
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-driven electron transfer in a lipid bilayer with mixed valence molecular wires†
Novitasari
Sinambela‡
 a,
Moritz
Nau‡
b,
Gernot
Haug
b,
Michael
Linseis
b,
Philip
Koblischek
a,
Rainer F.
Winter
*b and
Andrea
Pannwitz
*acde
a,
Moritz
Nau‡
b,
Gernot
Haug
b,
Michael
Linseis
b,
Philip
Koblischek
a,
Rainer F.
Winter
*b and
Andrea
Pannwitz
*acde
aInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
bDepartment of Chemistry, University of Konstanz, Universitätsstraße 10, 78464 Konstanz, Germany. E-mail: rainer.winter@uni-konstanz.de
cDepartment of Inorganic and Analytical Chemistry, Friedrich Schiller Universität Jena, Humboldtstr. 8, 07743 Jena, Germany. E-mail: andrea.pannwitz@uni-jena.de
dCenter for Energy and Environmental Chemistry Jena (CEEC), Friedrich-Schiller-Universität Jena, Philosophenweg 7a, 07743 Jena, Germany
eHelmholtz Institute for Polymers in Energy Applications Jena (HIPOLE Jena), Lessingstraße 12–14, 07743 Jena, Germany
First published on 18th February 2025
Abstract
Triarylamines (TAAs) are one of the most important classes of redox-active organic compounds, which are readily available from modular synthesis, thereby offering the possibility to easily adjust their intrinsic redox potentials. We present herein two bis(triarylamines) (BTAAs) with π-extended 2,7-diethynylfluorene or 2,2′-(1,3-butadiyne-1,4-diyl)-bis(7-ethynylfluorene) bridges and two benzoic acid headgroups per TAA and their (formally) mixed-valent radical cations. Owing to their amphiphilic character and favorable redox properties, these BTAAs are designed to serve as charge conduits through membranes. The lipid bilayer/BTAA systems are water-soluble, which allowed us to explore their photoactivity in aqueous solution and utilize their mixed valent form for membrane-mediated photoinduced electron transfer. Our findings will be relevant for constructing artificial nanoreactors for solar light energy conversion and light-driven redox chemistry in water.
Introduction
Light-driven long-range electron transfer is relevant for solar energy conversion in the natural photosynthesis of plants and photosynthetic bacteria, where photons drive the movement of electrons across biological membranes. Nature uses sophisticated assemblies of transmembrane protein complexes such as photosystem I and II (PSI and PSII) to convert NADP+ into NADPH on one side of the membrane and water into O2 on the other side of the membrane, as shown in Fig. 1. Previous artificial systems attempting to mimic light-driven transmembrane electron transfer across lipid bilayers employed embedded photosensitizers,1,2 along with electron carriers, to transfer electrons across membranes via diffusion of the photoreduced electron carrier.3–5 Additionally, there are some studies of light-driven transmembrane electron transfer using either sophisticated and synthetically challenging supramolecular barrels1 or rigid oligoaromatic molecular wires with varying levels of synthetic complexity.2,6 In the present study, we explore the concept of mixed valency, i.e. electron delocalization over two covalently connected redox centers,7 in a π-extended molecular wire for transmembrane electron transfer in lipid bilayers. These are multielectron processes that resemble the complexity of multielectron transfer in the photosynthesis system, which later on can be a future model for performing water-splitting and CO2 reduction. | ||
| Fig. 1 Comparison of natural and artificial photosynthesis pathways. NADPH = nicotinamide adenine dinucleotide diphosphate, NADP+ = nicotinamide adenine dinucleotide phosphate, PSII = photosystem II, PSI = photosystem I, ATP = adenosine triphosphate, ADP = adenosine diphosphate, and e− = electron. | ||
Triarylamines (TAAs) are one of the most important classes of redox-active organic compounds. One of their key advantages is their ready accessibility via modular synthesis, thereby offering the opportunity to attach one aryl substituent at a time and to generate derivatives with two or three different aryl substituents. This allows fine-tuning of their intrinsic redox potentials8–10 and the deliberate introduction of a broad range of chemical functionalities, e.g. for the construction of hybrids with two or more chemically different redox-active constituents11–21 or for rendering them water soluble.22–25 Once protected by substituents to prevent oxidatively induced coupling to benzidines26–29 or cyclization to carbazoles,30 one-electron oxidized triarylaminium ions are rather stable, even towards water and air.9,22,31 Bridged bis(triarylamines) (BTAAs), Ar1Ar2N-arylene-NAr1Ar2, typically undergo two or even more redox processes. Their associated mixed-valent radical cations generated by one-electron oxidation, i.e. the removal of an electron from only one of the available redox sites, are outstanding testbeds for probing the effects of the length, the chemical constitution and the conformation of the intervening π-conjugated (poly)arylene bridge and the chemical environment (solvent and counterions)11,32–35 on the extent of electronic coupling between the terminally appended redox sites.36–42 Their proven ability to facilitate efficient through-bond charge delocalization over long distances40,43–46 has also made them attractive for applications as single molecule wires in metal–molecule–metal nanojunctions. Recent studies on anchor group-modified bis(diarylamine)-capped oligophenylenes even revealed negative values of the attenuation factor β up to the terphenylene limit, implying that the electron transfer rate increases with increasing molecular length.47 The advantageous properties of BTAAs, i.e. their redox activity, modularity, and abundance, thus prompted us to utilize them as charge conduits through membranes. We here convey the results of our work on using BTAAs with polar head groups for this purpose.
Results and discussion
Design and synthesis
The molecular wires designed here have hydrophobic cores based on fluorene and alkyne units bridging the BTAA redox centers, which are functionalized with carboxylic acids or esters. The carboxylic acids serve as hydrophilic head groups to anchor the molecular wire on both sides of the lipid bilayer so that they span across the lipid bilayer (Fig. 2a). | ||
Fig. 2 (a) The structure of a bilayer membrane, built from an amphiphilic phospholipid. (b) Chemical structures and schematic syntheses of target bis(triarylamine) tetracarboxylic acids/methyl esters 1H/Me and 2H/Me. (i) PdCl2(PPh3)2, NEt3, CuI, THF, 70 °C, and overnight; 1Me → 1H: THF/H2O (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), LiOH, r.t., and overnight; (ii) PdCl2(PPh3)2, NEt3, CuI, THF, 70 °C, and two days; 2Me → 2H: THF/H2O (2:1), LiOH, r.t., and overnight. :1), LiOH, r.t., and overnight; (ii) PdCl2(PPh3)2, NEt3, CuI, THF, 70 °C, and two days; 2Me → 2H: THF/H2O (2:1), LiOH, r.t., and overnight. | ||
Fig. 2b summarizes the synthesis and chemical structures of the target BTAAs. Our synthetic routes afford the methyl esters of the envisioned tetracarboxylic acids and involve cascades of different C–N and C–C cross-coupling reactions, including a Glaser–Hey–Eglinton coupling in the case of 2Me.48 Ester cleavage subsequently provided free tetracarboxylic acids. Detailed synthetic protocols and methods are provided in the ESI (Schemes S1 and S2†), along with 1H, 13C{1H}, mass spectra, and the absorption spectra and molar extinction coefficients ε of the target BTAAs (ESI, Fig. S16 and S17†). The molecular design with two polar benzoic acid headgroups at each terminus and nonpolar aliphatic hexyl tails at the core segments was conceived to mimic classical lipid bilayer properties. The carboxylic functionalities render BTAAs 1H and 2H slightly soluble in aqueous media.
Electrochemistry and spectroscopy
The intended utilization of BTAAs 1H and 2H as transmembrane redox mediators puts their redox properties into focus. However, voltammetric characterization of free acids 1H and 2H was hindered by the limited solubility of the protonated 1H and 2H in aqueous media, while an increase in pH led to poorly reproducible voltammograms with ill-defined redox waves at Pt, Au and glassy carbon electrodes. We therefore resorted to esters 1Me and 2Me in the CH2Cl2/NBu4+ [BArF24]− (0.1 M) electrolyte. Fig. 3 displays cyclic voltammograms of 1Me (panel A) and 2Me (panel B). The very weakly coordinating [BArF24]− anion ([BArF24]− = [B{C6H3(CF3)2-3,5}4]−) is known for maximizing electrostatic repulsion, thereby maximizing the redox splitting ΔE1/2 between the individual redox couples and hence increasing the thermodynamic stability of the intermediate, mixed-valent radical cation with respect to disproportionation.49–51 The expected two oxidations of the TAA constituents nevertheless merge into a single composite wave with (average) half-wave potentials of E1/2(1Me) = 675 mV and E1/2(2Me) = 670 mV on the ferrocene/ferrocenium scale. Only in the case of the shorter 1Me can a small splitting into two separate waves be discerned. Computational simulation of the experimental voltammograms yielded a redox splitting of 80 mV in the case of 1Me and an even smaller value of only 45 mV for 2Me with roughly twice the spacer length. Simulated cyclic voltammograms are shown as blue circles in panels A (1Me) and B (2Me) of Fig. 3, superimposed with the experimental data. Details of the used software and the fitting procedures are given in the ESI,† and the parameters obtained by the simulations are provided in Table S4 of the ESI.† The half-wave potential splitting of 2 is barely larger than the statistical contribution of 2ln(2RT/F) = 36 mV to the free energy change for the stepwise oxidations of two non-interacting redox sites.52 Similar small half-wave potential separations were also reported for other BTAAs with large spatial extensions of the connecting linker (Table 1).39,44,53–55
 | ||
| Fig. 3 (A) Overlaid cyclic voltammogram of 1Me in CH2Cl2/NBu4+ [BArF24]− (0.1 M, T = 293 K (±3 K), v = 600 mV s−1, black solid line) and corresponding simulation (blue circles). (B) Overlaid cyclic voltammogram of 2Me in CH2Cl2/NBu4+ [BArF24]− (0.1 M, T = 293 K (±3 K), v = 600 mV s−1, black solid line) and corresponding simulation (blue circles). | ||
| E 1/2 avg 0/2+ (exp.) | E 1/2 0/+ (simulated) | E 1/2 +/2+ (simulated) | ΔE1/2 | |
|---|---|---|---|---|
| 1Me | 675 mV | 635 mV | 715 mV | 80 mV |
| 2Me | 670 mV | 640 mV | 685 mV | 45 mV |
The close proximity of the two redox waves makes it difficult to access and spectroscopically characterize and discriminate between the one- and two-electron-oxidized radical cations and dications as individual species, which is important in order to elucidate the effective oxidation state when using the present BTAAs as transmembrane redox mediators.
