
Quantification probing of available extra capacity: interfacial space-charge storage in FeOOH lithium-ion batteries†
Leqing
Zhang‡
a,
Zeyuan
Bu‡
a,
Haoyu
Fu
a,
Xiaoshan
Wang
*a,
Xianyi
Meng
a,
Meishuo
Liu
a,
Yakun
Zhou
a,
Xiang
Sui
a,
Guihuan
Chen
 a,
Qinghao
Li
a and
Qiang
Li
*ab
a,
Qinghao
Li
a and
Qiang
Li
*ab
aCollege of Materials Science and Engineering, College of Physics, Weihai Innovation Research Institute, Qingdao University, Qingdao 266071, China. E-mail: wangxiaoshan1@qdu.edu.cn; liqiang@qdu.edu.cn
bDepartment of Electrical and Computer Engineering, Institute for Quantum Computing, University of Waterloo, Ontario N2L 3G1, Canada
First published on 29th July 2024
Abstract
Extra capacity beyond a theoretically predicted value has been widely acknowledged in transition metal-based anodes for application in lithium batteries, but the precise definition of their charge storage behaviors remains elusive owing to the intricate and dynamic interior space. Herein, an urchin-like FeOOH nanostructure is fabricated to reveal the dynamic electrochemical evolution of extra capacity. It is found that electrons continually accumulate at the interface between Fe and the Li2O/LiOH matrix at a narrow discharge voltage range of 1–0 V, forming a space-charge storage region, thus enabling extra capacity besides the capacity from the reversible conversion reaction. Moreover, using advanced operando magnetometry technology, the significant interfacial space-charge storage behavior could be accurately quantified to be 239.1 mA h g−1. In contrast, the evolution of the partially reversible solid-state electrolyte interface (SEI) functions as a diminished additional source of capacity, significantly lowering the coulombic efficiency and stability of batteries. Further, by fully exploiting the potential of the reversible interfacial space-charge storage mechanism, a well-designed lithium-ion hybrid capacitor is constructed using a reduced FeOOH anode and commercial AC cathode, achieving a high energy density of 140.4 W h kg−1 and a remarkable capacity retention of ∼100% after 1000 cycles at 1 A g−1. This study offers a broad insight into the extended utilization of extra capacity, paving the way for a sustainable strategy in the development of new energy batteries.
Introduction
The relentless quest for enhanced performance in lithium-ion batteries serves as the primary motivation for the advancement of next-generation electrochemical devices, with capacity standing as a paramount criterion.1–4 Currently, besides the capacity that comes from conversion reactions, additional capacity exceeding theoretical limits, known as extra capacity, has been identified.5 Transition metal-based materials in lithium-ion battery anodes offer a promising avenue owing to their accommodation of this extra capacity. For example, it has been observed that extra capacities exceeding 136.9 mA h g−1 can be achieved in (CNT)/FeS2 after 100 cycles, and values as high as 428.3 mA h g−1 can be achieved in Fe2.76Sn0.24O4/rGO after the same number of cycles.6,7 Furthermore, Co2(OH)2CO3-GO nanocomposites demonstrate a contribution of extra capacity up to 249% after 500 cycles.8 These supplementary capacities not only improve the energy density and longevity of batteries but also play a crucial role in advancing the field of energy storage.Although the extra capacity has been acknowledged, its formation mechanism remains a topic of debate in academic circles.9 Various hypotheses, such as the solid electrolyte interphase (SEI) film establishment, interfacial space-charge storage, and structural alterations, may account for this phenomenon.5 In general, the formation of an SEI film typically occurs during the initial lithium insertion process, involving complex electrochemical and physical processes that are hard to observe. Nevertheless, the durability of an SEI film is limited by chemical reactions and mechanical stress, thus providing an insufficient explanation for the increased capacity observed in the low-voltage region.10–12 Enhanced by advanced material design and surface modification, the highly efficient and stable SEI layer has shown significant improvements in battery performance and lifespan.13 Moreover, Balaya et al. introduced a novel theoretical framework for RuO2 lithium batteries, highlighting interfacial storage capacity via charge separation between nano-metal and Li2O grains.14 This lithium storage mechanism is precisely stated as interfacial space-charge storage in the following discussion. However, the conclusive identification of electrons remains challenging due to the metastable nature of interfacial charge storage and the inherent limitations of electron-level detection techniques.15 Sources of extra capacity for electrode substance conversion remain an elusive area in current knowledge.
Operando magnetometry technology is an electron-level detection technology for the exploration of electrochemical physical interfaces.15,16 It is found to be sensitive in monitoring the charge transfer and phase evolution in electrochemical systems, which would facilitate a comprehensive understanding of the space charge distribution in the inaccessible electrode interfaces.9,17–28 Klinser et al. conducted in situ magnetic susceptibility detection in LixNi1/3Mn1/3Co1/3O2 cathodes to elucidate the charge compensation process of Ni2+/Ni3+/Ni4+ during the charging cycle.24 Further, for operando magnetometry in sodium-ion batteries, the incomplete reaction within FeS2 cores has been identified, indicating that the smaller iron nanoparticles reduced the kinetics.21 More importantly, in prior investigations into extra capacity, the designed in situ magnetometry has tracked the electronic structure evolution within typical Fe3O4 lithium-ion batteries, revealing substantial surface capacitance on metallic nanoparticles.9 This study directly confirmed the extra capacity associated with the interfacial space-charge storage mechanism, where spin-polarized electrons and lithium ions are individually stored at the interface of ionic and electronic conductors. However, the mechanisms underlying other roots of extra capacity, particularly in the function of SEI films, remain inadequately explored. To gain a more comprehensive understanding of various extra capacity mechanisms with potential applications, it is imperative to employ operando magnetometry technology for deeper insights into their complex interactions.
Herein, we delve into the underlying mechanisms of the extra capacity in an urchin-like FeOOH nanoparticle anode in LIBs and propose an effective way to benefit from it. Through the probing of operando magnetometry, a significant interfacial space-charge storage mechanism is determined to be responsible for the extra capacity. Within the narrow voltage range from 0 V to 1 V, a high extra capacity of 239.1 mA h g−1 is quantitatively observed, corresponding to the calculated value derived from spin-polarized charge analysis. However, the results of transmission electron microscopy (TEM) and impedance testing indicate that the additional capacity provided by the SEI film in FeOOH is irreversible, leading to capacity fading and low coulombic efficiency. In the designed Fe-reduced FeOOH//activated carbon (AC) asymmetric supercapacitors, a remarkable energy density of 140.4 W h kg−1 and a high-capacity retention of 99.6% after 1000 cycles at 1 A g−1 are achieved. These insights lay the groundwork for the development of next-generation anodes tailored for both high-capacity metal-based hydroxide batteries and supercapacitors.
