
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ growth of layered 1T-MoS2 onto carbon nanofibers as a strategy towards advanced hybrid materials for electrochemical energy storage and catalysis†
Felix
Boll
ab,
Micaela
Pozzati
c,
Matteo
Crisci
ab,
Bernd
Smarsly
 ab,
Teresa
Gatti
*bc and
Mengjiao
Wang
*c
ab,
Teresa
Gatti
*bc and
Mengjiao
Wang
*c
aInstitute of Physical Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392 Giessen, Germany
bCenter for Materials Research, Justus Liebig University, Heinrich-Buff-Ring 17, 35392 Giessen, Germany
cDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi, 10129 Turin, Italy. E-mail: mengjiao.wang@polito.it; teresa.Gatti@polito.it
First published on 9th April 2025
Abstract
Supercapacitors for electrochemical energy storage and electrocatalysts for hydrogen evolution reaction (HER) are pivotal in addressing global energy challenges. However, their widespread use is hindered by the limitations of the currently available functional materials. This study explores the in situ hydrothermal growth of 1T-phase MoS2 directly onto the surface of carbon nanofibers (CNFs), forming 3D hierarchical nanostructures with potential for both supercapacitor and HER applications. The effect of reaction time on the thickness of the 1T-MoS2 outer layer is systematically investigated, revealing a stepwise relationship between layer thickness and reaction time. The resulting 1T MoS2/CNFs hybrids, with varying MoS2 layer thicknesses, exhibit distinct specific capacitance behaviors under varying scan rates, due to differences in conductivity and ion diffusion distances. Post-electrochemical testing analysis reveals 1T-MoS2 inherent instability, which, rather than transitioning to the 2H-phase, predominantly oxidizes to form molybdenum oxides. Despite this limitation, the hybrids demonstrate promising performance in HER electrocatalysis, with thicker MoS2 layers offering more active sites, despite an unfavorable trade-off in conductivity. This study provides a mechanistic understanding of 1T-MoS2/CNFs nanohybrids in electrochemical applications, highlighting the interplay between MoS2 phase composition, thickness and electrochemical performance. Our findings underscore both the potential and challenges in optimizing these nanohybrids for enhanced energy storage and hydrogen evolution, paving the way for future advancements in multifunctional energy materials.
 Mengjiao Wang | Dr Mengjiao Wang is a research assistant at the Department of Applied Science and Technology of the Politecnico di Torino, Italy. Previously, she was a postdoctoral fellow at Istituto Italiano di Tecnologia, Italy and Center for Materials Research of the Justus-Liebig-Universität Gießen in Germany. She holds a PhD degree in science and technology of chemistry and materials from the University of Genoa. Her research focuses on renewable energy, photoelectrochemistry and heterojunction technology. She is a winner of the project “Bando TRAPEZIO – Paving the way to research excellence and talent attraction”, aiming at developing 2D heterojunctions for electrochemical applications. |
1 Introduction
Addressing the climate crisis has placed an urgent emphasis on innovative methods for energy production and storage. Despite significant advancements in energy generation, storage and transfer, driven by modern technology and research, ongoing discoveries continue to refine and optimize these processes. Efficient energy production and storage systems are indispensable, given the constant demand for reliable power sources. To overcome challenges in intermittent renewable energy production and, consequently required, energy storage, current technological efforts are directed towards developing advanced supercapacitors and electrocatalysts for the hydrogen evolution reaction (HER), seeing hydrogen as a sustainable energy carrier.1,2 However, the effectiveness of these systems is often limited by the availability of suitable functional materials. The key challenge in advancing these technologies lies in the discovery and development of novel nanomaterials with enhanced specific capacitance (Cs) for energy storage and/or superior catalytic activity for HER.Carbon-based materials have emerged as some of the most important electrode materials for supercapacitors, garnering continuous attention since several years. Currently, the development of one-dimensional carbon composite nanomaterials is a highly investigated approach to further enhance supercapacitor performance.3 This focus is primarily driven by the inherent advantages of 1D structures, which are believed to facilitate efficient electron transport and improve overall device functionality.4 Ongoing research is focused on improving carbon nanofibers (CNFs) to enhance their capacity and durability.5 The strategic design and selection of materials to combine with CNFs is a valuable tool to significantly boost electrochemical performance of this well-known nanocarbons. Materials such as multiwalled carbon nanotubes,6 graphene,7 MXenes,8 and NiCo2S4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 have been identified as effective additives for CNFs. These species not only improve charge storage capacity but also help maintain excellent conductivity, making them highly beneficial for advanced energy storage applications. In addition to the choice of components, the structure of hybrid materials plays a crucial role in optimizing performance. For supercapacitors incorporating CNFs, a 3D hierarchical structure has proven to be a promising design. This configuration enables effective integration of CNFs with other nanomaterials, such as MnO2,10 RuO2,11 NiCo2O4,12 NiCo2S4,9 and NiS,13 which can grow on the surface of CNFs. These structures can influence electron transfer and electrolyte diffusion, thereby improving overall performances as supercapacitors.
9 have been identified as effective additives for CNFs. These species not only improve charge storage capacity but also help maintain excellent conductivity, making them highly beneficial for advanced energy storage applications. In addition to the choice of components, the structure of hybrid materials plays a crucial role in optimizing performance. For supercapacitors incorporating CNFs, a 3D hierarchical structure has proven to be a promising design. This configuration enables effective integration of CNFs with other nanomaterials, such as MnO2,10 RuO2,11 NiCo2O4,12 NiCo2S4,9 and NiS,13 which can grow on the surface of CNFs. These structures can influence electron transfer and electrolyte diffusion, thereby improving overall performances as supercapacitors.
Such combinations have also shown potential for the HER. CNFs, as an excellent catalyst support, are frequently used to grow and disperse electrocatalysts as well. The growth of heterostructures from the CNF surface provides a larger surface area for HER compared to the use of the pure nanomaterial-based components.14–16 Furthermore, well-dispersed catalyst nanoparticles embedded within the CNF matrix reduce interfacial resistance between particles, enhancing both conductivity and stability. Additionally, the intrinsic conductivity of CNFs minimizes resistance in the HER process, further improving catalytic efficiency.17 Therefore, using CNFs as a conductive matrix to support other nanomaterials is an excellent strategy for producing hybrid species with enhanced electrochemical properties.
Meanwhile, the interest in 2D layered materials has rapidly expanded, with transition metal dichalcogenides (TMDCs) gaining attention in the scientific community as ideal candidates for applications such as energy storage and electrocatalysis.18–21 MoS2 is emerging as a viable new electrode material for supercapacitors and HER owing to a high conductivity and remarkable electrochemical activity.22–26 MoS2 is known as an excellent example of innovative approaches to energy production.27–29 Especially for capacitive and high-conductive applications, MoS2 in its metallic phase (the 1T-phase), is up to 107 times more conductive compared to the semiconducting 2H-phase. Additionally, induced S vacancies in the 1T-phase create beneficial active sites for hydrogen production. However, given its fascinating electrochemical characteristics, bare 1T-MoS2 frequently experiences poor CS. It is thus necessary to develop new approaches to raise CS of 1T-MoS2 electrode materials. Maximizing the accessible surface area for charge storage and increasing the number of active sites, along with their accessibility, is essential for optimizing the overall electrochemical properties. Consequently, the development of 1T-MoS2 with more active sites is both beneficial and strongly recommended.
