
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Constructing phase diagrams of block copolymers with A-block-(B-stat-C) architecture†
Britta
Weidinger‡
a,
Nadine
von Coelln‡
b,
Guohui
Yang
c,
Hermann
Nirschl
c,
Irene
Wacker
d,
Rasmus R.
Schröder
d,
Petra
Tegeder
 b and
Eva
Blasco
*a
b and
Eva
Blasco
*a
aInstitute for Molecular Systems Engineering and Advanced Materials, Universität Heidelberg, Im Neuenheimer Feld 225, 69120 Heidelberg, Germany. E-mail: eva.blasco@uni-heidelberg.de
bPhysikalisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany
cInstitute of Mechanical Process Engineering and Mechanics, Karlsruhe Institute of Technology (KIT), 76131 Karlsruhe, Germany
dBioQuant, Universität Heidelberg, Im Neuenheimer Feld 267, 69120 Heidelberg, Germany
First published on 20th September 2024
Abstract
Block copolymers (BCPs) are capable of self-organizing into precise nanoscale structures, generating interest across diverse fields such as drug delivery, catalysis, and lithography. Introducing new functionalities to BCPs through post-modification is a widely used approach. To further advance the design of functional BCPs, it is essential to understand how the incorporated functional groups affect their morphology. In this study, we focus on BCPs with an A-block-(B-statistical-C) architecture, where A is poly(styrene), B is poly(methyl methacrylate), and C is poly(2-hydroxyethyl methacrylate), which allows for the addition of photo-crosslinkable groups like methacrylates. In particular, we have synthesized a library of over twenty well-defined BCPs with different compositions and molecular weights using RAFT polymerization. Afterwards, we created a second library by introducing methacrylate groups through post-functionalization. The morphologies of these two sets of BCPs were carefully analyzed using small-angle X-ray scattering (SAXS), scanning electron microscopy (SEM), and infrared scanning near-field optical microscopy (IR-SNOM). By combining this data, we have built phase diagrams for both the original and functionalized copolymers and examined how post-functionalization affects their morphology. Our findings reveal that even small changes in the polymer composition significantly impact the morphological behavior.
1. Introduction
Block copolymers (BCPs) are able to spontaneously self-organize into precise nanoscale structures.1,2 Thus, these materials have attracted interest in fields ranging from drug delivery and catalysis to lithography.3–10 Many efforts have been made in the study of the phase behavior, both experimentally and theoretically.11–13 It is well-known that several parameters can influence the phase diagrams of BCPs. These include the Flory–Huggins interaction parameter (χ), the degree of polymerization (N) and the relative composition fractions (fx). Recently, some studies have also investigated the effect of dispersity. With increasing dispersity, an increase in domain spacing was reported, resulting in a polymer composition-dependent (de)stabilization of the microphase.14–16The morphology as well as the functionality can be fine-tuned using a post-modification process. For example, Feng et al. utilized the efficient thiol–ene click chemistry to post-functionalize poly(styrene-block-butadiene) with different thiol derivatives.17 The morphology of the obtained BCPs exhibited either cylindrical or lamellar structures, depending on the type and size of the thiol species incorporated. While a lamellar to cylindrical morphology change was observed for bulky thiols, the lamellar nanostructure was maintained when using less bulky thiols. In another work, McCallum et al. reported BCPs with switchable χ-parameters.18 In particular, BCPs consisting of poly(styrene) (PS) and a polysaccharide were employed, wherein the polar galactose units were protected with more hydrophobic acetonide groups. They demonstrated that the morphology could be switched from disordered (protected BCP) to either cylindrical or lamellar for the deprotected version or between different phases. Using a similar approach, Bosson et al. and Maekawa et al. made use of the para fluorine-thiol modification as an easy and mild method for the tailoring of functional BCP microstructures by varying the thiol functional side chains.19,20 Another interesting functionality is the incorporation of (photo)crosslinkable units. It has been demonstrated that the stability of the self-assembled BCP phases can be improved by both thermal as well as photocrosslinking.21–23 Furthermore, the post-functionalization of BCPs with photopolymerizable groups opens new possibilities for precise structuring and processing. Very recently, we have demonstrated the use of self-assembled BCPs for high-resolution two-photon 3D printing.24 In particular, we synthesized BCPs consisting of poly(styrene) (PS) and a polymethacrylate-based copolymer decorated with methacrylate groups. The BCPs were employed for 3D printing resulting in defined nano-ordered 3D structures exhibiting lamellar or cylindric morphologies, depending on the composition.