Results and discussion
Two-dimensional FeOOH is synthesized via a typical one-step hydrothermal method and illustrated by the structure characterization in Fig. 1. As depicted in Fig. 1a, the composition of FeOOH is confirmed by the powder X-ray diffraction (XRD) patterns of JCPDS No. 81-0463.29 The predominant peaks located at 21.2°, 33.2° and 36.6° are assigned to the (110), (130) and (111) planes, respectively. As is shown in the scanning electron microscopy (SEM) and the transmission electron microscopy images, FeOOH appears as urchin-like nanospheres featuring radial nanorods, with a length of ∼0.5 μm (Fig. 1b and c). In detail, the interplanar distance of FeOOH nanorods is measured to be 0.404 nm by high-resolution TEM (Fig. 1d and e), corresponding to the (110) plane. The high index crystal surface of FeOOH (110) contributes to the exposure of hydroxyl active sites, improving the electrochemical reactivity.30 The corresponding selected-area electron diffraction (SAED) pattern in Fig. 1f exhibits the circular diffraction of FeOOH nanoparticles, which is consistent with the XRD patterns. Subsequently, X-ray photoelectron spectroscopy (XPS) characterization (Fig. 1g and S1†) is further conducted to explore the detailed elemental composition and bonding characteristics of pure FeOOH. The high-resolution XPS spectra of Fe reveal two spin–orbit coupling peaks at 724.6 eV and 711.7 eV, assigned to the Fe3+ 2p1/2 and Fe3+ 2p3/2, respectively.31,32 In addition, the O 1s spectrum of FeOOH possesses two characteristic peaks for Fe–O–H and Fe–O–Fe, located at 531.6 eV and 530.3 eV, respectively.33 It is found that the integrated areas of the two peaks are equal in value, suggesting the high purity of the as-synthesized FeOOH.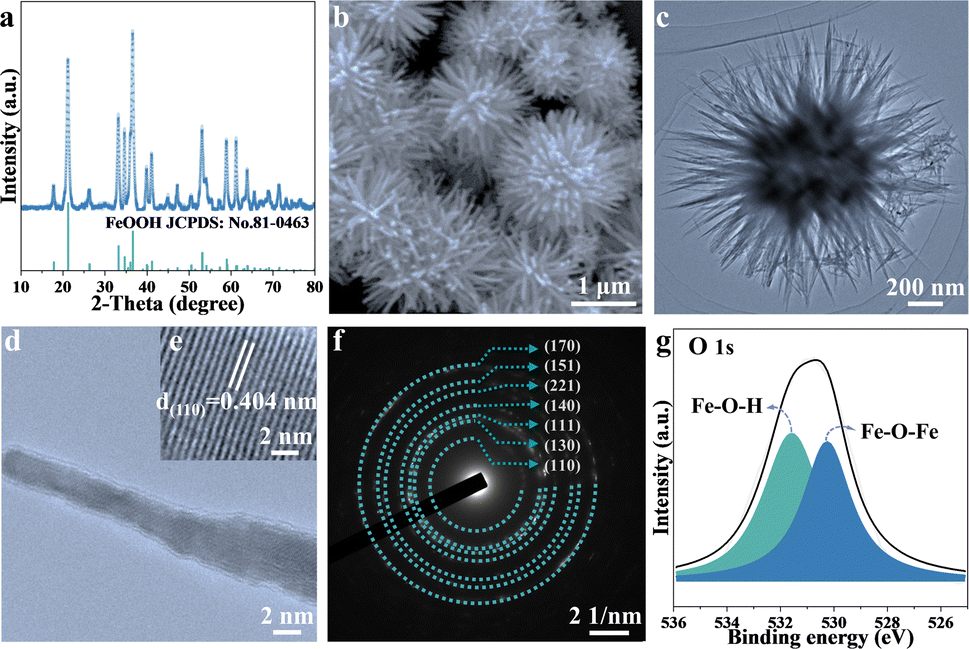 | ||
| Fig. 1 Structural characterization of FeOOH anodes. (a) XRD pattern. (b) SEM image. (c) TEM image. (d) HRTEM image of the nanorod and (e) lattice fringe inside. (f) SAED image. (g) The high resolution XPS of O 1s. | ||
As illustrated in Fig. S2,† the magnetic hysteresis curve exhibits paramagnetic characteristics. Compared with the robust ferromagnetism of Fe, the modest magnetization of 0.95 emu g−1 for the goethite α-FeOOH samples under a high magnetic field (3 T) is negligible, yet it provides a feasible approach for monitoring the evolution of iron in FeOOH LIBs based on the distinguished magnetization intensity mentioned above so that magnetic technology would be capable of revealing electrochemical behaviors.
The prepared FeOOH is assembled into half-coin cells with lithium counter electrodes, and their electrochemical performances are shown in Fig. S3.† The specific capacities of the FeOOH anode for the first charge and discharge are 1238.2 and 1838.4 mA h g−1, respectively (Fig. S3a†). Obviously, the capacity significantly exceeds the theoretical limit (903 mA h g−1), well consistent with the charge–discharge curves in previous reports. The rate capability of the FeOOH electrode at different current rates from 0.1 to 1 A g−1 is demonstrated in Fig. S3b.† It delivers a reversible capacity of 758.5 mA h g−1 at a high current density of 1 A g−1. When the current rate returns to 0.1 mA h g−1, nearly 100% of the original capacity reverts. Further, the FeOOH electrode maintained a high capacity of 847.5 mA h g−1 and 839.6 mA h g−1 over 200 cycles at 0.2 A g−1 and 1 A g−1, respectively, ensuring its prolonged usage and superior performance in various applications (Fig. S3c and S4†).
To figure out the Li-ion storage mechanisms in FeOOH, we employed operando magnetometry to monitor charge transfer during the electrochemical processes of operando pouch test cells, driven by cyclic voltammetry (CV) measurement. As demonstrated in the cyclic voltammetry profiles (Fig. 2a), operando cells exhibited nearly identical electrochemical behavior compared to coin cells.34 In the first discharge process, the FeOOH anode exhibited two prominent peaks at 1.68 V and 1.45 V, corresponding to the initiation of intercalation lithiation in the active sites and deep intercalation of lithium (Step I and Step II). Notably, the outstanding peak at 0.78 V could be attributed to the formation of the SEI layer and the conversion reaction (Step III). It is worth noting that SEI is commonly considered the donor of extra capacity but failed to quantitative due to the uncertain interface reaction.35,36 In the first charge scan, the decomposition peak of the SEI layer appeared at 1.06 V. And the following broad peak around 1.5 V was recognized as the oxidation of iron, following reversible pathways from Step III′ to Step I′.34,37 In the next cycles, the conversion reaction peak becomes less pronounced, correlating with the reduced and more uniform voltage plateaus observed in the galvanostatic discharge–charge curves (Fig. S3a†). This consistent trend may be attributed to structural alterations within the electrode. Due to the high consistency of CV and charge and discharge curves, the operando magnetic monitoring is coupled with CV scanning at 0.5 mV s−1 to bring about a more refined conversion reaction at the interface.
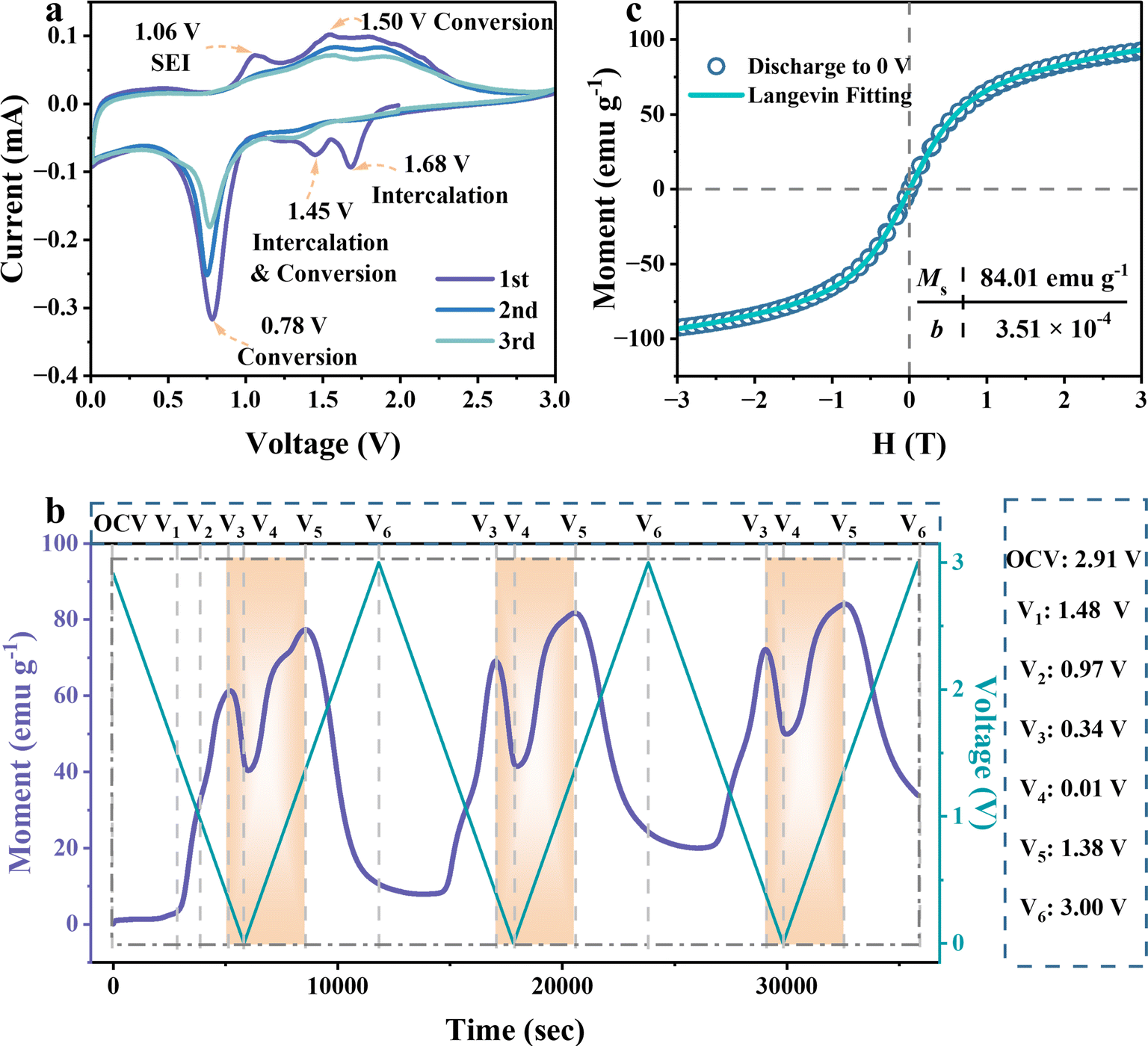 | ||
| Fig. 2 Electrochemical reaction mechanism of FeOOH LIBs. (a) CV curves for the first three cycles at a scan rate of 0.5 mV s−1 over the voltage range of 0.01–3 V. (b) Operando magnetic monitoring followed the voltage range of CV scanning under a magnetic field of 3 T. (c) Magnetic hysteresis loop with the Langevin fitting curve after the first full discharge measured at 300 K. | ||
The whole phase evolution of the FeOOH electrode during cycling is well and truly investigated by operando magnetometry. Fig. 2b presents CV curves alongside the time-sequenced magnetization of the FeOOH electrode, juxtaposed with the corresponding potential response observed throughout the initial three cycles. The periodic variation in magnetization illustrates a reversible and steady electrochemical process. Specifically, the voltages of the CV curves corresponding to distinct peaks and valleys in the real-time magnetization variation are labeled as V1 to V6. In the initial magnetic response, there is a negligible magnetization variation from open circuit voltage (OCV) to 1.48 V, indicating that the electrode structure of lithium-embedded FeOOH remains constant with negligible paramagnetism (Lix+yFeOOH). During the subsequent lithiation step from 1.48 V to 0.97 V, the pronounced increase in magnetization could be assigned to the accumulation of reduced Fe, since iron possesses strong magnetic properties. With the continuous generation of Fe particles, the metallic Fe keeps storing spin-polarized electrons, thus reducing the slope of the magnetization increase and reaching the first peak at 0.34 V. In particular, a rapid decline of magnetization occurs from 0.34 V to 0.01 V, implying that a large amount of charge persists to be stored in the reduced Fe nanocrystallite. As soon as the spin-polarized electrons are released from the metallic Fe, the rise trend of magnetization is resumed from 0.01 V to 1.38 V in the charge process. Generally, the first falling and then rising magnetization variation is highlighted in orange, revealing the reversibility of the extra capacity in FeOOH LIBs. Their intrinsic relationship will be discussed in the subsequent sections (Fig. 3c). Finally, the oxidation of Fe from 1.38 V to 3 V induces a rapid decline in magnetization. At the end of charging, the magnetization intensity cannot return to the initial negligible level, indicating the incomplete oxidation of ferromagnetic Fe.20,34,37 These operando magnetometry results are in good agreement with the electrochemical analysis process.
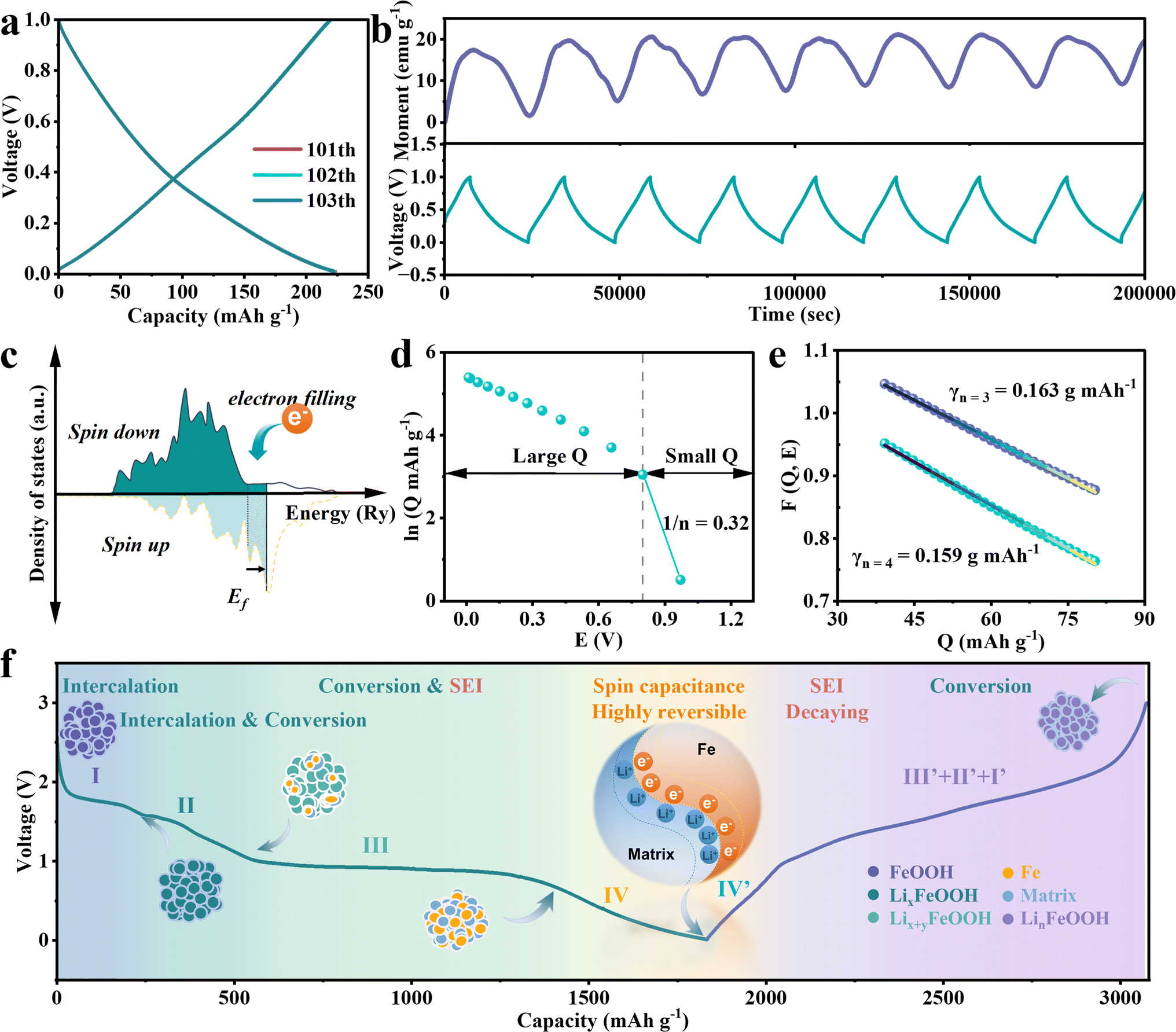 | ||
| Fig. 3 Reversible interface charge storage mechanism in an assigned voltage range of 0.01–1 V in FeOOH LIBs. (a) Galvanostatic charge–discharge curves after 100 cycles. (b) Constant operando magnetometry under an applied magnetic field of 3 T. (c) Schematic illustrating the spin-polarized density of states near the Fermi energy surface in reduced ferromagnetic metal grains. (d) Dependence of interfacial charge storage and lithium activity in Fe/matrix nanocomposites. (e) Thermodynamic fitting of the interfacial charge storage process. (f) Illustration of the space-charge interface responsible for extra lithium storage. | ||
To clarify the properties of reduced Fe, the magnetic hysteresis curve is collected after the first total discharge to 0 V. It shows an unsaturable magnetization without coercive force, revealing superparamagnetic behavior at the nanoscale (Fig. 2c). Fitted by the Langevin equation, the diameter of the resulting superparamagnetic Fe nanoparticles, produced by lithium-induced conversion, is calculated to be 2.6 nm (Section I in ESI†). The fitting value of saturated magnetization is 84.01 emu g−1, supporting that the enhanced magnetization intensity originates from the continuous reduction of superparamagnetic iron nanoparticles. Later, the transformation of FeOOH into smaller Fe nanoparticles and their dispersion in the Li2O/LiOH matrix are confirmed by the relevant SAED and HRTEM images (Fig. S5†). The diameter of Fe nanoparticles is measured to be 2 nm–5 nm, which is consistent with the Langevin fitting results. In this case, the interface between Fe nanoparticles and the Li2O/LiOH matrix would be expanded to facilitate the spin polarization coupling for extra capacity.