Significant beneficial aspects arise when carbon-based materials are interfaced with TMDCs such as MoS2 and WS2. Synergistic interactions at the carbon–TMDC interface help prevent aggregation, maintain surface area during electrochemical processes, increase the concentration of active sites, and enhance charge conductivity.30–32 Moreover, the capillary effects of CNFs play a vital role by facilitating electrolyte penetration and improving electrode wetting, further boosting performance. The combination of CNFs with MoS2 has been extensively studied and reported in the literature.33–36 However, most studies focus on incorporating semiconductive 2H-MoS2 with CNFs, while limited research has explored the fabrication of 1T-MoS2/CNFs hybrids and the study of their electrochemical performance in energy storage and electrocatalysis. Furthermore, a critical gap remains in understanding the influence of the carbon-to-MoS2 ratio on the electrochemical performance. Addressing this knowledge gap could provide valuable insights for optimizing these materials for energy applications. Additionally, to achieve strong bonding with carbon-based materials, the bottom-up hydrothermal method is more practical, as it allows the direct growth of 1T-MoS2 onto a carbon matrix. For instance, Niu et al. successfully synthesized ultrathin 1T–2H MoS2 heterostructures on CNFs using the hydrothermal method.33 Despite these advances, the synthesis process remains insufficiently explored and calls for further detailed investigation.
Building on previous studies, we investigate here the combination of 1T-MoS2 and CNFs to develop 1T-MoS2/CNFs hybrids for electrochemical applications. By testing the performance of these hybrids as supercapacitor materials and electrocatalysts, our goal was to understand how the structure and composition influence their functionality in these two important applications. We systematically examined the effects on MoS2 layer thickness over reaction time of the hydrothermal process used to grow it on the surface of CNFs and found that the MoS2 layer undergoes a stepwise growth with the reaction time. The resulting changes in phase composition and the MoS2-to-carbon ratio are analysed in relation to the performance of the hybrids in charge storage and HER. Understanding the functionality helps to provide valuable insights for the further optimization of nanocomposites for energy-related applications, enabling the precise fabrication of structures tailored to specific functions.
2 Results and discussion
2.1 Synthesis of 1T-MoS2/CNFs at different reaction times
CNFs were synthesized using a three-steps procedure which involves electrospinning of polyacrylonitrile (PAN), followed by stabilization at 325 °C in air and carbonization at 800 °C under N2, as reported in our recent work and recalled here in the Experimental for the sake of clarity (Fig. 1a).5 The synthesized samples are labelled as 1T-MoS2/CNFs and the corresponding reaction time. The resulting CNFs product was employed as a matrix for subsequent hydrothermal synthesis of MoS2, as illustrated in Fig. 1a. By varying the hydrothermal reaction times, MoS2 layers of different thicknesses were deposited onto the CNFs surface. The morphologies of these hybrid nanomaterials were characterized using scanning electron microscopy (SEM). As shown in Fig. 1a, the uncoated CNFs display a slender fiber structure with smooth surfaces and a diameter of 144 nm. Upon introducing MoS2 precursors and subjecting the CNFs to hydrothermal reactions, MoS2 begins to grow on the CNF surfaces, forming a 3D hierarchical architecture (Fig. 1b–i). This MoS2 growth significantly increases the surface roughness of the CNFs. In all samples containing MoS2, lamellar-like structures are observed extending perpendicular to the fiber surface. A higher-magnification SEM image (see ESI, Fig. S1†) further emphasizes the surface transformation compared to the original smooth CNFs. On top of 1T-MoS2/CNFs 18 h, MoS2 begins to form microspheres rather than continuing to grow along the fibers after 18 hours of reaction (Fig. 1h). By 24 hours, these MoS2 microspheres dominate, occupying much of the space between fibers (Fig. 1i). At this stage, nanoflakes composing the microspheres, a characteristic feature of hydrothermally synthesized MoS2, are prominently observed. It is reasonable to envision that the nucleation points for these structures arise from the pre-existing MoS2 formations within the solution, with the outer MoS2 layers likely having no direct contact with the CNFs core. | ||
| Fig. 1 (a) Schematic representation of the 1T-MoS2/CNFs nanohybrids production process and SEM image of pristine CNFs from the three-steps process. SEM of 1T-MoS2/CNFs samples with the reaction time of (b) 1 h, (c) 2 h, (d) 3 h, (e) 4 h, (f) 6 h, (g) 14 h, (h) 18 h, and (i) 24 h. Scale bar: 2 μm. (j) Relationship between the diameter of the hybrid samples and the reaction time. | ||
To determine the average fiber thickness, the diameters of 20 fibers were measured, and their standard deviations were calculated using pixel measurements from the SEM images (details provided in Fig. S2 and Table S1†). The average diameters of 1T-MoS2/CNFs hybrids are summarized in Fig. 1j. The results show a stepwise increase in MoS2 layer thickness as the reaction time progresses. After 1 h of reaction, the diameter of 1T-MoS2/CNFs 1 h sample increased to approximately 257 nm, indicating that surface reactions initiate early in the process. Between 2 and 4 h of reaction time, the fiber thickness reached a plateau of around 540 nm, and the MoS2 layer was ∼200 nm. During this period, significant surface changes were observed, with more MoS2 flakes forming and arranging themselves on the CNF surface. For 1T-MoS2/CNFs 6 h, the fiber diameter showed a sharp increase to approximately 1100 nm (MoS2 layer thickness of ∼480 nm), followed by stagnation in growth until the 18-hour mark. For 1T-MoS2/CNFs 24 h, the fiber diameter increased further to approximately 1600 nm (MoS2 layer thickness of ∼730 nm). The cause of the stagnation periods during growth is not yet fully understood. It is likely that consistent temperature and pressure conditions within the autoclave may influence the nucleation and growth dynamics of MoS2. During these plateau periods, a dynamic equilibrium might occur where newly formed MoS2 on the CNF surface undergoes simultaneous formation and dissolution, preventing further thickness increases. Additionally, it is possible that nucleation of MoS2 in the solution or on the autoclave walls becomes more favorable during these intervals, reducing deposition on the CNFs.
