PAzoMA5-b-PNIPAM226 (P2) were recorded for aqueous solutions and films before and after UV exposure (365 nm, 8W) (Fig. 4). The polymer exhibited one strong absorption band and one weak band; the bands are related to the π–π* and n–π* transition bands of the trans- and cis-azobenzene, respectively. The spectra changed after different irradiation times. The change in the UV-vis absorption spectra of the P2 in aqueous solution is shown in Fig. 4(A). The intensity of the π–π* transition band at about 338 nm decreased, whereas that of the n–π* transition band at about 450 nm gradually increased with prolonged irradiation time. The photostationary state was realized after irradiation for 12 min. In the dark, the trans-azobenzene content of the polymer was recovered completely.
 | ||
| Fig. 4 (A) Spectral changes in P2 in aqueous solution during UV irradiation (365 nm, 8W). Irradiation time: (a) 0 min, (b) 2 min, (c) 6 min, (d) 8 min, and (e) 12 min. (B) Spectral changes in P2 on quartz glass under UV irradiation. Irradiation time: (a) 0 min, (b) 2 min, (c) 6 min, and (d) 9 min. | ||
The P2 film was cast from the THF solution (1 wt%) onto a quartz surface, and dried at 50 °C in a vacuum oven to remove the solvent. When the P2 film was irradiated, the spectra on the film were similar to those in the aqueous solution; the intensity of the trans-isomer absorbance decreased, whereas that of the cis-isomer gradually increased. However, the absorbance at about 338 nm corresponding to the π–π* transition was blue-shifted on the solid film compared with the spectra in aqueous solution. In polar solvent, the π–π* absorption band of the aromatic compounds exhibited a red shift.
4. Conclusions
Well-defined diblock copolymers of NIPAM and azobenzene methacrylate monomer were successfully synthesized via ATRP and click chemistry. The copolymers exhibited thermo- and light-responsive properties as well as narrow polydispersity indexes. The copolymers P2–P4 exhibited low LCSTs in aqueous solution; the LCSTs strongly depended on the amount of incorporated azobenzene. Moreover, the reversible isomerization of the azobenzene groups in the copolymers influenced the LCST. After irradiation, the LCST values increased. In the temperature region between the LCST of the non-irradiated and the irradiated solutions, a change in the light-controlled reversible solubility was observed. The polymer P2 exhibited a typical trans-cis photoisomerization reaction under UV irradiation. In the film form, the trans-cis isomerization of the azobenzene chromophore resembled the light-responsiveness in solution. Various multifunctional polymers may be designed through the proposed synthetic strategy.Notes and references
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858–875 CrossRef CAS.
- B. Lindman and P. Alexandridis, in Amphiphilic block copolymers: self assembly and applications, Elsevier Science B.V., Dordrecht, The Netherlands, 1st edn, 2000, ch. 15, pp. 347–376 Search PubMed.
- J. Kopecek, Nature, 2002, 417, 388–391 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS.
- D. McIlroy, B. Barteau, J. Cany, P. Richard, C. Gourden, S. Conchon and B. Pitard, Mol. Ther., 2009, 17, 1473–1481 Search PubMed.
- L. Pinchuk, G. J. Wilson, J. J. Barry, R. T. Schoephoerster, J. M. Parel and J. P. Kennedy, Biomaterials, 2008, 29, 448–460 CrossRef CAS.
- E. S. Place, N. D. Evans and M. M. Stevens, Nat. Mater., 2009, 8, 457–470 CrossRef CAS.
- N. Hadjichristidis, S. Pispas and G. A. Floudas, in Block Copolymers: Synthetic Strategies, Physical Properities, and Applications, John Wiley &Sons, New York, 1st edn, 2002, ch. 21, pp. 383–406 Search PubMed.
- O. Diat and G. Gebel, Nat. Mater., 2008, 7, 13–14 CrossRef CAS.
- M. Lazzari, G. Liu and S. Lecommandoux, in Block Copolymers in Nanoscience, Wiley-VCH, Weinheim, 1st edn, 2006, ch. 15, pp. 337–366 Search PubMed.
- C. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Science, 2008, 332, 429–432 Search PubMed.
- A. Ryan, Nature, 2008, 456, 334–336 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. Yamazaki, J. M. Song, F. M. Winnik and J. L. Brash, Macromolecules, 1998, 31, 109–115 CrossRef CAS.
- X. Zhou, X. Ye and G. Zhang, J. Phys. Chem. B, 2007, 111, 5111–5115 CrossRef CAS.
- M. Nuopponen, J. Ojala and H. Tenhu, Polymer, 2004, 45, 3643–3650 CrossRef CAS.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS.
- H. G. Schild and D. A. Tirrell, Langmuir, 1991, 7, 1319–1324 CrossRef CAS.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341–348 CrossRef CAS.
- R. Kroeger, H. Menzel and M. L. Hallensleben, Macromol. Chem. Phys., 1994, 195, 2291–2298 CrossRef.
- H. Akiyama and N. Tamaoki, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5200–5214 CrossRef CAS.
- D. Kungwatchakun and M. Irie, Makromol. Chem. Rapid Commun., 1988, 9, 243–246 CrossRef CAS.
- H. Akiyama and N. Tamaoki, Macromolecules, 2007, 40, 5129–5132 CrossRef CAS.
- L. Chunhua, Z. Fang, Z. Zhaohui and P. Yuxing, J. Appl. Polym. Sci., 2008, 107, 2118–2125 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592–16596 CrossRef CAS.
- M. Irie and D. Kungwatchakun, Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci., 1992, 68, 127–132 Search PubMed.
- C. Luo, F. Zuo, X. Ding, Z. Zheng, X. Cheng and Y. Peng, J. Appl. Polym. Sci., 2008, 107, 2118–2125 CrossRef CAS.
- F. D. Jochum and P. Theato, Polymer, 2009, 50, 3079–3085 CrossRef CAS.
- K. Sugiyama and K. Sono, J. Appl. Polym. Sci., 2001, 81, 3056–3063 CrossRef CAS.
- K. Ishihara, N. Hamada, S. Kato and I. Shirohata, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 1551–1555 CrossRef.
- M. S. Kang and V. K. Gupta, J. Phys. Chem. B, 2002, 106, 4127–4132 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- J. A. Opsteen and J. M. C. van Hest, Chem. Commun., 2005, 57–59 RSC.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- M. Urien, H. Erothu, E. Cloutet, R. C. Hiorns, L. Vignau and H. Cramail, Macromolecules, 2008, 41, 7033–7040 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5699–5707 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6458–6465 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191–198 CrossRef CAS.
- J. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327–2329 CrossRef CAS.
- D. Quemener, T. P. Davis, C. Barner–Kowollik and M. H. Stenzel, Chem. Commun., 2006, 5051–5053 RSC.
- M. Ciampolini and N. Nardi, Inorg. Chem., 1966, 5, 41–44 CrossRef CAS.
- Jian Xu, Jing Ye and Shiyong Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- H. Ringsdorf and H. W. Schmidt, Makromol. Chem., 1984, 185, 1327–1334 CAS.
- Dehui Han, Xia Tong, Yi Zhao, Tigran Galstia and Yue Zhao, Macromolecules, 2010, 43, 3664–3671 CrossRef CAS.
- G. S. Hartley and R. J. W. LeFevre, J. Chem. Soc., 1939, 531–535 RSC.
| This journal is © The Royal Society of Chemistry 2011 |