The half-wave potentials of 1Me and 2Me are appreciably more positive than that of the triarylamine-derived mono- and dicarboxylic acids (4-MeO-C6H4)2N(C6H4-COOH-4) (E1/2 = 170 mV) and (4-MeOC6H4)2N(C6H3(COOH)2-3,5) (E1/2 = 90 mV) reported by Lahti et al.,22 which we ascribe to the absence of methoxy donor substituents (literature values were recalculated to the ferrocene/ferrocenium scale using the reported half-wave potential of the Cp2Fe0/+ redox couple of 553 mV in CHCl3 on the Ag/AgCl scale,56i.e. the conditions used by these authors). They are nevertheless ca. 150 mV lower than that of the 4-ethynylphenyl precursor (E1/2 = 820 mV in CH2Cl2vs. Cp2Fe0/+), indicating the extent of electron donation from the dihexyl-substituted fluorenyl core unit.
We next turn to the spectroscopic characterization of the oxidized forms of 1Me and 2Me as models of the oxidized BTAA tetracarboxylic acids that one expects to be the active species in transmembrane hole transfer. To generate these species, we applied UV-Vis/NIR spectroelectrochemistry (SEC), i.e. spectroscopic monitoring during in situ electrochemical oxidation upon incrementally increasing the applied potential to a value 250 mV positive of the convoluted overall two-electron wave. The latter experiments were conducted inside an optically transparent thin-layer electrolysis cell52 with 1,2-dichloroethane (DCE) as the solvent, using again the NBu4+ [BArF24]− (0.1 M) supporting electrolyte. The higher boiling point of the DCE solvent as compared to CH2Cl2 counteracts solvent evaporation in the vicinity of the working electrode during the experiments. Fig. 4 displays the evolution of the UV-Vis/NIR spectra alongside NIR spectra recorded with an IR/NIR spectrometer (for details, see the ESI, Section S3†) as insets; the latter has a superior sensitivity in the low-energy part of the NIR regime. Panels A and B of Fig. 4 show the results on 1Me, while those for 2Me are given in panels C and D.
 | ||
| Fig. 4 UV-Vis/NIR spectroelectrochemistry of 1Me (left, panels A and B) and 2Me (right, panels C and D) with the first oxidation shown as blue to orange curves (top, panels A and C) and the second oxidation shown as orange to green curves (bottom, panels B and D), respectively, in CH2Cl2 with 0.1 M NBu4+ [BArF24]− as the supporting electrolyte. IR/NIR spectra are provided in insets in panels A to D. An artifact due to the experimental setup is marked with an asterisk. | ||
In both cases, the overall two-electron oxidation proceeds in separate steps via an intermediate species (orange lines), which has a characteristic broad NIR band at 1405 nm (1Me˙+) or 1400 nm respectively (2Me˙+, see panels A and C of Fig. 4 and the corresponding insets). As the oxidation progresses further, the NIR band intensifies and shifts to higher energies (i.e. lower wavelengths) of 1250 nm for both 1Me2+ and 2Me2+. These observations are indicative of the stepwise formation of first the mixed-valent radical cations and then the dications of BTAAs with large, π-extended linkers.14,39,53,57 Other spectroscopic changes are the gradual and continuous growth of a Vis/NIR band, which is located at 715 nm for 1Me and at 705 nm for 2Me. This band corresponds to a π–π* transition of a triarylaminium radical cation and is characteristic of the TAA˙+ motif. This assignment agrees with the observed rough doubling in intensity upon the second oxidation, which generates an additional triarylaminium chromophore. Also, both oxidized compounds feature one additional band of similar intensity at 550 nm each.
In interpreting the results, one should notice that the close proximity of the individual E1/2 values of 1Me and 2Me implies that the intermediate mixed-valent radical cations have only limited stability with respect to disproportionation to the neutral starting compounds and the dioxidized forms, both with isovalent TAA redox sites. This is expressed by the only modest values for the comproportionation equilibrium constant Kc for the comproportionation (cf.eqn (1)) of ca. 23 (1Me) or only 6 (2Me).
 | (1) |
In eqn (1), R is the universal gas constant, F Faraday's constant, and ΔE1/2 the half-wave potential separation between the two consecutive redox processes. The individual Kc values indicate that a maximum of 70% or 55% of the mixed-valent radical cations are formed as such after the passage of one equivalent of charge, with the remaining 30% or 45% present as equimolar mixtures of the neutral and dioxidized forms. The overlapping NIR bands of the radical cations and the dications in the NIR unfortunately preclude us from deconstructing the spectra and calculating the spectra of the pristine, intermediate radical cations, but it is nevertheless clear that both species have distinct absorptions in the NIR region. The high intensity of the NIR absorption of the radical cations is at first surprising when considering the small half-wave potential splittings and the large N⋯N distances of 23.15 Å in 1Me˙+ or 36.65 Å in 2Me˙+, as obtained from quantum chemical calculations (see below). The intensity gain and the blue-shift upon the second oxidation let us assign the prominent NIR band to charge-transfer (CT) excitation from the respective fluorene linker to the TAA˙+ acceptor(s) rather than to a pure TAA → TAA˙+ intervalence charge-transfer (IVCT) excitation, as the latter would be expected to be specific for the mixed-valent radical cations and to vanish as the oxidation proceeds further to the dications.
To shed more light on the electronic structures of one- and two-electron oxidized 1Me+/2+ and 2Me+/2+, we resorted to density functional quantum-chemical calculations at the B3LYP+35% exact Hartree–Fock exchange def2SVP level of theory. The latter has proven to provide adequate descriptions of the electronic structures of mixed-valent BTAAs by compensating for the inherent tendency of the DFT method to delocalize charges in extended π-conjugated systems.37,58–60 Our calculations employed slightly truncated models with methyl instead of hexyl substituents, which are henceforth denoted as 1Me′ and 2Me′. Solvent effects were considered within the framework of the polarizable continuum model, applying the standard parameters for CH2Cl2. Further details as well as coordinates of the geometry-optimized structures are provided in Section S11 of the ESI.†
Our calculations indeed provided intense transitions in the NIR for both the one- and the two-electron oxidized forms 1Me′˙+/2 and 2Me′˙+/2+, at energies close to the experimental values. Charge density losses on the individual TAA+ sites and the fluorenyl segments of the linkers were evaluated by natural bond orbital (NBO) analysis (see Fig. 5).61 According to these results, both radical cations possess localized electronic structures where the unipositive charge is confined to one specific TAA˙+ entity. In contrast to other BTAAs with electron-rich para-anisyl (An) substituents NAn2, the most electron-rich phenyl group of 1Me and 2Me is the one bonded to the fluorenyl linker. This causes the occupied frontier MOs to spread onto the linker rather than being strictly confined to the periphery.