Elemental analysis using energy-dispersive X-ray spectroscopy (EDX) was performed on the fiber surface to further investigate the coverage of MoS2 on CNFs. The EDX results for samples at different reaction times, ranging from the initial stage to 4 hours, are shown in Fig. 2. The C and Mo content are presented to compare the ratio of MoS2 to CNFs on the surface. For 1T-MoS2/CNFs 1 h, the C signal remains strong, while the Mo signal begins to appear. As the reaction time increases from 2 h to 4 h, the C signal weakens while the Mo signal strengthens. Quantitatively, after 1 h of reaction, the C content is approximately 84 wt%, while Mo and S account for around 5 wt% (Table S2†). This indicates that only a small amount of MoS2 has bound to the CNF surface at this stage, which is consistent with the slight increase in fiber diameter observed in Fig. 1b. As the reaction time increases, the C content gradually decreases, while the Mo content increases. For 1T-MoS2/CNFs 2 h, the C content drops significantly to 7.7 wt%, while Mo and S rise to 84.7 wt%. After this point, as the reaction time exceeds 2 h, the C content continues to decrease slightly to around 4%, and MoS2 becomes the dominant material on the CNF surface. This indicates that the density of MoS2 sheets on the CNF surface increases rapidly within the first 2 hours, while after 2 hours, the growth of MoS2 sheets appears to level off. This stagnation likely reflects a dynamic equilibrium, where the formation and decomposition of MoS2 on the CNF surface reach balance. Additionally, the N signal primarily originates from the CNFs, showing a similar trend to that of C. N-containing species can provide chemically active sites, which are beneficial for electrochemical processes.37 As the MoS2 layer thickens after 6 hours, the growing MoS2 layer effectively shields the underlying CNF material, making it difficult to assess the MoS2 coverage using EDX at this stage.
 | ||
| Fig. 2 EDX mapping of C and Mo in the 1T-MoS2/CNFs hybrids. C mapping of 1T-MoS2/CNFs with the reaction time of (a) 1 h, (b) 2 h, (c) 3 h and (d) 4 h. Mo mapping of 1T-MoS2/CNFs with the reaction time of (e) 1 h, (f) 2 h, (g) 3 h and (h) 4 h. Scale bar: 2.5 μm. | ||
The Raman spectra of the hybrid samples, shown in Fig. 3, were recorded to identify the components of the nanohybrids. No vibrational modes related to Mo species were observed on 1T-MoS2/CNFs 1 h. Only the D-band and G-band of carbon, located at 1353 cm−1 and 1585 cm−1 respectively, were present, indicating that only a small amount of Mo composites had formed at this early stage (Fig. 3a). Starting at 2 h of reaction, the formation of Mo composites became detectable in the Raman spectra, with vibrational modes related to Mo species appearing in the range of 100–1000 cm−1 (Fig. 3b). At 150 cm−1 and 338 cm−1, the J1 and J3 vibrational modes associated with the 1T-phase of MoS2 are observed.26 Vibrational modes at 378 cm−1 and 405.5 cm−1 correspond to the in-plane (E2g) and out-of-plane (A1g) vibrations of the 2H MoS2 phase.20,38–41 Lattice modes related to MoO3 are observed at 125 cm−1, corresponding to the out-of-plane B2g deformation mode of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) MoO and OMo bonds.42–44 A peak at 284 cm−1 represents the bending mode of the MoO vibration, while the peak at 819.5 cm−1 corresponds to Ag modes of MoO3, associated with stretching vibrations of O atoms in the O–Mo–O bond. Additional weak peaks at 214.5 cm−1 and 667.5 cm−1 are attributed to the Ag deformation mode and the stretching B3g mode of MoO3. All these characteristic peaks are present in the 1T-MoS2/CNFs hybrid samples except for the 1T-MoS2/CNFs 1 h. This suggests that 1T-MoS2, 2H-MoS2, and MoO3 coexist in the 1T-MoS2/CNFs hybrids. As the reaction time increases, the peaks related to MoS2 and MoO3 become more pronounced, while the intensity of the carbon-related peaks diminishes, indicating that the CNF surface is progressively less exposed.
MoO and OMo bonds.42–44 A peak at 284 cm−1 represents the bending mode of the MoO vibration, while the peak at 819.5 cm−1 corresponds to Ag modes of MoO3, associated with stretching vibrations of O atoms in the O–Mo–O bond. Additional weak peaks at 214.5 cm−1 and 667.5 cm−1 are attributed to the Ag deformation mode and the stretching B3g mode of MoO3. All these characteristic peaks are present in the 1T-MoS2/CNFs hybrid samples except for the 1T-MoS2/CNFs 1 h. This suggests that 1T-MoS2, 2H-MoS2, and MoO3 coexist in the 1T-MoS2/CNFs hybrids. As the reaction time increases, the peaks related to MoS2 and MoO3 become more pronounced, while the intensity of the carbon-related peaks diminishes, indicating that the CNF surface is progressively less exposed.
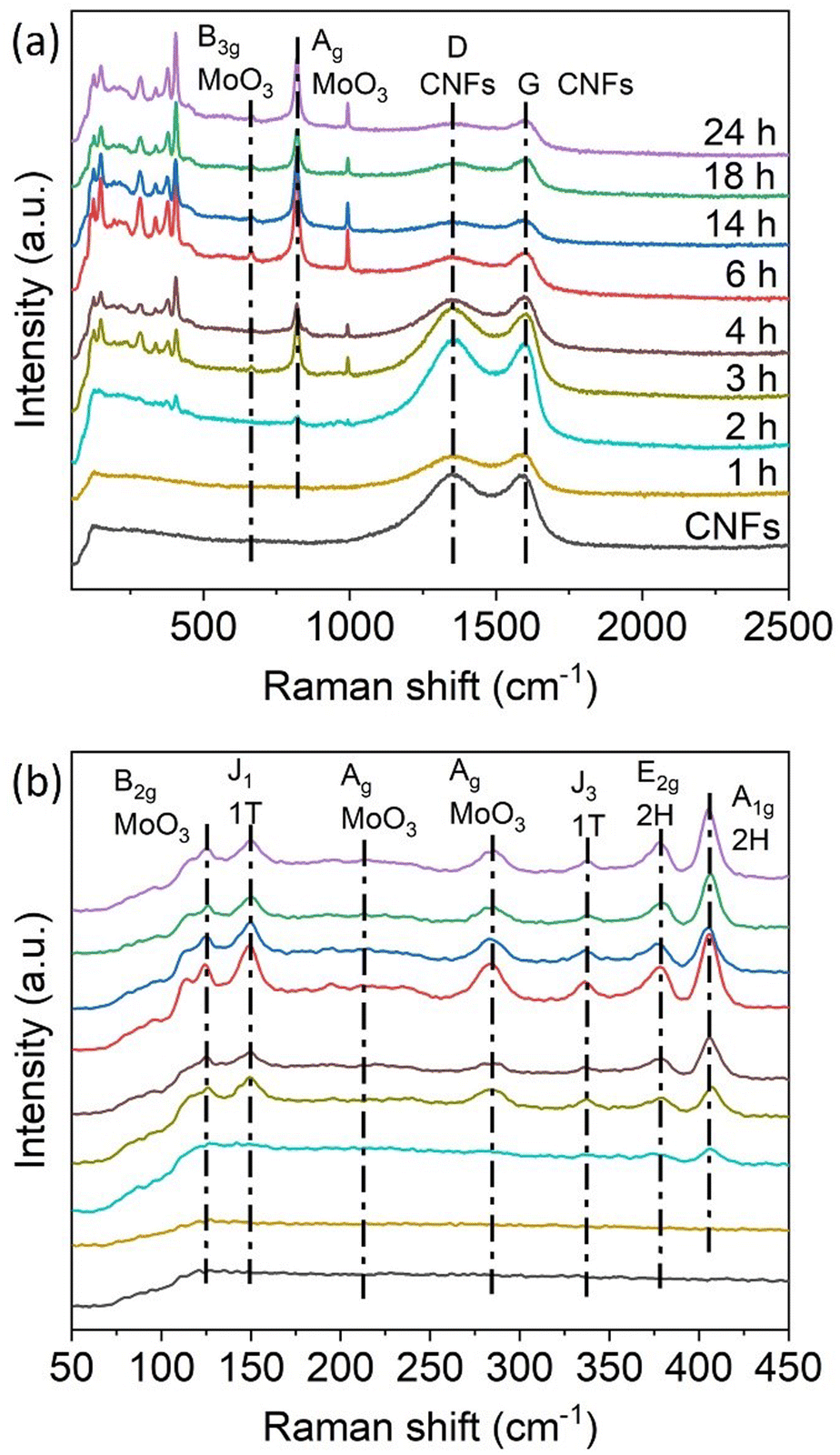 | ||
| Fig. 3 (a) Raman spectra of the 1T-MoS2/CNFs hybrids labelled with the reaction time. (b) Magnification of the Raman spectra of the hybrids in the range of 50–450 cm−1. | ||
To further confirm the simultaneous presence of 1T-MoS2, 2H-MoS2, and MoO3, X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) measurements were conducted on typical samples, including 1T-MoS2/CNFs 2 h, 1T-MoS2/CNFs 6 h, and 1T-MoS2/CNFs 24 h. As shown in Fig. 4a, the Mo 3d region of the XPS spectrum displays distinct signals, which were fitted using three components for Mo and one for S, with the S 2s peak (225.8 eV) overlapping in this region. The peaks at 228.1 eV and 231.3 eV correspond to Mo(1T) 3d5/2 and Mo(1T) 3d3/2, respectively. Peaks at higher binding energies, 229.1 eV and 232.2 eV, are attributed to Mo(2H) 3d5/2 and Mo(2H) 3d3/2. Additionally, peaks at 232.7 eV and 235.6 eV correspond to MoO3 3d5/2 and MoO3 3d3/2, indicating the presence of MoOx, which forms during synthesis. These MoO3-related peaks are visible in Fig. 4b but disappear in Fig. 4c, suggesting a transformation of MoO3 to MoS2. The XPS results clearly demonstrate that the MoS2 coverage consists of two MoS2 phases on the surface of 1T-MoS2/CNFs 24 h: predominantly 1T-phase MoS2, with a minor amount of 2H-phase MoS2. For 1T-MoS2/CNFs 2 h and 1T-MoS2/CNFs 6 h, MoO3 is also detected. Two trends emerge with increasing reaction time: first, a significant increase in the metallic 1T-phase, which rises from 57.25% after 2 hours to 81.52% after 6 hours, and further increases to 87.09% after 24 hours; and second, a decrease in MoO3 content, which drops from 23.02% to zero over the same period (Table S3.†). By comparing the XPS spectra of these three samples, it is clear that the increase in the 1T-phase is not solely due to a phase transition from 2H to 1T but it also involves the conversion of MoOx to sulfides. It is likely that MoO3 forms initially and is later volcanized, leading to the formation of MoS2. In contrast to the Raman data from the sample 1T-MoS2/CNFs 24 h, no MoO3 is detected in the XPS spectrum of the same sample. This discrepancy might be attributed to the use of an intense 532 nm laser during Raman measurements, which provides sufficient energy to produce some MoO3 on the surface of the material.45
 | ||
| Fig. 4 Mo 3d high-resolution XPS spectra of (a) 1T-MoS2/CNFs 2 h, (b) 1T-MoS2/CNFs 6 h and (c) 1T-MoS2/CNFs 24 h. (d) XRD patterns of CNFs and the representative 1T-MoS2/CNFs hybrids. | ||
The XRD traces in Fig. 4d illustrate the crystalline properties of CNFs, 1T-MoS2/CNFs 2 h, 1T-MoS2/CNFs 6 h, and 1T-MoS2/CNFs 24 h. The pure CNFs exhibit two broad and weak reflexes at 2θ = 24°, corresponding to the (002) plane of graphitic crystallites.46 The reflexes of 1T-MoS2/CNFs 2 h align well with the 2H-MoS2 phase (JCPDS: 37-1492). However, for 1T-MoS2/CNFs 6 h and 1T-MoS2/CNFs 24 h, characteristic reflexes of the 1T-phase appear at 9.27° for the (002) plane and 18.17° for the (004) plane, indicating that the MoS2 coating on the CNFs in these two samples primarily exists in the 1T-phase. A slight peak shift towards higher 2θ angles compared to literature values is observed, suggesting a reduction in interlayer spacing relative to previously reported values, as lower 2θ angles typically correspond to wider interlayer spacings in crystalline materials.47 When comparing the results from Raman spectroscopy, XPS, and XRD, it is evident that the phase configuration and surface characteristics of the 1T-MoS2/CNFs hybrids differ significantly. XRD analysis reveals distinct phase formation in Mo-based composites, showing that the primary crystal structure differs from the surface composition. Initially, the reaction favors the formation of 2H-MoS2, while 1T-MoS2 emerges through a phase transformation of 2H-MoS2 and direct monomer-based formation, as indicated by the exclusive presence of dominant 1T-MoS2 phase in 1T-MoS2/CNFs 6 h and 1T-MoS2/CNFs 24 h. Additionally, no clear XRD signals corresponding to MoOx are detected, implying that MoOx species are predominantly amorphous and primarily located on the surface of the Mo-based composites. On the other hand, on the surface of the Mo composites, a mixture of different phases is consistently observed, with a clear trend indicating phase transformation from 2H-MoS2 and MoOx to 1T-MoS2 over the reaction time (Fig. 4a–c).
2.2 Supercapacitive performance
Based on the previous characterization, representative 1T-MoS2/CNFs 2 h, 1T-MoS2/CNFs 6 h, and 1T-MoS2/CNFs 24 h were selected for electrochemical measurements, which correspond to different thicknesses of MoS2 coverage and varying Mo species ratios on the surface. The electrochemical performance of the MoS2/CNFs hybrids was first evaluated in a Swagelok-type electrochemical cell, using 1 M H2SO4 as the electrolyte and a SiO2 Whatman membrane as the separator. For comparison, the electrochemical performance of pure CNFs was also tested. Fig. 5a shows the cyclic voltammetry (CV) curves of pure CNFs and 1T-MoS2/CNFs hybrids at 10 mV s−1 within the potential window of 0–0.8 V. The CV curves of all electrodes, except for 1T-MoS2/CNFs 24 h, display nearly rectangular shapes without distinct redox peaks, indicating electrical double layer capacitance (EDLC). In contrast, the CV curves of 1T-MoS2/CNFs 24 h show quasi-rectangular shapes with a pair of weak but distinguishable redox peaks at 0.4 V and 0.6 V, indicating faradaic redox pseudocapacitance associated with both surface and bulk faradaic mechanisms. Notably, the current and integral area of the CV curve for 1T-MoS2/CNFs 2 h is significantly larger than those of the other electrodes, suggesting enhanced specific capacitance due to the positive synergistic effects between the ultrathin MoS2 sheets on the surface and the CNF skeleton. Fig. 5b shows the galvanostatic charge/discharge (GCD) curves of the samples at a current density of 1 A g−1. The nonlinear behaviour of the charge/discharge curves of the hybrids confirms the involvement of redox processes due to hybridization. The longer charge/discharge times of 1T-MoS2/CNFs 2 h compared to pure CNFs further demonstrate the improved capacitance of 1T-MoS2/CNFs 2 h. In contrast, the shorter charge/discharge times of 1T-MoS2/CNFs 6 h and 1T-MoS2/CNFs 24 h highlight the poorer performance of these two samples. It is noteworthy that 1T-MoS2/CNFs 24 h exhibited a longer charge/discharge time compared to 1T-MoS2/CNFs 6 h, with an asymmetrical plateau during the charging process. This suggests the presence of an irreversible reaction and a possible phase change.48Fig. 5c and Table S4† present the CS of all samples at different scan rates. As expected, the CS of all samples decreases as the scan rate increases. The 1T-MoS2/CNFs 2 h electrode achieves the highest CS, with a value of 29.7 F g−1 at 10 mV s−1, outperforming the other samples (∼20 F g−1). However, at higher scan rates (above 200 mV s−1), pure CNFs (8 F g−1 at 500 mV s−1) outperform 1T-MoS2/CNFs 2 h (5 F g−1 at 500 mV s−1). In stark contrast, 1T-MoS2/CNFs 6 h and 1T-MoS2/CNFs 24 h show significantly lower CS, with performance dropping below 2 F g−1 after 200 mV s−1. This is likely due to the inferior electrical conductivity and the thicker, aggregated structure of 1T-MoS2 in these samples. However, between 1T-MoS2/CNFs 6 h and 1T-MoS2/CNFs 24 h samples, 1T-MoS2/CNFs 24 h shows slightly higher Cs. Based on these observations, we infer that the enhancement of the capacitance of the hybrids varies with the thickness of the 1T-MoS2 layer. In 1T-MoS2/CNFs 2 h, the MoS2 layer, with a thickness of ∼200 nm, achieves an optimal balance between high conductivity, fast ion transport, and active site availability. However, as the MoS2 layer thickens to 480 nm in 1T-MoS2/CNFs 6 h, conductivity and ion accessibility decrease. When the MoS2 layer becomes excessively thick in 1T-MoS2/CNFs 24 h, charge storage increases slightly, but this enhancement is primarily attributed to faradaic reactions from 1T-MoS2, as evidenced by the redox peaks and plateau observed in Fig. 5a and b. | ||
| Fig. 5 (a) CV curves of the as-prepared 1T-MoS2/CNFs hybrids and CNFs at 10 mV s−1. (b) Galvanostatic charge/discharge curves of 1T-MoS2/CNFs hybrids and pristine CNFs. (c) CS of 1T-MoS2/CNFs hybrids and bare CNFs as a function of scan rate. (d) Nyquist plots of the 1T-MoS2/CNFs hybrids and the bare CNFs. (e) Capacitance retention based on discharge capacity and coulombic efficiency versus cycle number for 1T-MoS2/CNFs 2 h. | ||
An electrochemical impedance spectroscopy (EIS) test was performed to evaluate the electrical conduction and ion transfer kinetics of the as-prepared electrodes. The comparative EIS results for the electrodes are shown in Fig. 5d and Table S5.† In comparison, the charge-transfer resistance (Rct) of the 1T-MoS2/CNFs hybrids is noticeably higher than that of the bare CNFs, indicating slower charge transfer kinetics across the capacitor materials/electrolyte interface (Table S5†). As the MoS2 layer thickness increases, the Rct becomes larger, which reduces conductivity and hampers ion transfer kinetics. Additionally, the straight lines observed in the low-frequency region suggest that faster ion transfer occurs when the MoS2 layer is thinner or absent. This indicates that the absence of MoS2 or a thinner MoS2 layer facilitates faster ion transport. Durability tests were conducted over 5000 cycles to assess the performance retention of 1T-MoS2/CNFs 2 h at 1 A g−1 (Fig. 5e). Initially, a sharp decline in discharge capacity was observed during the early cycles, followed by a gradual decrease that eventually stabilized at approximately 87% retention. This reduction in capacitance is likely due to a decrease in the active surface area, possibly resulting from compositional changes in the material, as further explained in Fig. 6 description. The coulombic efficiency, defined as the percentage ratio of the electrode's discharge capacity to its charge capacity, was 88.3% in the first cycles. It then increased and stabilized above 93% after 30 cycles. This initial rise in coulombic efficiency may be attributed to the phase transition of MoS2, which likely slows down the charge capacity in the early cycles, as confirmed by the subsequent XPS analysis.
 | ||
| Fig. 6 Mo 3d high-resolution XPS spectra of (a) 1T-MoS2/CNFs 2 h, (b) 1T-MoS2/CNFs 6 h and (c) 1T-MoS2/CNFs 24 h after the GCD test. | ||
High-resolution XPS measurements were conducted to examine the oxidation states of Mo within the nanohybrids after the capacitance test. The distinctive peaks corresponding to 1T-MoS2, 2H-MoS2, and MoO3 are observed in all three hybrids. However, the ratio of these phases has significantly changed compared to the pre-capacitance test samples. Specifically, the proportion of the 1T-phase has decreased dramatically, while the amount of MoO3 has increased significantly. These findings indicate that the conductive 1T-phase has been converted into either the less conductive 2H-MoS2 or MoO3, both of which negatively impact electrochemical performance.
These results clearly demonstrate that hybrids synthesized with different reaction times exhibit varying thicknesses of 1T-MoS2 on CNFs, which in turn affects their capacitance. CNFs coated with 1T-MoS2 layer enhance capacitance at lower scan rates due to the synergistic interaction between CNFs and 1T-MoS2. This effect is most pronounced when 2 h of reaction time produces a thin 1T-MoS2 layer of ∼200 nm, which facilitates ion diffusion without compromising conductivity. However, hybrids synthesized with longer reaction times, which form thicker MoS2 layers, exhibit reduced capacitance. As the MoS2 thickness increases, charges become trapped at the surface, as the thick metallic 1T-phase prevents efficient charge displacement toward the CNF core. Although the faradaic reaction within MoS2 contributes to capacitance, its effect is negligible compared to the EDLC of CNFs. Additionally, the 1T-phase is metastable and tends to convert into the less conductive 2H-phase or fully oxidize to MoO3, which further increases resistance and hinders charge transfer.