In order to further expand and rationally design functional and printable BCPs with defined nanostructure, the investigation of the effect of incorporation of photocrosslinkable groups on the morphology is crucial. To this aim, we present herein an in-depth study of this type of BCPs having an A-block-(B-statistical-C) architecture, where A is poly(styrene), B is poly(methylmethacrylate) and C poly(2-hydroxyethyl methacrylate) in the parent copolymers, which can be used for the introduction of a photocrosslinkable group such as methacrylate (Fig. 1). In particular, we synthesized a library of more than twenty well-defined BCPs with various compositions and molecular weights using RAFT polymerization. Subsequent post-functionalization for the introduction of methacrylate groups led to a second library. The morphologies in bulk for the two libraries were carefully characterized by small-angle X-ray scattering (SAXS), scanning electron microcopy (SEM) and infrared scanning near-field optical microscopy (IR-SNOM). By combining all obtained data, we were able to construct phase diagrams for the “two families” (parent copolymers and functionalized copolymers), as well as to study the influence of the post-functionalization on morphology. Importantly, we show that even small changes in the degree of functionalization influence the morphology.
 | ||
| Fig. 1 Overview of the workflow: (I) a library of BCPs with A-block-(B-stat-C) architecture consisting of PS (yellow) and a polymethacrylate-based copolymer (blue) decorated with hydroxyl groups is synthesized using RAFT polymerization. Post-functionalization of the hydroxyl groups with methacrylate groups (red) enables a second library of photocrosslinkable BCPs; (II) the morphologies in bulk are characterized by SAXS, SEM and IR-SNOM. (III) Phase diagrams of the synthesized BCPs are generated. Special attention is paid to the effect of the incorporation of photocrosslinkable groups. | ||
2. Results and discussion
Synthesis of block copolymers
The synthetic strategy chosen for the synthesis of the BCPs with an A-block-(B-stat-C) architecture depicted in Fig. 2 is based on the procedure reported in our recent publication24 and described in sections 1.1–1.3 of the ESI.† First, RAFT copolymerization of methyl methacrylate (M) and 2-hydroxyethyl methacrylate (H) was carried out using cyanopropyldithiobenzoate as the chain transfer agent. All synthesized macro chain transfer agents (see Table S1†) were characterized by nuclear magnetic resonance (NMR) spectroscopy and gel permeation chromatography (GPC) (see Fig. S1–S24†). Subsequently, a chain extension with styrene (S) led to the formation of the desired block copolymers having hydroxyl groups as side chains in the polymethacrylic block. In order to study the effect of the functional groups on the self-assembly, several compositions of the blocks as well as molecular weights were targeted. The details of the resulting 24 BCPs are summarized in Table 1. The BCPs are named according to their composition and molecular weights S1−f–MHf–Mn, where “S” is styrene, “M” methyl methacrylate, “H” 2-hydroxyethyl methacrylate, “f” is the fraction of the methacrylate-based block and Mn is the molecular weight in kDa. The degree of polymerization varied from 310 to 889 and the fraction of the methacrylate block varied from 0.25 to 0.91. The “H” content in the methacrylate blocks was kept constant at around 20 mol%. For all synthesized BCPs, a good control over the molecular weight was accomplished with dispersities between 1.06–1.39 (see Table S2†) and all BCPs were characterized by NMR spectroscopy and GPC (see Fig. S25–S72†). | ||
| Fig. 2 Synthetic strategy for the preparation of BCPs via sequential RAFT-controlled radical polymerization followed by post-functionalization to generate the two libraries with A-block-(B-stat-C) architecture. | ||
| S1−f–MHf–Mn | f | N | Morphology | S1−f–MH*f–Mn | Morphology* |
|---|---|---|---|---|---|
| S0.75–MH0.25–66 | 0.25 | 628 | Cylinder | S0.75–MH*0.25–66 | Lamella |
| S0.68–MH0.32–33 | 0.32 | 310 | Lamella | S0.68–MH*0.32–33 | Disorder |
| S0.66–MH0.34–70 | 0.34 | 662 | Lamella | S0.66–MH*0.34–70 | Cylinder |
| S0.56–MH0.44–52 | 0.44 | 495 | Lamella | S0.56–MH*0.44–52 | Lamella |
| S0.54–MH0.46–45 | 0.46 | 430 | Lamella | ||
| S0.53–MH0.47–94 | 0.47 | 889 | Lamella | S0.53–MH*0.47–94 | Lamella |
| S0.52–MH0.48–67 | 0.48 | 636 | Lamella | S0.52–MH*0.48–67 | Lamella |
| S0.50–MH0.50–91 | 0.50 | 868 | S0.50–MH*0.50–91 | Lamella | |
| S0.50–MH0.50–45 | 0.50 | 429 | Lamella | S0.50–MH*0.50–45 | Lamella |
| S0.49–MH0.50–92 | 0.51 | 879 | Lamella | ||
| S0.46–MH0.54–81 | 0.54 | 768 | Lamella | S0.46–MH*0.54–81 | Cylinder/lamella |
| S0.45–MH0.55–42 | 0.55 | 399 | Lamella | S0.45– MH50%*0.55–42 | Cylinder/lamella |
| S0.42–MH0.58–89 | 0.58 | 845 | Lamella | S0.42–MH*0.58–89 | Cylinder |
| S0.42–MH0.58–57 | 0.58 | 539 | Lamella | S0.42–MH*0.58–57 | Cylinder |
| S0.39–MH0.61–35 | 0.61 | 333 | Lamella | ||
| S0.37–MH0.63–65 | 0.63 | 615 | Gyroid | S0.37–MH*0.63–65 | Cylinder |
| S0.31–MH0.69–48 | 0.69 | 451 | Cylinder | S0.31–MH*0.69–48 | Cylinder |
| S0.28–MH0.72–67 | 0.72 | 634 | Cylinder | S0.28–MH*0.72–67 | Cylinder |
| S0.28–MH0.72–29 | 0.72 | 275 | Cylinder | S0.28–MH*0.72–29 | Cisorder |
| S0.27–MH0.73–70 | 0.73 | 662 | Cylinder | S0.27–MH*0.73–70 | Cylinder |
| S0.16–MH0.84–45 | 0.84 | 425 | Disorder | ||
| S0.16–MH0.84–43 | 0.84 | 401 | Disorder | S0.16–MH*0.84–43 | Disorder |
| S0.12–MH0.88–40 | 0.88 | 375 | Disorder | ||
| S0.09–MH0.91–39 | 0.91 | 364 | Disorder |
To generate the second library of BCPs equipped with photocrosslinkable moieties, i.e., methacrylate groups, the synthesized BCPs containing hydroxyl groups were esterified with methacryloyl chloride and are named as S1−f–MH*f–Mn. The functionalization of the hydroxyl groups is indicated by *. The conversion was monitored by NMR spectroscopy (see Fig. S73–S90†). The obtained BCPs are also included in Table 1, next to their corresponding “parent” copolymers.
Morphology analysis
As a next step, the morphology of all the synthesized BCPs was investigated. For this purpose, thick films were prepared using a solvent-annealing process (see details in the experimental section). Thermal annealing was discarded to avoid undesired thermal crosslinking, especially for the BCPs containing methacrylate groups. The self-assembled films were characterized by SAXS, SEM as well as IR-SNOM after sectioning in ultrathin films. A summary of the assigned morphologies is presented in Table 1. For both libraries, morphologies ranging from lamella to cylinder and gyroid were found. The complete overview of the microscope images and SAXS scattering plots can be found in the ESI (see Fig. S91–S111†).In this study, we paid special attention to the effect of the incorporated methacrylate groups. While lamella was the morphology most frequent for the S1−f–MHf–Mn family containing hydroxyl groups in the side chains, interesting changes in morphology were observed when post-functionalizing with methacrylate groups (S1−f–MH*f–Mn) (Fig. 3). For example, a clear lamellar morphology was observed for S0.42–MH0.58–89 as one would expect with a fraction close to 0.5 (Fig. 3a). After post-functionalization, however, the morphology changed to a cylindrical morphology. These morphologies were visualized using SEM and IR-SNOM at 1152 cm−1 (Fig. 3b), as well as confirmed by SAXS measurements showing reflections apparent at q, √4q* and √7q* (Fig. 3c). Another example of morphology change was observed for S0.37–MH0.63–65 and S0.37–MH*0.63–65 with a higher polymethacrylate block fraction of 0.63 (Fig. 3d and e). While gyroid structures were assigned for S0.37–MH0.63–65, due to a characteristic shoulder at √(4/3)q* in the diffraction pattern (Fig. 3f),25 for the post-functionalized polymer a cylindrical morphology was confirmed by the SEM images in combination with the diffraction pattern. IR-SNOM near-field optical imaging was employed to gain further information on the obtained morphologies. Illuminating the sample at an independently addressable absorption band of the methacrylate block at 1152 cm−1 (C–O–C stretching vibration) allows to distinguish between the different block copolymer units and to image phase segregation via spectroscopic contrast.26,27 Regions with high optical phase value (red) can be assigned to the methacrylate block, while those with low optical phase value (blue) correspond to the PS block. Importantly, various morphologies, which are consistent with SEM and SAXS measurements were characterized. Further SNOM phase images of the unfunctionalized BCPs with different morphologies can be found in Fig. 3g. For S0.75–MH0.25–66, the PS phase serves as matrix surrounding polymethacrylate-based cylinders. In the case of S0.52–MH0.48–67, a clear lamellar morphology was imaged. Increasing the fraction of the “MH” block further, PS cylinders in a polymethacrylate-based matrix were observed for S0.31–MH0.69–48. While the atomic force microscopy (AFM) height images of the different BCPs show similar unspecific topographic structures that do not clearly indicate the morphology, the corresponding IR-SNOM phase images reveal pronounced optical contrasts enabling a clear identification of morphologies.
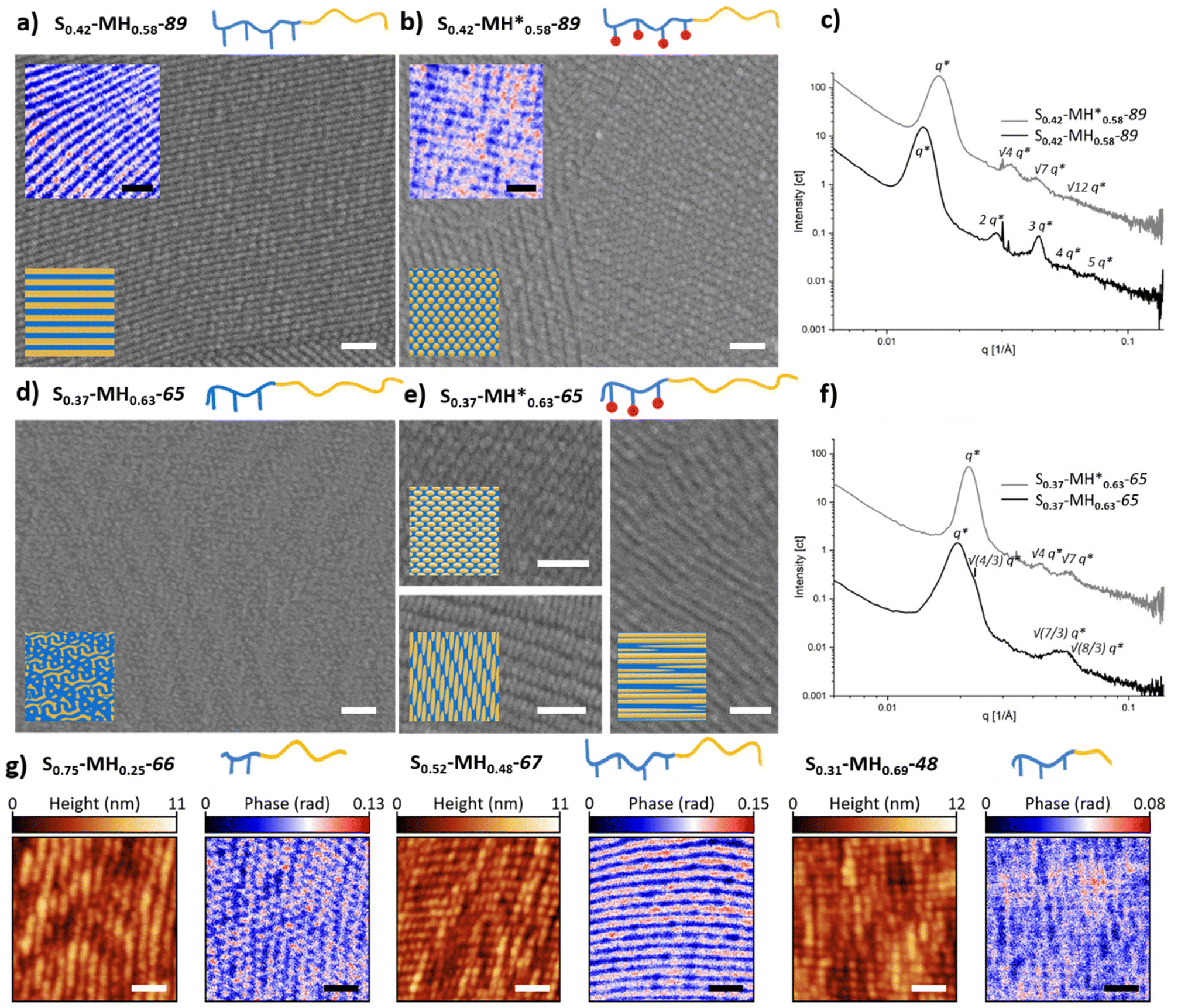 | ||
| Fig. 3 Exemplary SEM and IR-SNOM images of different BCPs before (a and d) and after (b and e) functionalization. SEM images are shown in grayscale and IR-SNOM phase images, mapped at an independently addressable absorption band of the methacrylate block at 1152 cm−1, in blue-to-red scale. Scale bar = 200 nm. 3D renderings of the sectioned planes and complete phase images with corresponding AFM images can be found in Fig. S112 and S113.† (c) and (f) SAXS spectra. (g) AFM height images and corresponding IR-SNOM phase images mapped at 1152 cm−1 for different morphologies of unfunctionalized BCPs. | ||
To further get insights into the effect of the composition of the BCPs, a detailed analysis of the domain spacings (d) was carried out. Several studies have confirmed that for a linear diblock copolymer the scaling behavior in the strong segregation limit is d ∼ MW2/3, which is in agreement with the theoretical prediction.28 In our case, d values were determined from the scattering maxima of the SAXS spectra (dsaxs = 2π/q) and from SEM images (dSEM). It should be noted that in the case of a cylindrical morphology, we observed large deviations from the fit, especially after functionalization with methacrylate groups (see Table S3 and Fig. S114†). Thus, we concentrated on the BCPs exhibiting lamellar morphologies. Fig. 4 shows the domain spacings determined by SAXS for the lamellar structures as a function of the molecular weight obtained by GPC. By fitting the data to a power law (d = a*MWb), we found that while the relationships between dSAXS and the molecular weight are as expected close to d ∼ MW2/3, a less clear scaling behavior was observed in the case of dSEM (see Fig. S115 and Table S3†). Similar effects were also observed by Richards and Thomason.28 This was attributed to the high dependency of the SEM analysis on the orientation of the lamellae in the sections depending on the angle with respect to the knife edge, while the thickness of the section is also influential, which is most important for the lamellar domains. We also observed that for dSAXS the mean value scaling exponent is smaller for the functionalized polymers. This could be due to the change in χ and/or architectural asymmetry by incorporation of methacrylate groups. Asymmetry effects were also reported by Liberman et al.29 when investigating the self-assembly of linear-bottlebrush BCPs with varying degrees of architectural asymmetry by changing the side-chain length.
 | ||
| Fig. 4 Double logarithmic plot of the domain spacing against the molecular weight for the lamellar morphology determined from dSAXS The dark grey lines are the power law fits to d = a*MWb. | ||
Last, the data obtained for the two libraries was employed for the construction of phase diagrams. For this purpose, the fraction of the methacrylate block (molar ratio) was plotted as a function of the degree of polymerization (N) (Fig. 5). For better visualization, we have marked the phase boundaries in different colors. A first clear observation is that the “lamellar window” becomes smaller for the functionalized polymers, while the “cylindrical window” is larger. Additionally, for some polymers with an ordered morphology in the original state, the morphology becomes disordered upon functionalization. It should be noted that due to the limited number of experimental sample points in this study, it is possible that further phases exist, such as spheres that were not detected in the SEM or SAXS. However, it is clearly demonstrated that the post-functionalization has an effect on the morphology obtained which can be reasoned with a change in χ and volume fraction upon the functionalization of the hydroxyl groups with methacrylate groups.
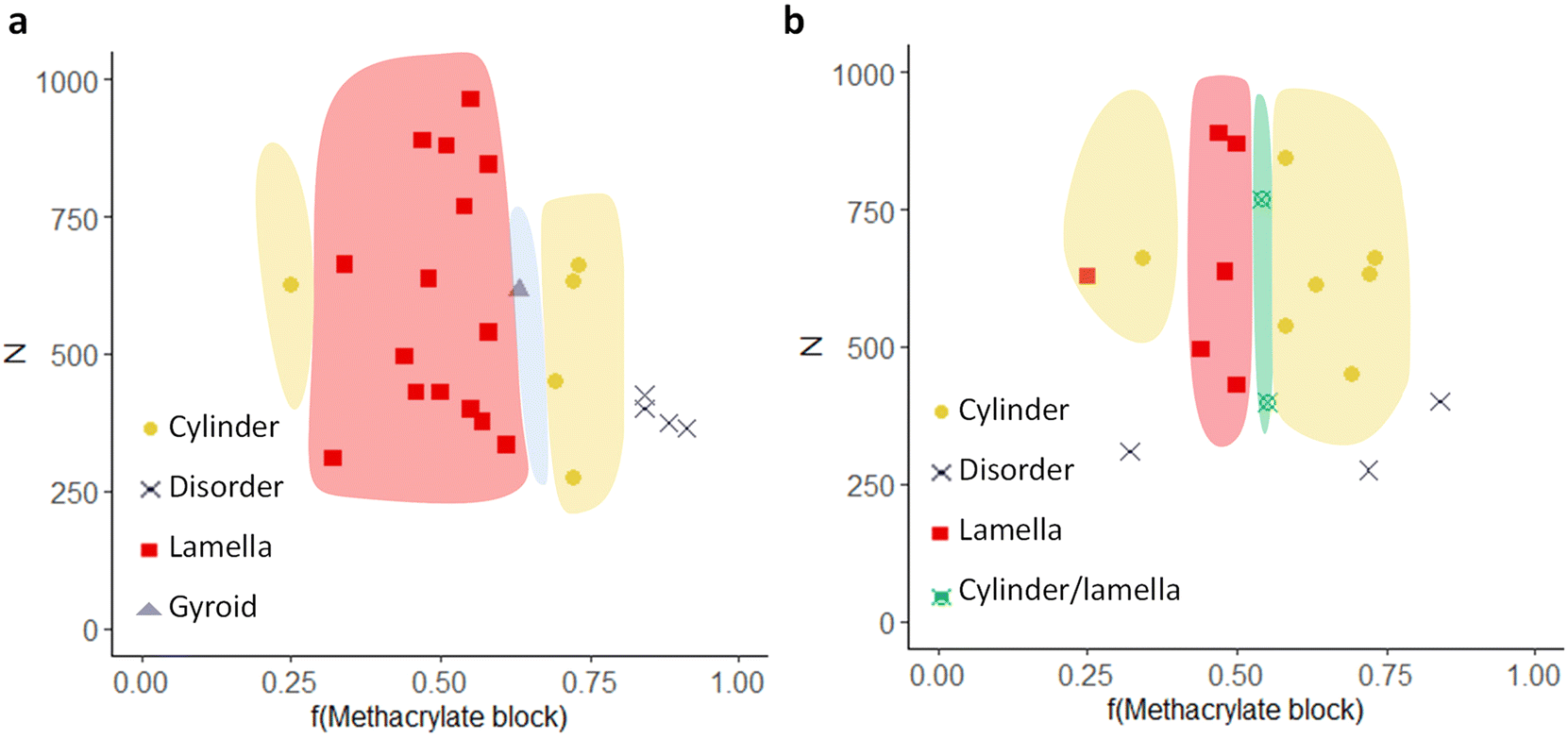 | ||
| Fig. 5 Phase diagrams of (a) the precursor BCPs and (b) the functionalized BCPs. The phase boundaries are drawn as guides. | ||
3. Conclusion
To summarize, two libraries of A-block-(B-stat-C) architectures have been synthesized containing hydroxyl and methacrylate functional groups, respectively. Insights into the complex phase behavior have been gained via a careful study of the morphologies by combining SAXS, SEM and IR-SNOM characterization. While the lamellar morphology is most common for the parent copolymers containing hydroxyl groups in the side chains, interesting changes in morphology were observed upon post-functionalization with methacrylate groups. The influence of the domain spacing for the lamellar polymers has been analyzed, and the fit was close to the predicted value d ∼ MW2/3, especially for the precursor polymer. Additionally, the decrease of domain spacing/size upon functionalization confirms the assumption that the Flory–Huggins interaction parameter (χ) is decreasing when introducing the less polar methacrylate group in comparison to the more polar free hydroxyl group. Last, phase diagrams were generated. A narrower lamellar and broader cylindrical window for the functionalized polymer species was found, demonstrating a clear effect of the functional group on the morphology. Thus, this study paves the way towards a rational design of functional BCPs with defined morphologies.4. Experimental section/methods
Chemicals and materials
Chemicals and solvents were supplied from either Sigma-Aldrich or Fisher Scientific unless otherwise mentioned.Synthesis and structure characterization
To synthesize the block copolymers, the sequential RAFT polymerization was performed following an approach modified from Varadhajaran and Delaittre.30 The synthetic details are provided in the ESI.†All synthesized compounds were characterized with 1H nuclear magnetic resonance (NMR) spectroscopy (Bruker Avance III 300, Bruker Avance III 600 or Bruker Avance Neo 700). Gel permeation chromatography (GPC) measurements were performed on a Shimadzu Nexera LC-40 system (with LC-40D pump, autosampler SIL-40C, DGU-403 (degasser), CBM-40 (controlling unit), column oven CTO-40C, UV-detector SPD40 and RI-detector RID-20A).
The system was equipped with 4 analytical GPC-columns (PSS): 1 × SDV precolumn 3 μm 8 × 50 mm, 2 × SDV column 3 μm 1000 Å 8 × 300 mm, 1 × SDV column 3 μm 10 × 104 Å 8 × 300 mm.
The measurements were performed in THF at a flow speed of 1 mL min−1 at a temperature of 40 °C. Chromatograms were analysed using the LabSolutions (Shimadzu) software. Calibration was performed against different polymethylmethacrylate standards (800–2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200000 Da, PSS) or PS standards (370–2520000 Da, PSS).
200000 Da, PSS) or PS standards (370–2520000 Da, PSS).
Self-assembly procedure
Films were cast from a chloroform solution by slow evaporation of the solvent over the course of days. After removal of the glass vial by immersing the film into liquid nitrogen and gently tapping with a hammer, the free-standing films were obtained.Ultramicrotome
Self-assembled bulk films were cut into small pieces and embedded in Epon (prepared by mixing 26.2 g glycid ether 100, 14.8 g dodecenylsuccinic acid anhydride, 9.2 g methyl-5-norbornene-2,3-dicarboxylic anhydride and 1.2 g benzyldimethylamine; all chemicals purchased from SERVA) and polymerized for 2 days at 62 °C.The resin blocks were trimmed to expose the structures. Ultrathin (100–120 nm) cross sections were cut using a PowerTome PC ultramicrotome (RMC Boeckeler) and placed on pieces of silicon wafer.
Scanning electron microscope (SEM)
The sections were imaged in a field-emission scanning electron microscope (Ultra 55, Carl Zeiss Microscopy) at a primary electron energy of 1.5 keV, either with or without post-staining in RuO4 vapour (0.5% in H2O, Polysciences Inc) for 30 min.Small angle X-ray scattering
SAXS experiments were performed using a Xeuss 2.0 Q-Xoom (Xenocs SA, Grenoble, France) instrument, equipped with a Genix3D Cu ULC (ultra-low divergence) micro focus source of Cu Kα with an energy of 8.04 keV, a wavelength of 1.5406 Å, and a Pilatus3 R 300K detector (Dectris Ltd, Baden, Switzerland). Solid films were measured without substrate. The 2D scattering patterns were obtained using a sample-to-detector distance of 2500 mm, resulting in the range of accessible scattering vector (q) from 0.003 to 0.14 Å−1. To obtain 1D plots of the intensity I(q) versus q with q = 4πsin(2θ/2)/λ, where 2θ is the scattering angle and λ is the wavelength of the Cu Kα source, an azimuthal integration of the scattering patterns was used. The measurement time was set to 600 s.Infrared scanning near-field optical microscopy (IR-SNOM)
The measurements were performed on unstained ultrathin cross sections utilizing a neaSNOM microscope from attocube systems AG. Near-field optical amplitude and phase images together with the sample's topography were acquired employing a pseudo-heterodyne detection scheme.31 Image acquisition was conducted for areas of sizes between 1 × 1 μm2 and 3 × 3 μm2 with a maximum pixel size of 10 nm and a minimum integration time of 6.6 ms per pixel. Measurements were performed in tapping mode with a platinum–iridium coated probe (Arrow-NCPt). The probe was illuminated with a mid-infrared quantum cascade laser (Daylight solutions) at a power setting of 1.5 mW. Signal detection was performed using a mercury cadmium telluride detector, with demodulation at the third harmonic of the tapping frequency for background suppression.Data availability
Data for this article, including raw data of the characterization of the materials and images are available at HeiData repository (https://heidata.uni-heidelberg.de/dataverse/blasco).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Council) under Germany's Excellence Strategy for the Excellence Cluster “3D Matter Made to Order” (EXC-2082/1-390761711) and the Carl Zeiss Foundation for financial support. The authors thank E.R. Curticean and C. Huck for the support with SEM and SNOM measurements, respectively.References
- I. W. Hamley, Nanotechnology, 2003, 14, R39 CrossRef.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 7641 CrossRef.
- J. Y. Cheng, C. A. Ross, E. L. Thomas, H. I. Smith and G. J. Vancso, Appl. Phys. Lett., 2002, 81, 3657 CrossRef.
- L. Wan, R. Ruiz, H. Gao, K. C. Patel, T. R. Albrecht, J. Yin, J. Kim, Y. Cao and G. Lin, ACS Nano, 2015, 9, 7506 CrossRef PubMed.
- F. M. Cabrini, M. Champeau and M. G. de Oliveira, J. Appl. Polym. Sci., 2020, 137, 49056 CrossRef CAS.
- C. Boucher-Jacobs, M. Rabnawaz, J. S. Katz, R. Even and D. Guironnet, Nat. Commun., 2018, 9, 841 CrossRef PubMed.
- J. Zhou, C. Zhang and Y. Wang, Polym. Chem., 2019, 10, 1642 RSC.
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265 CrossRef CAS.
- M. Karayianni and S. Pispas, J. Polym. Sci., 2021, 59, 1874 CrossRef CAS.
- H. Cabral, K. Miyata, K. Osada and K. Kataoka, Chem. Rev., 2018, 118, 6844 CrossRef CAS.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32 CrossRef CAS.
- M. Svensson, P. Alexandridis and P. Linse, Macromolecules, 1999, 32, 637 CrossRef CAS.
- T. M. Bennett, K. S. Jack, K. J. Thurecht and I. Blakey, Macromolecules, 2016, 49, 205 CrossRef CAS.
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2005, 38, 8803 CrossRef CAS.
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2007, 40, 8050 CrossRef CAS.
- D. T. Gentekos, J. Jia, E. S. Tirado, K. P. Barteau, D.-M. Smilgies, R. A. DiStasio and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 4639 CrossRef CAS PubMed.
- H. Feng, M. Dolejsi, N. Zhu, P. J. Griffin, G. S. W. Craig, W. Chen, S. J. Rowan and P. F. Nealey, Adv. Funct. Mater., 2022, 32, 2206836 CrossRef CAS.
- F. McCallum, J. Zhao, M. D. Hossain, J. A. Kaitz, J. F. Cameron, P. Trefonas III, I. Blakey, H. Peng and A. K. Whittaker, Macromolecules, 2023, 56, 2651 CrossRef CAS.
- K. Bosson, P. Marcasuzaa, A. Bousquet, G. E. M. Tovar, V. Atanasov and L. Billon, Eur. Polym. J., 2022, 179, 111560 CrossRef.
- S. Maekawa, T. Seshimo, T. Dazai, K. Sato, K. Hatakeyama-Sato, Y. Nabae and T. Hayakawa, Nat. Commun., 2024, 15, 5671 CrossRef PubMed.
- I. Kawarazaki, M. Hayashi, K. Yamamoto and A. Takasu, ChemistrySelect, 2020, 5, 2842 CrossRef.
- E. Kim, C. Shin, H. Ahn, D. Y. Ryu, J. Bang, C. J. Hawker and T. P. Russell, Soft Matter, 2008, 4, 475 RSC.
- M. B. Sims, B. Zhang, Z. M. Gdowski, T. P. Lodge and F. S. Bates, Macromolecules, 2022, 55, 3317 CrossRef.
- B. Weidinger, G. Yang, N. von Coelln, H. Nirschl, I. Wacker, P. Tegeder, R. R. Schröder and E. Blasco, Adv. Sci., 2023, 10, 2302756 CrossRef CAS.
- D. A. Hajduk, P. E. Harper, S. M. Gruner, C. C. Honeker, G. Kim, E. L. Thomas and L. J. Fetters, Macromolecules, 1994, 27, 4063 CrossRef CAS.
- G. Duan, C. Zhang, A. Li, X. Yang, L. Lu and X. Wang, Nanoscale Res. Lett., 2008, 3, 118 CrossRef CAS.
- I. Wacker, R. Curticean, D. Ryklin, B. Weidinger, F. Mayer, L. Huang, J. Hoffmann, M. Islam, N. Von Coelln, T. Schmitt, C. Huck, P. Tegeder, F. Feist, J. A. Kammerer, C. Barner-Kowollik, M. Wegener, E. Blasco, U. Gengenbach and R. R. Schröder, Adv. Funct. Mater., 2024, 34, 2302025 CrossRef CAS.
- R. W. Richards and J. L. Thomason, Macromolecules, 1983, 16, 982 CrossRef CAS.
- L. Liberman, M. L. Coughlin, S. Weigand, F. S. Bates and T. P. Lodge, Macromolecules, 2022, 55, 2821 CrossRef CAS.
- D. Varadharajan and G. Delaittre, Polym. Chem., 2016, 7, 7488 RSC.
- N. Ocelic, A. Huber and R. Hillenbrand, Appl. Phys. Lett., 2006, 89, 101124 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00872c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |