
Nanoporous YVO4 as a luminescent host for probing molecular encapsulation†
Milena Lima
Brito
a,
Steven
Huband
b,
Marc
Walker
 b,
Richard I.
Walton
c and
Paulo C.
de Sousa Filho
*a
b,
Richard I.
Walton
c and
Paulo C.
de Sousa Filho
*a
aDepartment of Inorganic Chemistry, Institute of Chemistry, University of Campinas (Unicamp), R. Monteiro Lobato, 270, 13083-970, Campinas, São Paulo, Brazil. E-mail: pcsfilho@unicamp.br
bDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
cDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK
First published on 11th August 2023
Abstract
Control of phase separation of VO43− and rare earth precursors in reverse microemulsions afforded ∼35 nm YVO4 nanoparticles with functionalisable ∼7 ± 3 nm nanopores. Doping by Eu3+ allowed luminescent probing of interfacial crystallisation while xylenol orange absorption showed molecular encapsulation in particle cavities. This provides potential multifunctional systems combining UV-Vis-NIR luminescence and (photo)active molecules for optical sensing.
Strategies for the association of multiple properties in a single structure are a challenge for the development of new cutting-edge nanomaterials.1 In particular, light-driven responses of nanoparticles, which include thermometry,2 chemical sensing,3 and localized activation of chemical reactions,4 can be combined to afford multiple actions against a local chemical environment.5 Rare earths (RE) fulfil lifetime, antiphotobleaching, and linewidth requirements for luminescent sensing,6 and RE vanadates are chemically versatile solids providing both ultraviolet (UV)- and near infrared (NIR)-excited emissions,7 as well as catalytic potential towards biomimetic reactions.8 Hollow or nanoporous particles9 with accessible cavities for functionalisation are promising structures to further improve the range of applications of this class of solids towards multifunctional materials. Engineered cavities can act as nanocontainers for encapsulation and transport of molecules of interest such as dyes and drugs, which can be selectively activated via the light emitted by the particles. In turn, these systems offer novel prospects for theranostic applications, where different selected wavelengths can provide orthogonal sensing and photoactivation of encapsulated molecules.4,10 The synthesis of porous and hollow particles generally follow soft template routes9via the growth of the inorganic phase over liquid–liquid or liquid–air discontinuities. Regular (i.e., oil-in-water) and reverse (i.e., water-in-oil) microemulsions consist of thermodynamically stable systems providing nanometric liquid–liquid interfaces, which can further enable phase separation of different precursors into polar and non-polar domains for a controlled crystallisation of the inorganic product.11 Although this has been widely explored to produce hollow metal and oxide nanoparticles,12 a limited number of examples demonstrate the possibility of functionalisation of the inner surface of RE-based luminescent particles,13 especially of vanadates. We have therefore investigated the formation of tetragonal REVO4 hollow particles in reverse microemulsions containing separated RE3+ and VO43− precursors in dodecane and water phases, respectively (ESI,† Section S1 and Fig. S1–S3). We produced Eu3+-doped particles to probe spectroscopically the crystallisation process, and Yb3+/Er3+-doped particles to confirm the potentiality of the products for mutifunctional NIR-excited luminescent sensing.