2.3 HER performance
Given the unique structure and the increased exposure of active sites in the 1T-MoS2/CNFs hybrids, these are promising candidates for electrocatalysis in the HER. To evaluate their electrocatalytic activity, linear sweep voltammetry (LSV) measurements were conducted in a 0.5 M H2SO4 solution using a glassy carbon working electrode, Ag/AgCl reference electrode, and Pt counter electrode setup. Fig. 7 presents the LSV curves of bare CNFs and the hybrids. The bare CNFs show a large overpotential of 0.66 V at 5 mA cm−2, whereas the 1T-MoS2/CNFs samples exhibit significantly lower overpotentials, with the 1T-MoS2/CNFs 6 h showing the smallest overpotential of 0.2 V at 5 mA cm−2 (Table S6†). This demonstrates that the addition of the 1T-MoS2 layer substantially reduces the overpotential for HER. Fig. 7b shows the Tafel plots derived from the LSV curves for all samples. The Tafel slopes for the 1T-MoS2/CNFs hybrids range between 67 mV dec−1 and 75 mV dec−1, compared to a much higher Tafel slope of 312 mV dec−1 for bare CNFs. This indicates that the hybrids exhibit significantly faster HER kinetics than bare CNFs. Therein, the electrochemical surface area (ECSA)-normalized polarization curves of both CNFs and 1T-MoS2/CNFs samples further confirm that the 1T-MoS2 coating enhances the intrinsic HER activity of CNFs (Fig. 7c and Table S7†). Therefore, although the availability of electrochemical active sites on CNFs contribute to catalytic performance, they are not the primary factor responsible for the improved activity. Instead, the active sites on 1T-MoS2 are mainly responsible for HER. The long-term stability of 1T-MoS2/CNFs 6 h was tested by chronoamperometry at an applied voltage of −0.25 V vs. RHE for 5.5 h, and this also showed that there was a gradual reduction from 10.6 mA cm−2 to 8.7 mA cm−2 in the current density before reaching stability. This might be associated with the desorption of some catalyst from the glassy carbon substrate during operation and not to an intrinsic material degradation (Fig. 7d). | ||
| Fig. 7 (a) LSV, (b) Tafel slope and (c) ECSA-normalized polarization curves of pristine CNFs and of the 1T-MoS2/CNFs hybrids. (d) Chronoamperometry plot of 1T-MoS2/CNFs 6 h, plotted at a constant volt of −2.5 V vs. RHE. | ||
Overall, our results suggest that CNFs and 1T-MoS2/CNFs hybrids show potential for both supercapacitor and electrocatalytic applications. However, their performance in these two electrochemical uses varies significantly. For supercapacitors, pure CNFs and CNFs partially covered with MoS2 (1T-MoS2/CNFs 2 h) outperform the other samples. Notably, the 1T-MoS2/CNFs 2 h hybrid exhibits higher CS than pure CNFs at low scan rates, due to the synergistic effect between the two components. However, at high scan rates, the diffusion layer thickness of 1T-MoS2/CNFs 2 h decreases, leading to lower capacitance compared to pure CNFs. As the hydrothermal reaction time increases beyond 6 hours, the MoS2 layer thickens significantly, nearly covering the CNF core and reducing its contact with the electrolyte. Since pure CNFs have better conductivity and surface area (Fig. 6b and c) than the hybrids, the capacitance of bare CNFs remains superior. Furthermore, the conversion of 1T-MoS2 to the less conductive 2H-MoS2 and MoO3 reduces charge transfer speed and worsens the capacitance performance.
For electrocatalytic HER, low resistance and high ECSA can enhance performance, but they are not the main contributors. 1T-MoS2/CNFs 6 h performs as the best catalyst for HER. The related MoS2 thickness strikes a balance between good conductivity and a sufficient number of active sites for HER. Unlike pure CNFs, which have a higher ECSA but less effective HER performance, the 1T-MoS2 layer in the hybrids provides effective active sites for H* intermediate adsorption and H2 desorption, leading to lower overpotentials and improved HER activity.49 While CNFs have high ECSA and low resistance, the charges adsorbed on pure CNFs do not efficiently reduce the HER reaction barrier, resulting in a higher overpotential compared to the hybrids.
3 Conclusions
In summary, we have designed 3D hierarchical MoS2/CNFs structure through a combination of electrospinning/stabilization/carbonization and hydrothermal approaches. The thickness of the MoS2 grown on the surface of CNFs shows a stepwise relationship with reaction time. This might be due to some dynamic balance during the examined different reaction times that leads to the formation and decomposition of MoS2 under specific pressure and concentration of the precursors. Representative samples with different thickness of the MoS2 layer on the CNFs surface were used for electrochemical tests. Benefiting from the high electrical conductivity of CNFs and synergistic effect between CNFs and a thinner MoS2 layer, the SC performance under low scan rate of 1T-MoS2/CNFs 2 h results improved compared to that of bare CNFs. On the other hand, the hybrids with a thick MoS2 layer exhibit low CS, which result from the low conductivity of the MoS2 layer and poor phase transfer in 1T-MoS2.The hybrids display enhanced electrocatalytic activity for HER compared to the pure CNFs, and 1T-MoS2/CNFs 6 h has the optimized MoS2 thickness to result in the lowest overpotential of 0.2 V at 5 mA cm−2 in acidic electrolyte. The optimized sample provides the balance of conductivity and number of active sites. Our strategy to combine MoS2 with CNFs sheds new light on the development of transition-metal chalcogenides/nanocarbon hybrids for high-performance energy systems, which will have an impact on future sustainable and low-cost devices for powering electronics and the IoT.
4 Experimental
4.1 Materials
Chemicals and solvents including PAN, dimethyl formamide (DMF) ethanol (EtOH), H2SO4 and CN2H4S were purchased from Sigma Aldrich and used as received, unless otherwise specified. The ammonium molybdate (para) tetrahydrate (NH4)6Mo7O24 4H2O was purchased from Alfa Aesar with a purity of 99%. Milli-Q water was used as a solvent for the preparation of the electrolytes for the electrochemical measurements.4.2 Preparation of CNFs
For CNFs production, 1.131 g PAN (MW = 150.000) are dissolved in 15 mL DMF under continuous stirring between 300 and 500 rpm to obtain an 8 wt% solution of PAN. Afterwards, the polymer solution is electrospun using a self-constructed electrospinning setup. Two high-voltage power sources (Scientific Instruments, TSI-HV) are used to setup a potential between a pre-treated needle tip and a collecting electrode. An injection needle with an inner diameter of 1.2 mm is used for insertion of the polymer solution into the electrospinning chamber. As the fiber-collecting electrode a rotating metallic drum with an inner diameter of 5 cm was connected to the other power source. Both electrodes were positioned with a 12 cm distance to each other. Before this process the drum roll is wrapped with aluminium foil and then rotates at 65 rpm during deposition. The polymer solution was injected using a syringe pump from HAVARD APPARATUS, PHD 2000 Infusion. The flow rate was set to 0.25 mL h−1 that the insertion of 15 mL is done within 60 h. The collecting electrode was set to a potential of −2 kV while at the needle tip a potential of 10.75 kV was applied. Additionally, the electrospinning setup is placed in a box enabling the control of the relative humidity of 30–35% by a N2 flow. Afterwards, the produced nanofibers were cut into pieces of 2 × 2 cm2 and stabilized in air at 325 °C for 2 h with a heating rate of 2 °C h−1. After cooling down, the nanofibers were carbonized in a tube furnace under N2 atmosphere. The N2 flow was fixed at 100 mL min−1. Again, using a heating rate of 2 °C h−1 to heat the samples up to 800 °C, which was hold for 1 h.4.3 Preparation of 1T-MoS2/CNFs hybrids
After cooling down, the CNFs were coated with a layer of hydrothermally produced MoS2. Therefore, 1.6 g of CN2H4S were dissolved in 20 mL deionized H2O acting as S source. As Mo source, H32Mo7N6O28 was dissolved as well and the solution was stirred for 60 min. Afterwards, the solution was transferred into a Teflon autoclave reactor (V = 45 mL) reaching a filling factor of 44.4% (Fig. 1a). To the solution two pieces of CNFs were inserted into the reactor and closed tightly. The autoclave reactor was put into a furnace at 200 °C and a reaction time of 1 h, 2 h, 3 h, 4 h, 6 h, 14 h, 18 h and 24 h was used to produce 1T-MoS2/CNFs hybrids having different amounts of MoS2 grown onto the surface of CNFs. After reaction, the reactor was cooled down to room temperature under ambient conditions. The samples are carefully removed from the reactor and rinsed with water to clean the samples from residual solution. After three times of washing with H2O, the hybrids were washed in EtOH for three times. As a final step, the washed hybrids were inserted into a vacuum drying furnace for 18 hours at 40 °C to remove residual solvents.4.4 Morphological and physico-chemical characterizations
SEM images were acquired using a Zeiss Gemini SEM 560 with an acceleration voltage of 3 kV and a 20.0 μm aperture size. A secondary electron detector was used to capture the images. EDS measurements were conducted with an Oxford Instruments Ultimax detector, employing an acceleration voltage of 5 kV and a 75.0 μm aperture size. Raman spectra were collected using a Bruker Senterra Infinity 1 spectrometer over a range of 50–2500 cm−1, equipped with an optical microscope for sample alignment at a magnification factor of 50×. A 532 nm excitation laser was used for the measurements. XRD data were obtained with an Anton Paar XRDynamic 500 using a Cu Kα radiation source (λ = 0.154178 nm) operated at 40 kV and 40 mA. The incident beam angle was set to 0.5°. XPS measurements were performed using a PHI 5000 VersaProbe IV Scanning ESCA Microprobe (Physical Electronics) equipped with a monochromated Al Kα X-ray source (1486.6 eV). The X-ray source operated in high-power mode with a beam diameter of 200 μm, a power of 50 W, and a lamp voltage of 15 kV. Samples were prepared by filling a Teflon sample cap and securing it to the XPS holder with insulating tape. Detailed spectra were collected with a step size of 0.2 eV, a dwell time of 50 ms, and an analyzer pass energy of 27 eV, while survey spectra were measured at a pass energy of 127 eV. The chamber pressure during measurements was maintained between 10−7 Pa and 10−6 Pa. Data analysis was conducted using CasaXPS software. The MoS2 3d orbitals were fitted using an asymmetric Gaussian–Lorentzian function (A(0.35, 0.4, 0) GL(20)), while the MoOx 3d and S 2p orbitals were fitted with SGL(80) and GL(10) functions, respectively.4.5 Electrochemical characterization
For supercapacitive test, symmetrical Swagelok-cells were built using 1 M H2SO4 solution as electrolyte. As separator SiO2 fibrous membranes from Whatman were used. CVs were recorded using an Autolab galvanostatic and potentiostatic in the range from 0–0.8 V with scan steps of 2.44 mV at different scan rates of 10 mV s−1, 20 mV s−1, 50 mV s−1, 100 mV s−1, 150 mV s−1, 200 mV s−1, 250 mV s−1, and 500 mV s−1. Sample stabilization was done ranging from −0.1–0.1 V below and above the open circuit potential for 20 cycles. The sample cycling was done 6 times to ensure the equilibrium state of samples and only the polygon area of the last cycle was used for calculating the specific capacitance. The GCD tests were conducted at various current density of 1 A g−1. EIS measurements were done using the Autolab including the impedance module, from the frequency range of 0.1 Hz up to 100.000 Hz in combination with a Swagelok electrochemical cell.For HER test, the electrocatalyst inks containing 5.0 mg mL−1 of the ground samples, and 30 μL mL−1 of Nafion were prepared. The inks were first sonicated for 1 h in a water bath, then drop casted on a glassy carbon with a diameter of 3 mm. The electrochemical cell was set up using the previously prepared GC as a working electrode, a Pt wire as the counter electrode, an Ag/AgCl electrode as a reference electrode. A 0.5 M H2SO4 solution (pH 0) was used as electrolytes. All the measurements were performed at room temperature (298 K). At the beginning of each measurement, CV with a scan rate of 50 mV s−1 were performed to stabilize the samples. Then, LSV measurements were measured at a scan rate of 10 mV s−1.
Author contributions
Conceptualization, F.B., M.W. and T.G.; methodology, F.B., M.C., and M.P.; formal analysis, M.W., M.P., M.C. and F.B.; resources, T.G.; data curation, M.W.; writing—original draft preparation, F.B.; writing—review and editing, M.W. and T.G.; visualization, M.W.; supervision, T.G. and M.W.; project administration, T.G.; funding acquisition, M.W. and T.G. All authors have read and agreed to the published version of the manuscript.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
F. B., M. C., B. S. and T. G. thank the European Commission for the project LIGHT CAP (grant agreement no. [101017821]). T. G. acknowledges the support of the European Research Council for the project JANUS BI (grant agreement no. [101041229]). M. W. and T. G. also thank Fondazione Compagnia di San Paolo for financial support through the “Bando TRAPEZIO – Paving the way to research excellence and talent attraction”. T. G. is also further grateful to Compagnia di San Paolo for the support through the Starting Grant ERC program. We acknowledge the help of Sara Domenici and Dr Daniele Versaci for their assistance in electrochemical experiments.References
- R. Gupta, P. Sahni, S. Sharma, A. Roy and A. K. Pal, Sustainable Energy Fuels, 2022, 6, 1891–1922 RSC.
- Y. Zhou, H. Qi, J. Yang, Z. Bo, F. Huang, M. S. Islam, X. Lu, L. Dai, R. Amal, C. H. Wang and Z. Han, Energy Environ. Sci., 2021, 14, 1854–1896 RSC.
- Y. Peng, X. Zhang, R. Sun, X. Zhang, C. Ge and Y. Liu, J. Mater. Chem. A, 2024, 12, 32566–32592 RSC.
- L. F. Chen, Y. Lu, L. Yu and X. W. Lou, Energy Environ. Sci., 2017, 10, 1777–1783 RSC.
- F. Boll, M. Crisci, L. Merola, F. Lamberti, B. Smarsly and T. Gatti, Adv. Energy Sustainable Res., 2023, 4, 2300121 CrossRef CAS.
- Q. Guo, X. Zhou, X. Li, S. Chen, A. Seema, A. Greiner and H. Hou, J. Mater. Chem., 2009, 19, 2810–2816 RSC.
- T. Kshetri, D. T. Tran, D. C. Nguyen, N. H. Kim, K. tak Lau and J. H. Lee, Chem. Eng. J., 2020, 380, 122543 CrossRef CAS.
- A. S. Levitt, M. Alhabeb, C. B. Hatter, A. Sarycheva, G. Dion and Y. Gogotsi, J. Mater. Chem. A, 2018, 7, 269–277 RSC.
- Y. Liu, G. Jiang, S. Sun, B. Xu, J. Zhou, Y. Zhang and J. Yao, J. Alloys Compd., 2018, 731, 560–568 CrossRef CAS.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Kang, Electrochim. Acta, 2012, 75, 213–219 CrossRef CAS.