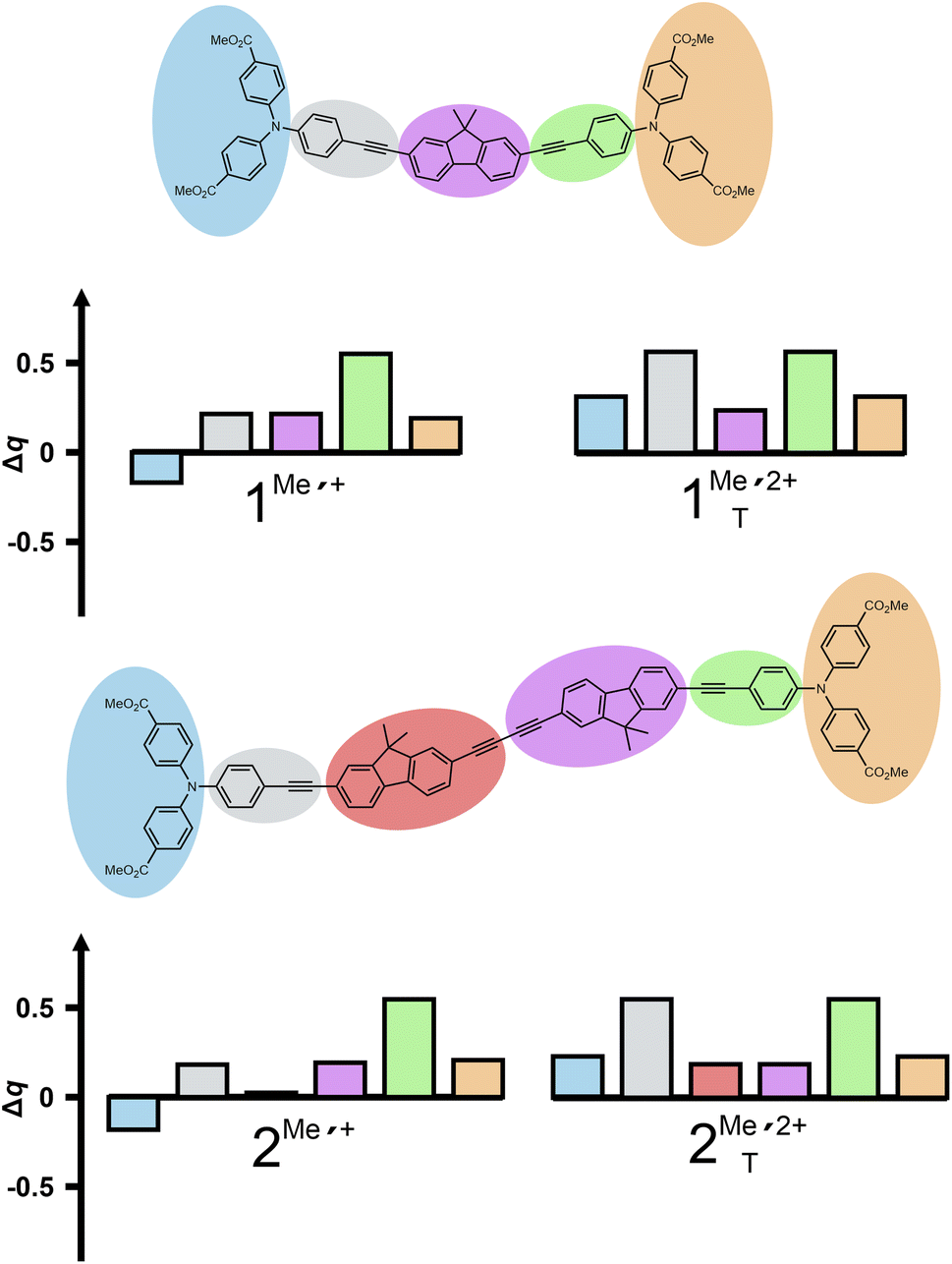 | ||
| Fig. 5 Calculated electron density losses upon oxidation of model complexes 1Me′ and 2Me′ to their corresponding one- (left) and two-electron oxidized forms (triplet state), based on natural bond orbital (NBO) analysis. Δq refers to the charge difference of the respective molecular region of the radical monocations (left panels) and the respective dications in their triplet ground state (right panels) relative to the corresponding neutral state. The individual fragments are color-coded according to the graphical representation in the top panels. | ||
The calculated NIR transitions in radical cation 2Me′˙+ are depicted in panel A of Fig. 6, along with graphical representations of the involved molecular orbitals and the corresponding electron density difference maps (EDDMs; blue color = electron density loss and red color = electron density gain). Computational results for its shorter homolog 1Me′˙+ can be found in Fig. S18 of the ESI.† For both compounds, our calculations predict two separate transitions at low energies as highly intense bands (1Me˙+: λcalc = 1036 nm/1675 nm, f = 0.28/1.07; 2Me˙+: λcalc = 1212 nm/1550 nm, f = 0.46/0.84). They both target β-LUSO, which is localized on the oxidized TAA unit, and emanate from the first three occupied orbitals of the β-manifold, i.e. β-HOSO to β-HOSO-2. The latter donor MOs are either localized on the other TAA unit or spread over the entire π-conjugated backbone, including the ester-functionalized diarylamine entities. Both NIR excitations therefore assume bridge-to-TAA+ CT, augmented with IVCT contributions. The absorption bands of one-electron oxidized 1Me˙+ and 2Me˙+ generally agree well with the results of investigations on other BTAAs with similarly π-extended linkers.39,44,54,55
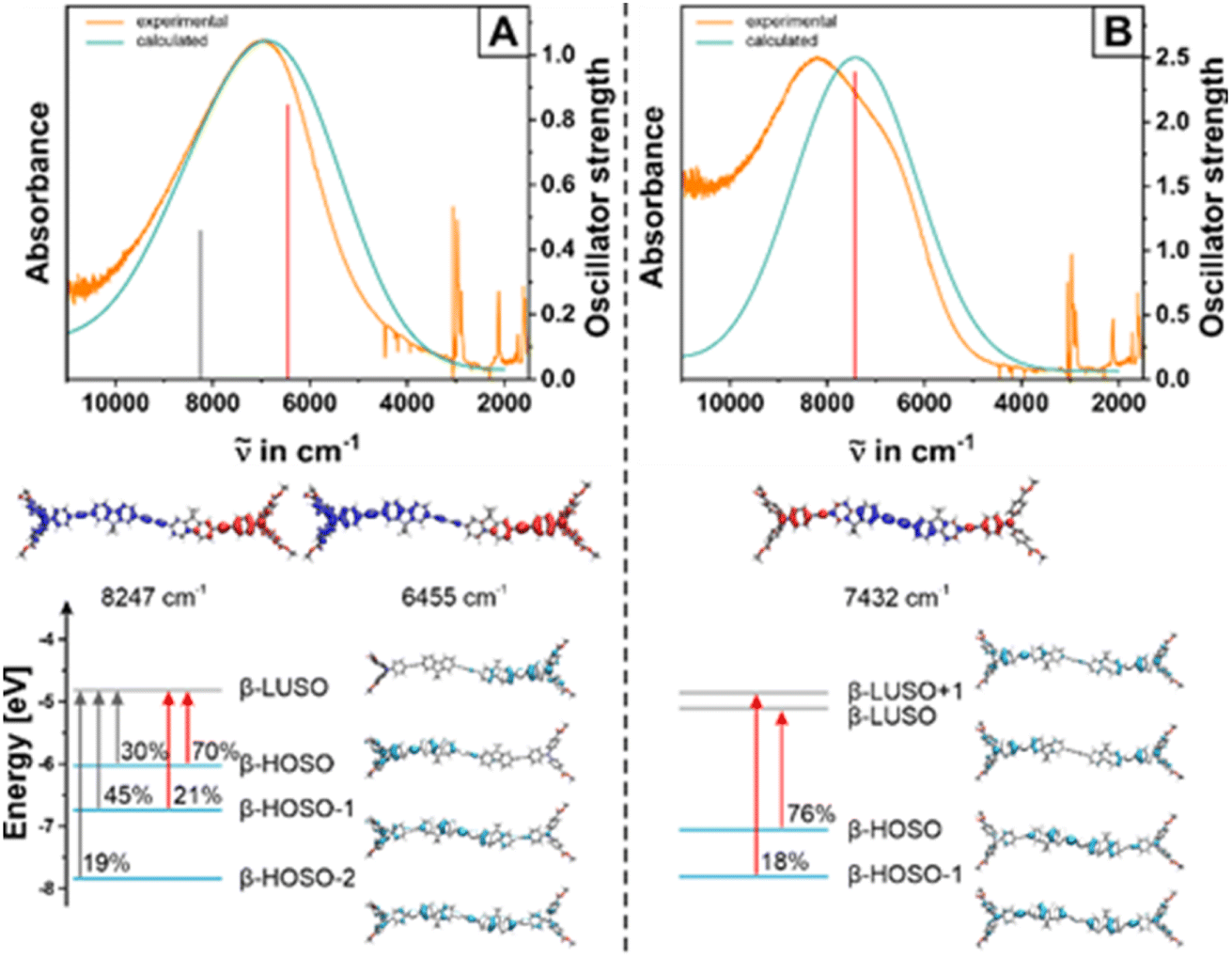 | ||
| Fig. 6 Top: Comparison between experimental NIR spectra of 2Me+ (orange line in panel A) and 2Me2+ (green line in panel B) in 1,2-CH4Cl2 (0.1 M NBu4+ [BArF24]− as the supporting electrolyte) and TD-DFT calculated electronic spectra of model compound 2Me′+ (panel A, turquoise line) and 2Me′2+ (panel B, violet). Computed transitions are provided as red and grey vertical bars. Bottom: Contour diagrams of the acceptor and donor MOs involved in the NIR transitions together with their contributions to the respective transition and corresponding electron density difference maps (EDDMs). Blue color indicates electron density loss and red color an increase in electron density. Isosurface values for the MO plots are set as ±0.02. | ||
In contrast to the plethora of BTAAs with shorter linkers, but in line with important bridge-to-TAA˙+ contributions,39,54,55 the NIR transitions of 1Me˙+ and 2Me˙+ intensify during further oxidation to the dications while experiencing a shift to higher energies and shorter wavelengths respectively, from 1405 nm to 1250 nm for 1Me˙+/2+ (λcalc = 1183 nm, f = 1.82), and from 1400 nm to 1220 nm for 2Me˙+/2+ (λcalc = 1345 nm, f = 2.39); see panels A and B of Fig. 6. The dications were computed to prefer the triplet state by 54.0 kJ mol−1 (1Me′2+) or 42.5 kJ mol−1 (2Me′2+) over the closed and open-shell singlet states, which were calculated to be energetically degenerate. Panels A, B in Fig. 6 and S18 of the ESI† demonstrate the excellent agreement between computed and experimental spectra. A complete collection of all computed electronic absorption bands of 1Me′n+ and 2Me′n+ (n = 0, 1, 2, the latter in their triplet ground and excited singlet states) along with the corresponding TD-DFT calculated transitions on their methyl-substituted models, contour plots of the contributing orbitals, and computed oscillator strengths are provided in the ESI (see Table S5†). On oxidation of the second TAA entity, the character of the NIR electronic excitation assumes pure bridge-to-TAA˙+ CT character.