To further investigate the oxidation states of iron, we conducted a series of characterization tests. The quasi-in situ XRD patterns of the electrodes (Fig. S6†) exhibit distinct patterns at varying lithiation states. The voltage indications align precisely with those shown in Fig. 2b. These patterns reveal that upon discharging to 1.5 V, the material transforms from a crystalline into an amorphous state during subsequent cycles. This observation agreed with previous reports.34,38 Furthermore, only trace amounts of characteristic Fe peaks were detected in the following discharge, consistent with operando magnetic monitoring. Corresponding high-resolution XPS of Fe species has witnessed their variation in valence (Fig. S7†). Specifically, on discharging to 1.48 V, Fe3+ is gradually reduced to Fe2+, located at 709.6 eV and 723.1 eV, respectively.39 On discharging to 0.97 V, Fe0 exhibits a characteristic peak at 706.8 eV and 719.5 eV and completely dominates the spectrum at the end of the discharge.22 However, after charging, the signals of Fe0 and Fe2+ still persist, indicating incomplete reversible electrode reduction reactions (Fig. S8†). In summary, the XPS findings align with the demonstrated electrochemical conversion mechanism from operando magnetometry.
The electronic structure and the local Fe environments during cycling between 0 V and 3 V are monitored by operando EPR at room temperature (Fig. S9†). The initial material shows a signal at g = 2.004 of lithium metal with the absence of Fe. This is likely due to strong superexchange interactions between Fe3+ ions at room temperature, resulting in an overall EPR-silent state. Upon lithiation to 1.42 V, only a slight drop in lithium metal signal intensity was observed. Subsequently, with deep discharging, a noticeable broad peak appeared, which originated from the delocalized electron in the reduced iron nanoparticles. During charging, the g-factor of delocalized electrons of Fe shifted to g = 2.092, denoted as trivalent.40,41 Upon full charging, the EPR intensity of the iron oxide state gradually increased, indicating that the electrode structure evolved with the oxidation of iron. Although in situ EPR results demonstrated changes in the electronic structure during the electrochemical process, it was difficult to perform a qualitative analysis at room temperature, especially in the oxidation state of iron after charging. Therefore, the quasi-in-situ MT testing was used to clarify the aforementioned issues (Fig. S10†). According to the Curie–Weiss law, the fitted effective magnetic moment of the charged material is 3.88 μB. Considering the low-spin Fe3+ and high-spin Fe2+, the calculated proportions of Fe3+ and Fe2+ are 42.3% and 57.7% (see Methods in Section VIII), respectively, close to their proportions in XPS (Fig. S8†). In conclusion, after charging, Fe was not completely oxidized to FeOOH but transformed into LinFeOOH.34
Based on the CV and operando magnetic analysis, the oxidation of Fe and the decomposition of SEI both exist in the voltage range above 1 V, which would complicate the elucidation of the mechanism underlying the observed extra capacity. Hence, a narrow voltage range from 0.01 V to 1 V is selected for further investigation of extra capacity. As is shown in Fig. 3a, after 100 cycles of discharge/charge processes, subsequent discharge/charge curves completely overlap for the next three times, accompanied by a coulombic efficiency of approximately 98.4% (Fig. S11†). The average highly reversible capacity reaches 237.1 mA h g−1 after 200 cycles (Fig. S12†). No reaction plateau is observed in these curves, indicating the domination of the capacitive behavior connected with extra capacity in this applied voltage range. Aiming to obtain a fully reduced electrode for the following operando magnetometry management within the low voltage of 0–1 V, the FeOOH LIBs underwent an adequate discharge process at a small current density of 0.05 A g−1. The operando magnetometry curves (Fig. 3b) vary monotonically as the voltage changes in the same direction. The magnetization of the electrode descends during discharging and then rises during charging. This result confirmed that the accumulation of charge on Fe nanoparticles contributes to the significant difference in the magnetization intensity. The characteristic of spin-splitting orbital energy levels at the surface of ferromagnetic metal grains imparts rich magnetic information to the electrode material, coupled with electron properties (Fig. 3c). The essence of ferromagnetism lies in the net magnetic moment, that is, the difference in magnetic moment between spin-up and spin-down electrons. During the discharge from 0.01 V to 1 V, the surplus electrons occupy the 3d orbital of iron, thereby shifting the Fermi level of iron towards higher energy states.9 The numerical preponderance of spin-up electrons over spin-down electrons in this process is noteworthy. This results in a continual diminution of the net magnetic moment of the iron particles, aligning with the experimental observations of a decrease in magnetization intensity. During charging, the opposite process occurs where electrons are removed. Thus, the magnetization exhibited a reversible decrease and increase in the voltage of the interfacial charge zone, inspired by reversible e−/Li+ storage. Considering the reported theoretical model of interfacial space-charge storage, we proposed that there is an indispensable interfacial space-charge mechanism in the FeOOH anode, which is the origin of extra capacity. An abundance of electrons can be accommodated and evacuated in the spin-splitting bands of the metallic Fe. Simultaneously, lithium ions undergo adsorption and desorption along the inner side of the Li2O/LiOH matrix, which is in contact with and adjacent to the above Fe nanoparticles. This behavioral mechanism induces excellent spin-polarized surface capacitance in Fe/matrix nanocomposites, as shown in the schematic diagram in Fig. 3f. To further evaluate the extra capacity contribution from this interfacial space capacitance mechanism, a series of theoretical computations combined with thermodynamic fitting are employed. The interfacial spin-polarized charge storage Q can be quantified by the change of magnetization ΔM (Section II in ESI†).42 We estimate a spin-polarized capacity (Q) of 185.3 mA h g−1–225.4 mA h g−1 with a magnetization of 17.4 emu g−1 in the voltage range of 0.01–1 V (Fig. 3b). The calculated value of the theoretical spin-polarized capacity is consistent with the experimental capacity of 239.1 mA h g−1 (Fig. 3b). Later, the thermodynamics evidence of the surface charge storage is shown in Fig. 3d and e. By computing the slope of the small Q region in the ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Q vs. E curve (Fig. 3d and Section III in ESI†), the concentration of conduction electrons n is calculated to be 3.125. This magnitude is in the expectation range of 3 < n < 4 when interfacial space-charge storage remains dominant.43 As for perfect linearization in the large Q region, the fitting γ between 0.163 g (mA h)−1 and 0.159 g (mA h)−1 is consistent with the ideal value on the order of ∼1.0 (Fig. 3e). The aforementioned results suggest that the liberation of electrons from the 3d bands of iron dominates the thermodynamics in low-voltage storage.
Q vs. E curve (Fig. 3d and Section III in ESI†), the concentration of conduction electrons n is calculated to be 3.125. This magnitude is in the expectation range of 3 < n < 4 when interfacial space-charge storage remains dominant.43 As for perfect linearization in the large Q region, the fitting γ between 0.163 g (mA h)−1 and 0.159 g (mA h)−1 is consistent with the ideal value on the order of ∼1.0 (Fig. 3e). The aforementioned results suggest that the liberation of electrons from the 3d bands of iron dominates the thermodynamics in low-voltage storage.