- S. Jeon, J. H. Jeong, H. Yoo, H. K. Yu, B. H. Kim and M. H. Kim, ACS Appl. Nano Mater., 2020, 3, 3847–3858 CrossRef CAS.
- G. Zhang and X. W. D. Lou, Sci. Rep., 2013, 3, 1–6 Search PubMed.
- J. Xu, L. Zhang, G. Xu, Z. Sun, C. Zhang, X. Ma, C. Qi, L. Zhang and D. Jia, Appl. Surf. Sci., 2018, 434, 112–119 CrossRef CAS.
- L. Wang, W. He, D. Yin, H. Zhang, D. Liu, Y. Yang, W. Yu and X. Dong, Renewable Sustainable Energy Rev., 2023, 181, 113354 CrossRef CAS.
- S. Surendran, S. Shanmugapriya, A. Sivanantham, S. Shanmugam and R. Kalai Selvan, Adv. Energy Mater., 2018, 8, 1800555 CrossRef.
- Y. Chen, G. Tian, Z. Ren, K. Pan, Y. Shi, J. Wang and H. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 13841–13849 CrossRef CAS PubMed.
- M.-Q. Wang, C. Ye, H. Liu, M. Xu and S.-J. Bao, Angew. Chemie, 2018, 130, 1981–1985 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- M. W. Iqbal, K. Shahzad, R. Akbar and G. Hussain, Microelectron. Eng., 2020, 219, 111152 CrossRef CAS.
- J. Theerthagiri, R. A. Senthil, B. Senthilkumar, A. Reddy Polu, J. Madhavan and M. Ashokkumar, J. Solid State Chem., 2017, 252, 43–71 CrossRef CAS.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147–150 CrossRef CAS PubMed.
- S. Palencia-Ruiz, D. Uzio, C. Legens, D. Laurenti and P. Afanasiev, Appl. Catal., A, 2021, 626, 118355 CrossRef CAS.
- C. Nagaraju, C. V. V. M. Gopi, J. W. Ahn and H. J. Kim, New J. Chem., 2018, 42, 12357–12360 RSC.
- E. G. Da Silveira Firmiano, A. C. Rabelo, C. J. Dalmaschio, A. N. Pinheiro, E. C. Pereira, W. H. Schreiner and E. R. Leite, Adv. Energy Mater., 2014, 4, 1–8 Search PubMed.
- Z. Liu, L. Zhao, Y. Liu, Z. Gao, S. Yuan, X. Li, N. Li and S. Miao, Appl. Catal., B, 2019, 246, 296–302 CrossRef CAS.
- D. Wang, X. Zhang, S. Bao, Z. Zhang, H. Fei and Z. Wu, J. Mater. Chem. A, 2017, 5, 2681–2688 RSC.
- X. Gan, L. Y. S. Lee, K. Y. Wong, T. W. Lo, K. H. Ho, D. Y. Lei and H. Zhao, ACS Appl. Energy Mater., 2018, 1, 4754–4765 CrossRef CAS.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- L. Zhang, W. Fan and T. Liu, Nanoscale, 2016, 8, 16387–16394 RSC.
- H. Wu, X. Chen, C. Qian, H. Yan, C. Yan, N. Xu, Y. Piao, G. Diao and M. Chen, Small, 2020, 16, 1–10 CrossRef PubMed.
- F. Boll, M. Fadda, M. Happel, M. Crisci, A. Athanassiou, B. Smarsly, F. Bella, F. Lamberti, G. Perotto and T. Gatti, ACS Appl. Energy Mater., 2024, 7, 4733–4744 CrossRef CAS.
- H. Niu, Z. Zou, Q. Wang, K. Zhu, K. Ye, G. Wang, D. Cao and J. Yan, Chem. Eng. J., 2020, 399, 125672 CrossRef CAS.
- A. Li, Y. Hu, M. Yu, X. Liu and M. Li, Int. J. Hydrogen Energy, 2017, 42, 9419–9427 CrossRef CAS.
- C. Zhang, Z. Wang, S. Bhoyate, T. Morey, B. L. Neria, V. Vasiraju, G. Gupta, S. Palchoudhury, P. K. Kahol, S. R. Mishra, F. Perez and R. K. Gupta, C, 2017, 3, 33 Search PubMed.
- F. N. I. Sari and J. M. Ting, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS PubMed.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef CAS.
- B. Adilbekova, Y. Lin, E. Yengel, H. Faber, G. Harrison, Y. Firdaus, A. El-Labban, D. H. Anjum, V. Tung and T. D. Anthopoulos, J. Mater. Chem. C, 2020, 8, 5259–5264 RSC.
- B. Chakraborty, H. S. S. R. Matte, A. K. Sood and C. N. R. Rao, J. Raman Spectrosc., 2013, 44, 92–96 CrossRef CAS.
- Y. Wang, C. Cong, C. Qiu and T. Yu, Small, 2013, 9, 2857–2861 CrossRef CAS PubMed.
- M. Wen, X. Chen, Z. Zheng, S. Deng, Z. Li, W. Wang and H. Chen, J. Phys. Chem. C, 2021, 125, 765–773 CrossRef CAS.
- L. Seguin, M. Figlarz, R. Cavagnat and J. C. Lassègues, Spectrochim. Acta, Part A, 1995, 51, 1323–1344 CrossRef.
- J. Z. Ou, J. L. Campbell, D. Yao, W. Wlodarski and K. Kalantar-Zadeh, J. Phys. Chem. C, 2011, 115, 10757–10763 CrossRef CAS.
- X. Zhang, X. F. Qiao, W. Shi, J. Bin Wu, D. S. Jiang and P. H. Tan, Chem. Soc. Rev., 2015, 44, 2757–2785 RSC.
- J. Fu, H. Qiao, D. Li, L. Luo, K. Chen and Q. Wei, Sensors, 2014, 14, 3543–3556 CrossRef CAS PubMed.
- G.-H. Nam, Q. He, X. Wang, Y. Yu, J. Chen, K. Zhang, Z. Yang, D. Hu, Z. Lai, B. Li, Q. Xiong, Q. Zhang, L. Gu, H. Zhang, G. Nam, Q. He, J. Chen, D. Hu, Z. Lai, H. Zhang, X. Wang, Q. Xiong, Y. Yu, K. Zhang, Q. Zhang, Z. Yang, L. Gu and B. Li, Adv. Mater., 2019, 31, 1807764 CrossRef PubMed.
- M. Z. Iqbal, S. Khan, A. M. Alsharari, M. Shakil, A. M. Afzal, A. Kumar, N. Badi, V. Mishra and A. Dahshan, J. Phys. Chem. Solids, 2025, 200, 112585 CrossRef CAS.
- K. Qi, X. Cui, L. Gu, S. Yu, X. Fan, M. Luo, S. Xu, N. Li, L. Zheng, Q. Zhang, J. Ma, Y. Gong, F. Lv, K. Wang, H. Huang, W. Zhang, S. Guo, W. Zheng and P. Liu, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00711a |
| This journal is © The Royal Society of Chemistry 2025 |