In summary, the highly intense NIR absorptions of one- and two-electron oxidized BTAAs 1Me˙+/2+ and 2Me˙+/2+ can be traced to bridge-to-TAA˙+ CT augmented by TAA-to-TAA˙+ IVCT excitations in the radical cations. The energetically low-lying bridge-to-TAA˙+ excitations and the bridge participation in the frontier MOs make 1Me˙+ and 2Me˙+ (and by inference the one-electron oxidized free carboxylic acids 1H˙+ and 2H˙+) interesting candidates for transmembrane electron transfer when incorporated into a lipid bilayer.
Membrane integration
Incorporation of BTAAs in the lipid membrane bilayers was performed with 1H or 2H. At neutral pH, their molecular structures mimic the amphiphilicity of the phospholipid bilayer best by having charged carboxylates at each end of the linear and rigid molecular wire. The charged ends interact with the polar part of the membrane at the water interface, while the entire linker, including the inner phenyl rings, stays inside the hydrophobic part of the membrane. This secures the integration of 1H or 2H into the membrane (Fig. 7) of vesicles. In this study, we used sub-micrometer-sized liposome vesicles for all spectroscopy and electron transfer studies. We also prepared micrometer-sized giant vesicles for microscopic visualization. All vesicles were prepared with lipid bilayers based on 2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) because DPPC bilayers are known to be impermeable to larger water-soluble molecules at room temperature.62,63 Along with the main phospholipid DPPC, we added 1 mol% of 14:0 PEG2000 PE to the lipid bilayer membranes to increase the stability of the liposome, as this lipid creates steric bulk and thereby prevents aggregation of the vesicles (see Fig. 7 for the structures of DPPC and 14:0 PEG2000 PE).5,64,65 | ||
| Fig. 7 Integration of 1H or 2H into a DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) liposome and a molecular dynamic simulation snapshot of (a) 1H and (b) 2H in DPPC after 100 ns simulation time. The lipid bilayer membrane is omitted for clarity. | ||
Molecular dynamics (MD) simulations of both molecular wires after 100 ns simulation time are shown in Fig. 7a and b. The MD simulations were done using YASARA and the AMBER force field;66,67 the details can be found in ESI S4, Fig. S19 and S20.† These MD simulations suggest that both molecular wires 1H and 2H assemble with the lipid bilayer, but 1H assembles at the membrane–water interface, while 2H can align in a transmembrane fashion. We explain these results as follows: in 2H, the N⋯N distance is 33.66 Å in the phospholipid bilayer (similar to the N⋯N distance of 36.65 Å in 2Me˙+ determined by DFT, see above). The hydrophilic carboxylate groups are even further apart from each other and can therefore easily span a lipid bilayer membrane, being exposed to the membrane's water interfaces, as can be seen in Fig. 7b. Lipid bilayer membranes typically have a thickness of 30 to 50 Å.68,69 For the shorter BTAA 1H, the N⋯N distance is 23.48 Å (similar to the N⋯N distance of 23.15 Å in 1Me˙+ determined by DFT, see above). The distance of the hydrophilic carboxylic groups in 1H is 28.51 Å, which is too short to span across the membrane and which can lead to the disruption of the membrane by 1H.
In order to study how the respective molecular wire affects the membrane, we conducted a calcein leakage test using liposomes. In this test, we quantified the amount of calcein, a fluorescent dye, leaked out of the liposomes into the surrounding solution. For detailed information, please refer to ESI S5, Fig. S21.†70,71 We tested both the modified and unmodified DPPC liposomes for three days with and without irradiation using a 470 nm LED. The measurements with irradiation revealed that the modified DPPC liposome with 1H released 93% of calcein, while the modified DPPC liposome with 2H only released 4.5% of calcein. These observations align with our MD simulations, which predicted that the modified liposome with 1H would disrupt the membrane more effectively than 2H; as 2H is longer in size, it has a higher possibility of aligning in the transmembrane position in the membrane and would not disrupt the membrane.
UV-Vis absorption and emission spectroscopy were used to characterize the photophysical properties of 1H and 2H before and after membrane integration (Fig. 8a and b and Table 2). When measured in a mixture of DCM and MeOH (1:1) (Fig. 8a and b, solid black line), 1H and 2H showed absorption bands at 366 nm and 385 nm, respectively. This indicates that the respective LUMO of 2H is lower in energy than that of 1H due to its extended π-system. The absorption spectra shift to the blue for both structures in the membrane environment as can be seen in Fig. 8a and b and in Table 2. While 1H experienced only a 4 nm blue shift, the absorbance of 2H experienced a pronounced blue-shift by ∼20 nm. The very minor blue-shift for 1H might be explained by Tyndall scattering of the liposome suspension, similar to other chromophores in liposomes with minor solvent-dependent absorbance.75 The very pronounced blue-shift of 2H is likely due to the lower polarity and rigidity around the compound in a lipid bilayer environment.72–74 This observation is in line with the MD simulation, which showed that the entire extended π-system of 2H is located in the lipophilic part of the membrane, which leads to destabilizing the LUMO of 2H, resulting in a more pronounced blue shift in the UV-Vis absorption.
 | ||
| Fig. 8 (a) UV-Vis and emission spectra of 1H and (b) UV-Vis and emission spectra of 2H in a mixture of dichloromethane (DCM):methanol (MeOH) (1:1) and in DPPC liposomes with a composition of DPPC:(14:0 PEG2000 PE):1H or 2H = 100:1:1, in phosphate buffer at pH = 7.0; (c) confocal microscopy image of 2H in a giant vesicle with a composition of 100:1:1 of DPPC:14:0 PEG2000PE:2H in phosphate buffer at pH = 7.0 after size exclusion chromatography, upon fixation in agarose hydrogel. | ||
The emission spectra of 1H and 2H have their maximum at 422 nm and 430 nm respectively in DCM:CH2Cl2 (1:1), as indicated by the solid green line in Fig. 8a and b. Both compounds have a shoulder at a higher wavelength in their emission spectra, suggesting the existence of unresolved vibrational energy levels in both compounds. Upon incorporation into the membrane, the maximum is slightly red-shifted by 13 nm and 15 nm for 1H and 2H respectively. However, this apparent red shift might also be partly induced by the broadened absorption and respective inner filter effects (Fig. 8a and b, dashed green line).
Furthermore, to show that 2H is successfully integrated into the lipid membrane, we prepared a giant multilamellar vesicle of 2H with the same methodology as that used for preparing the liposomes and performed confocal microscopy. We excited the giant multilamellar vesicle with a laser at λ = 405 nm and detected the fluorescence of the vesicle in the region of 420–650 nm. A double half-moon-shaped emission profile was observed in the microscopic image (Fig. 8c), typical for molecules with a preferred alignment in the lipid membrane.65,72,75 It shall also be noted that, apart from the half-moon effect, the molecules are overall evenly distributed within the membrane and no larger macroscopic phase separation can be observed.76
Electron transfer at the membrane
To study the photoactive properties of 1H and 2H toward electron transfer across the lipid bilayer, we started our studies on photoinduced transmembrane electron transfer by preparing a DPPC liposome solution containing 1 mol% of 1H or 2H in the membrane. The water-soluble electron donor NADH was encapsulated, and the water-soluble electron acceptor (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) (XTT) was added to the bulk solution of the liposome, yielding a composition of NADH/1H/XTT or NADH/2H/XTT (Fig. 9a). A similar system was also prepared with encapsulated XTT and NADH at the bulk solution of liposomes, yielding a composition of XTT/2H/NADH. NADH was chosen due to its water solubility and favorable absorption and emission properties.77 XTT was selected because it is a well-known compound to quantify intracellular reducing agents in biological assays.78 The details of sample preparation can be found in the ESI Section S6 and Fig. S22.† Based on dynamic light scattering, the liposomes measure 130–140 nm in diameter for all types of samples (the empty liposome with only 1H or 2H integrated and with the encapsulated NADH or XTT; see ESI Section S7 and Fig. S23†). | ||
| Fig. 9 (a) Schematic illustration of the targeted photoinduced electron transfer at the lipid bilayer with encapsulation of NADH, integration of the chromophore, and presence of XTT in the bulk solution, (b) subtracted temporal evolution of UV-Vis spectra from t = 0 min with sample composition NADH/2H/XTT, (c) number of Fz molecules formed under irradiation with 460 nm and 370 nm light sources over time for NADH/1H/XTT, NADH/2H/XTT, and XTT/2H/NADH, (d) Stern–Volmer plots of liposome samples with a composition of 100:1:1 of DPPC:14:0 PEG2000 PE:2H with different concentrations of encapsulated NADH, and (e) Stern–Volmer plots of liposome samples with a composition of 100:1:1 of DPPC:14:0 PEG2000 PE:2H with different concentrations of XTT in bulk solution. All samples were prepared with 10 mM phosphate buffer at pH 7.0 under ambient conditions. The grey bar corresponds to the used concentration in photoinduced electron transfer experiments. | ||
Upon light irradiation, we observed a gradual increase of the absorption band at λmax = 470 nm, corresponding to the formation of formazan (Fz), the reduced form of XTT. At the same time, the NAD+ absorption band at 260 nm increased, the latter corresponding to the oxidized form of NADH and indicating that photoinduced electron transfer had occurred (Fig. 9b). The extent of electron transfer was quantified via the intensity of the Fz absorption band (Fig. 9c).