Considering the electrochemical behavior of FeOOH lithium-ion batteries, we implemented a comprehensive analytical approach. Initially, the galvanostatic intermittent titration technique (GITT) is applied to assess the kinetic parameters and diffusion characteristics of the batteries within 0–3 V (Fig. S13a†). The diffusion coefficient of Li+ decreases after 1.5 V, indicating that the kinetics of the conversion reaction is slower than the intercalation reaction. Notably, benefitting from the spin-capacitance behavior, the diffusion coefficient increases in a low voltage range, representing a faster charge transfer dynamics.44–46 This was followed by a detailed capacitance analysis to elucidate the pseudo-capacitive contributions. The CV curves at different scan rates in 0–3 V are examined (Fig. S13b†). The capacitive contribution ratio grows from 28.1% to 48.6% for 0.2 mV s−1 and 1 mV s−1, respectively (Fig. S13c†). To further confirm the pseudo-capacitance contribution by interfacial space-charge storage, a similar dynamics test of the space charge storage is performed within 0–1 V in reduced FeOOH LIBs. All the CV curves exhibit typical overlapping rectangle-like curves, indicating a reliable capacitance behavior dominated by interfacial space-charge storage (Fig. S14a†). Moreover, the extra capacity of interfacial space-charge storage is up to 73.7% at a large scan rate of 5 mV s−1 (Fig. S14b and S14c†). Furthermore, we employed b-value fitting to quantify the charge-transfer processes, enhancing our understanding of the reaction mechanisms at the electrode–electrolyte interface (Fig. S13d†). These values suggest that the electrochemical reaction kinetics are primarily governed by diffusion-limited behavior at cathodic and anodic conditions, as indicated by b values close to 0.5. Conversely, at 0.2 V and 0.5 V, the higher b values near 1 suggest a transition to surface-controlled capacitive behavior in the interfacial space-charge region.47
TEM images of the SEI film after the initial and second full discharge/charge revealed the quantified evolution of the SEI film. As the cycling proceeded, a smooth SEI film with a thickness of ∼6.8 nm was formed during the initial discharge (Fig. 4a), whereas a rough film with a thickness of 2.4 nm was observed after charging (Fig. 4b). It indicated that the transition of the SEI film is not fully reversible, which can significantly deteriorate the coulombic efficiency. After the second discharge and charge, the SEI film thickness became uneven and decreased to 4.9 nm (Fig. 4c) and 2.3 nm (Fig. 4d), respectively, caused by electrolyte erosion and oxidation. Note that the SEI film was primarily formed during the initial discharge, contributing significantly to the enormous additional capacity of FeOOH lithium-ion batteries. However, the irreversibility of SEI leads to capacity fading and a low coulombic efficiency of 77.3% for the first cycle. To further analyze the composition of the SEI film, XPS characterization was performed on the electrode material after discharge. As depicted in Fig. 4e and f, the components observed at approximately 290.8, 288.3, 285.7, 283.9, and 283.0 eV in the C 1s spectrum can be attributed to CO3, O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, C–H, and C–C bonds in the SEI film.13,28 With an increase in cycling, the relative content of C–O and O–CO in the C 1s XPS spectrum gradually increased, while CO3 dropped. It could be attributed to the decreased content of LiOH by the electrode structure change in cycling, because the LiOH matrix can catalyze lithium ethylene di-carbonate components in SEI to lithium ethylene mono-carbonate, resulting in more Li2CO3.48 While Li2CO3 serves as an ionic conductor, excessive accumulation within the SEI layer could potentially compromise its uniformity and stability.49 Therefore, the formation of SEI film can provide significant additional capacity but is irreversible, leading to reduced coulombic efficiency and poor cycling performance. A robust and reversible SEI film is anticipated to be a promising direction for future research endeavors.
O, C–O, C–H, and C–C bonds in the SEI film.13,28 With an increase in cycling, the relative content of C–O and O–CO in the C 1s XPS spectrum gradually increased, while CO3 dropped. It could be attributed to the decreased content of LiOH by the electrode structure change in cycling, because the LiOH matrix can catalyze lithium ethylene di-carbonate components in SEI to lithium ethylene mono-carbonate, resulting in more Li2CO3.48 While Li2CO3 serves as an ionic conductor, excessive accumulation within the SEI layer could potentially compromise its uniformity and stability.49 Therefore, the formation of SEI film can provide significant additional capacity but is irreversible, leading to reduced coulombic efficiency and poor cycling performance. A robust and reversible SEI film is anticipated to be a promising direction for future research endeavors.
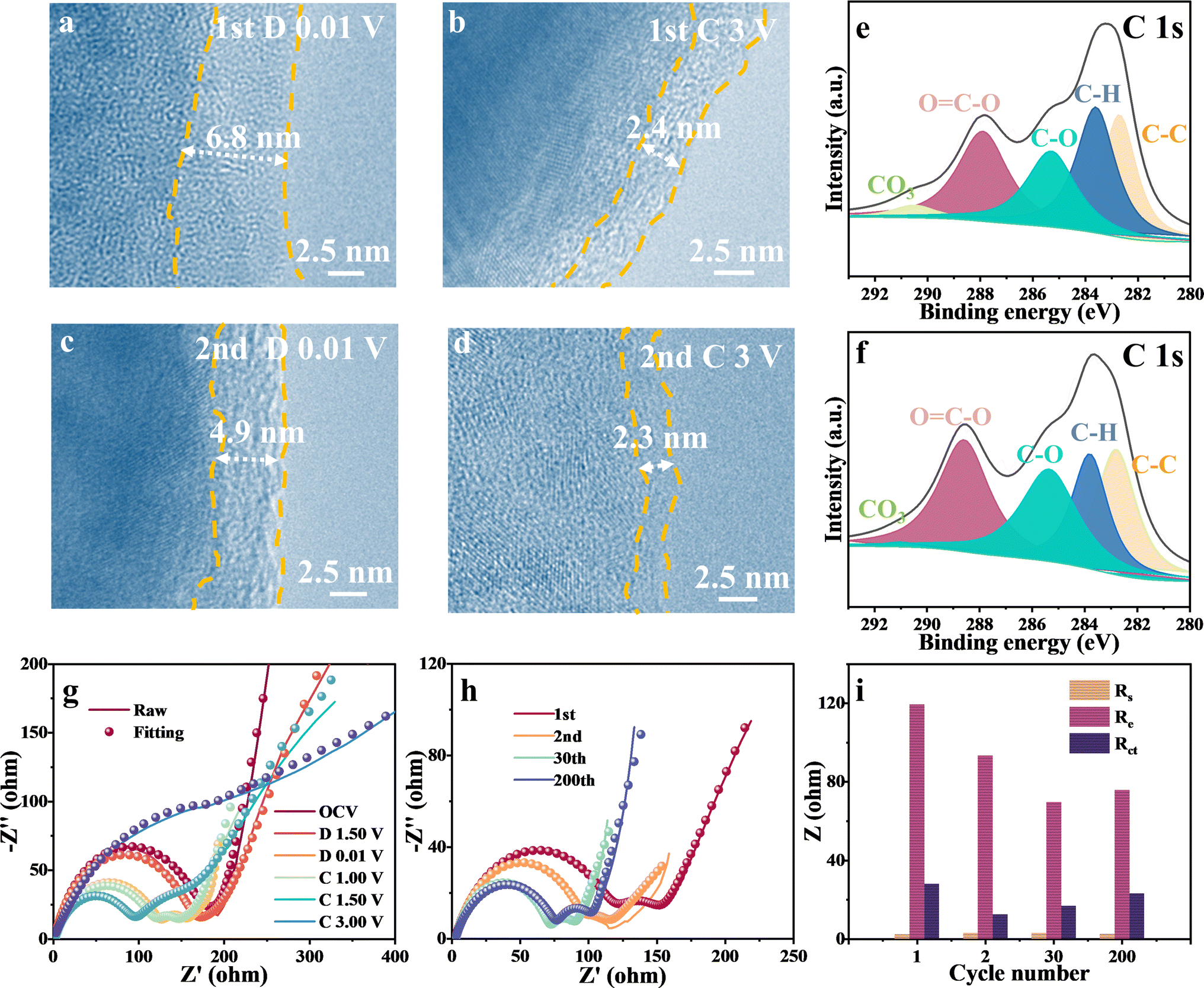 | ||
| Fig. 4 SEI and dynamic characterization of FeOOH LIBs. TEM images after the first discharge to (a) 0.01 V and (b) 3 V as well as the second discharge to (c) 0.01 V and (d) 3 V. (e) and (f) Corresponding high-resolution XPS C 1s spectra for (a) and (c), respectively. Nyquist plots and fitting plots (g) during discharge and charge and (h) after various cycles. (i) Impedance comparison. | ||
To investigate the SEI interface response and the phenomenon of reverse capacity decay, electrochemical impedance spectroscopy (EIS) was employed in FeOOH LIBs under different charge/discharge states (Fig. 4g and h). The equivalent circuit diagram is presented in Fig. S15.† During discharge from 1.2 V to 0.01 V, a distinct semicircle curve appeared in the high-frequency range, exhibiting typical SEI characteristics. Upon charging, there was no significant change in impedance until up to 1.5 V, in which the SEI-related impedance (Re) decreased to 89.6 Ω, indicating the decomposition of SEI film. Additionally, as the metal iron particles partially oxidized, the charge transfer impedance (Rct) began to increase, and the two impedances overlapped into a single semicircle after charging. The evolution of SEI film was further detected by the high-resolution XPS of O 1s (Fig. S16†). Upon discharging to 0 V, three characteristic peaks located at 532.8 eV, 531.3 eV and 529.2 eV were attributable to CO, Li2CO3/ROCO2Li and LiOH/Li2O.28 The SEI composition remained stable at 1 V until O–H and O–Fe bonds emerged at the end of the charge. A comparison of impedance and fitting parameters during discharge to 0.01 V at increasing cycles is shown in Fig. 4i. With increasing cycles, Re decreased significantly, suggesting irreversible degradation of the SEI film. However, a slight increase in Re was observed after 200 cycles. The unstable SEI film could account for the reverse capacity decay, which may be related to electrode material pulverization or SEI film cracking.5 These characterization results indicate that the SEI film in FeOOH LIBs exhibits incomplete reversibility, adversely affecting the electrochemical performance of the battery.
Inspired by the promising reversible interfacial capacitance, a reduced FeOOH//AC asymmetric supercapacitor is designed to carry it forward. The principle of the reduced FeOOH//AC lithium-ion capacitor (LIC) is shown in Fig. 5a. The double-layer cathode AC can absorb the anion PF6− quickly, while the pseudo-capacitor anode can store the cation Li+ and improve the specific capacity of the device. However, considering that the interfacial space-charge mechanism chemical kinetics in the pseudo-capacitor anode is far superior to the redox dynamics, we optimize the electrochemical voltage window of the capacitor, and only the interfacial space-charge effect in the anode is selectively retained. Thus, we preliminary took a set of measurements to determine the capacitive voltage range of 1.5–3.5 V (Fig. 5b and S17†). The quasi-rectangular shape cyclic voltammograms of the LIC demonstrate the dominant behavior of capacitance at both high and low scan rates.50,51 The galvanostatic charge/discharge curves for the device reveal the discharge time growth with the expansion of the voltage range from 3.1 V to 3.5 V (Fig. 5c). In the meantime the corresponding energy density is remarkably improved to 140.4 W h kg−1, achieving one of the best electrochemical performances in previous reports (Fig. S18a and Table S1†).52–62 Accordingly, the potential window of 1.5–3.5 V is chosen to further evaluate the electrochemical performance. The galvanostatic charge/discharge (Fig. S18b†) shows nearly symmetric triangular-shaped curves at different current densities from 0.1 to 2 A g−l, implying the combination of the faradaic process and the non-faradaic process. The presence of the non-faradaic process would provide extra capacitance for the capacitor.63,64 Furthermore, the long-term cycling test is conducted at 1 A g−1 over 1000 cycles and reveals a high-capacity retention of 99.6% (Fig. 5d). Superior to the traditional type, our home-designed LIC combines the virtues of double-layer capacitance and the spin-polarized capacitance of a Li-ion battery to enhance output energy capabilities.
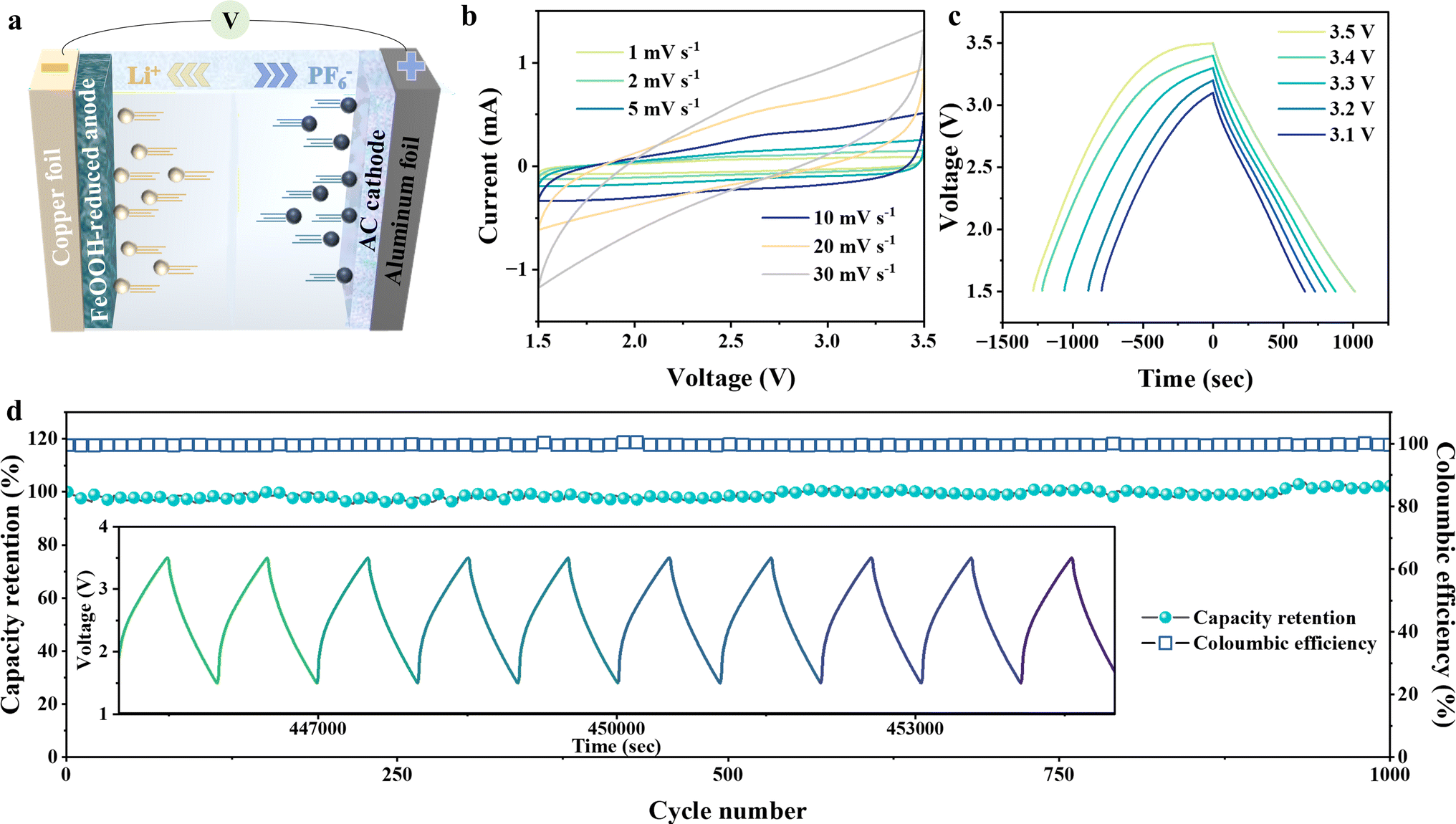 | ||
| Fig. 5 Designed application of interface capacitance in the asymmetric reduced FeOOH//AC lithium-ion capacitor. (a) Illustration of the capacitance behavior in the reduced FeOOH//AC LIC. (b) CV curves at various scan rates. (c) Galvanostatic charge/discharge voltage profiles across different voltage ranges at 0.2 A g−1. (d) Long-term cycling performance and corresponding coulombic efficiency within a voltage range of 1.5–3.5 V at 1 A g−1. The inset shows galvanostatic charge/discharge curves at a constant current density of 1 A g−1. | ||
Conclusions
In conclusion, beyond the perceived lithium storage mechanism involving intercalation–conversion reactions, the ultra-high extra capacity from interfacial space charge storage is deeply investigated on a well-defined FeOOH electrode by operando magnetometry technology, and the dynamic evolution is illustrated in Fig. 3f. Using typical thermodynamic and kinetic modeling analysis, the interfacial space-charge storage mechanism, especially in spin-polarization capacitance behavior, is determined at the interface between Fe nanoparticles and the matrix from 0 V to 1 V, revealing remarkable reversibility at 237.1 mA h g−1 even after 200 cycles. It is the first time to make a quantification of the interfacial space-charge storage effects in such an electrode system. We also found that the incomplete reversibility of the SEI film poses a severe detriment to the charge conversion efficiency and durability of the battery. Further, with engineering by the interface spin-capacitance, the novel LIC devices achieve excellent capacity retention with a high energy density of 140.4 W h kg−1. The findings would provide an advanced method to reveal the underlying mechanism in the closed electrochemical space and make a breakthrough in the quantitative analysis of available extra capacity.Experimental section
Material preparation
Firstly, 0.48 g iron(II) sulfate heptahydrate (FeSO4·7H2O) was dissolved in a mixed solution (40 mL H2O and 5 mL glycerin), followed by vigorous stirring for 60 minutes to form a homogeneous solution. Then the solution was transferred into a 50 mL Teflon-lined stainless-steel autoclave and a hydrothermal process was carried out that lasted 12 hours at 110 °C. After natural cooling, the precipitate was collected by filtration, washed with deionized water and ethanol, and dried at 60 °C overnight, respectively. Finally, a yellowish-brown solid, denoted as FeOOH, was obtained.Characterization
The crystal structure of the samples was characterized by X-ray diffraction (XRD) using a Bruker Discover 8 Diffractometer with Cu-Kα radiation (λ = 1.5406 Å). The valence states and surface compositions were analyzed by X-ray photoelectron spectroscopy (XPS) on an Axis Ultra DLD spectrometer equipped with a Mg Kα source (hν = 1283.3 eV). The morphology of the as-prepared samples was examined using a JEOL JSM-7100F field emission scanning electron microscope (SEM) and transmission electron microscope (TEM).Electrochemical measurements
The electrochemical evaluation was conducted with CR2032 coin cells by applying lithium metal as an anode. Active electrodes were made from homogeneous slurries containing 70 wt% active material, 20 wt% conductive carbon Super P, and 10 wt% carboxymethyl cellulose (CMC) in deionized water, and subsequently coated onto copper foil. The electrolyte consisted of 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), and ethyl methyl carbonate (EMC) in a volume ratio of 1:1:1. All CR2032 coin-type cells and soft-packaging cells for magnetometry were assembled in an argon-filled glovebox. Galvanostatic charge/discharge performance and cyclic voltammetry (CV) curves were measured using a Neware battery system (CT-ZWJ-4′S-T-1U) and an electrochemical workstation (CHI660E), respectively. The reversible specific capacities were calculated based on the mass loading of the anode materials. A pure Super P electrode was prepared from homogeneous slurries containing 70 wt% Super P and 30 wt% CMC in deionized water. Activated carbon (AC) was mixed with conductive carbon Super P and CMC in a weight ratio of 7:2:1 to form the cathode of supercapacitors. Before testing in a low-voltage range and the assembly of supercapacitors, the FeOOH LIBs underwent a reduction process, specifically by discharging them to 0.01 V to form Fe/matrix interfaces. The reduced FeOOH//AC asymmetric supercapacitors were constructed using preactivated cathode and anode materials with a mass ratio of 1:1.5.
Magnetic characterization
The magnetic properties were measured using a physical property measurement system (PPMS, Quantum Design) at a temperature of 300 K. The magnetism was assessed in response to the electrochemical processes of soft-packaging cells driven by CV measurements or galvanostatic charge/discharge under a magnetic field of 3 Tesla. All specific mass magnetization intensities were quantified based on the mass of FeOOH.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
All authors have approved the final version of the manuscript. Leqing Zhang: investigation & methodology, data curation, software, writing – original draft, and writing – review & editing. Zeyuan Bu and Xiaoshan Wang: formal analysis, funding acquisition, and writing – review & editing. Haoyu Fu and Xianyi Meng: investigation, methodology, validation and visualization. Meishuo Liu and Yakun Zhou and Xiang Sui: data curation and software. Guihuan Chen and Qinghao Li: formal analysis and supervision. Qiang Li: conceptualization, formal analysis, funding acquisition supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (92372127, 22179066) and the Natural Science Foundation of Shandong, China (202210060028, ZR2021QE061) and the Postdoctoral Fellowship Program of CPSF under Grant (GZC20231188).References
- J. Xiao, F. Shi, T. Glossmann, C. Burnett and Z. Liu, Nat. Energy, 2023, 8, 329–339 CrossRef.
- Y. Zhong, M. Yang, X. Zhou, Y. Luo, J. Wei and Z. Zhou, Adv. Mater., 2015, 27, 806–812 CrossRef CAS PubMed.
- S. Sahoo, R. Kumar, E. Joanni, R. K. Singh and J.-J. Shim, J. Mater. Chem. A, 2022, 10, 13190–13240 RSC.
- K. Chen and D. Xue, J. Mater. Chem. A, 2016, 4, 7522–7537 RSC.
- H. Kim, W. Choi, J. Yoon, J. H. Um, W. Lee, J. Kim, J. Cabana and W. S. Yoon, Chem. Rev., 2020, 120, 6934–6976 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, Y. Chen, M. Yousaf, H. Wu, A. Cao and R. P. S. Han, Adv. Funct. Mater., 2019, 29, 1807467 CrossRef.
- J. H. Um, K. Palanisamy, M. Jeong, H. Kim and W. S. Yoon, ACS Nano, 2019, 13, 5674–5685 CrossRef CAS PubMed.
- S. Zhao, Z. Wang, Y. He, H. Jiang, Y. W. Harn, X. Liu, C. Su, H. Jin, Y. Li, S. Wang, Q. Shen and Z. Lin, Adv. Energy Mater., 2019, 9, 1901093 CrossRef.
- Q. Li, H. Li, Q. Xia, Z. Hu, Y. Zhu, S. Yan, C. Ge, Q. Zhang, X. Wang, X. Shang, S. Fan, Y. Long, L. Gu, G. X. Miao, G. Yu and J. S. Moodera, Nat. Mater., 2021, 20, 76–83 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- F. A. Soto, Y. Ma, J. M. Martinez de la Hoz, J. M. Seminario and P. B. Balbuena, Chem. Mater., 2015, 27, 7990–8000 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- L. Su, J. Hei, X. Wu, L. Wang and Z. Zhou, Adv. Funct. Mater., 2017, 27, 1605544 CrossRef.
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621–625 CrossRef CAS.
- X. Li, L. Zhang, H. Liu, Q. Li and Y. Hou, Adv. Energy Mater., 2023, 13, 23300927 Search PubMed.
- Z. Zhao and Q. Li, Renewables, 2023, 1, 169–189 CrossRef.
- Z. Li, H. Liu, Z. Zhao, Q. Zhang, X. Fu, X. Li, F. Gu, H. Zhong, Y. Pan, G. Chen, Q. Li, H. Li, Y. Chen, L. Gu, K. Jin, S. Yan, G. X. Miao, C. Ge and Q. Li, Adv. Mater., 2023, 35, e2207353 CrossRef PubMed.
- H. Li, Z. Hu, Q. Xia, H. Zhang, Z. Li, H. Wang, X. Li, F. Zuo, F. Zhang, X. Wang, W. Ye, Q. Li, Y. Long, Q. Li, S. Yan, X. Liu, X. Zhang, G. Yu and G. X. Miao, Adv. Mater., 2021, 33, e2006629 CrossRef PubMed.
- Q. Xia, X. Li, K. Wang, Z. Li, H. Liu, X. Wang, W. Ye, H. Li, X. Teng, J. Pang, Q. Zhang, C. Ge, L. Gu, G. Miao, S. Yan, H. Hu and Q. Li, Chem. Mater., 2022, 34, 5852–5859 CrossRef CAS.
- L. Zhang, Q. Xia, Z. Li, Y. Han, X. Xu, X. Zhao, X. Wang, Y. Pan, H. Li and Q. Li, Chin. Phys. Lett., 2022, 39, 028202 CrossRef.
- Z. Li, Y. Zhang, X. Li, F. Gu, L. Zhang, H. Liu, Q. Xia, Q. Li, W. Ye, C. Ge, H. Li, H. Hu, S. Li, Y. Long, S. Yan, G. Miao and Q. Li, J. Am. Chem. Soc., 2021, 143, 12800–12808 CrossRef CAS PubMed.
- F. Zhang, Z. Li, Q. Xia, Q. Zhang, C. Ge, Y. Chen, X. Li, L. Zhang, K. Wang, H. Li, L. Gu, S. Yan, G. Miao and Q. Li, Matter, 2021, 4, 3605–3620 CrossRef CAS.
- D. Eum, B. Kim, J. Song, H. Park, H. Jang, S. J. Kim, S. Cho, M. H. Lee, J. H. Heo, J. Park, Y. Ko, S. K. Park, J. Kim, K. Oh, D. Kim, S. J. Kang and K. Kang, Nat. Mater., 2022, 21, 664–672 CrossRef CAS PubMed.
- G. Klinser, S. Topolovec, H. Kren, S. Koller, W. Goessler, H. Krenn and R. Würschum, Appl. Phys. Lett., 2016, 109, 213901 CrossRef.
- G. Klinser, R. Zettl, M. Wilkening, H. Krenn, I. Hanzu and R. Wurschum, Phys. Chem. Chem. Phys., 2019, 21, 20151–20155 RSC.
- H. Liu, F. Gu, X. Sang, Y. Han, F. Zou, Z. Li, Y. Qin, L. Cai, Y. Pan, Q. Cao, G. Miao and Q. Li, Phys. Rev. Appl., 2023, 19, 054022 CrossRef CAS.
- X. Li, J. Su, Z. Li, Z. Zhao, F. Zhang, L. Zhang, W. Ye, Q. Li, K. Wang, X. Wang, H. Li, H. Hu, S. Yan, G. Miao and Q. Li, Sci. Bull., 2022, 67, 1145–1153 CrossRef CAS PubMed.
- Z. Zhao, W. Ye, F. Zhang, Y. Pan, Z. Zhuo, F. Zou, X. Xu, X. Sang, W. Song, Y. Zhao, H. Li, K. Wang, C. Lin, H. Hu, Q. Li, W. Yang and Q. Li, Chem. Sci., 2023, 14, 12219–12230 RSC.
- K. Li, Z. Guo, Q. Sun, X. Dai, Y. Li, K. Yao, X. Liu, Z. Bao, J. Rao and Y. Zhang, Chem. Eng. J., 2023, 454, 140223 CrossRef CAS.
- M. Yang, Y. X. Li, M. Jiang, P. H. Li, S. H. Chen, J. H. Liu, C. H. Lin, X. J. Huang and W. Q. Liu, Small, 2020, 16, e1906830 CrossRef PubMed.
- A. K. Paidi, W. B. Park, P. Ramakrishnan, S. H. Lee, J. W. Lee, K. S. Lee, H. Ahn, T. Liu, J. Gim, M. Avdeev, M. Pyo, J. I. Sohn, K. Amine, K. S. Sohn, T. J. Shin, D. Ahn and J. Lu, Adv. Mater., 2022, 34, e2202137 CrossRef PubMed.
- H. Liang, H. Zhang, L. Zhao, Z. Chen, C. Huang, C. Zhang, Z. Liang, Y. Wang, X. Wang, Q. Li, X. Guo and H. Li, Chem. Eng. J., 2022, 427, 131481 CrossRef CAS.
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, Chem. Mater., 2002, 2, 186–191 CrossRef.
- L. Yu, S. Xi, C. Wei, W. Zhang, Y. Du, Q. Yan and Z. Xu, Adv. Energy Mater., 2015, 5, 1401517 CrossRef.
- L. Yu, L. P. Wang, S. Xi, P. Yang, Y. Du, M. Srinivasan and Z. J. Xu, Chem. Mater., 2015, 27, 5340–5348 CrossRef CAS.
- U. G. Nielsen, Y. Paik, K. Julmis, M. A. Schoonen, R. J. Reeder and C. P. Grey, J. Phys. Chem. B, 2005, 109, 18310–18315 CrossRef CAS PubMed.
- X. Hua, A. S. Eggeman, E. CastilloMartinez, R. Robert, H. S. Geddes, Z. Lu, C. J. Pickard, W. Meng, K. M. Wiaderek, N. Pereira, G. G. Amatucci, P. A. Midgley, K. W. Chapman, U. Steiner, A. L. Goodwin and C. P. Grey, Nat. Mater., 2021, 20, 841–850 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- T. Dong, W. Yi, T. Deng, T. Qin, X. Chu, H. Yang, L. Zheng, S. J. Yoo, J. G. Kim, Z. Wang, Y. Wang, W. Zhang and W. Zheng, Energy Environ. Mater., 2022, 6, e12262 CrossRef.
- P. M. Vinaya Teja, A. Ramesh Babu, P. Srinivasa Rao, R. Vijay and D. Krishna Rao, J. Phys. Chem. Solids, 2013, 74, 963–970 CrossRef CAS.
- B. Hu, C. Li, F. Ge and M. Shen, J. Electrochem., 2022, 28, 2108421 Search PubMed.
- J. M. Rondinelli, M. Stengel and N. A. Spaldin, Nat. Nanotechnol., 2008, 3, 46–50 CrossRef CAS PubMed.
- L. Fu, C. Chen, D. Samuelis and J. Maier, Phys. Rev. Lett., 2014, 112, 208301 CrossRef.
- F. Zuo, H. Zhang, Y. Ding, Y. Liu, Y. Li, H. Liu, F. Gu, Q. Li, Y. Wang, Y. Zhu, H. Li and G. Yu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2314362120 CrossRef CAS PubMed.
- H. L. Fan, G. Y. Zhou, J. L. Li, Y. Y. Zhao, L. Bai, H. Q. Chang, R. G. Zheng, Z. Y. Wang, Y. G. Liu and H. Y. Sun, Small, 2023, 19, e2300490 CrossRef PubMed.
- M. Yang, Y. Zhong, J. Ren, X. Zhou, J. Wei and Z. Zhou, Adv. Energy Mater., 2015, 5, 1500550 CrossRef.
- Y. Liu, Z. Sun, X. Sun, Y. Lin, K. Tan, J. Sun, L. Liang, L. Hou and C. Yuan, Angew Chem. Int. Ed. Engl., 2020, 59, 2473–2482 CrossRef CAS PubMed.
- X. Xie, C. S. Smith, M. Wen, H. D. Patel, S. M. Blau and K. A. Persson, J. Am. Chem. Soc., 2021, 143, 13245–13258 CrossRef CAS PubMed.
- B. Han, Z. Zhang, Y. Zou, K. Xu, G. Xu, H. Wang, H. Meng, Y. Deng, J. Li and M. Gu, Adv. Mater., 2021, 33, 2100404 CrossRef CAS PubMed.
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS PubMed.
- L. Jin, X. Guo, C. Shen, N. Qin, J. Zheng, Q. Wu, C. Zhang and J. P. Zheng, J. Power Sources, 2019, 441, 227211 CrossRef CAS.
- C. Liu, B. Wang, L. Xu, K. Zou, W. Deng, H. Hou, G. Zou and X. Ji, ACS Appl. Mater. Interfaces, 2023, 15, 5387–5398 CrossRef CAS PubMed.
- H. Zhou, L. Lin, Z. Sui, H. Wang and B. Han, ACS Appl. Mater. Interfaces, 2023, 15, 12161–12170 CrossRef CAS PubMed.
- S. Tao, J. Cai, Z. Cao, B. Song, W. Deng, Y. Liu, H. Hou, G. Zou and X. Ji, Adv. Energy Mater., 2023, 13, 2301653 CrossRef CAS.
- C. Han, L. Xu, H. Li, R. Shi, T. Zhang, J. Li, C. Wong, F. Kang, Z. Lin and B. Li, Carbon, 2018, 140, 296–305 CrossRef CAS.
- Z. Bo, Z. Zheng, Y. Huang, Z. Huang, P. Chen, J. Yan, K. Cen, H. Yang and K. K. Ostrikov, Chem. Eng. J., 2023, 473, 145183 CrossRef CAS.
- L. Wang, X. Zhang, C. Li, Y. Xu, Y. An, W. Liu, T. Hu, S. Yi, K. Wang, X. Sun, Y. Gong, Z. Wu and Y. Ma, Chem. Eng. J., 2023, 468, 143507 CrossRef CAS.
- W. Wei, L. Wang, C. Liang, W. Liu, C. Li, Y. An, L. Zhang, X. Sun, K. Wang, H. Zhang, X. Zhang and Y. Ma, Chem. Eng. J., 2023, 474, 145788 CrossRef CAS.
- A. Brandt and A. Balducci, Electrochim. Acta, 2013, 108, 219–225 CrossRef CAS.
- Y. Peng, H. Liu, Y. Li, Y. Song, C. Zhang and G. Wang, J. Colloid Interface Sci., 2021, 596, 130–138 CrossRef CAS PubMed.
- L. Wang, X. Zhang, Y. Kong, C. Li, Y. An, X. Sun, K. Wang and Y. Ma, Rare Met., 2024, 43, 2150–2160 CrossRef CAS.
- X. Lan, X. Liu, T. Meng, S. Yang, Y. Shen and X. Hu, Small Methods, 2023, 7, 2201290 CrossRef CAS PubMed.
- Z. Wang, Z. Yi, S. Yu, Y. Fan, J. Li, L. Xie, S. Zhang, F. Su and C. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 24497–24508 CrossRef CAS PubMed.
- Z. Pan, H. Zhi, Y. Qiu, J. Yang, L. Xing, Q. Zhang, X. Ding, X. Wang, G. Xu, H. Yuan, M. Chen, W. Li, Y. Yao, N. Motta, M. Liu and Y. Zhang, Nano Energy, 2018, 46, 266–276 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02174f |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2024 |