Considering both size distributions and dynamic light scattering (DLS) polydispersity indices (ESI,† Section S2.1 and Fig. S3), the microemulsions with water/surfactant ratios (WO, ESI,† Section S1) WO = 20 and WO = 25 induced the interface precipitation of the vanadate phase; WO = 25 was further investigated. The formation kinetics of REVO4 solids from aqueous precursors usually follows a non-classic nucleation/growth mechanism,14 being strongly dependent on the pH because of vanadate speciation and formation of non-crystalline metastable intermediates. Highly basic pH results in higher concentrations of VO43−(aq), but also induces competitive precipitation of kinetically stable RE hydroxides.14,15 Conversely, acidic metavanadate ((VO3)nn−) solutions show a smaller availability of free VO43− and may lead to the parallel precipitation of polyvanadates.16
We therefore evaluated initially the effect of the pH (= 7, 11.5 or 13) of the aqueous cores on the interface crystallization of REVO4 in reverse microemulsions with WO = 25 (ESI,† Section S2.1 and Fig. S4). At strongly basic conditions (pH = 13), the reaction mixture containing organic RE oleate precursors and the aqueous vanadate solution remained clear after 80 h. Although the formation of primary grains of tetragonal nanocrystalline (Y,Eu)VO4 phase could be observed by wide-angle X-ray scattering (WAXS, Fig. S5, ESI†), no solid product was isolated from the reaction mixture. If the pH of the aqueous phase was brought to 7 using NaVO3 as the vanadate source, the particles underwent an uncontrolled growth and only large micrometric aggregates were recovered after 24 h (Fig. S6, ESI†). Therefore, the optimal conditions to assure an interfacial crystallisation of the REVO4 phase in the reverse microemulsions comprised the use of a Na3VO4 solution with pH adjusted to 11.5. Such conditions led to the formation of ∼35 nm spheroid particles containing multiple cavities (Fig. 1(A)–(D) and Fig. S7, ESI†). The nanoporous nature of the products was probed by small-angle X-ray scattering profiles (SAXS, Fig. 1(E), (G)–(I) and ESI†), which revealed a pore distribution centred at ∼5 ± 3 nm. WAXS profiles (Fig. 1(F)) confirmed the formation of tetragonal (I41/amd) nanocrystallites with (200) coherence lengths of ∼7–9 nm. Other characterisation of the particles is shown on the ESI† (Section S2.2). The presence of solvent and surfactant residues that induce partial aggregation in aqueous suspension (Fig. 1(A)–(D), TEM, and Fig. 1(G)–(I), DLS) is confirmed by X-ray photoelectron spectroscopy (XPS, Fig. 2(A)–(D)). The C1s peaks confirmed the presence of high amounts of aliphatic (284.6 eV) and partially oxidised (C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COO, 285.8, 287.0, 288.5 eV) carbon species. Also, minor amounts of carbonates (290.0 eV) were detected due to partial carbonation of the Na3VO4 precursor, as observed for REVO4 particles prepared by aqueous coprecipitation.14,15 The interfacial crystallisation enabled the encapsulation of dye molecules in the particle cavities, which showed no effect on the general morphologic and structural properties of the final REVO4 solids (Fig. 1(E)–(I)) besides an increased degree of aggregation (Fig. 1(G)–(I) and ESI,† Section S2.2). Regarding the REVO4 phase, Y3d and V2p signals (Fig. 2(A) and (C)) showed metal sites with oxygen deficiency, as shown by the side-peaks in the Y3d doublet (3d3/2, 159.9 eV and 3d5/2, 157.9 eV), and by V2p peaks due to V(IV) (2p1/2, 524.9 eV, 2p3/2, 517.7 eV). Compositional analysis of XPS spectra (Fig. 2(D)) showed a high Y/V non-stoichiometry (Y/V = 1.7), attesting that the proposed synthesis afforded (Y,Eu)VO4 particles with a vanadium-deficient surface because of carbonates and hydroxides,15 also showing a high density of partially reduced metal sites due to oxygen vacancies. Together these results demonstrate the possibility of preparation of REVO4 nanoparticles in water-in-n-dodecane reverse microemulsions formed by nanoaggregates of reverse micelles. The phase separation of RE3+ and VO43− precursor results in the interfacial crystallisation over these templates upon diffusion of RE oleates and VO43− ions along the surfactant film, yielding nanoporous REVO4 particles (Fig. 2(E)). Given the high chemical and structural similarity of RE vanadates, this method can be easily adapted to produce nanoporous particles composed by other lanthanides to offer multiple luminescent properties.
O, COO, 285.8, 287.0, 288.5 eV) carbon species. Also, minor amounts of carbonates (290.0 eV) were detected due to partial carbonation of the Na3VO4 precursor, as observed for REVO4 particles prepared by aqueous coprecipitation.14,15 The interfacial crystallisation enabled the encapsulation of dye molecules in the particle cavities, which showed no effect on the general morphologic and structural properties of the final REVO4 solids (Fig. 1(E)–(I)) besides an increased degree of aggregation (Fig. 1(G)–(I) and ESI,† Section S2.2). Regarding the REVO4 phase, Y3d and V2p signals (Fig. 2(A) and (C)) showed metal sites with oxygen deficiency, as shown by the side-peaks in the Y3d doublet (3d3/2, 159.9 eV and 3d5/2, 157.9 eV), and by V2p peaks due to V(IV) (2p1/2, 524.9 eV, 2p3/2, 517.7 eV). Compositional analysis of XPS spectra (Fig. 2(D)) showed a high Y/V non-stoichiometry (Y/V = 1.7), attesting that the proposed synthesis afforded (Y,Eu)VO4 particles with a vanadium-deficient surface because of carbonates and hydroxides,15 also showing a high density of partially reduced metal sites due to oxygen vacancies. Together these results demonstrate the possibility of preparation of REVO4 nanoparticles in water-in-n-dodecane reverse microemulsions formed by nanoaggregates of reverse micelles. The phase separation of RE3+ and VO43− precursor results in the interfacial crystallisation over these templates upon diffusion of RE oleates and VO43− ions along the surfactant film, yielding nanoporous REVO4 particles (Fig. 2(E)). Given the high chemical and structural similarity of RE vanadates, this method can be easily adapted to produce nanoporous particles composed by other lanthanides to offer multiple luminescent properties.
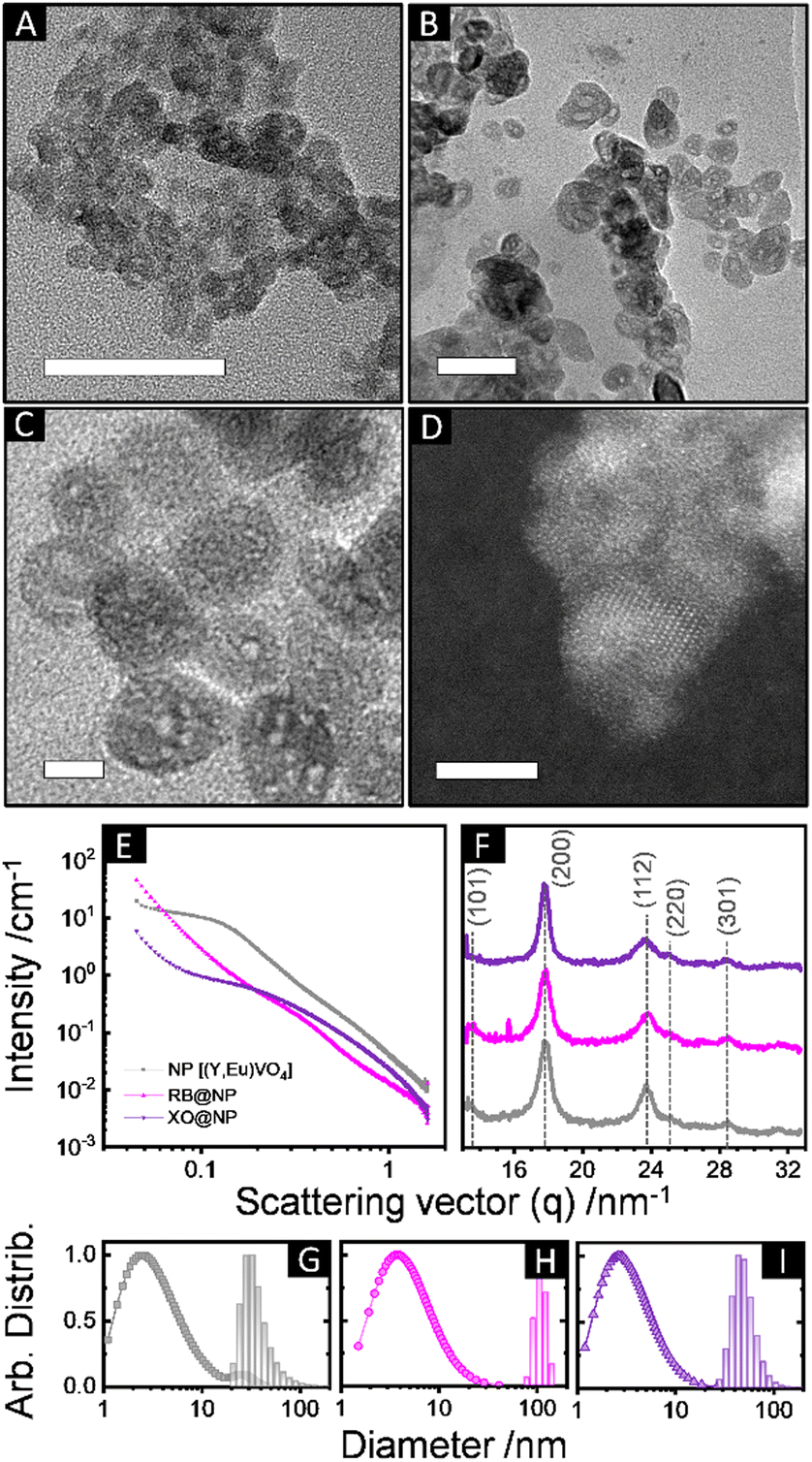 | ||
| Fig. 1 (A)–(D) Transmission electron microscopy (TEM) images of (Y,Eu)VO4 nanoparticles synthesised in reverse microemulsions (WO = 25, pH = 11.5). Scale bars correspond to 50 nm in (A) and (B), 10 nm in (C), and 5 nm in (D, HRTEM). (E) SAXS and (F) WAXS profiles of non-modified (Y,Eu)VO4 nanoparticles (NP, grey), and particles synthesised at the same conditions containing encapsulated rose Bengal (RB@NP, rose), or xylenol orange (XO@NP, purple) dyes. (G)–(I) Size distributions obtained from SAXS (in volumes, points) and DLS (in number, bars) of (G) NP [SAXS: Dmi = 6.7 ± 2.4 nm, Dmii = 28 ± 7 nm; DLS: DH = 34 ± 8 nm, PDI = 0.4], (H) RB@NP [SAXS: Dm = 8.7 ± 3.5 nm; DLS: DH = 110 ± 15 nm, PDI = 0.86], and (I) XO@NP [SAXS: DH = 5.8 ± 2.2 nm; DLS: DH = 51 ± 13 nm, PDI = 0.6]. The colour pattern of (E), (F) was kept for (G)–(I). Dm and DH stand for mean SAXS-modelled and hydrodynamic (DLS) diameters, respectively, obtained from lognormal fits. SAXS distributions account mostly for the porosity of the particles; DLS distributions refer to hydrodynamic diameters of particles and aggregates in suspension (ESI,† Section S2.2). | ||
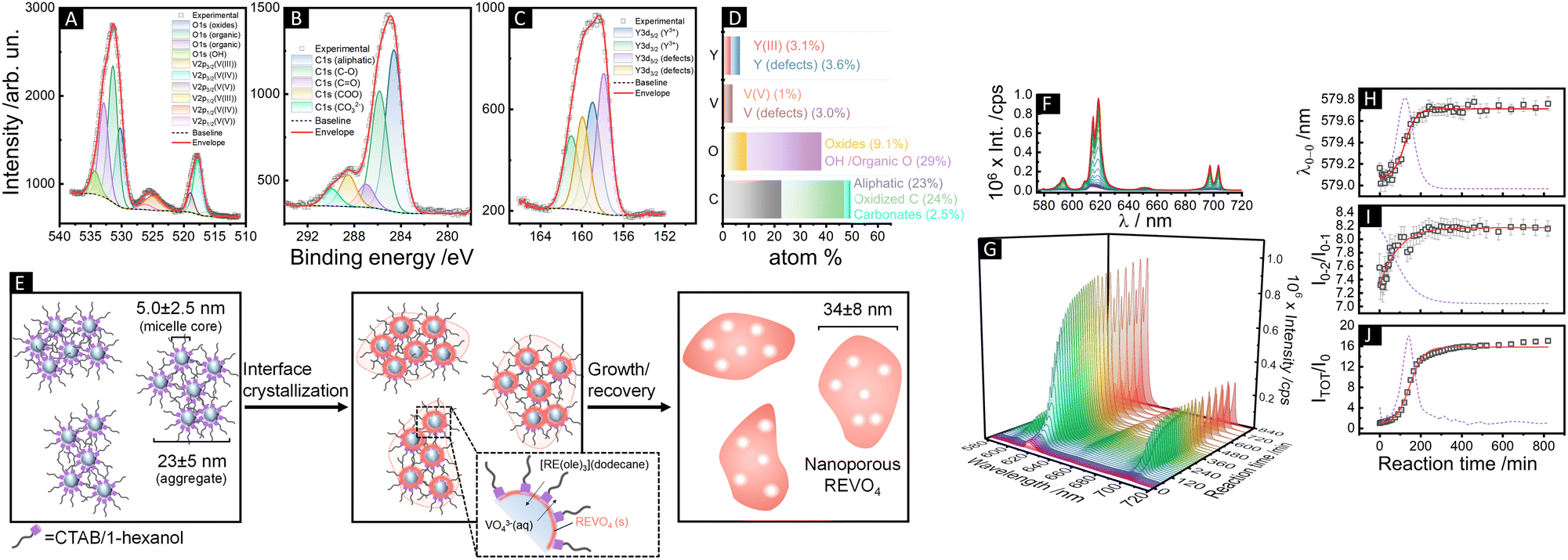 | ||
| Fig. 2 (A)–(C) XPS spectra of (Y,Eu)VO4 nanoparticles obtained by reverse microemulsion synthesis (WO = 25, pH = 11.5) showing the (A) O1s and V2p, (B) C1s, and (C) Y3d peaks. Survey spectra are shown in Fig. S10 (ESI†). (D) Compositional results (% mol) obtained from XPS spectra (Eu and residual Si are not shown for clarity). (E) Schematic illustration of the formation of nanoporous REVO4 particles by interface crystallization over micellar aggregates in water-in-n-dodecane reverse microemulsions. (F)–(J) In situ luminescence monitoring of the formation of (Y,Eu)VO4 particles in water-in-n-dodecane reverse microemulsions (WO = 25, 25 °C, pH = 11.5). Emission spectra (λexc = 288 nm) shown as (F) superposed lines and (G) wavelength vs. time vs. intensity profiles. (H) Position of the 5D0 → 7F0 barycentre (λ0–0), (I) intensity ratio between the 5D0 → 7F2 and 5D0 → 7F1 transitions (I02/I01), and (J) the total integrated intensity normalised with respect to the integrated intensity at t = 0 (ITOT/I0). The red solid lines and the dotted violet lines correspond to sigmoidal fits of the experimental data and the respective first derivatives. Kinetic data involving different reaction conditions are shown in ESI† (Section S2.3 and Fig. S12). | ||
The obtained (Y,Eu)VO4 particles showed the characteristic 5D0 → 7FJ (J = 0–4) Eu3+ emissions upon 280 nm excitation due to 1T1,2 ← 1A1 absorption followed by VO43− → Eu3+ energy transfer.7,17 The luminescent intensities and the Eu3+spectral profiles in the oleate precursors and REVO4 products are remarkably different (Fig. S10, ESI†), enabling application of Eu3+ luminescence spectroscopy to follow the crystallisation of the REVO4 phase (Fig. 2(F)–(J) and ESI,† Section S2.3). While the total luminescence intensities (ITOT/I0) are known to be correlated to the number and crystalline quality of the nanocrystals,14,18 here we demonstrate that additional qualitative kinetic information can be accessed by the ratio between the Eu3+ 5D0 → 7F2 and 5D0 → 7F1 intensities (I02/I01) and by the barycentre of the 5D0 → 7F0 transition (λ0–0), which account for the evolution of microsymmetry around Eu3+ ions (ESI,† Section S2.3).
Such parameters reflect the degree of formation of nanocrystals from precursors, as well as amorphous-to-crystalline conversions of intermediates. The luminescence vs. time profiles (Fig. 2(F) and (G)) show an initial induction period of low luminescence, followed by a steep increase in intensities due to fast nucleation and growth of nanocrystals. The λ0–0 and I02/I01 parameters became constant after 280 min of reaction at the optimised conditions (pH = 11.5 and 25 °C), indicating no further alterations of microsymmetry around Eu3+ ions after this period (Fig. 2(H) and (I)). Conversely, the total intensities (Fig. 2(J)) still increase at low rates from 280 to 800 min of reaction, which is ascribed to the improvement of the crystalline quality of the particles due to redissolution of smaller grains without significant alteration of the chemical environment around Eu3+.17 Different conditions of temperature and pH (Fig. S12, ESI†) resulted in weaker Eu3+ emissions and led to uncontrolled morphology (pH = 7, 25 °C) or to unrecoverable products with incomplete consumption of precursors (pH = 11.5, 10 °C or pH = 13, 25 °C). The induction times before burst nucleation and growth in the different conditions (shown by logarithmic plots in Fig. S13, ESI†) correspond to the transformation of homogeneous precursors diffusing along the surfactant film to generate pre-nucleation clusters towards the formation of nuclei and nanocrystallites. The increase in the pH of the aqueous phase resulted in a decrease of the induction period from ∼100 min (pH = 7) to ∼50 min (pH = 11.5) and <10 min (pH = 13.5) due to higher availability of free VO43− anions in comparison to protonated (e.g., HxVO4(3−x)−) or condensed (e.g., (VO3)nn−) species at high pH.16 However, at pH = 13 the total intensity starts to decrease after ∼60 min, indicating the consumption of the initial RE/VO43− clusters and primary grains to form non-luminescent species such as hydroxides or hydroxo complexes. In this case (pH = 13) no evolution of the I02/I01 parameter is observed (I02/I01 < 7, Fig. S12Q, ESI†), suggesting that most of the RE ions remain as oleates or as kinetically stable hydroxides/hydroxo complexes, and only a minor amount of REVO4 primary grains was formed at the end of the reaction (Fig. S6, ESI†). At pH = 7, vanadate species are mainly present as (VO3)nn−, and the lower availability of VO43− imposes a longer induction period (∼100 min) and a slower growth rate (Fig. S12J–N, ESI†). This accounts for the competitive formation of kinetically stable metavanadates (e.g., REn(VO3)3n) which are further converted into the REVO4 phase in a slow step. Interestingly, the reaction at low temperature (10 °C, pH = 11.5) resulted in a shorter induction period (10–20 min) in comparison to the room temperature conditions, contrary to the expected behaviour. This can be tentatively ascribed to the preferential concentration of anionic groups of the aqueous phase in the vicinity of the cationic surfactant film due to the reduced thermal diffusion of the solubilizates.
Particles obtained in optimised conditions also showed NIR-excited luminescence despite their rather low processing temperature (Fig. S14, ESI†). Although photon upconversion in high phonon energy (>600 cm−1) lattices is often limited to highly crystalline particles,19 nanoporous (Y0.78Yb0.20Er0.02)VO4 clearly exhibited the characteristic green Er3+ emissions (2H11/2 → 4I15/2, 525 nm; 4S3/2 → 4I15/2, 545 nm) under λexc = 980 nm,7 with low contribution of emissions of the 4F9/2 state in the red. The Er3+-doped particles changed their emission profile after dispersion in water (Fig. S13B, ESI†), with a reduced contribution of the upper 2H11/2 state in comparison to the 4S3/2 state. This is possibly due to a laser-induced local self-heating of the particles, which is further confirmed by the linear dependence of the emission intensity ratio of the thermally coupled 2H11/2 and 4S3/2 states with the pump power (Fig. S14E, ESI†).20 The bi-logarithmic upconversion power dependence of the Yb3+/Er3+ couple showed negative deviations from the expected 2-photon slope (2H11/5: 1.67 ± 0.03; 4S3/2: 1.34 ± 0.03, Fig. S13C and D, ESI†), due to the high Yb/Er ratio21 and to competitive thermal population of 2H11/2 and non-radiative relaxation of 4S3/2.20 These results indicate that the Er3+-doped particles not only can provide thermally responsive emissions but also act as localised nanoheaters, with the additional advantage of showing functionalisable nanocavities.
To further demonstrate this possibility, we performed the precipitation reactions in the presence of xylenol orange (XO) and rose Bengal (RB) dyes previously dissolved in the aqueous phase (Fig. 1, ESI,† Sections S1.3 and S2.4). Encapsulated RB was easily removed by washing, but the presence of the dye could be verified by XPS (Fig. S15, ESI†). XO shows a stronger coordination towards RE3+ and a pH-responsive absorption, so this dye was used as a probe to attest encapsulation (i.e. XO@NP). For comparison, XO was also grafted in equivalent amounts at the external surface (i.e. NP@XO Fig. S16–S18, ESI†) of identical (Y,Eu)VO4 nanoparticles (ESI,† Section S1.3 and S2.4). Even though XO is a pH indicator, NP@XO and XO@NP suspensions in different pHs (3–12) showed no alteration in the absorption profile of the dye (Fig. S17, ESI†), confirming the high affinity of the dye towards the oxide surfaces. After 30 days of exposure (ESI,† Section S2.4, ESI†) at these pHs, XO was partially extracted from the surfaces especially at high pHs, but a more prominent release is observed for the NP@XO system in comparison to XO@NP (Fig. S18 and S19(A)–(B), ESI†). If XO@NP and NP@XO systems are exposed to pH = 11 at 50 °C and ultrasound to force the release of XO, the amount of removed XO is about 10x lower in the first hour of extraction for the encapsulated system, and always lower after 5 washing cycles (Fig. S19(C)–(F), ESI†). This ultimately confirms that for XO@NP the dye molecules are effectively occluded in the cavities of the nanoparticles due to the reverse microemulsion synthesis. Also, low density nature of the particles allowed the removal of the encapsulated dye at controlled conditions, thus enabling us to envisage these systems as nanocontainers for controlled release of different molecules.
In summary, we demonstrated the reverse microemulsion formation of nanoporous REVO4 particles with downshift or upconversion luminescence, with an inner surface that can be functionalised with dye molecules. Luminescence spectroscopy combined to multiple techniques was used as a tool to probe the interfacial crystallisation, and the use of dyes confirmed the possibility of molecule encapsulation for further development of new multifunctional systems for luminescent sensing. We thank CAPES, CNPq (405048/2021-1, 310654/2022-0), FAEPEX, and FAPESP (2022/03442-3, 2018/08334-9), and University of Warwick for funding and access to instrumentation. We also thank CA-IQSC-USP and LNNano/CNPEM for TEM.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Skripka, D. Mendez-Gonzalez, R. Marin, E. Ximendes, B. del Rosal, D. Jaque and P. Rodrígues-Sevilla, Nanoscale Adv., 2021, 3, 6310–6329 RSC; (b) B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef CAS PubMed.
- S. Premcheska, M. Lederer and A. M. Kaczmarek, Chem. Commun., 2022, 58, 4288–4307 RSC.
- W. Deng and E. M. Goldys, Analyst, 2014, 139, 5321–5334 RSC.
- (a) Z. Zhang, M. K. G. Jayakumar, X. Zheng, S. Shikha, Y. Zhang, A. Bansal, D. J. J. Poon, P. L. Chu, E. L. L. Yeo, M. L. K. Chua, S. K. Chee and Y. Zhang, Nat. Commun., 2019, 10, 1–12 CrossRef PubMed; (b) M. Tang, X. Zhu, Y. Zhang, Z. Zhang, Z. Zhang, Q. Mei, J. Zhang, M. Wu, J. Liu and Y. Zhang, ACS Nano, 2019, 13, 10405–10418 CrossRef CAS PubMed.
- J. Krämer, R. Kang, L. M. Grimm, L. De Cola, P. Picchetti and F. Biedermann, Chem. Rev., 2022, 122, 3459–3636 CrossRef PubMed.
- (a) G. Lin and D. Jin, ACS Sens., 2021, 6, 4272–4282 CrossRef CAS PubMed; (b) G. Sun, Y. Xie, L. Sun and H. Zhang, Nanoscale Horiz., 2021, 6, 766–780 RSC.
- R. V. Perrella and P. C. de Sousa Filho, Dalton Trans., 2020, 49, 911–922 RSC.
- N. Singh, S. K. NaveenKumar, M. Geethika and G. Mugesh, Angew. Chem., Int. Ed., 2021, 60, 3121–3130 CrossRef CAS PubMed.
- S. F. Soares, T. Fernandes, A. L. Daniel da Silva and T. Trindade, Proc. R. Soc. A, 2019, 475, 20180677 CrossRef PubMed.
- X. Wei, H. Chen, H. P. Tham, N. Zhang, P. Xing, G. Zhang and Y. Zhao, ACS Appl. Nano Mater., 2018, 1, 3663–3672 CrossRef CAS.
- A. K. Ganguli, A. Ganguly and S. Vaidya, Chem. Soc. Rev., 2010, 39, 474–485 RSC.
- S. Wolf and C. Feldmann, Angew. Chem., Int. Ed., 2016, 55, 15728–15752 CrossRef CAS PubMed.
- (a) J. Jung-König, M. Sanhaji, R. Popescu, C. Seidl, E. Zittel, U. Schepers, D. Gerthsen, I. Hilger and C. Feldmann, Nanoscale, 2017, 9, 8362–8372 RSC; (b) M. Wang, Y. Zhang, M. Ng, A. Skripka, T. Cheng, X. Li, K. K. Bhakoo, A. Y. Chang, F. Rosei and F. Vetrone, Chem. Sci., 2020, 11, 6653–6661 RSC.
- (a) B. Fleury, M. A. Neouze, J. M. Guigner, N. Menguy, O. Spalla, T. Gacoin and D. Carriere, ACS Nano, 2014, 8, 2602–2608 CrossRef CAS PubMed; (b) M. A. Neouze, A. P. Freitas, R. K. Ramamoorthy, R. Mohammedi, E. Larquet, S. Tusseau-Nenez, D. Carrière and T. Gacoin, Langmuir, 2020, 36, 9124–9131 CrossRef CAS PubMed; (c) A. P. Freitas, R. K. Ramamoorthy, M. Durelle, E. Larquet, I. Maurin, F. Testard, C. Chevallard, T. Gacoin and D. Carriere, Nano Lett., 2022, 22, 29–35 CrossRef CAS PubMed.
- R. V. Perrella, M. Walker, T. W. Chamberlain, R. I. Walton and P. C. de Sousa Filho, Nano Lett., 2022, 22, 3569–3575 CrossRef CAS PubMed.
- D. C. Crans and A. S. Tracey, in Vanadium Compounds: Chemistry, Biochemistry, and Therapeutic Applications, ed. A. S. Tracey and D. C. Crans, ACS, Washington D.C., 1998, ACS Symposium Series, vol. 711, ch. 1, pp. 2–29 Search PubMed.
- (a) G. Guida, S. Huband, M. Walker, R. I. Walton and P. C. de Sousa Filho, Nanoscale, 2021, 13, 4931–4945 RSC; (b) K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- G. Schwartz, U. Hananel, L. Avram, A. Goldbourt and G. Markovich, J. Am. Chem. Soc., 2022, 144, 9451–9457 CrossRef CAS PubMed.
- A. Nadort, J. Zhao and E. M. Goldys, Nanoscale, 2016, 8, 13099–13130 RSC.
- A. H. Li, Z. J. Sun and Q. Lü, J. Nanopart. Res., 2013, 15, 1–10 Search PubMed.
- V. T. Vera, D. M. Gonzalez, D. J. R. Ramos, A. Igalla, M. Laurenti, R. C. Caceres, E. L. Cabarcos, E. Díaz and J. R. Retama, J. Mater. Chem. C, 2021, 9, 8902–8911 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional data, FTIR, TGA, ζ potential, XPS, XRD, Raman, SEM, luminescence, and UV-Vis spectra. See DOI: https://doi.org/10.1039/d3cc03501h |
| This journal is © The Royal Society of Chemistry 2023 |