We chose 370 nm and 470 nm LED light sources to irradiate the assembled liposome systems NADH/1H/XTT and NADH/2H/XTT. The applied LED light sources have a full-width at half maximum (FWHM) of ±24 nm at λmax = 373 nm for the 370 nm LED and FWHM ±22 nm at λmax = 464 nm for the 470 nm LED light source. This leads to the excitation of 1H and 2H at either the absorption maximum or in the tail of the absorption (see ESI S8, Fig. S25†). Photoexcitation with the 470 nm LED excited the molecular wire only and excluded the simultaneous excitation of NADH, which has an absorption maximum at 340 nm.77,79 In the case of the 370 nm LED, both the molecular wire and NADH are simultaneously excited.
Based on the extinction coefficient of Fz, it was calculated in the case of NADH/2H/XTT that 1.00 nmol of Fz formed after three hours with the 470 nm LED irradiation. This corresponds to 4.4 electron transfers per molecule of 2H. In the case of 2H with a 370 nm LED, 0.71 nmol of Fz was formed, which is equal to 1.4 nmol of electrons and 3.1 electron transfers per 2H molecule. This lower performance by the 370 nm light source may be due to the simultaneous absorption of light by both the molecular wire and NADH, as the NADH also absorbs some of the light at 370 nm (see ESI S8, Fig. S25†). In the case of NADH/1H/XTT and after three hours of irradiation with a 470 nm or 370 nm LED, the Fz yield is 0.77 nmol and 0.78 nmol, respectively. The number of Fz is similarly low with both light sources for 1H, probably due to the lower spectral overlaps of the absorbance of 1H with the 470 nm LED compared to 2H (see ESI S8, Fig. S25†). In a previous study, we demonstrated that the light-driven conversion of the water-soluble substrate can be significantly accelerated within the inner aqueous liposome compartment compared to the aqueous bulk.77 We therefore tested the reverse location of the XTT and NADH and generated XTT/2H/NADH liposomes. In this system, we observed that by compartmentalization of XTT instead of NADH, we could almost double the production of Fz within three hours, yielding 1.80 nmol, which corresponds to 3.6 nmol electron transfer and 8 electrons transferred per molecule of 2H. This observation indicates that electron transfer from XTT is a limitation that can be minimized through encapsulation of XTT within the inner aqueous compartment (see ESI S9† for absorption growth of Fz over time, Fig. S26–S28†).
To better understand the photoinduced electron transfer dynamics in NADH/2H/XTT liposomes, we performed a Stern–Volmer luminescence quenching study of 2H liposomes in the presence of various concentrations of NADH in the inner compartment and XTT in the bulk, respectively. In the Stern–Volmer experiment, the excited state reaction dynamics of 2H can be resolved by using steady-state and time-resolved emission spectroscopy, by plotting the intensities and the lifetimes as  and
and  vs. quencher concentrations [Q],80 where I0 and I are the emission intensities in the absence and presence of the quencher, and τ0 and τ are the excited state lifetimes in the absence and presence of the quencher, respectively.
vs. quencher concentrations [Q],80 where I0 and I are the emission intensities in the absence and presence of the quencher, and τ0 and τ are the excited state lifetimes in the absence and presence of the quencher, respectively.
 | (2) |
The slope from the linear regression of  vs. [Q] is the Stern–Volmer constant Ksv. By applying eqn (2), the quenching constant kq can be derived. After the linear fitting of both plots, NADH and XTT showed characteristic static quenching behavior with the lifetime remaining independent of the quencher concentration. Quenching by encapsulated NADH takes place with kq = (8.2 ± 0.7)1010 M−1 s−1. This value is one order of magnitude faster than that in a related study with encapsulated NADH and a positively charged transmembrane molecular wire6 and might indicate that a stronger ground state encounter complex is formed between the here investigated 2H and NADH. Similarly, the quenching by XTT from the bulk takes place with kq = (1.71 ± 0.1)1013 M−1 s−1 (Table 3). This value is three orders of magnitudes higher than typical collision-based quenching constants, which are on the order of 1011 and it is two orders of magnitude higher than in a similar case between XXT and a positively charged chromophore within the DPPC lipid bilayer.6 Therefore, the kq values obtained for both quenchers indicate the quenching of 2H due to a strong association with the quencher already in the ground state.76,81 We hypothesize that such strong complex formation is partly due to additional hydrogen bond formation by the carboxylate functionality of 2H with the quencher. The concentrations of NADH and XTT that were applied during the full electron transfer reaction with NADH/2H/XTT are shown in the highlighted region in the Stern–Volmer quenching plot in Fig. 9d and e. The luminescence and kinetic traces can be found in ESI Section S10 and Fig. S30–S33.†
vs. [Q] is the Stern–Volmer constant Ksv. By applying eqn (2), the quenching constant kq can be derived. After the linear fitting of both plots, NADH and XTT showed characteristic static quenching behavior with the lifetime remaining independent of the quencher concentration. Quenching by encapsulated NADH takes place with kq = (8.2 ± 0.7)1010 M−1 s−1. This value is one order of magnitude faster than that in a related study with encapsulated NADH and a positively charged transmembrane molecular wire6 and might indicate that a stronger ground state encounter complex is formed between the here investigated 2H and NADH. Similarly, the quenching by XTT from the bulk takes place with kq = (1.71 ± 0.1)1013 M−1 s−1 (Table 3). This value is three orders of magnitudes higher than typical collision-based quenching constants, which are on the order of 1011 and it is two orders of magnitude higher than in a similar case between XXT and a positively charged chromophore within the DPPC lipid bilayer.6 Therefore, the kq values obtained for both quenchers indicate the quenching of 2H due to a strong association with the quencher already in the ground state.76,81 We hypothesize that such strong complex formation is partly due to additional hydrogen bond formation by the carboxylate functionality of 2H with the quencher. The concentrations of NADH and XTT that were applied during the full electron transfer reaction with NADH/2H/XTT are shown in the highlighted region in the Stern–Volmer quenching plot in Fig. 9d and e. The luminescence and kinetic traces can be found in ESI Section S10 and Fig. S30–S33.†
| Quencher | K sv from Io/I (M−1) | k q (M−1 s−1) | τ 0 (ns) | Mechanism |
|---|---|---|---|---|
| NADH/2H | 70.8 ± 6.2 | (8.2 ± 0.7)1010 | 0.86 | Static |
| 2H/XTT | (14.7 ± 0.7)103 | (1.7 ± 0.1)1013 | 0.86 | Static |
Conclusion
In summary, we demonstrate that the molecular BTAAs 1H and 2H can form mixed-valence species upon one-electron oxidation and that the amphiphilic BTAAs 1H and 2H can be integrated into the lipid bilayer of liposomes for light-driven electron transfer from NADH to the acceptor XTT or vice versa. While the shorter version 1H disrupts the lipid bilayer significantly, the long molecule 2H integrates well into the bilayer structure, spanning the membrane. Both compounds are redox-active and generate stable one- and two-electron oxidized cations, thereby enabling transmembrane electron transfer. The mechanism of membrane-mediated electron transfer is likely promoted via a mixed valence radical cation state of the one-electron oxidized molecular wire, delocalizing an unpaired electron across the entire molecule. The findings of this study provide a design principle for artificial nanoreactors and are highly relevant for the construction of light-driven redox chemistry systems in water with applications in solar light energy conversion.Data availability
The data supporting the findings of the article can be found in the ESI.† The raw data of photocatalysis will be made available via Chemotion.Author contributions
N. S. and M. N. contributed equally. N. S. performed the membrane integration, luminescence quenching experiments, transmembrane electron transfer experiments and molecular dynamics simulations. M. N. performed the spectroscopic characterization, cyclic voltammetry (including the simulations), spectroelectrochemistry measurements with data analysis, and all (TD-)DFT calculations on the model compounds, supervised by M. L., R. F. W. and A. P., R. F. W. and A. P. devised the project. P. K. conducted one chemical synthesis. G. H. conducted most chemical syntheses. All authors contributed to scientific discussions and the preparation of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. N. thanks the Fonds der Chemischen Industrie for financial support. The authors acknowledge support from the state of Baden-Württemberg and the Deutsche Forschungsgemeinschaft through bwHPC, access to the supercomputing facilities of JUSTUS2 (grant number 40/575-1 FUGG), and the core facilities of the University of Konstanz for NMR spectroscopic and mass spectrometric measurements as well as the platform for mass spectrometry at Friedrich-Schiller-University Jena. A. P. gratefully acknowledges financial support from the Vector Stiftung (project number P2019-0110) as well as the Carl-Zeiss-Foundation and the Deutsche Forschungsgemeinschaft (DFG), project TRR234 “CataLight” project number 364549901, project B8. NMR at Ulm University was recorded on a spectrometer funded by DFG project number 445471845. The confocal images were recorded at the Core Facility for Confocal and Multiphoton Microscopy at Ulm University which is supported by the DFG through project number 91BGG INST 381/39-1.References
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstanberg, N. Banetji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Photoproduction of Proton Gradients with π-Stacked Fluorophore Scaffolds in Lipid Bilayers, Science, 2006, 313(5783), 84–86, DOI:10.1126/science.1126524.
- A. Perez-Velasco, V. Gorteau and S. Matile, Rigid Oligoperylenediimide Rods: Anion–π Slides with Photosynthetic Activity, Angew. Chem., Int. Ed., 2008, 47(5), 921–923, DOI:10.1002/anie.200703749.
- T. A. Moore, D. Gust, P. Mathis, J.-C. Mialocq, C. Chachaty, R. V. Bensasson, E. J. Land, D. Doizi, P. A. Liddell, W. R. Lehman, G. A. Nemeth and A. L. Moore, Photodriven Charge Separation in a Carotenoporphyrin–Quinone Triad, Nature, 1984, 307(5952), 630–632, DOI:10.1038/307630a0.
- P. Seta, E. Bienvenue, A. L. Moore, P. Mathis, R. V. Bensasson, P. Liddell, P. J. Pessiki, A. Joy, T. A. Moore and D. Gust, Photodriven Transmembrane Charge Separation and Electron Transfer by a Carotenoporphyrin–Quinone Triad, Nature, 1985, 316(6029), 653–655, DOI:10.1038/316653a0.
- B. Limburg, J. Wermink, S. S. van Nielen, R. Kortlever, M. T. M. Koper, E. Bouwman and S. Bonnet, Kinetics of Photocatalytic Water Oxidation at Liposomes: Membrane Anchoring Stabilizes the Photosensitizer, ACS Catal., 2016, 6(9), 5968–5977, DOI:10.1021/acscatal.6b00151.
- N. Sinambela, R. Jacobi, D. Sorsche, L. Gonzalez and A. Pannwitz, Photoinduced Electron Transfer across Phospholipid Bilayers in Anaerobic and Aerobic Atmospheres, ChemRxiv, December 3, 2024, preprint, DOI:10.26434/chemrxiv-2024-x7wgx.
- U. Fuerholz, H. B. Buergi, F. E. Wagner, A. Stebler, J. H. Ammeter, E. Krausz, R. J. H. Clark, M. J. Stead and A. Ludi, The Creutz-Taube Complex Revisited, J. Am. Chem. Soc., 1984, 106(1), 121–123, DOI:10.1021/ja00313a025.
- R. I. Walter, Substituent Effects on the Properties of Stable Aromatic Free Radicals. The Criterion for Non-Hammett Behavior, J. Am. Chem. Soc., 1966, 88(9), 1923–1930, DOI:10.1021/ja00961a014.
- S. Dapperheld, E. Steckhan, K.-H. G. Brinkhaus and T. Esch, Organic Electron Transfer Systems, II Substituted Triarylamine Cation-Radical Redox Systems – Synthesis, Electrochemical and Spectroscopic Properties, Hammet Behavior, and Suitability as Redox Catalysts, Chem. Ber., 1991, 124(11), 2557–2567, DOI:10.1002/cber.19911241127.
- S. Amthor, B. Noller and C. Lambert, UV/Vis/NIR Spectral Properties of Triarylamines and Their Corresponding Radical Cations, Chem. Phys., 2005, 316(1), 141–152, DOI:10.1016/j.chemphys.2005.05.009.
- A. Heckmann, C. Lambert, M. Goebel and R. Wortmann, Synthesis and Photophysics of a Neutral Organic Mixed-Valence Compound, Angew. Chem., Int. Ed., 2004, 43(43), 5851–5856, DOI:10.1002/anie.200460495.
- G. Grelaud, M. P. Cifuentes, T. Schwich, G. Argouarch, S. Petrie, R. Stranger, F. Paul and M. G. Humphrey, Multistate Redox-Active Metalated Triarylamines, Eur. J. Inorg. Chem., 2012, 2012(1), 65–75, DOI:10.1002/ejic.201100747.
- W. Polit, T. Exner, E. Wuttke and R. Winter, Vinylruthenium-Triarylamine Conjugates as Electroswitchable Polyelectrochromic NIR Dyes, BioInorg. React. Mech., 2012, 8, 85–105, DOI:10.1515/irm-2012-0005.
- C. Hassenrück and R. F. Winter, Manipulation and Assessment of Charge and Spin Delocalization in Mixed-Valent Triarylamine–Vinylruthenium Conjugates, Inorg. Chem., 2017, 56(21), 13517–13529, DOI:10.1021/acs.inorgchem.7b02186.
- S. Breimaier and R. F. Winter, Electrochemical and Spectroscopic Studies on Triarylamine-Polychlorotriphenylmethyl Dyads with Particularly Strong Triarylamine Donors, Eur. J. Org Chem., 2021, 2021(33), 4690–4700, DOI:10.1002/ejoc.202100476.
- C.-J. Yao, R.-H. Zheng, Q. Shi, Y.-W. Zhong and J. Yao, 1,4-Benzene-Bridged Covalent Hybrid of Triarylamine and Cyclometalated Ruthenium: A New Type of Organic–Inorganic Mixed-Valent System, Chem. Commun., 2012, 48(45), 5680–5682, 10.1039/C2CC32471G.
- J.-H. Tang, J.-Y. Shao, Y.-Q. He, S.-H. Wu, J. Yao and Y.-W. Zhong, Transition from a Metal-Localized Mixed-Valence Compound to a Fully Delocalized and Bridge-Biased Electrophore in a Ruthenium–Amine–Ruthenium Tricenter System, Chem.–Eur. J., 2016, 22(30), 10341–10345, DOI:10.1002/chem.201601806.
- J. Zhang, S.-Z. Guo, Y.-B. Dong, L. Rao, J. Yin, G.-A. Yu, F. Hartl and S. H. Liu, Multistep Oxidation of Diethynyl Oligophenylamine-Bridged Diruthenium and Diiron Complexes, Inorg. Chem., 2017, 56(2), 1001–1015, DOI:10.1021/acs.inorgchem.6b02809.
- K. E. Linton, M. A. Fox, L.-O. Pålsson and M. R. Bryce, Oligo(p-Phenyleneethynylene) (OPE) Molecular Wires: Synthesis and Length Dependence of Photoinduced Charge Transfer in OPEs with Triarylamine and Diaryloxadiazole End Groups, Chem.–Eur. J., 2015, 21(10), 3997–4007, DOI:10.1002/chem.201406080.
- Y.-P. Ou, J. Zhang, A. Wang, A. Yuan, C. Yin and S. H. Liu, Rutheniumethynyl-Triarylamine Organic–Inorganic Mixed-Valence Systems: Regulating Ru-N Electronic Coupling by Different Aryl Bridge Cores, Chem.–Asian J., 2020, 15(20), 3338–3349, DOI:10.1002/asia.202000879.
- Z. Lin, Z. Li, W. Xiao, L. Kong, J. Xu, Y. Xia, X. Zhu, F. Zhang and Y.-P. Ou, Terpyridine Ruthenium–Triarylamine Asymmetrical Mixed-Valence Systems: Syntheses, (Spectro) Electrochemistry and Theoretical Calculations, J. Organomet. Chem., 2023, 993, 122708, DOI:10.1016/j.jorganchem.2023.122708.
- H. Murata and P. M. Lahti, Synthesis and Oxidation of Triarylamine Derivatives Bearing Hydrogen-Bonding Groups, J. Org. Chem., 2007, 72(13), 4974–4977, DOI:10.1021/jo070318a.
- G. Sforazzini, A. Kahnt, M. Wykes, J. K. Sprafke, S. Brovelli, D. Montarnal, F. Meinardi, F. Cacialli, D. Beljonne, B. Albinsson and H. L. Anderson, Synthesis and Photophysics of Coaxial Threaded Molecular Wires: Polyrotaxanes with Triarylamine Jackets, J. Phys. Chem. C, 2014, 118(8), 4553–4566, DOI:10.1021/jp500624q.
- P. Thamaraiselvi, N. Duraipandy, M. S. Kiran and S. Easwaramoorthi, Triarylamine Rhodanine Derivatives as Red Emissive Sensor for Discriminative Detection of Ag+ and Hg2+ Ions in Buffer-Free Aqueous Solutions, ACS Sustain. Chem. Eng., 2019, 7(11), 9865–9874, DOI:10.1021/acssuschemeng.9b00417.
- C. A. Swamy P, A. V. Raveendran, N. Sivakrishna and R. P. Nandi, Triarylborane-Triphenylamine Based Luminophore for the Mitochondria Targeted Live Cell Imaging and Colorimetric Detection of Aqueous Fluoride, Dalton Trans., 2022, 51(40), 15339–15353, 10.1039/D2DT01887J.
- R. R. Nelson and R. N. Adams, Anodic Oxidation Pathways of Substituted Triphenylamines. II. Quantitative Studies of Benzidine Formation, J. Am. Chem. Soc., 1968, 90(15), 3925–3930, DOI:10.1021/ja01017a004.
- R. F. Nelson and R. H. Philp, Electrochemical and Spectroscopic Studies of Cation Radicals. 4. Stopped-Flow Determination of Triarylaminium Radical Coupling Rate Constants, J. Phys. Chem., 1979, 83(6), 713–716, DOI:10.1021/j100469a015.
- K. Sreenath, C. V. Suneesh, V. K. Ratheesh Kumar and K. R. Gopidas, Cu(II)-Mediated Generation of Triarylamine Radical Cations and Their Dimerization. An Easy Route to Tetraarylbenzidines, J. Org. Chem., 2008, 73(8), 3245–3251, DOI:10.1021/jo800349n.
- M. R. Talipov, M. M. Hossain, A. Boddeda, K. Thakur and R. Rathore, A Search for Blues Brothers: X-Ray Crystallographic/Spectroscopic Characterization of the Tetraarylbenzidine Cation Radical as a Product of Aging of Solid Magic Blue, Org. Biomol. Chem., 2016, 14(10), 2961–2968, 10.1039/c6ob00140h.
- R. Reynolds, L. L. Line and R. F. Nelson, Electrochemical Generation of Carbazoles from Aromatic Amines, J. Am. Chem. Soc., 1974, 96(4), 1087–1092, DOI:10.1021/ja00811a024.
- N. G. Connelly and W. E. Geiger, Chemical Redox Agents for Organometallic Chemistry, Chem. Rev., 1996, 96(2), 877–910, DOI:10.1021/cr940053x.
- B. Mladenova, D. R. Kattnig, C. Kaiser, J. Schäfer, C. Lambert and G. Grampp, Investigations of the Degenerate Intramolecular Charge Exchange in Symmetric Organic Mixed Valence Compounds: Solvent Dynamics of Bis(Triarylamine)Paracyclophane Redox Systems, J. Phys. Chem. C, 2015, 119(16), 8547–8553, DOI:10.1021/acs.jpcc.5b01386.
- J. Schäfer, M. Holzapfel, B. Mladenova, D. Kattnig, I. Krummenacher, H. Braunschweig, G. Grampp and C. Lambert, Hole Transfer Processes in Meta- and Para-Conjugated Mixed Valence Compounds: Unforeseen Effects of Bridge Substituents and Solvent Dynamics, J. Am. Chem. Soc., 2017, 139(17), 6200–6209, DOI:10.1021/jacs.7b01650.
- S. Dümmler, W. Roth, I. Fischer, A. Heckmann and C. Lambert, Excited-State Dynamics in a Neutral Organic Mixed-Valence Compound, Chem. Phys. Lett., 2005, 408(4), 264–268, DOI:10.1016/j.cplett.2005.04.045.
- K. Tahara, T. Nakakita, A. A. Starikova, T. Ikeda, M. Abe and J. Kikuchi, Small Anion-Assisted Electrochemical Potential Splitting in a New Series of Bistriarylamine Derivatives: Organic Mixed Valency across a Urea Bridge and Zwitterionization, Beilstein J. Org. Chem., 2019, 15(1), 2277–2286, DOI:10.3762/bjoc.15.220.
- J. Hankache and O. S. Wenger, Organic Mixed Valence, Chem. Rev., 2011, 111(8), 5138–5178, DOI:10.1021/cr100441k.
- M. Kaupp, M. Renz, M. Parthey, M. Stolte, F. Würthner and C. Lambert, Computational and Spectroscopic Studies of Organic Mixed-Valence Compounds: Where Is the Charge?, Phys. Chem. Chem. Phys., 2011, 13(38), 16973–16986, 10.1039/C1CP21772K.
- A. Heckmann and C. Lambert, Organic Mixed-Valence Compounds: A Playground for Electrons and Holes, Angew. Chem., Int. Ed., 2012, 51(2), 326–392, DOI:10.1002/anie.201100944.
- C. Lambert and G. Nöll, The Class II/III Transition in Triarylamine Redox Systems, J. Am. Chem. Soc., 1999, 121(37), 8434–8442, DOI:10.1021/ja991264s.
- K. Lancaster, S. A. Odom, S. C. Jones, S. Thayumanavan, S. R. Marder, J.-L. Brédas, V. Coropceanu and S. Barlow, Intramolecular Electron-Transfer Rates in Mixed-Valence Triarylamines: Measurement by Variable-Temperature ESR Spectroscopy and Comparison with Optical Data, J. Am. Chem. Soc., 2009, 131(5), 1717–1723, DOI:10.1021/ja808465c.
- M. Uebe and A. Ito, Intramolecular Charge Transfer in Kekulé- and Non-Kekulé-Bridged Bis(Triarylamine) Radical Cations: Missing Key Compounds in Organic Mixed-Valence Systems, Chem.–Asian J., 2019, 14(10), 1692–1696, DOI:10.1002/asia.201900036.
- M. Uebe, K. Kaneda, S. Fukuzaki and A. Ito, Bridge-Length-Dependent Intramolecular Charge Transfer in Bis(Dianisylamino)-Terminated Oligo(p-Phenylene)s, Chem.–Eur. J., 2019, 25(68), 15455–15462, DOI:10.1002/chem.201903667.
- K. E. Linton, M. A. Fox, L.-O. Pålsson and M. R. Bryce, Oligo(p-phenyleneethynylene) (OPE) Molecular Wires: Synthesis and Length Dependence of Photoinduced Charge Transfer in OPEs with Triarylamine and Diaryloxadiazole End Groups, Chem.–Eur. J., 2015, 21(10), 3997–4007, DOI:10.1002/chem.201406080.
- A. Heckmann, S. Amthor and C. Lambert, Mulliken–Hush Analysis of a Bis(Triarylamine) Mixed-Valence System with a N⋯N Distance of 28.7 Å, Chem. Commun., 2006,(28), 2959–2961, 10.1039/b604603g.
- C. Lambert and V. Kriegisch, Heterogeneous Electron Transfer Processes in Self-Assembled Monolayers of Amine Terminated Conjugated Molecular Wires, Langmuir, 2006, 22(21), 8807–8812, DOI:10.1021/la061404t.
- L. G. Heinz, O. Yushchenko, M. Neuburger, E. Vauthey and O. S. Wenger, Tetramethoxybenzene Is a Good Building Block for Molecular Wires: Insights from Photoinduced Electron Transfer, J. Phys. Chem. A, 2015, 119(22), 5676–5684, DOI:10.1021/acs.jpca.5b03649.
- L. Li, J. Z. Low, J. Wilhelm, G. Liao, S. Gunasekaran, C. R. Prindle, R. L. Starr, D. Golze, C. Nuckolls, M. L. Steigerwald, F. Evers, L. M. Campos, X. Yin and L. Venkataraman, Highly Conducting Single-Molecule Topological Insulators Based on Mono- and Di-Radical Cations, Nat. Chem., 2022, 14(9), 1061–1067, DOI:10.1038/s41557-022-00978-1.
- X. Shi, M. Liu, L. Li, J. Zhang, H. Li, Z. Huang, W. Zhang, Z. Zhang, N. Zhou and X. Zhu, Efficient Synthesis of Discrete Oligo(Fluorenediacetylene)s toward Chain-Length-Dependent Optical and Structural Properties, Polym. Chem., 2021, 12(17), 2598–2605, 10.1039/D1PY00165E.
- A. Nafady, T. T. Chin and W. E. Geiger, Manipulating the Electrolyte Medium to Favor Either One-Electron or Two-Electron Oxidation Pathways for (Fulvalendiyl)Dirhodium Complexes, Organometallics, 2006, 25(7), 1654–1663, DOI:10.1021/om051101e.
- A. K. Diallo, C. Absalon, J. Ruiz and D. Astruc, Ferrocenyl-Terminated Redox Stars: Synthesis and Electrostatic Effects in Mixed-Valence Stabilization, J. Am. Chem. Soc., 2011, 133(3), 629–641, DOI:10.1021/ja109380u.
- U. Pfaff, A. Hildebrandt, D. Schaarschmidt, T. Hahn, S. Liebing, J. Kortus and H. Lang, Di- and Triferrocenyl (Hetero)Aromatics: Synthesis, Characterization, (Spectro-)Electrochemistry, and Calculations, Organometallics, 2012, 31(19), 6761–6771, DOI:10.1021/om300511v.
- R. F. Winter, Half-Wave Potential Splittings ΔE1/2 as a Measure of Electronic Coupling in Mixed-Valent Systems: Triumphs and Defeats, Organometallics, 2014, 33(18), 4517–4536, DOI:10.1021/om500029x.
- S. Barlow, C. Risko, S.-J. Chung, N. M. Tucker, V. Coropceanu, S. C. Jones, Z. Levi, J.-L. Brédas and S. R. Marder, Intervalence Transitions in the Mixed-Valence Monocations of Bis(Triarylamines) Linked with Vinylene and Phenylene–Vinylene Bridges, J. Am. Chem. Soc., 2005, 127(48), 16900–16911, DOI:10.1021/ja054136e.
- S. Barlow, C. Risko, S. A. Odom, S. Zheng, V. Coropceanu, L. Beverina, J.-L. Brédas and S. R. Marder, Tuning Delocalization in the Radical Cations of 1,4-Bis[4-(Diarylamino)Styryl]Benzenes, 2,5-Bis[4-(Diarylamino)Styryl]Thiophenes, and 2,5-Bis[4-(Diarylamino)Styryl]Pyrroles through Substituent Effects, J. Am. Chem. Soc., 2012, 134(24), 10146–10155, DOI:10.1021/ja3023048.
- C. Lambert, S. Amthor and J. Schelter, From Valence Trapped to Valence Delocalized by Bridge State Modification in Bis(Triarylamine) Radical Cations: Evaluation of Coupling Matrix Elements in a Three-Level System, J. Phys. Chem. A, 2004, 108(31), 6474–6486, DOI:10.1021/jp048449s.
- I. Noviandri, K. N. Brown, D. S. Fleming, P. T. Gulyas, P. A. Lay, A. F. Masters and L. Phillips, The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer, J. Phys. Chem. B, 1999, 103(32), 6713–6722, DOI:10.1021/jp991381+.
- K.-T. Wong, T. Hsi Hung, S. C. Kao, C. Hsien Chou and Y. Oliver Su, Synthesis and Properties of Novel Bis(Triarylamines) Based on a 3,3′-Diphenyl-2,2′-Bithiophene Core, Chem. Commun., 2001, 0(17), 1628–1629, 10.1039/B103194P.
- M. Renz, K. Theilacker, C. Lambert and M. Kaupp, A Reliable Quantum-Chemical Protocol for the Characterization of Organic Mixed-Valence Compounds, J. Am. Chem. Soc., 2009, 131(44), 16292–16302, DOI:10.1021/ja9070859.
- M. Renz and M. Kaupp, Predicting the Localized/Delocalized Character of Mixed-Valence Diquinone Radical Anions. Toward the Right Answer for the Right Reason, J. Phys. Chem. A, 2012, 116(43), 10629–10637, DOI:10.1021/jp308294r.
- F. S. Gogesch, S. Schwab, A. Rehse, M. Linseis, A. Baksi, G. H. Clever and R. F. Winter, Diruthenium Complexes with π-Extended Bridging Bis(Alkenyl)Arylene Ligands, A Derived Metallamacrocycle, and Their Oxidized Forms, Organometallics, 2023, 42(21), 3085–3098, DOI:10.1021/acs.organomet.3c00337.
- E. D. Glendening, C. R. Landis and F. Weinhold, Natural Bond Orbital Methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(1), 1–42, DOI:10.1002/wcms.51.
- E. Wu, K. Jacobson and D. Papahadjopoulos, Lateral Diffusion in Phospholipid Multibilayers Measured by Fluorescence Recovery after Photobleaching, Biochemistry, 1977, 16(17), 3936–3941, DOI:10.1021/bi00636a034.
- K. Watanabe, K. Moriya, T. Kouyama, A. Onoda, T. Minatani, S. Y. Takizawa and S. Murata, Photoinduced Transmembrane Electron Transport in DPPC Vesicles: Mechanism and Application to a Hydrogen Generation System, J. Photochem. Photobiol., A, 2011, 221(1), 113–122, DOI:10.1016/j.jphotochem.2011.04.034.
- H. Song, A. Amati, A. Pannwitz, S. Bonnet and L. Hammarström, Mechanistic Insights into the Charge Transfer Dynamics of Photocatalytic Water Oxidation at the Lipid Bilayer–Water Interface, J. Am. Chem. Soc., 2022, 144(42), 19353–19364, DOI:10.1021/jacs.2c06842.
- N. Sinambela, R. Jacobi, D. Hernández-Castillo, E. Hofmeister, N. Hagmeyer, B. Dietzek, L. González and A. Pannwitz, Alignment and Photooxidation Dynamics of a Perylene Diimide Chromophore in Lipid Bilayers, Mol. Syst. Des. Eng., 2023, 8(7), 842–852, 10.1039/D2ME00243D.
- E. Krieger, T. Darden, S. B. Nabuurs, A. Finkelstein and G. Vriend, Making Optimal Use of Empirical Energy Functions: Force-Field Parameterization in Crystal Space, Proteins: Struct., Funct., Genet., 2004, 57(4), 678–683, DOI:10.1002/prot.20251.
- E. Krieger, J. E. Nielsen, C. A. E. M. Spronk and G. Vriend, Fast Empirical pKa Prediction by Ewald Summation, J. Mol. Graphics Modell., 2006, 25(4), 481–486, DOI:10.1016/j.jmgm.2006.02.009.
- D. Marsh, Handbook of Lipid Bilayers, CRC Press, 2013, DOI:10.1201/b11712.
- D. Drabik, G. Chodaczek, S. Kraszewski and M. Langner, Mechanical Properties Determination of DMPC, DPPC, DSPC, and HSPC Solid-Ordered Bilayers, Langmuir, 2020, 36(14), 3826–3835, DOI:10.1021/acs.langmuir.0c00475.
- S. Dutta, B. Watson, S. Mattoo and J.-C. Rochet, Calcein Release Assay to Measure Membrane Permeabilization by Recombinant Alpha-Synuclein, Bio-Protoc., 2020, 10(14), e3690, DOI:10.21769/BioProtoc.3690.
- B. Maherani, E. Arab-Tehrany, A. Kheirolomoom, D. Geny and M. Linder, Calcein Release Behavior from Liposomal Bilayer; Influence of Physicochemical/Mechanical/Structural Properties of Lipids, Biochimie, 2013, 95(11), 2018–2033, DOI:10.1016/j.biochi.2013.07.006.
- L. E. Garner, J. Park, S. M. Dyar, A. Chworos, J. J. Sumner and G. C. Bazan, Modification of the Optoelectronic Properties of Membranes via Insertion of Amphiphilic Phenylenevinylene Oligoelectrolytes, J. Am. Chem. Soc., 2010, 132(29), 10042–10052, DOI:10.1021/ja1016156.
- B. Wang, M. Wang, A. Mikhailovsky, S. Wang and G. C. Bazan, A Membrane-Intercalating Conjugated Oligoelectrolyte with High-Efficiency Photodynamic Antimicrobial Activity, Angew. Chem., Int. Ed., 2017, 56(18), 5031–5034, DOI:10.1002/anie.201701146.
- C. M. Legaspi, R. E. Stubbs, M. Wahadoszaman, D. J. Yaron, L. A. Peteanu, A. Kemboi, E. Fossum, Y. Lu, Q. Zheng and L. J. Rothberg, Rigidity and Polarity Effects on the Electronic Properties of Two Deep Blue Delayed Fluorescence Emitters, J. Phys. Chem. C, 2018, 122(22), 11961–11972, DOI:10.1021/acs.jpcc.7b12025.
- A. Pannwitz, H. Saaring, N. Beztsinna, X. Li, M. A. Siegler and S. Bonnet, Mimicking Photosystem I with a Transmembrane Light Harvester and Energy Transfer-Induced Photoreduction in Phospholipid Bilayers, Chem.–Eur. J., 2021, 27(9), 3013–3018, DOI:10.1002/chem.202003391.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer US, Boston, MA, 2006, DOI:10.1007/978-0-387-46312-4.
- R. E. P. Nau, J. Bösking and A. Pannwitz, Compartmentalization Accelerates Photosensitized NADH to NAD+ Conversion, ChemPhotoChem, 2022, 6(11), e202200158, DOI:10.1002/cptc.202200158.
- M. W. Sutherland and B. A. Learmonth, The Tetrazolium Dyes MTS and XTT Provide New Quantitative Assays for Superoxide and Superoxide Dismutase, Free Radical Res., 1997, 27(3), 283–289, DOI:10.3109/10715769709065766.
- S. Fukuzumi, M. Ishikawa and T. Tanaka, Mechanisms of Photo-Oxidation of NADH Model Compounds by Oxygen, J. Chem. Soc., Perkin Trans. 2, 1989,(8), 1037–1045, 10.1039/P29890001037.
- V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, John Wiley & Sons, 2014 Search PubMed.
- Y. Rahimi, A. Goulding, S. Shrestha, S. Mirpuri and S. K. Deo, Mechanism of Copper Induced Fluorescence Quenching of Red Fluorescent Protein, DsRed, Biochem. Biophys. Res. Commun., 2008, 370(1), 57–61, DOI:10.1016/j.bbrc.2008.03.034.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional references. See DOI: https://doi.org/10.1039/d4se01752h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |